 Giada Sebastiani
Giada Sebastiani Nicole Wilkinson
Nicole Wilkinson Kostas Pantopoulos
Kostas Pantopoulos- 1Department of Medicine, McGill University, Montreal, QC, Canada
- 2Division of Gastroenterology, Royal Victoria Hospital, Montreal, QC, Canada
- 3Lady Davis Institute for Medical Research, Jewish General Hospital, Montreal, QC, Canada
The iron regulatory hormone hepcidin limits iron fluxes to the bloodstream by promoting degradation of the iron exporter ferroportin in target cells. Hepcidin insufficiency causes hyperabsorption of dietary iron, hyperferremia and tissue iron overload, which are hallmarks of hereditary hemochromatosis. Similar responses are also observed in iron-loading anemias due to ineffective erythropoiesis (such as thalassemias, dyserythropoietic anemias and myelodysplastic syndromes) and in chronic liver diseases. On the other hand, excessive hepcidin expression inhibits dietary iron absorption and leads to hypoferremia and iron retention within tissue macrophages. This reduces iron availability for erythroblasts and contributes to the development of anemias with iron-restricted erythropoiesis (such as anemia of chronic disease and iron-refractory iron-deficiency anemia). Pharmacological targeting of the hepcidin/ferroportin axis may offer considerable therapeutic benefits by correcting iron traffic. This review summarizes the principles underlying the development of hepcidin-based therapies for the treatment of iron-related disorders, and discusses the emerging strategies for manipulating hepcidin pathways.
Introduction
Iron is an essential constituent of hemoglobin, myoglobin and several other proteins, but also potentially toxic due to its redox reactivity that promotes oxidative stress (Papanikolaou and Pantopoulos, 2005). Balanced iron homeostasis is required to satisfy metabolic needs and minimize the risk of toxic side effects. More than two thirds of body iron (3–5 g in adults) is used in hemoglobin of red blood cells (RBCs) (Gkouvatsos et al., 2012). Erythroblasts require a daily supply of ~20–30 mg and non-erythroid cells another ~5 mg of iron, which is provided by plasma transferrin (Figure 1). The transferrin iron pool does not exceed ~3 mg at steady state and turns over >10 times/day. It is mostly replenished with iron recycled from senescent RBCs by tissue macrophages. The contribution of dietary iron absorption (1–2 mg/day) is minimal in healthy individuals, and mostly compensates for non-specific iron losses due to cell desquamation or bleeding. Iron stored in hepatocytes can be mobilized for erythropoiesis under conditions of iron deficiency.
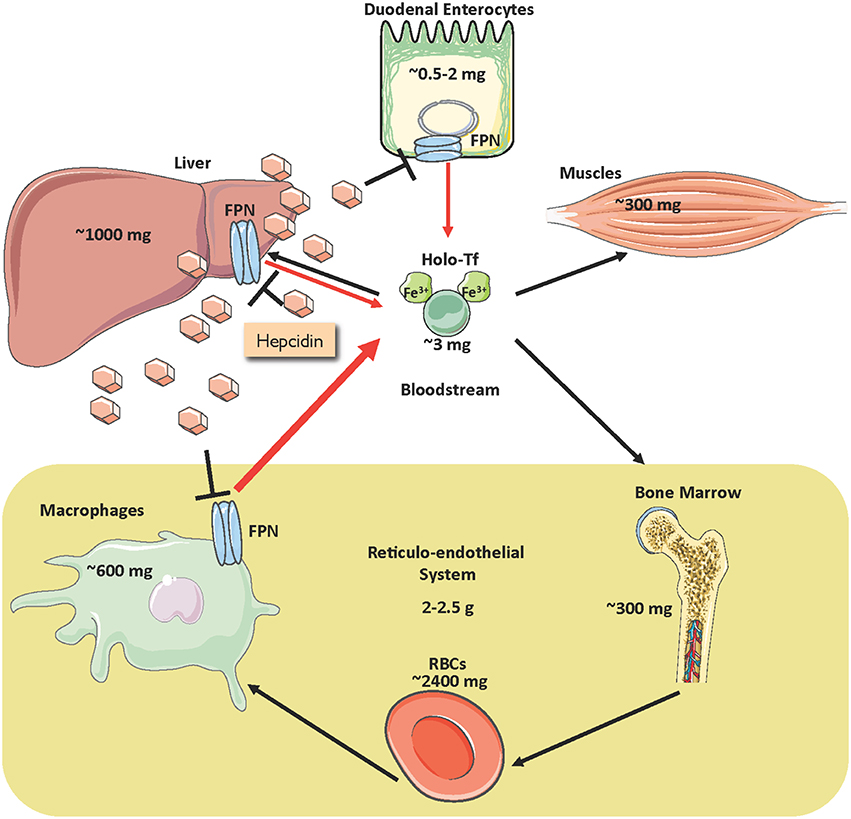
Figure 1. Distribution of iron in the adult human body and regulation of iron traffic. Circulating iron is bound to transferrin (holo-Tf) and delivered to tissues (black arrows). Holo-Tf is primarily replenished by iron recycled from tissue macrophages (thick red arrow), but also by dietary iron absorbed by duodenal enterocytes (thin red arrow). Under conditions of iron deficiency, iron stored in hepatocytes can also be mobilized (thin red arrow). Iron efflux to the bloodstream is inhibited by the liver-derived peptide hormone hepcidin, which binds to the iron exporter ferroportin (FPN) and promotes its degradation.
Iron efflux to the bloodstream is physiologically restrained by hepcidin, an antimicrobial peptide and master hormonal regulator of systemic iron metabolism (Ganz, 2013). Hepcidin is synthesized in hepatocytes and secreted to the bloodstream for binding to the iron exporter ferroportin (FPN) in target cells, primarily macrophages and enterocytes, and to some extent hepatocytes (Figure 1). The binding of hepcidin promotes internalization and lysosomal degradation of ferroportin (Drakesmith et al., 2015). As a result, recycled iron remains in macrophages, while dietary iron absorption through the intestine is inhibited.
Hepcidin expression is predominantly modulated by iron, inflammation and erythropoiesis (Figure 2), but also responds to other stimuli such as endoplasmic reticulum stress, oxidative stress, gluconeogenesis, gonadal hormones, growth factors and hypoxia (Ganz and Nemeth, 2015; Kim and Nemeth, 2015; Pietrangelo, 2016; Wang and Babitt, 2016). Increased serum or tissue iron levels (reflected in transferrin saturation or expression of hepatic BMP6, respectively) promote hepcidin induction via BMP/SMAD signaling. Inflammatory IL-6 triggers hepcidin induction via IL-6/STAT signaling, in crosstalk with the BMP/SMAD pathway. Activin B is another inflammatory cytokine that activates hepcidin via non-canonical BMP/SMAD signaling. Increased erythropoietic activity orchestrated by erythropoietin leads to hepcidin suppression via erythroferrone (ERFE) and other cytokines. It is not well understood how ERFE suppresses hepcidin, but it appears to attenuate BMP/SMAD signaling (Nai et al., 2016). Misregulation of hepcidin is etiologically linked or contributes to iron-related disorders (listed in Table 1), in which pharmacological targeting of the hepcidin/ferroportin axis to restore physiological hepcidin levels is expected to offer therapeutic benefits (Poli et al., 2014c; Ruchala and Nemeth, 2014; Schmidt and Fleming, 2014; Rochette et al., 2015; Blanchette et al., 2016; Liu et al., 2016). Pathological implications of hepcidin misregulation are schematically outlined in Figure 3 and discussed below.
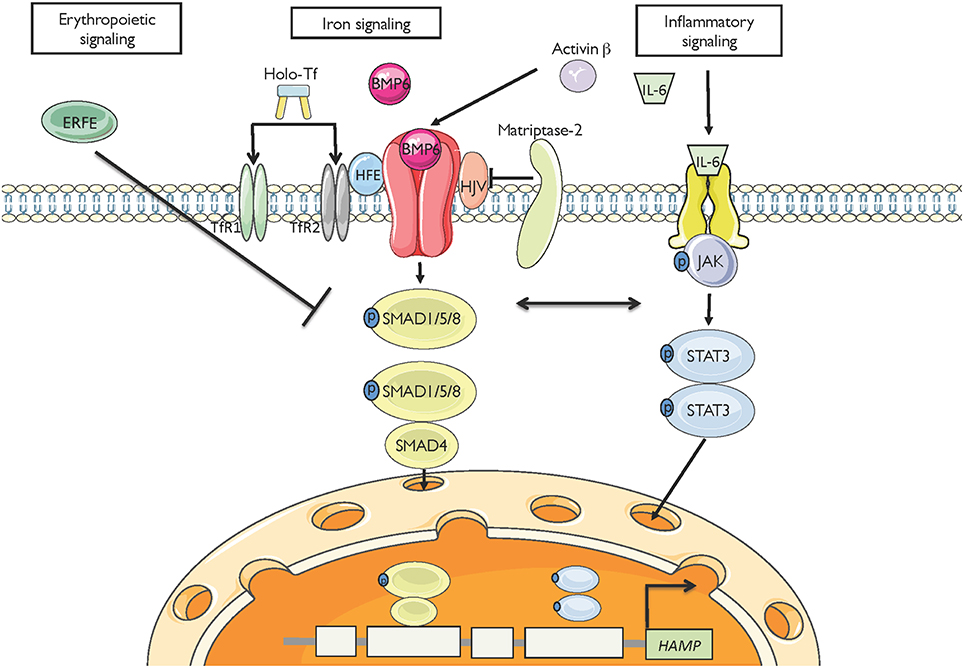
Figure 2. Major pathways for hepcidin regulation. High serum iron levels or hepatic iron stores (reflected in BMP6) induce hepcidin mRNA transcription via the BMP/SMAD signaling cascade. The inflammatory cytokines IL-6 and activin B induce hepcidin mRNA transcription via JAK/STAT and non-canonical BMP/SMAD signaling, respectively. High erythropoietic drive (reflected in ERFE) suppresses hepcidin transcription, likely via interference with BMP/SMAD signaling.
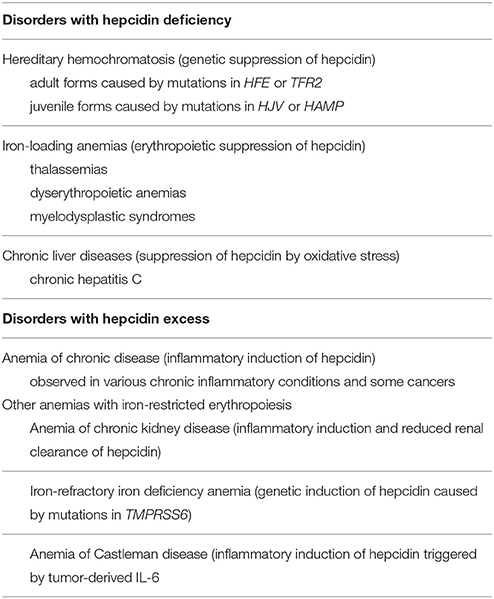
Table 1. Disorders associated with misregulation of hepcidin.
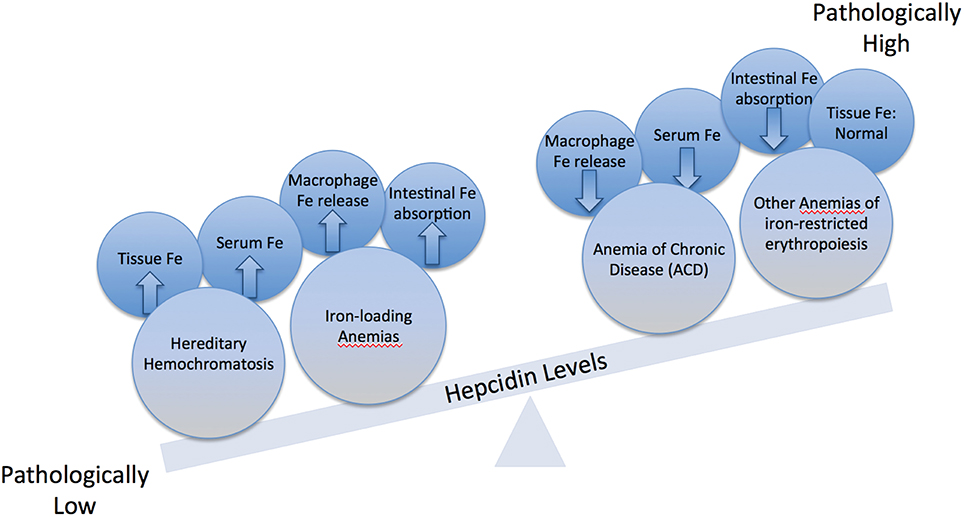
Figure 3. Imbalance in hepcidin expression. Physiological hepcidin responses correlate with healthy body iron metabolism. Pathologically low hepcidin expression occurs in hereditary hemochromatosis (HH) and in iron-loading anemias; this leads to hyperferremia and parenchymal tissue iron overload due to increased iron efflux to the bloodstream from macrophages and intestinal enterocytes. Pathologically high hepcidin expression occurs in the anemia of chronic disease (ACD) and other anemias of iron-restricted erythropoiesis; this leads to hypoferremia and decreased iron availability for erythropoiesis due to iron sequestration in macrophages.
Disorders of Hepcidin Deficiency
Hereditary hemochromatosis (HH) is an endocrine disorder of systemic iron overload that is caused by hepcidin insufficiency (Pietrangelo, 2015; Powell et al., 2016). It constitutes the most frequent genetic disorder in populations of Northern European ancestry and is characterized by chronic hyperabsorption of dietary iron (up to 8–10 mg/day) and unrestricted release of iron from macrophages. These responses lead to hyperferremia, gradual saturation of transferrin and buildup of redox-active non-transferrin bound iron (NTBI), which is deposited within tissue parenchymal cells. HH patients fail to mount appropriate iron-dependent hepcidin induction due to mutations in upstream regulators of iron signaling to hepcidin (HFE, TfR2 or HJV; see Figure 2) or disruption of the hepcidin gene (HAMP). Patients with non-classical ferroportin disease, carrying mutations in ferroportin that prevent hepcidin binding, develop clinical features of HH due to hepcidin resistance (Mayr et al., 2010).
HH is genetically heterogeneous and its severity correlates with the degree of inhibition in hepcidin induction relative to body iron stores. The prevalent HFE-related variant exhibits a relatively milder phenotype due to residual hepcidin responsiveness. Rare variants (TFR2-, HJV- or HAMP-related) are associated with more profound iron overload and hepcidin inactivation. Disruption of either HJV or HAMP genes leads to early onset juvenile HH, the most severe form of the disease.
Clinical complications of adult HH develop after the fourth decade of life and include liver pathology (fibrosis, cirrhosis, hepatocellular cancer), diabetes, skin hyperpigmentation, arthritis and osteoporosis. Juvenile HH patients usually present with hypogonadism in their early 20s and develop fatal cardiomyopathy before the age of 30 if untreated. The standard of care for all forms of HH is reduction of iron burden via therapeutic phlebotomy (Sivakumar and Powell, 2016). This approach is effective and cheap. However, some patients are intolerant, or have low acceptance and compliance to a life-long treatment, or present contraindications (severe heart disease or anemia). These patients are good candidates for new therapies based on restoration of appropriate hepcidin levels.
Hepcidin deficiency is common in hematological disorders associated with ineffective erythropoiesis, such as thalassemias, dyserythropoietic anemias and myelodysplastic syndromes, and contributes to iron overload (Ginzburg and Rivella, 2011; Camaschella and Nai, 2016). Ineffective erythropoiesis is characterized by massive expansion of bone marrow erythroblasts due to decreased production of mature RBCs. This creates a high demand for iron, which leads to suppression of hepcidin in spite of systemic iron overload. Evidently, the negative erythropoietic signals dominate over the positive iron signals under these conditions. Erythropoietic suppression of hepcidin triggers iron overload in non-transfused patients with mild forms of iron loading anemias, and aggravates secondary iron overload in transfused patients (Ginzburg and Rivella, 2011). Restoration of hepcidin could prevent iron overload in the former and improve the efficacy of iron chelation therapy in the latter.
Inhibition of hepcidin expression also contributes to iron overload observed in chronic hepatitis C and other chronic liver diseases (Sebastiani and Pantopoulos, 2011; Pietrangelo, 2016). This is mainly attributed to oxidative stress mechanisms, which appear to override hepcidin-inducing inflammatory signals. Eradication of hepatitis C virus with direct-acting antiviral (DAA) drugs (Zopf et al., 2016) is expected to restore hepcidin expression without need for further interventions.
Disorders of Hepcidin Excess
Excessive hepcidin expression is commonly observed in chronic inflammatory conditions due to infectious or autoimmune disorders or cancer (Weiss, 2015; Wang and Babitt, 2016). Inflammatory induction of hepcidin is primarily mediated by IL-6 and leads to hypoferremia due to ferroportin degradation and iron sequestration in tissue macrophages. Remodeling of iron metabolism by hepcidin-independent mechanisms may further exacerbate this phenotype. Thus, lipopolysaccharide (LPS) and interferon-γ (IFN-γ) inhibit iron efflux from monocytes by decreasing ferroportin expression (Ludwiczek et al., 2003), while the Toll-like receptor 2 and 6 (TLR2/6) ligands FSL1 or PAM3CSK4 trigger hypoferemia in mice by suppressing ferroportin transcription in tissue macrophages (Guida et al., 2015).
The acute hypoferremic response is considered to be protective against infection by depriving bacteria from iron, and may also be enhanced by antimicrobial activities of hepcidin. However, persistent chronic hypoferremia restricts iron availability for erythropoiesis (Ganz and Nemeth, 2015). Together with immune-driven reduced proliferation and life span of RBCs, the diversion of iron traffic contributes to pathogenesis of the anemia of chronic disease (ACD), or anemia of inflammation, the most frequent anemia among hospitalized patients (Weiss, 2015). ACD is typically normocytic/normochromic and unassociated with a reduction in body iron stores, but may be confounded by true iron deficiency due to chronic blood losses and/or scarcity or malabsorption of dietary iron. ACD patients with true iron deficiency exhibit reduced hepcidin levels and a microcytic/hypochromic phenotype.
Correction of ACD improves quality of patients' life. The best strategy is the successful treatment of the primary underlying cause. When this is not possible, ACD is often managed with erythropoiesis-stimulating agents (ESAs), combined or not with oral or intravenous iron administration or RBC transfusions. Nevertheless, these approaches are not always efficacious because hepcidin overexpression blunts responses to ESAs and maintains iron unavailable to erythroblasts. Therefore, they could be complemented by strategies to lower hepcidin levels, thereby mitigating erythropoietic iron-restriction.
Patients with chronic kidney disease (CKD) accumulate high hepcidin levels in the bloodstream due to reduced renal clearance, but also due to inflammatory induction of hepcidin transcription (Tsuchiya and Nitta, 2013). This is associated with iron-restricted erythropoiesis and contributes to anemia. Moreover, it negatively affects therapy with ESAs and oral or intravenous iron. Thus, hepcidin-lowering strategies could improve therapeutic outcomes.
Genetic inactivation of the TMPRSS6 gene leads to unrestricted hepcidin production in spite of low body iron stores, which underlies the pathogenesis of iron-refractory iron deficiency anemia (IRIDA) (Heeney and Finberg, 2014). TMPRSS6 encodes matriptase-2, a transmembrane serine protease that negatively regulates BMP/SMAD signaling to hepcidin (see Figure 2). It appears that hepcidin overexpression is the sole driver of IRIDA, which is microcytic and hypochromic. High hepcidin levels render IRIDA patients unresponsive to oral and partially responsive to intravenous iron therapy. Here, correction of hepcidin excess would not only improve responsiveness to iron therapy, but also provide an etiologic cure.
Pediatric patients with Castleman disease develop a chronic inflammatory anemia with IRIDA-like features. Castleman disease is caused by tumors overproducing IL-6, which in turn promotes an inflammatory state and excessive hepcidin expression (Arlet et al., 2010). The ensuing anemia is refractory to oral iron therapy and can be reversed by resection of the tumor, which eliminates the hepcidin inducer (IL-6). Pharmacological reduction of hepcidin could offer another option for anemia management.
All above-described conditions constitute disorders of systemic hepcidin excess, where high circulating hepcidin levels are derived from hepatocytes. Nevertheless, different cell types in several tissues can also produce hepcidin locally, and this may have profound pathophysiological ramifications. For instance, hepcidin generated by breast or prostate cancer epithelial cells promotes iron retention via autocrine degradation of ferroportin, which in turn favors survival and growth (Pinnix et al., 2010; Tesfay et al., 2015). Therefore, targeted delivery of hepcidin antagonists to hepcidin-overexpressing tumors may sensitize them to anti-cancer therapies.
Hepcidin is also produced in response to inflammation or other signals by heart cardiomyocytes (Merle et al., 2007) and by brain astrocytes and microglia (Urrutia et al., 2013). Considering that the hepcidin/ferroportin axis is crucial for cardiac function (Lakhal-Littleton et al., 2015) and that inflammatory induction of hepcidin causes iron accumulation in nervous system cells (Urrutia et al., 2013), localized targeting of hepcidin excess in the heart or brain may be important in the context of cardiovascular or neurodegenerative disorders.
Pharmacological Restoration of Hepcidin
The therapeutic potential of restoring hepcidin levels in iron overload states is highlighted by genetic studies in mice. Thus, transgenic expression of hepcidin prevented iron overload in Hfe−∕− (Nicolas et al., 2003) and Hbbth3∕+ (Gardenghi et al., 2010) mice, models of HH and β-thalassemia intermedia, respectively. Likewise, genetic disruption of Tmprss6 enhanced BMP/SMAD signaling, increased endogenous hepcidin expression and prevented iron overload in Hfe−∕− (Finberg et al., 2011) and Hbbth3∕+ mice (Nai et al., 2012). Moreover, manipulation of hepcidin levels by these strategies also improved erythropoiesis in Hbbth3∕+ mice (Gardenghi et al., 2010; Nai et al., 2012). It should be noted that disruption of both Tmprss6 alleles offered optimal correction of iron overload but also caused microcytic anemia in Hfe−∕− mice (Finberg et al., 2011), suggesting that titration of hepcidin levels within a physiological window is imperative to prevent adverse effects of hepcidin excess.
The protective effects of Tmprss6 ablation in the Hfe−∕− and Hbbth3∕+ backgrounds rendered this gene a candidate pharmacological target for therapeutic hepcidin induction. In fact, suppression of hepatic Tmprss6 by using RNAi (Schmidt et al., 2013) or antisense oligonucleotides (Guo et al., 2013a) increased hepcidin expression in wild type, Hfe−∕− and Hbbth3∕+ mice. Importantly, it reduced systemic iron overload in Hfe−∕− and Hbbth3∕+ mice, and ameliorated anemia in Hbbth3∕+ mice. The development of oligonucleotide therapeutics against target genes in the liver is an active area of research (Sehgal et al., 2013). Limitations are related to potential off-target effects and toxicity, pharmacokinetic problems of delivery and clearance from the bloodstream, as well as high cost.
Administration of recombinant BMP6 was shown to improve iron signaling to hepcidin and correct systemic iron homeostasis in Hfe−∕− mice, even though endogenous BMP6 expression is appropriately induced in these animals (Corradini et al., 2010). Nevertheless, prolonged BMP6 application caused peritoneal calcifications, indicative of lack of target specificity, which may lead to further pleiotropic side effects.
Cell culture studies and chemical screens identified several small molecules capable of stimulating hepcidin transcription in vitro, such as genistein (Zhen et al., 2013), SB204741, daunorubicin, ethacridine, phenazopyridine, 9-aminoacridine, amlexanox, lansoprazole, leflunomide, ipriflavone, AS252424, pterostilbene, AG1296, GTP14564, SU6668, leflunomide, 10058-F4 and vorinostat (Gaun et al., 2014). An RNAi screen identified sorafenib, wortmannin, rapamycin and metformin as inducers of hepcidin mRNA expression (Mleczko-Sanecka et al., 2014). These drugs affect different pathways, including growth factor signaling, anti-inflammatory signaling, DNA repair and apoptosis. Their exact mechanisms of action are not well understood and their capacity to control hepcidin expression in vivo has not been examined. Broad target specificity may disqualify many of the above-described drugs for pharmacological applications to stimulate hepcidin.
Another small molecule screen identified three steroid molecules as hepcidin inducers (Li et al., 2016). Progesterone, epitiostanol, and mifepristone were shown to stimulate hepcidin transcription independently of the BMP/SMAD and IL-6/STAT3 pathways, by a mechanism requiring progesterone receptor membrane component-1 (PGRMC1). Administration of progesterone to women in the context of standard fertility treatments resulted in hepcidin induction (Li et al., 2016). Contrary to progesterone, 17β-estradiol inhibits hepcidin transcription via an estrogen-responsive promoter element (Yang et al., 2012). Testosterone likewise inhibits hepcidin but operates by enhancing negative epidermal growth factor receptor (EGFR) signaling (Latour et al., 2014), and by competing positive BMP signaling via androgen receptor binding to SMAD proteins (Guo et al., 2013b). Hepcidin-inducing steroids or drugs that lower 17β-estradiol or testosterone could be applied for pharmacological restoration of hepcidin. Nevertheless, potential long-term side effects of steroids should be considered.
Hepcidin supplementation therapy would provide a straightforward approach, devoid of inherent limitations associated with the targeting of upstream regulators. However, the chemical synthesis of hepcidin or its expression as recombinant peptide is costly, while appropriate folding to the biologically active form is complicated by the presence of 8 cysteine residues within the 25 amino acids of the mature peptide forming 4 disulfide bridges (Figure 4). Moreover, synthetic or recombinant hepcidin is rapidly cleared in the circulation and therefore pharmacological concentrations cannot be sustained. Encapsulation of hepcidin into biocompatible nanocarriers suitable for controlled release of therapeutics (Cheng et al., 2015) could theoretically address this issue. A formulation of hepcidin developed by La Jolla Pharmaceutical Company (LJPC-401) is currently undergoing Phase 1 clinical trials with patients at risk of iron overload (http://lajollapharmaceutical.com/2015/10/la-jolla-pharmaceutical-company-doses-first-patient-in-phase-1-clinical-trial-of-ljpc-401-in-patients-at-risk-of-iron-overload/).
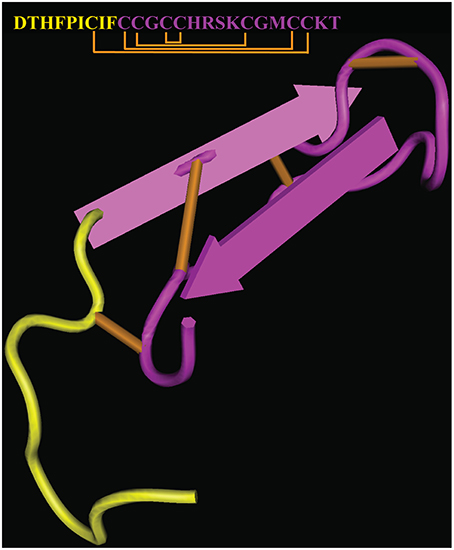
Figure 4. Crystal structure and sequence of human hepcidin. The nine N-terminal amino acids involved in binding to ferroportin are highlighted in yellow (PDB ID 1M4F). Disulfide bonds are highlighted in orange.
Biochemical studies revealed that hepcidin interacts with ferroportin via its N-terminal region (highlighted in Figure 4), and that C-terminal amino acid residues are dispensable. This led to the development of mini-hepcidin analogs containing a minimal core of 7–9 N-terminal amino acids with a single cysteine, which suffice to trigger ferroportin internalization and degradation in cells and mice (Preza et al., 2011). Substitution of natural with modified amino acids and introduction of polyethylene glycol (PEG) and hydrophobic linkers (palmitic acid) improved stability and pharmacological properties of mini-hepcidins. Cyclization may increase stability but appears to reduce biological activity (Chua et al., 2015). Treatment with an engineered mini-hepcidin mimetic (PR65) prevented iron overload in iron-depleted hepcidin-deficient Hamp−∕− mice, but was less efficient in correcting iron traffic when these mice were already iron overloaded (Ramos et al., 2012). High doses of PR65 caused anemia, emphasizing the importance of titration. The mini-hepcidin pro-drug M009 is metabolized to active mini-hepcidin M004 and was recently shown to improve erythropoiesis in Hbbth3∕+ mice and in a mouse model of polycythemia vera (Casu et al., 2016). Another mini-hepcidin derivative developed by Merganser Biotech (M012) initiated a clinical trial in February 2016 (http://merganserbiotech.com/2016/02/24/merganser-biotech-inc-initiates-first-clinical-trial/).
Interestingly, administration of the mini-hepcidin mimetic PR73 protected Hamp−∕− mice against infection with the siderophilic bacterium Vibrio vulnificus (Arezes et al., 2015). PR73 caused hypoferremia to these animals in spite of tissue iron overload, which restricted circulating iron from V. vulnificus and thereby inhibited its growth. Considering that infections with extracellular siderophilic pathogens can be lethal to HH patients (Frank et al., 2011), these data highlight another possible application of hepcidin agonists. Finally, the adverse effects of hepcidin inactivation in mouse models of malaria infection (Portugal et al., 2011) or microbial sepsis suggest a therapeutic potential of hepcidin agonists in the context of these pathologies (Zeng et al., 2015).
Pharmacological Reduction of Hepcidin
Targeting of hepcidin pathways is expected to correct defects triggered by excessive hepcidin expression. Supporting evidence is provided by studies with multicentric Castleman disease patients who were treated with therapeutic monoclonal antibodies against IL-6 receptor (tocilizumab) or IL-6 (siltuximab). These treatments reduced hepcidin levels and improved anemia (Song et al., 2010; Casper et al., 2015). Tocilizumab administration also lowered hepcidin and improved anemia in rheumatoid arthritis patients (Isaacs et al., 2013; Song et al., 2013) and in a monkey arthritis model (Hashizume et al., 2010). Similar results were obtained when rheumatoid arthritis patients were treated with antibodies against TNFα (golimumab or infliximab), possibly as an indirect result of concomitant suppression of IL-6 (Doyle et al., 2013; Song et al., 2013).
Studies with cell and mouse models showed that downstream inhibition of the IL-6/STAT3 signaling pathway by using small molecule inhibitors of STAT3 (curcumin, AG490 and PpYLKTK) likewise decreases hepcidin (Jiao et al., 2009; Fatih et al., 2010; Zhang et al., 2011). However, further development of this class of drugs as hepcidin inhibitors is hindered by lack of specificity (all STAT3 inhibitors), competing iron chelating properties (curcumin) or poor pharmacokinetics (AG490 and PpYLKTK).
As the IL-6/STAT3 and BMP/SMAD signaling pathways are tightly connected (Wang and Babitt, 2016), targeting the latter has the potential to efficiently antagonize hepcidin induction under inflammatory conditions. One strategy exploited the high affinity of BMPs to heparin, a glycosaminoglycan that is clinically applied as anticoagulant. Thus, heparin was shown to inhibit hepcidin expression in cells and mice by sequestering BMPs, while patients treated with heparin for prevention of deep vein thrombosis had reduced serum hepcidin levels (Poli et al., 2011). Because the anticoagulant properties of heparin are undesired for interventions to reduce hepcidin excess, glycol-split non-anticoagulant heparins were tested and found to be equally effective as hepcidin inhibitors; moreover they improved anemia in mouse models of inflammation (Poli et al., 2014a). Highly sulfated heparins with low anticoagulant activity likewise inhibited hepcidin in cells and mice (Poli et al., 2014b). Preservation of high sulfation grade and molecular weight are critical for their hepcidin inhibitory activity (Asperti et al., 2016). Glycol-split and highly sulfated heparins are good candidates for pharmacological targeting of hepcidin in clinical settings and are amenable to further development. Their efficacy and safety profile needs to be established in randomized controlled trials.
HJV is a BMP co-receptor that enhances iron signaling to hepcidin (Babitt et al., 2006). Its neutralization by using humanized monoclonal HJV antibodies (developed by Abbvie) reduced hepcidin expression in rats and cynomolgus monkeys (Böser et al., 2015). Moreover, they corrected anemia via hepcidin inhibition in rat and mouse models of ACD, and in Tmprss6−∕− mice (Kovac et al., 2016). A secreted soluble HJV form (sHJV) functions as a competitive inhibitor of BMP binding to BMP receptors (Lin et al., 2005). Administration of sHJV.Fc, a fusion of sHJV with the Fc fragment of immunoglobulin G, reduced hepcidin and ameliorated anemia in a rat model of ACD (Theurl et al., 2011). Two clinical trials sponsored by Ferrumax Pharmaceuticals Inc aimed to use sHJV.Fc (FMX-8) for the treatment of patients with renal disease, but were not completed due to inability to recruit patients meeting eligibility criteria (ClinicalTrials.gov Identifiers: NCT01873534 and NCT02228655).
Preliminary data of hepcidin inhibition by targeting upstream regulators of the BMP/SMAD and IL-6/STAT3 pathways with oligonucleotide therapeutics were reported a few years ago (Akinc et al., 2011), but the current stage of development of these technologies is unknown.
Small molecule inhibitors of the BMP/SMAD pathway have been tested and shown to act as hepcidin-lowering agents. Thus, dorsomorphin, an inhibitor of the type I BMP receptors ALK2, ALK3, and ALK6, prevented hepcidin induction by IL-6 in cells and induced hyperferremia due to hepcidin suppression in mice (Yu et al., 2008). Furthermore, the dorsomorphin derivative LDN-193189, antagonized inflammatory induction of hepcidin in cells and mitigated anemia in rat (Theurl et al., 2011) and mouse (Steinbicker et al., 2011; Mayeur et al., 2015) models of ACD, and in a rat model of kidney disease (Sun et al., 2013). Interestingly, suppression of hepcidin by LDN-193189 was associated with anti-atherogenic responses in apoE−∕− mice, which were reversed by exogenous hepcidin (Saeed et al., 2012). These findings suggest that pharmacological targeting of hepcidin may be valuable beyond the context of improving erythropoiesis. Nevertheless, dorsomorphin and LDN-193189 are unlikely to be considered for clinical application due to lack of target specificity (Boergermann et al., 2010). TP-0184, a specific ALK2 inhibitor developed by Tolero Pharmaceuticals, was recently shown to block inflammatory induction of hepcidin in mouse models of ACD and cancer-induced anemia (Peterson et al., 2015). Further studies are required to assess its suitability for clinical applications.
Anemia in elderly persons with chronic inflammatory conditions (Perlstein et al., 2011) and in CKD patients (Zughaier et al., 2014), is often associated with vitamin D deficiency. Biochemical studies showed that binding of 1,25-dihydroxy-vitamin D to its receptor directly suppresses hepcidin transcription by binding to a promoter element (Bacchetta et al., 2014). Importantly, pilot clinical studies demonstrated that vitamin D supplementation can decrease hepcidin levels in healthy volunteers (Bacchetta et al., 2014) and early stage CKD patients (Zughaier et al., 2014). These findings underline the potential pharmacological value of vitamin D as a hepcidin-lowering agent, which needs to be further explored in animal models and randomized controlled trials.
Direct inhibition of hepcidin by neutralizing antibodies or other inhibitory molecules offers further opportunities for mitigating the adverse effects of hepcidin overexpression. Neutralizing hepcidin antibodies were shown to modulate iron metabolism in mice and cynomolgus monkeys; moreover, they improved erythropoiesis and responses to ESA therapy in mouse models of ACD (Sasu et al., 2010; Cooke et al., 2013). LY2787106, a monoclonal hepcidin antibody developed by Eli Lily and Company was evaluated in a Phase 1 clinical trial in patients with cancer-associated anemia (ClinicalTrials.gov Identifier: NCT01340976). The antibody treatment was well tolerated and resulted in transient iron mobilization and increased reticulocyte count relative to baseline (Vadhan-Raj et al., 2015).
Neutralization of hepcidin is also possible by using relatively high molecular weight antagonists. Pieris Pharmaceuticals Inc generated PRS-080, a PEGylated anticalin protein that specifically binds to hepcidin and inhibits its activity. Following successful initial testing in vitro and in mice (Hohlbaum et al., 2011), the pharmacokinetic properties and safety profile of PRS-080 were assessed in a clinical trial with healthy volunteers (ClinicalTrials.gov Identifier: NCT02340572). The results were encouraging and a further trial is planned with anemic CKD patients (Moebius et al., 2015).
NOXXON Pharma AG generated NOX-H94 (Lexaptepid Pegol), a Spiegelmer hepcidin antagonist. This PEGylated non-natural mirror-image L-oligoribonucleotide binds with high affinity to hepcidin and operates as a specific inhibitor. NOX-H94 administration improved inflammation-related anemia in cynomolgus monkeys (Schwoebel et al., 2013). Moreover, NOX-H94 prevented hypoferremia in experimental human endotoxemia with LPS-injected volunteers (Van Eijk et al., 2014) (ClinicalTrials.gov Identifier: NCT01522794). NOX-H94 is currently being evaluated in another three Phase 1 and Phase 2 clinical trials on patients with cancer-related anemia, ACD or CKD (ClinicalTrials.gov Identifiers: NCT01691040, NCT01372137, and NCT02079896).
A chemical screen for small molecule hepcidin antagonists identified fursultiamine, a thiol-reactive thiamine derivative that binds to ferroportin on C326 and thereby precludes the binding of hepcidin (Fung et al., 2013). This drug is available over-the-counter for treating vitamin B1 deficiency. While it efficiently protected ferroportin against hepcidin in cultured cells, it failed to exhibit in vivo anti-hepcidin activity, possibly due to rapid plasma turnover. These results suggest that fursultiamine could only be further considered as a hepcidin-lowering agent if modified to stable derivatives, or delivered in formulations that enable sustained controlled release. Nevertheless, they also show that blocking the hepcidin-binding site of ferroportin offers another option for pharmacological targeting the hepcidin/ferroportin axis. Along these lines, Eli Lily and Company developed a monoclonal ferroportin antibody (LY2928057), which increased iron mobilization in cynomolgus monkeys by binding to ferroportin and thereby inhibiting access to hepcidin (Witcher et al., 2013). LY2928057 is currently being evaluated in two Phase 1 clinical trials with healthy volunteers and anemic CKD patients (ClinicalTrials.gov Identifiers: NCT01330953 and NCT01991483).
Conclusions
Hepcidin is a master hormonal regulator of systemic iron homeostasis. Under physiological conditions, its expression remains within a relatively narrow window. Misregulation of hepcidin is associated with a broad spectrum of disorders ranging from iron overload states to anemias with iron-restricted erythropoiesis. Correction of hepcidin levels can provide an etiologic cure to some of these disorders (HH, IRIDA), or offer therapeutic benefits to others (thalassemia, ACD). Several agonists and antagonists of hepcidin have been developed. The ones that have been validated in preclinical or clinical settings are summarized in Table 2 (inducers/mimetics) and Table 3 (antagonists). These drugs act at different levels through the hepcidin/ferroportin axis. Promising candidates are currently being further evaluated in randomized controlled trials. A major challenge for using hepcidin therapeutics is to maintain physiological concentrations of circulating hepcidin, and thereby avoid a shift from hepcidin deficiency to excess and vice versa.
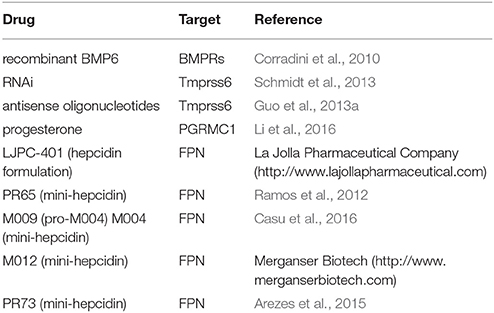
Table 2. Hepcidin inducers and mimetics validated in vivo.
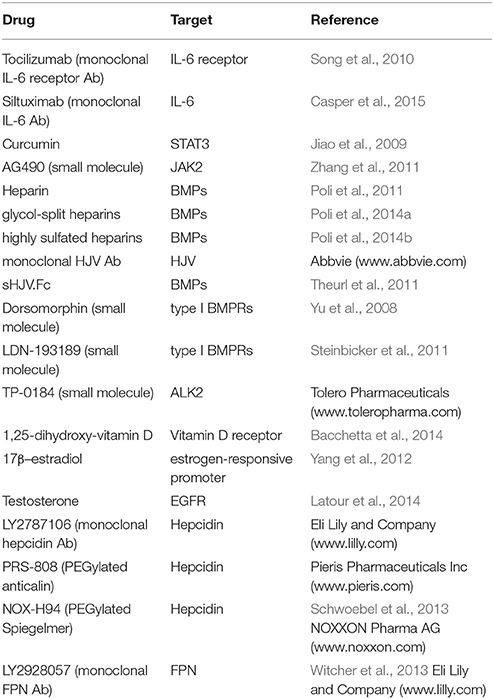
Table 3. Hepcidin antagonists validated in vivo.
Author Contributions
GS and NW contributed to writing the manuscript. KP wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by a grant from the Canadian Institutes for Health Research (CIHR; MOP-86514).
References
Akinc, A., Chan-Daniels, A., Sehgal, A., Foster, D., Bettencourt, B. R., Hettinger, J., et al. (2011). “Targeting the hepcidin pathway with RNAi therapeutics for the treatment of Anemia,” in 53th Annual Meeting of the American Society for Hematology (ASH) (San Diego, CA), Abstract 688.
Arezes, J., Jung, G., Gabayan, V., Valore, E., Ruchala, P., Gulig, P. A., et al. (2015). Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe 17, 47–57. doi: 10.1016/j.chom.2014.12.001
Arlet, J. B., Hermine, O., Darnige, L., Ostland, V., Westerman, M., Badoual, C., et al. (2010). Iron-deficiency anemia in Castleman disease: implication of the interleukin 6/hepcidin pathway. Pediatrics 126, e1608– e1612. doi: 10.1542/peds.2010-1123
Asperti, M., Naggi, A., Esposito, E., Ruzzenenti, P., Di Somma, M., Gryzik, M., et al. (2016). High sulfation and a high molecular weight are important for anti-hepcidin activity of heparin. Front. Pharmacol. 6:316. doi: 10.3389/fphar.2015.00316
Babitt, J. L., Huang, F. W., Wrighting, D. M., Xia, Y., Sidis, Y., Samad, T. A., et al. (2006). Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 38, 531–539. doi: 10.1038/ng1777
Bacchetta, J., Zaritsky, J. J., Sea, J. L., Chun, R. F., Lisse, T. S., Zavala, K., et al. (2014). Suppression of iron-regulatory hepcidin by vitamin D. J. Am. Soc. Nephrol. 25, 564–572. doi: 10.1681/ASN.2013040355
Blanchette, N. L., Manz, D. H., Torti, F. M., and Torti, S. V. (2016). Modulation of hepcidin to treat iron deregulation: potential clinical applications. Expert Rev. Hematol. 9, 169–186. doi: 10.1586/17474086.2016.1124757
Boergermann, J. H., Kopf, J., Yu, P. B., and Knaus, P. (2010). Dorsomorphin and LDN-193189 inhibit BMP-mediated Smad, p38 and Akt signalling in C2C12 cells. Int. J. Biochem. Cell Biol. 42, 1802–1807. doi: 10.1016/j.biocel.2010.07.018
Böser, P., Seemann, D., Liguori, M. J., Fan, L., Huang, L., Hafner, M., et al. (2015). Anti-repulsive guidance molecule C (RGMc) antibodies increases serum iron in rats and cynomolgus monkeys by hepcidin downregulation. AAPS J. 17, 930–938. doi: 10.1208/s12248-015-9770-4
Camaschella, C., and Nai, A. (2016). Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 172, 512–523. doi: 10.1111/bjh.13820
Casper, C., Chaturvedi, S., Munshi, N., Wong, R., Qi, M., Schaffer, M., et al. (2015). Analysis of inflammatory and anemia-related biomarkers in a randomized, double-blind, placebo-controlled study of siltuximab (Anti-IL6 Monoclonal Antibody) in patients with multicentric castleman disease. Clin. Cancer Res. 21, 4294–4304. doi: 10.1158/1078-0432.CCR-15-0134
Casu, C., Oikonomidou, P. R., Chen, H., Nandi, V., Ginzburg, Y., Prasad, P., et al. (2016). Minihepcidin peptides as disease modifiers in mice affected by beta-thalassemia and polycythemia vera. Blood. doi: 10.1182/blood-2015-10-676742. [Epub ahead of print].
Cheng, C. J., Tietjen, G. T., Saucier-Sawyer, J. K., and Saltzman, W. M. (2015). A holistic approach to targeting disease with polymeric nanoparticles. Nat. Rev. Drug Discov. 14, 239–247. doi: 10.1038/nrd4503
Chua, K., Fung, E., Micewicz, E. D., Ganz, T., Nemeth, E., and Ruchala, P. (2015). Small cyclic agonists of iron regulatory hormone hepcidin. Bioorg. Med. Chem. Lett. 25, 4961–4969. doi: 10.1016/j.bmcl.2015.03.012
Cooke, K. S., Hinkle, B., Salimi-Moosavi, H., Foltz, I., King, C., Rathanaswami, P., et al. (2013). A fully human anti-hepcidin antibody modulates iron metabolism in both mice and nonhuman primates. Blood 122, 3054–3061. doi: 10.1182/blood-2013-06-505792
Corradini, E., Schmidt, P. J., Meynard, D., Garuti, C., Montosi, G., Chen, S., et al. (2010). BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology 139, 1721–1729. doi: 10.1053/j.gastro.2010.07.044
Doyle, M. K., Rahman, M. U., Frederick, B., Birbara, C. A., De Vries, D., Toedter, G., et al. (2013). Effects of subcutaneous and intravenous golimumab on inflammatory biomarkers in patients with rheumatoid arthritis: results of a phase 1, randomized, open-label trial. Rheumatology 52, 1214–1219. doi: 10.1093/rheumatology/kes381
Drakesmith, H., Nemeth, E., and Ganz, T. (2015). Ironing out Ferroportin. Cell Metab. 22, 777–787. doi: 10.1016/j.cmet.2015.09.006
Fatih, N., Camberlein, E., Island, M. L., Corlu, A., Abgueguen, E., Detivaud, L., et al. (2010). Natural and synthetic STAT3 inhibitors reduce hepcidin expression in differentiated mouse hepatocytes expressing the active phosphorylated STAT3 form. J. Mol. Med. 88, 477–486. doi: 10.1007/s00109-009-0588-3
Finberg, K. E., Whittlesey, R. L., and Andrews, N. C. (2011). Tmprss6 is a genetic modifier of the Hfe-hemochromatosis phenotype in mice. Blood 117, 4590–4599. doi: 10.1182/blood-2010-10-315507
Frank, K. M., Schneewind, O., and Shieh, W. J. (2011). Investigation of a researcher's death due to septicemic plague. N. Engl. J. Med. 364, 2563–2564. doi: 10.1056/NEJMc1010939
Fung, E., Sugianto, P., Hsu, J., Damoiseaux, R., Ganz, T., and Nemeth, E. (2013). High-throughput screening of small molecules identifies hepcidin antagonists. Mol. Pharmacol. 83, 681–690. doi: 10.1124/mol.112.083428
Ganz, T. (2013). Systemic iron homeostasis. Physiol. Rev. 93, 1721–1741. doi: 10.1152/physrev.00008.2013
Ganz, T., and Nemeth, E. (2015). Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 15, 500–510. doi: 10.1038/nri3863
Gardenghi, S., Ramos, P., Marongiu, M. F., Melchiori, L., Breda, L., Guy, E., et al. (2010). Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J. Clin. Invest. 120, 4466–4477. doi: 10.1172/JCI41717
Gaun, V., Patchen, B., Volovetz, J., Zhen, A. W., Andreev, A., Pollastri, M. P., et al. (2014). A chemical screen identifies small molecules that regulate hepcidin expression. Blood Cells Mol. Dis. 53, 231–240. doi: 10.1016/j.bcmd.2014.06.002
Ginzburg, Y., and Rivella, S. (2011). beta-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood 118, 4321–4330. doi: 10.1182/blood-2011-03-283614
Gkouvatsos, K., Papanikolaou, G., and Pantopoulos, K. (2012). Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta 1820, 188–202. doi: 10.1016/j.bbagen.2011.10.013
Guida, C., Altamura, S., Klein, F. A., Galy, B., Boutros, M., Ulmer, A. J., et al. (2015). A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood 125, 2265–2275. doi: 10.1182/blood-2014-08-595256
Guo, S., Casu, C., Gardenghi, S., Booten, S., Aghajan, M., Peralta, R., et al. (2013a). Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J. Clin. Invest. 123, 1531–1541. doi: 10.1172/JCI66969
Guo, W., Bachman, E., Li, M., Roy, C. N., Blusztajn, J., Wong, S., et al. (2013b). Testosterone administration inhibits hepcidin transcription and is associated with increased iron incorporation into red blood cells. Aging Cell 12, 280–291. doi: 10.1111/acel.12052
Hashizume, M., Uchiyama, Y., Horai, N., Tomosugi, N., and Mihara, M. (2010). Tocilizumab, a humanized anti-interleukin-6 receptor antibody, improved anemia in monkey arthritis by suppressing IL-6-induced hepcidin production. Rheumatol. Int. 30, 917–923. doi: 10.1007/s00296-009-1075-4
Heeney, M. M., and Finberg, K. E. (2014). Iron-Refractory Iron Deficiency Anemia (IRIDA). Hematol. Oncol. Clin. North Am. 28, 637–652. doi: 10.1016/j.hoc.2014.04.009
Hohlbaum, A. M., Trentman, S., Gille, H., Allesdorfer, A., Belaiba, R. S., Huelsmeyer, M., et al. (2011). “Discovery and preclinical characterization of a novel hepcidin antagonist with tunable PK/PD properties for the treatment of anemia in different patient populations,” in 53th Annual Meeting of the American Society for Hematology (ASH) (San Diego, CA), Abstract 687.
Isaacs, J. D., Harari, O., Kobold, U., Lee, J. S., and Bernasconi, C. (2013). Effect of tocilizumab on haematological markers implicates interleukin-6 signalling in the anaemia of rheumatoid arthritis. Arthritis Res. Ther. 15, R204. doi: 10.1186/ar4397
Jiao, Y., Wilkinson, J. T., Di, X., Wang, W., Hatcher, H., Kock, N. D., et al. (2009). Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood 113, 462–469. doi: 10.1182/blood-2008-05-155952
Kim, A., and Nemeth, E. (2015). New insights into iron regulation and erythropoiesis. Curr. Opin. Hematol. 22, 199–205. doi: 10.1097/MOH.0000000000000132
Kovac, S., Boser, P., Cui, Y., Ferring-Appel, D., Casarrubea, D., Huang, L., et al. (2016). Anti-hemojuvelin antibody corrects anemia caused by inappropriately high hepcidin levels. Haematologica 101, e173. doi: 10.3324/haematol.2015.140772
Lakhal-Littleton, S., Wolna, M., Carr, C. A., Miller, J. J., Christian, H. C., Ball, V., et al. (2015). Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. U.S.A. 112, 3164–3169. doi: 10.1073/pnas.1422373112
Latour, C., Kautz, L., Besson-Fournier, C., Island, M. L., Canonne-Hergaux, F., Loreal, O., et al. (2014). Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology 59, 683–694. doi: 10.1002/hep.26648
Li, X., Rhee, D. K., Malhotra, R., Mayeur, C., Hurst, L. A., Ager, E., et al. (2016). Progesterone receptor membrane component-1 regulates hepcidin biosynthesis. J. Clin. Invest. 126, 389–401. doi: 10.1172/JCI83831
Lin, L., Goldberg, Y. P., and Ganz, T. (2005). Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood 106, 2884–2889. doi: 10.1182/blood-2005-05-1845
Liu, J., Sun, B., Yin, H., and Liu, S. (2016). Hepcidin: a promising therapeutic target for iron disorders: a systematic review. Medicine 95, e3150. doi: 10.1097/MD.0000000000003150
Ludwiczek, S., Aigner, E., Theurl, I., and Weiss, G. (2003). Cytokine-mediated regulation of iron transport in human monocytic cells. Blood 101, 4148–4154. doi: 10.1182/blood-2002-08-2459
Mayeur, C., Kolodziej, S. A., Wang, A., Xu, X., Lee, A., Yu, P. B., et al. (2015). Oral administration of a bone morphogenetic protein type I receptor inhibitor prevents the development of anemia of inflammation. Haematologica 100, e68–e71. doi: 10.3324/haematol.2014.111484
Mayr, R., Janecke, A. R., Schranz, M., Griffiths, W. J., Vogel, W., Pietrangelo, A., et al. (2010). Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J. Hepatol. 53, 941–949. doi: 10.1016/j.jhep.2010.05.016
Merle, U., Fein, E., Gehrke, S. G., Stremmel, W., and Kulaksiz, H. (2007). The iron regulatory peptide hepcidin is expressed in the heart and regulated by hypoxia and inflammation. Endocrinology 148, 2663–2668. doi: 10.1210/en.2006-1331
Mleczko-Sanecka, K., Roche, F., Da Silva, A. R., Call, D., D'Alessio, F., Ragab, A., et al. (2014). Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood 123, 1574–1585. doi: 10.1182/blood-2013-07-515957
Moebius, U., Feuerer, W., Fenzl, E., Van Swelm, R., Swinkels, D. W., and Hohlbaum, A. (2015). “A phase I study investigating the safety, tolerability, pharmacokinetics and pharmacodynamic activity of the hepcidin antagonist PRS-080#022. Results from a randomized, placebo controlled, double-blind study following single administration to healthy subjects,” in 57th Annual Meeting of the American Society for Hematology (ASH) (Orlando, FL), Abstract 536.
Nai, A., Pagani, A., Mandelli, G., Lidonnici, M. R., Silvestri, L., Ferrari, G., et al. (2012). Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood 119, 5021–5029. doi: 10.1182/blood-2012-01-401885
Nai, A., Rubio, A., Campanella, A., Gourbeyre, O., Artuso, I., Bordini, J., et al. (2016). Limiting hepatic Bmp-Smad signaling by matriptase-2 is required for erythropoietin-mediated hepcidin suppression in mice. Blood 127, 2327–2336. doi: 10.1182/blood-2015-11-681494
Nicolas, G., Viatte, L., Lou, D. Q., Bennoun, M., Beaumont, C., Kahn, A., et al. (2003). Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat. Genet. 34, 97–101. doi: 10.1038/ng1150
Papanikolaou, G., and Pantopoulos, K. (2005). Iron metabolism and toxicity. Toxicol. Appl. Pharmacol. 202, 199–211. doi: 10.1016/j.taap.2004.06.021
Perlstein, T. S., Pande, R., Berliner, N., and Vanasse, G. J. (2011). Prevalence of 25-hydroxyvitamin D deficiency in subgroups of elderly persons with anemia: association with anemia of inflammation. Blood 117, 2800–2806. doi: 10.1182/blood-2010-09-309708
Peterson, P. A., Soh, K. K., Lee, Y. S., Kim, W., Whatcott, C. J., Siddiqui-Jain, A., et al. (2015). “ALK2 Inhibition via TP-0184 abrogates inflammation-induced hepcidin expression and is a potential therapeutic for anemia of chronic disease,” in 57th Annual Meeting of the American Society for Hematology (ASH), (Orlando, FL), Abstract 273.
Pietrangelo, A. (2015). Genetics, genetic testing, and management of hemochromatosis: 15 years since hepcidin. Gastroenterology 149, 1240–1251. doi: 10.1053/j.gastro.2015.06.045
Pietrangelo, A. (2016). Iron and the liver. Liver Int. 36(Suppl. 1), 116–123. doi: 10.1111/liv.13020
Pinnix, Z. K., Miller, L. D., Wang, W., D'agostino, R. Jr., Kute, T., Willingham, M. C., et al. (2010). Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2, 43ra56. doi: 10.1126/scitranslmed.3001127
Poli, M., Asperti, M., Naggi, A., Campostrini, N., Girelli, D., Corbella, M., et al. (2014a). Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo. Blood 123, 1564–1573. doi: 10.1182/blood-2013-07-515221
Poli, M., Asperti, M., Ruzzenenti, P., Mandelli, L., Campostrini, N., Martini, G., et al. (2014b). Oversulfated heparins with low anticoagulant activity are strong and fast inhibitors of hepcidin expression in vitro and in vivo. Biochem. Pharmacol. 92, 467–475. doi: 10.1016/j.bcp.2014.09.007
Poli, M., Asperti, M., Ruzzenenti, P., Regoni, M., and Arosio, P. (2014c). Hepcidin antagonists for potential treatments of disorders with hepcidin excess. Front. Pharmacol. 5:86. doi: 10.3389/fphar.2014.00086
Poli, M., Girelli, D., Campostrini, N., Maccarinelli, F., Finazzi, D., Luscieti, S., et al. (2011). Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood 117, 997–1004. doi: 10.1182/blood-2010-06-289082
Portugal, S., Carret, C., Recker, M., Armitage, A. E., Goncalves, L. A., Epiphanio, S., et al. (2011). Host-mediated regulation of superinfection in malaria. Nat. Med. 17, 732–737. doi: 10.1038/nm.2368
Powell, L. W., Seckington, R. C., and Deugnier, Y. (2016). Haemochromatosis. Lancet. doi: 10.1016/S0140-6736(15)01315-X. [Epub ahead of print].
Preza, G. C., Ruchala, P., Pinon, R., Ramos, E., Qiao, B., Peralta, M. A., et al. (2011). Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J. Clin. Invest. 121, 4880–4888. doi: 10.1172/JCI57693
Ramos, E., Ruchala, P., Goodnough, J. B., Kautz, L., Preza, G. C., Nemeth, E., et al. (2012). Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood 120, 3829–3836. doi: 10.1182/blood-2012-07-440743
Rochette, L., Gudjoncik, A., Guenancia, C., Zeller, M., Cottin, Y., and Vergely, C. (2015). The iron-regulatory hormone hepcidin: a possible therapeutic target? Pharmacol. Ther. 146, 35–52. doi: 10.1016/j.pharmthera.2014.09.004
Ruchala, P., and Nemeth, E. (2014). The pathophysiology and pharmacology of hepcidin. Trends Pharmacol. Sci. 35, 155–161. doi: 10.1016/j.tips.2014.01.004
Saeed, O., Otsuka, F., Polavarapu, R., Karmali, V., Weiss, D., Davis, T., et al. (2012). Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 299–307. doi: 10.1161/ATVBAHA.111.240101
Sasu, B. J., Cooke, K. S., Arvedson, T. L., Plewa, C., Ellison, A. R., Sheng, J., et al. (2010). Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 115, 3616–3624. doi: 10.1182/blood-2009-09-245977
Schmidt, P. J., and Fleming, M. D. (2014). Modulation of hepcidin as therapy for primary and secondary iron overload disorders: preclinical models and approaches. Hematol. Oncol. Clin. North Am. 28, 387–401. doi: 10.1016/j.hoc.2013.11.004
Schmidt, P. J., Toudjarska, I., Sendamarai, A. K., Racie, T., Milstein, S., Bettencourt, B. R., et al. (2013). An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood 121, 1200–1208. doi: 10.1182/blood-2012-09-453977
Schwoebel, F., Van Eijk, L. T., Zboralski, D., Sell, S., Buchner, K., Maasch, C., et al. (2013). The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood 121, 2311–2315. doi: 10.1182/blood-2012-09-456756
Sebastiani, G., and Pantopoulos, K. (2011). Disorders associated with systemic or local iron overload: from pathophysiology to clinical practice. Metallomics 3, 971–986. doi: 10.1039/c1mt00082a
Sehgal, A., Vaishnaw, A., and Fitzgerald, K. (2013). Liver as a target for oligonucleotide therapeutics. J. Hepatol. 59, 1354–1359. doi: 10.1016/j.jhep.2013.05.045
Sivakumar, M., and Powell, L. W. (2016). Management of human factors engineering-associated hemochromatosis: a 2015 update. World J. Hepatol. 8, 395–400. doi: 10.4254/wjh.v8.i8.395
Song, S. N., Iwahashi, M., Tomosugi, N., Uno, K., Yamana, J., Yamana, S., et al. (2013). Comparative evaluation of the effects of treatment with tocilizumab and TNF-alpha inhibitors on serum hepcidin, anemia response and disease activity in rheumatoid arthritis patients. Arthritis Res. Ther. 15, R141. doi: 10.1186/ar4323
Song, S. N., Tomosugi, N., Kawabata, H., Ishikawa, T., Nishikawa, T., and Yoshizaki, K. (2010). Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman disease. Blood 116, 3627–3634. doi: 10.1182/blood-2010-03-271791
Steinbicker, A. U., Sachidanandan, C., Vonner, A. J., Yusuf, R. Z., Deng, D. Y., Lai, C. S., et al. (2011). Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 117, 4915–4923. doi: 10.1182/blood-2010-10-313064
Sun, C. C., Vaja, V., Chen, S., Theurl, I., Stepanek, A., Brown, D. E., et al. (2013). A hepcidin lowering agent mobilizes iron for incorporation into red blood cells in an adenine-induced kidney disease model of anemia in rats. Nephrol. Dial. Transplant. 28, 1733–1743. doi: 10.1093/ndt/gfs584
Tesfay, L., Clausen, K. A., Kim, J. W., Hegde, P., Wang, X., Miller, L. D., et al. (2015). Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. 75, 2254–2263. doi: 10.1158/0008-5472.CAN-14-2465
Theurl, I., Schroll, A., Sonnweber, T., Nairz, M., Theurl, M., Willenbacher, W., et al. (2011). Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood 118, 4977–4984. doi: 10.1182/blood-2011-03-345066
Tsuchiya, K., and Nitta, K. (2013). Hepcidin is a potential regulator of iron status in chronic kidney disease. Ther. Apher. Dial. 17, 1–8. doi: 10.1111/1744-9987.12001
Urrutia, P., Aguirre, P., Esparza, A., Tapia, V., Mena, N. P., Arredondo, M., et al. (2013). Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 126, 541–549. doi: 10.1111/jnc.12244
Vadhan-Raj, S., Abonour, R., Goldman, J. W., Smith, D. A., Slapak, C. A., Ytiu, R. V., et al. (2015). “Phase 1 study of a hepcidin antagonist, LY2787106, in cancer-associated anemia,” in 57th Annual Meeting of the American Society for Hematology (ASH) (Orlando, FL), Abstract 537.
Van Eijk, L. T., John, A. S., Schwoebel, F., Summo, L., Vauleon, S., Zollner, S., et al. (2014). Effect of the antihepcidin Spiegelmer lexaptepid on inflammation-induced decrease in serum iron in humans. Blood 124, 2643–2646. doi: 10.1182/blood-2014-03-559484
Wang, C. Y., and Babitt, J. L. (2016). Hepcidin regulation in the anemia of inflammation. Curr. Opin. Hematol. 23, 189–197. doi: 10.1097/MOH.0000000000000236
Weiss, G. (2015). Anemia of chronic disorders: new diagnostic tools and new treatment strategies. Semin. Hematol. 52, 313–320. doi: 10.1053/j.seminhematol.2015.07.004
Witcher, D. R., Leung, D., Hill, K. A., De Rosa, D. C., Xu, J., Manetta, J., et al. (2013). “LY2928057, an antibody targeting ferroportin, is a potent inhibitor of hepcidin activity and increases iron mobilization in normal cynomolgus monkeys,” in 55th Annual Meeting of the American Society for Hematology (ASH) (New Orleans, LA), Abstract 3433.
Yang, Q., Jian, J., Katz, S., Abramson, S. B., and Huang, X. (2012). 17beta-Estradiol inhibits iron hormone hepcidin through an estrogen responsive element half-site. Endocrinology 153, 3170–3178. doi: 10.1210/en.2011-2045
Yu, P. B., Hong, C. C., Sachidanandan, C., Babitt, J. L., Deng, D. Y., Hoyng, S. A., et al. (2008). Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 4, 33–41. doi: 10.1038/nchembio.2007.54
Zeng, C., Chen, Q., Zhang, K., Chen, Q., Song, S., and Fang, X. (2015). Hepatic hepcidin protects against polymicrobial sepsis in mice by regulating host iron status. Anesthesiology 122, 374–386. doi: 10.1097/ALN.0000000000000466
Zhang, S. P., Wang, Z., Wang, L. X., and Liu, S. J. (2011). AG490: an inhibitor of hepcidin expression in vivo. World J. Gastroenterol. 17, 5032–5034. doi: 10.3748/wjg.v17.i45.5032
Zhen, A. W., Nguyen, N. H., Gibert, Y., Motola, S., Buckett, P., Wessling-Resnick, M., et al. (2013). The small molecule, genistein, increases hepcidin expression in human hepatocytes. Hepatology 58, 1315–1325. doi: 10.1002/hep.26490
Zopf, S., Kremer, A. E., Neurath, M. F., and Siebler, J. (2016). Advances in hepatitis C therapy: what is the current state - what come's next? World J. Hepatol. 8, 139–147. doi: 10.4254/wjh.v8.i3.139
Keywords: iron metabolism, hemochromatosis, anemia, inflammation, erythropoiesis
Citation: Sebastiani G, Wilkinson N and Pantopoulos K (2016) Pharmacological Targeting of the Hepcidin/Ferroportin Axis. Front. Pharmacol. 7:160. doi: 10.3389/fphar.2016.00160
Received: 23 April 2016; Accepted: 31 May 2016;
Published: 21 June 2016.
Edited by:
Gaetano Cairo, University of Milan, ItalyReviewed by:
Raffaella Gozzelino, Chronic Diseases Research Center (CEDOC), PortugalZvi Ioav Cabantchik, Hebrew University of Jerusalem, Israel
Paolo Arosio, University of Brescia, Italy
Copyright © 2016 Sebastiani, Wilkinson and Pantopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kostas Pantopoulos, a29zdGFzLnBhbnRvcG91bG9zQG1jZ2lsbC5jYQ==