
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 22 December 2015
Sec. Translational and Clinical Endocrinology
Volume 6 - 2015 | https://doi.org/10.3389/fendo.2015.00187
 Erika Peverelli1
Erika Peverelli1 Donatella Treppiedi1
Donatella Treppiedi1 Elena Giardino1
Elena Giardino1 Eleonora Vitali2
Eleonora Vitali2 Andrea G. Lania3
Andrea G. Lania3 Giovanna Mantovani1*
Giovanna Mantovani1*
Pituitary tumors, that origin from excessive proliferation of a specific subtype of pituitary cell, are mostly benign tumors, but may cause significant morbidity in affected patients, including visual and neurologic manifestations from mass-effect, or endocrine syndromes caused by hormone hypersecretion. Dopamine (DA) receptor DRD2 and somatostatin (SS) receptors (SSTRs) represent the main targets of pharmacological treatment of pituitary tumors since they mediate inhibitory effects on both hormone secretion and cell proliferation, and their expression is retained by most of these tumors. Although long-acting DA and SS analogs are currently used in the treatment of prolactin (PRL)- and growth hormone (GH)-secreting pituitary tumors, respectively, clinical practice indicates a great variability in the frequency and entity of favorable responses. The molecular basis of the pharmacological resistance are still poorly understood, and several potential molecular mechanisms have been proposed, including defective expression or genetic alterations of DRD2 and SSTRs, or an impaired signal transduction. Recently, a role for cytoskeleton protein filamin A (FLNA) in DRD2 and SSTRs receptors expression and signaling in PRL- and GH-secreting tumors, respectively, has been demonstrated, first revealing a link between FLNA expression and responsiveness of pituitary tumors to pharmacological therapy. This review provides an overview of the known molecular events involved in SS and DA resistance, focusing on the role played by FLNA.
Pituitary tumors are generally benign tumors that represent 10–25% of all intracranial neoplasms, and may cause visual field deficits and neurologic manifestations from mass effect, and/or endocrine syndromes caused by excessive pituitary hormone secretion, signs and symptoms depending from specific pituitary cell subtype origin of the adenoma. Prolactin (PRL)-secreting tumors are the most common of all functional pituitary tumors and cause amenorrhea, infertility, and galactorrhea in females, and impotence or infertility in males. Excessive secretion of growth hormone (GH) by tumorigenic somatotroph cells is the cause of gigantism during childhood and acromegaly in adults, with significant morbidity due to clinical complications involving cardiovascular, respiratory, and metabolic systems. Adenocorticotroph hormone (ACTH)-secreting tumors cause Cushing’s disease (hypercortisolism), and TSH-secreting tumors present with signs and symptoms of hyperthyroidism. Non-functioning pituitary tumors (NFPAs) are hormonally inactive, and patients with this tumor type often present with neurological symptoms due to the mass effect.
Pituitary tumors frequently preserve responsiveness to hypophysiotropic factors, including dopamine (DA) and somatostatin (SS), ubiquitous peptides that physiologically inhibit hormone secretion and cell proliferation at both the pituitary and the periphery levels, and thus are considered as molecules with therapeutical potential (1).
The inhibitory actions of DA in pituitary tumors are mediated by DA receptor subtype 2 (DRD2) [reviewed in Ref. (2)] that inhibits both synthesis and secretion of PRL, by coupling with inhibitory heterotrimeric G proteins, Gi and Go, that in turn inhibit adenylyl cyclase and calcium channels. Moreover, DRD2 exerts antiproliferative effects through regulation of ERK1/2 activity (Figure 1). An alternative splicing generates two isoforms of this receptor, D2S (short) and D2L (long), possessing additional 29 residues in the third intracellular loop. DRD2 agonists, among which cabergoline is the most effective and best tolerated, are first-line therapy for prolactinomas as they are effective in controlling clinical symptoms, PRL levels, and tumor volume (3) and are used to a lesser extent in the treatment of ACTH-secreting tumors (4) and NFPAs (5).
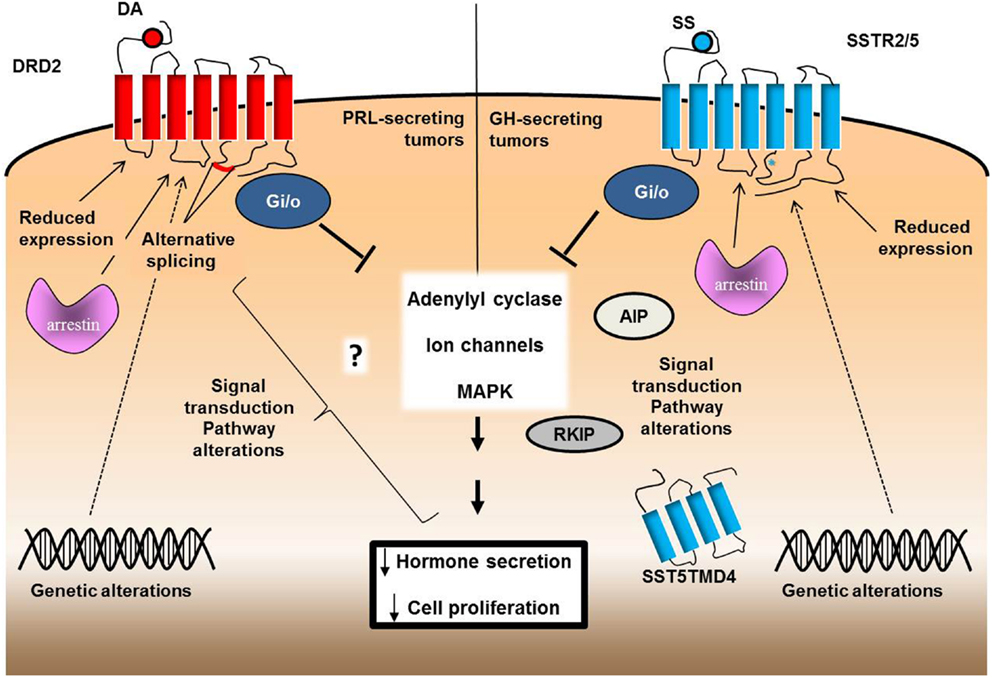
Figure 1. Schematic representation of the possible molecular mechanisms involved in drug resistance of pituitary tumors. DA-resistance in PRL-secreting tumors might be related to a defective expression of DRD2 or to an altered expression of specific splice variants. Genetic alterations of DRD2 or possible alterations in the molecules involved in receptor internalization or signal transduction culminating in the inhibitory action on PRL secretion and cell proliferation might also be involved. SS-resistance of GH-secreting tumors, besides to reduced SSTR2 and/or SSTR5 expression, rarely associated with mutations of SSTR2 and SSTR5 genes, but it has been correlated with the expression of truncated variatns of SSTR5 (SST5TMD4). Asterisk indicates the only mutational change found in the SSTR5 gene (R240W, in the third intracellular loop). Alterations in signal transduction may include G proteins, AIP, or arrestins.
Somatostatin binds to a family of G-proteins coupled receptors (SSTR1-5), among which subtypes 2 and 5 are expressed at high density in most GH-secreting pituitary tumors (6, 7). By coupling with multiple PTX-sensitive G proteins, SSTRs inhibit adenylyl cyclase activity and some subtypes reduce calcium entry by modulating L-type Ca2+ and K+ channels, all these events being involved in the reduction of hormone secretion [reviewed in Ref. (8)] (Figure 1). Both SSTR2 and SSTR5 mediate the antiproliferative effects of SS by tyrosine phosphatase activation and ERK1/2 phosphorylation inhibition, respectively, whereas only SSTR2 and SST3 subtypes mediate apoptotic effects (9–12). SS analogs, such as long-acting octreotide and lanreotide, are currently used in the treatment of pituitary tumors, particularly GH- and TSH-secreting tumors (13, 14).
Despite the demonstrated inhibitory effects on cell proliferation and hormone secretion of DA and SS analogs, a subset of patients (10% of patients with prolactinoma and 30% of acromegalic patients) are resistant to cabergoline and octreotide, respectively, while the large majority of patients with NFPA and ACTH-secreting pituitary tumors are unresponsive to both drugs (15–17).
Pharmacological resistance of PRL-secreting tumors is generally defined as failure to normalize PRL levels, and to achieve tumor size reduction of at least 50%, although this definition is still controversial and a variety of definitions of DA-resistance have been used (15, 18).
As far as acromegalic patients were concerned, definition of pharmacological resistance to SS analogs includes both biochemical remission (normal IGF1, random GH <1 μg/l, and a nadir GH after 75-g oral glucose tolerance test <0.4 μg/l) and tumor shrinkage (<20% of baseline volume) (17, 19).
Drug resistance of pituitary tumors might play a major impact on mortality, morbility, and quality of life. Moreover, prolactinomas resistance to cabergoline has been associated with invasive and more aggressive tumors, and with the increased risk to develop a carcinoma (15, 20).
Intensive investigations in the past years have discovered different molecular alterations possibly involved in this variable responsiveness to DA and SS, including defective expression or genetic alterations of DRD2 and SSTRs, alterations in their signaling pathways, and cytoskeleton proteins involvement (Figure 1) but much remains to be clarified about this complex phenomenon.
Generally, a reduced expression of functional DA and SS receptors in pituitary tumors has been associated with a poor response.
In particular, this event has been documented in prolactinomas, where a reduced density of DRD2 has been found in DA-resistant tumors vs. DA-sensitive tumors (21, 22). Lower expression of a specific isoform of DRD2, D2S (23, 24), or D2L (25) has also been documented in resistant vs. sensitive tumors.
More controversial is the association of SS resistance with the reduction of SSTRs expression, since data from literature suggest on the one hand that some GH-secreting tumors are resistant to therapy despite high SSTR2 expression, and on the other hand that the in vivo GH suppression and tumor shrinkage induced by SS analogs correlates with SSTR2 mRNA and protein levels (26–34).
Anyway, the molecular events responsible for reduced DRD2 and SSTRs expression are still largely unknown. Both genetic alterations and posttranslational mechanisms have been examined, and recently a key role for cytoskeleton proteins-mediated regulation has emerged, as discussed below.
Published studies searching for genetic alterations of DRD2 and SSTRs have demonstrated that mutations in their coding sequence and/or loss of heterozigosity (LOH) in loci where they are located are very rare events.
In particular, no mutation of the DRD2 gene has been found in 79 prolactinomas (35), and a recently whole-exome sequencing on bromocriptine-resistant and -responsive prolactinomas demonstrated no sequence variants in DRD2 gene (36, 37). By genotyping about 200 patients with prolactinomas and controls for common DRD2 polymorphisms, DA-resistance has been correlated only to a common polymorphism, a synonymous cytosine to thymine transition at position 957, a change that has been associated with susceptibility to migraine and schizophrenia, and that might alter the predicted mRNA folding, leading to a decrease in mRNA stability and translation (38).
Similarly to DRD2, no mutations of SSTR2 and SSTR5 genes are usually found in resistant patients (39, 40). In fact, the only mutational change so far reported involving a SSTR is the germline R240W mutation in the SSTR5 gene, that was found in one acromegalic patient resistant to octreotide (41).
This missense mutation is located in the third intracellular loop of SST5, a critical region for G protein coupling, and functional studies have demonstrated that mutant R240W receptor failed to mediate the inhibition of GH release and cell proliferation induced by SSTR5 agonist, although it retained the ability to inhibit intracellular cAMP levels, as the result of a loss of coupling with GoA protein but not with the other G proteins activated by wild type SSTR5, e.g., Gi1, i2, i3 and GoB (11, 42).
Polymorphic variants in SSTR2 gene seem to have no role in determining SS resistance of GH-secreting tumors. Two single nucleotide substitutions detected in the SSTR2 promoter sequence at positions −83 and −57 from the major transcription initiation site were not associated with differences in clinical characteristics, hormonal profile, or responsiveness to SS (43). Regarding SSTR5 gene, it has been found that T allele of rs34037914 single nucleotide polymorphism predisposes to resistance to antiproliferative effects of SS, increased aggressiveness, and post-surgical reoccurrence of pituitary tumors (44). However, LOH at the SSTR5 gene locus, located at chromosome 16p13.3, is a rare phenomenon, occurring in about 10% of pituitary tumors (45) and no mutational change in the retained allele was found in the GH-secreting tumor with LOH at the SSTR5 locus, LOH resulting associated with a normal responsiveness to octreotide (45).
Finally, the expression of a truncated variant of SSTR5 (SST5TMD4) that shows a preferential intracellular localization but is able to mediate signal transduction (46) has been found to correlate with a reduced ability of octreotide at normalizing hormone secretion in poorly responsive tumors in vivo (47) and with aggressive features, including SS-resistance and tumor invasiveness (48, 49). High levels of SST5TMD4 are correlated with poor responsiveness to SS analogs even in the presence of a high expression of SSTR2 (49), suggesting a dominant-negative effect of this truncated isoform of SSTR5 on SSTR2-mediated signaling.
Beside alterations in SS and DA receptors, defects in the downstream signaling pathways activated by these receptors might be involved in the pharmacological resistance of pituitary tumors. The hypothesis of post-receptor alterations involved in resistance to SS analogs is supported by the dissociation between the antisecretory and antiproliferative effects of SS analogs observed in vivo in some acromegalic patients and in in vitro experiments in tumoral somatotroph cells where SS analogs exert antiproliferative effects without inhibition of GH secretion (50–52). Since SSTRs couple to different members of the inhibitory G proteins family to inhibit hormone secretion and cells proliferation, it is possible to hypothesize that alteration in the coupling with a specific G protein, or in a specific downstream effector, could determine the lack of activation of a specific pathway without affecting the other signaling cascades. In this regard, it has been shown that SSTR5 exerts antiproliferative and antisecretory effects in somatotroph by coupling with the inhibitory G protein GoA, independently of cAMP reduction, suggesting a dissociation between these pathways (11, 42).
Few data are available about a possible role of G proteins alterations in pituitary tumor resistance to pharmacologic treatment with DA or SS analogs. A reduction in Gi2 protein, but not Go and Gs, expression has been described in cabergoline-resistant with respect to sensitive prolactinomas (53). Although a low expression of Gi1–3 proteins has been reported in GH-secreting tumors (54), a possible correlation with SS responsiveness has not been analyzed. The alpha subunit of stimulatory G proteins deserves particular attention, since somatic activating mutations in its gene (GNAS, also called gsp oncogene) are found in 30–40% of GH-secreting pituitary tumors. It is worth noting that gsp-positive tumors are characterized by an increased responsivity to treatment with SS analogs, a feature up to now unexplained since no increase of SS receptors have been found in these tumors (40, 55–57).
Alterations in SSTRs signal transduction might be the cause of typical octreotide-resistance observed in acromegalic patients bearing germline mutations in aryl hydrocarbon receptor interacting protein (AIP) gene (58) or expressing low levels of AIP, even in the absence of mutations. AIP is a tumor-suppressor gene, which has been shown to induce tumor shrinkage via ZAC1 (59). Moreover, it has been shown that AIP inactivation leads to pituitary tumorigenesis through defective Gi signaling (60). It is of interest to note that low AIP expression does not associate with resistance to pasireotide (61), the SS analog with the highest affinity for SSTR5, suggesting that AIP is not involved in the signal transduction of this specific SSTR subtype.
Genetic analysis of AIP in 50 non-familial acromegalic patients resistant to treatment with SS analogs revealed a low, but non-negligible, prevalence of AIP germline mutations (62), suggesting a role for AIP alterations in SS-resistance of sporadic GH-secreting adenomas.
Fougner et al. reported an association of Raf kinase inhibitory protein (RKIP) expression with responsiveness of GH-secreting tumors to SS analogs. RKIP regulates Raf1 kinase activity, and consequently MAPK signaling, a pathway involved in mediating the antiproliferative effects of SS, and furthermore inhibits G protein receptor kinase 2, possibly affecting SSTRs internalization and degradation (63).
Beside activation of coupled G proteins, GPCRs are known to directly interact through their intracellular loops with cytoplasmic and surface proteins involved in GPCRs stabilization, desensitization, internalization, and signal transduction. Among these proteins, beta-arrestin 1 and 2 are scaffold proteins involved both in desensitization and signal transduction of several GPCRs, including SSTRs and DRD2 (64–66). Low expression of β-arrestin 1, but not β-arrestin 2, in GH- and PRL-secreting pituitary tumors correlates with a reduced recycling rate of SSTR2 and better SS analogs response in terms of GH suppression, both in vitro and in vivo (67).
A lesser amount of data is available on possible DRD2 signal transduction alterations. TGF-β/Smad pathway, that may in part mediate the antiproliferative effects of DA, was found down-regulated in DA-resistant prolactinomas compared to normal human anterior pituitaries (68). A recent whole-exome sequencing analysis of six bromocriptine-resistant and six responsive prolactinomas, in addition to exclude genetic variants of DRD2 gene, revealed sequence variants associated with 10 genes selected as potentially involved in resistance, among which PRB3 (36) and PRDM2 (37) were lower in resistant prolactinomas than in the responsive tumors, but the mechanisms involved are unknown. Finally, recent studies have identified a specific miRNA expression profile associated with bromocriptine-resistant prolactinoma. Among the differentially expressed miRNAs, mir-93 directly affected p21 expression in MMQ cells by targeting the 3′-UTR (69).
Recently, cytoskeleton has emerged as novel player implicated in the complex mechanisms of pharmacological resistance of pituitary tumors to DA and SS.
Cell cytoskeleton not only plays a fundamental role in cell morphology maintenance, cell migration and adhesion, cell division, and cytoplasmic organelles localization and movement but it also has a role in extracellular signal transduction and in regulation of the activity of several receptors. The three main structural components of the cytoskeleton are microtubules, intermediate filaments, and microfilaments that originate from the polymerization of different monomers. These polymers that are characterized by structural and functional differences undergo continual turnover and rearrangement and specifically bind different families of partner proteins. Microfilaments, the thinnest of these filaments, are involved in cell morphology and motility and consist of actin filaments (F-actin) that are polymerized from monomeric globular actin (G-actin). Many families of actin-binding proteins that share an actin binding domain are involved in the regulation of polymerization/depolymerization processes and in crosslinking F-actin in bundles and networks. Among these, filamins (FLN) are actin crosslinking proteins that not only form orthogonal networks of F-actin but are also scaffold proteins able to bind different partner proteins.
The family of filamins consists of three homologous high-molecular weight proteins, filamin A, B, and C (FLNA, FLNB, and FLNC) that are encoded by different genes located on chromosome X, 3 and 7, respectively. These three isoforms of FLN in mammals show a strong homology in the entire coding sequences and possess highly conserved genomic organization (70).
Specifically, human FLNA, mapping to Xq28, is the first actin filament cross-linking protein identified in non-muscle cells (71) and is the most abundant filamin isoform in adults. FLNA as well as FLNB are ubiquitously expressed, contrary to FLNC, whose expression is restricted to skeletal and cardiac muscle. Several studies suggest that FLNs are essential for normal human development. Indeed, mutations in the respective genes cause a wide range of developmental malformations of the brain, bone, and heart with moderate to lethal consequences (72). In particular, FLNA mutations cause periventricular nodular heterotopia, a brain malformation due to abnormal neuronal migration, in which a subset of neurons fails to migrate into the developing cerebral cortex (73) or a wide spectrum of congenital malformations, such as otopalotodigital syndrome, frontometaphyseal dysplasia, and Melnick Needles syndrome (74).
The main role of FLNA, deriving from its ability to bind actin and omodimerize, is to stabilize F-actin network in a three-dimensional structure, but its functions are not limited to a structural role. Indeed, FLNA can bind many different partner proteins, including transmembrane proteins, such as several GPCRs, integrins, and ion channels, allowing their anchorage to actin cytoskeleton (75), but also intracellular signaling molecules, kinases, and transcription factors [revised in Ref. (76)], playing as important scaffold for signal transduction.
All these interactions are regulated by FLNA phosphorylation, proteolysis, mechanical forces, competition, and multimerization of partners.
These multiple functions of FLNA derive from its unique structure, schematically represented in Figure 2. FLNA is composed of two subunits of 280 kDa each that self-assemble, each monomer possesses an actin-binding domain (ABD) at the N-terminus, that consists of two calponin homology domains, followed by 24 immunoglobulin (Ig)-like repeats of about 96 amino acid residues. Two calpain-sensitive hinge regions (H1 and H2) separate the 24 repeats in a rod-1 domain (repeats 1–15), rod-2 domain (repeats 16–23), and repeat 24. A secondary ABD of lower affinity is located in the rod-1 domain, whereas rod-2 is involved in the interaction with partner proteins. The repeat 24 is the self-association domain that mediates FLNA homodimerization, allowing the formation of V-shaped flexible structures that results in the perpendicular cross-linking of actin filaments.
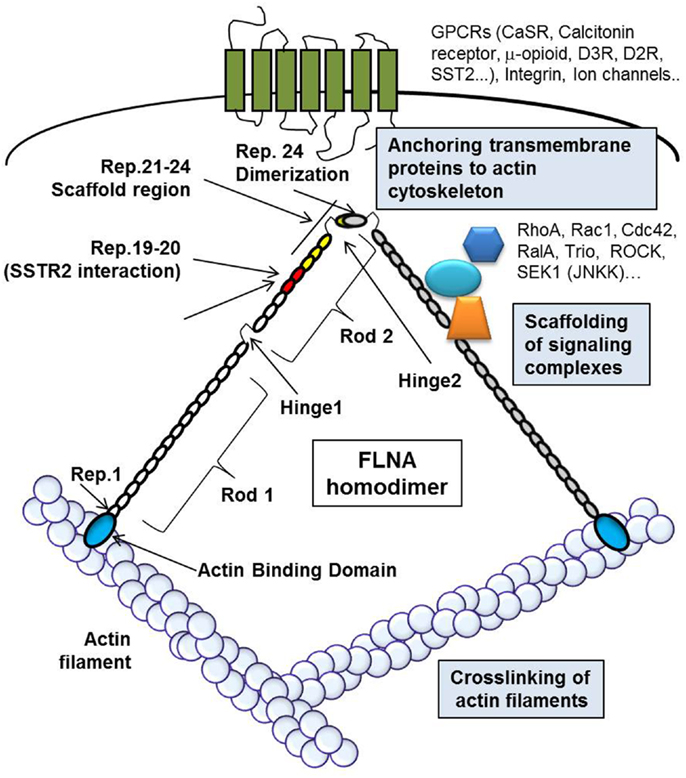
Figure 2. The figure schematically represents FLNA structure and cellular functions. The actin-binding domain (ABD, blue circle) is located at the N-terminus of each monomer of FLNA. The circles represent the Ig-like repeats of FLNA, in particular red circles represent repeats 19–20, involved in the interaction with DRD2 and SSTR2, and yellow circles represent the repeats 21–24, known as scaffold region for signal transduction molecules. The repeat 24 is the self-association domain that allows homodimer formation. FLNA crosslinks actin filaments, binds different transmembrane proteins, anchoring them to the actin cytoskeleton, and interacts with a variety of proteins involved in signal transduction.
The diversity of the FLN family is increased by alternative splicing of FLN mRNA. Alternative poly(A) signals are employed and in some variants, there are internal deletions in some of the domains. In particular, previous studies have reported and alternative splicing of sequences encoding a region of eight amino acids in repeat 15 of FLNA, and an internal deletion of 41 amino acids between repeats 19 and 20 (residues 2127–2167) that identify FLNA variant-1 (77) that is widely expressed at low levels (70).
The interaction of FLNA with DRD2 was first demonstrated by Li and colleagues by a yeast two-hybrid screen and protein binding assays (78). Interestingly, this binding increased coupling efficiency of DRD2 to adenylate cyclase and plays a role in cell surface receptor clustering. Almost simultaneously, another study using the third intracellular loop of the DRD2 as bait in a yeast two-hybrid approach to screen a human brain cDNA library, confirmed a specific DRD2/FLNA association, better defining the specific regions of FLNA (repeat 19) and DRD2 (aminoacids 211–241 in the N-terminal region of the third intracellular loop) involved (79). FLNA and DRD2 colocalized in cell cultures of rat striatum. The authors found that DRD2 was predominantly intracellular in M2 cells, a human melanoma cell line does not express FLNA, whereas it localized at the plasma membrane in A7 cells, the same line stably transfected with FLNA, suggesting that FLNA is required for the cell surface localization of DRD2, as also demonstrated by using a dominant negative truncated form of FLNA (repeats 18–19, containing the DRD2, but not the actin, binding domain) (80).
Similar effects of FLNA on DRD2 regulation were also found in PRL-secreting pituitary tumors (Figure 3). First, it has been shown that in DA-resistant tumors the reduction of DRD2, previously associated with resistance, is accompanied by a low level of FLNA (81). In vitro experiments starting from this experimental observation demonstrated that alterations of FLNA levels in primary cultured prolactinoma cells by gene silencing or overexpression resulted in corresponding modifications of DRD2 levels, demonstrating that these two events are causally related. Furthermore, reduction of PRL release and ERK1/2 phosphorylation mediated by DRD2 agonist were impaired after FLNA silencing, whereas DA-resistant prolactinomas lacking FLNA recovered responsiveness when transfected with FLNA (81).
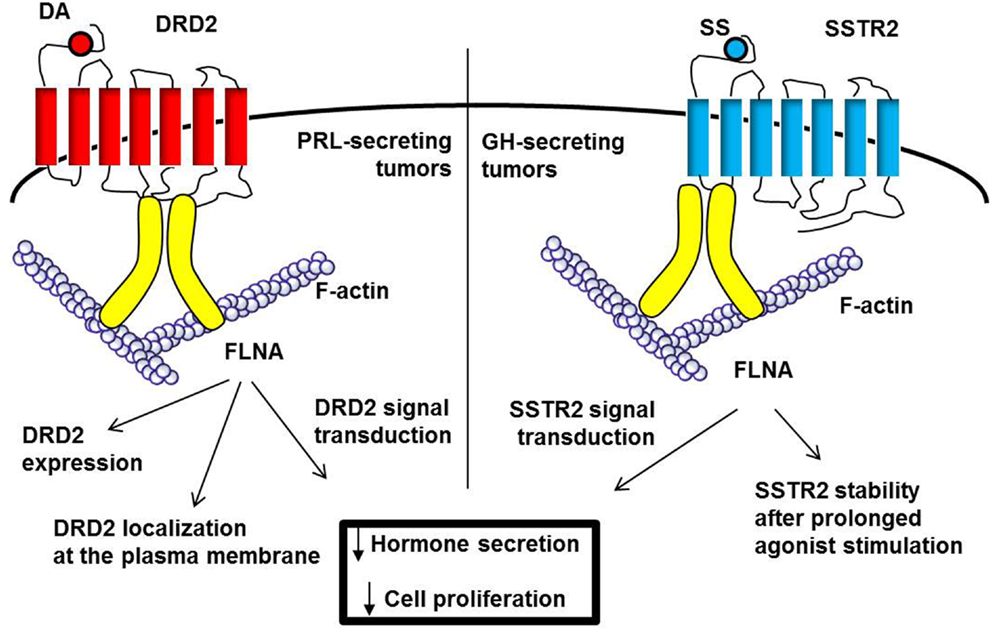
Figure 3. FLNA is required for DRD2 and SSTR2 expression and signal transduction. FLNA, schematically represented in yellow, bind DRD2 (third intracellular loop) and SSTR2 (first intracellular loop). In PRL-secreting tumors (left), FLNA binding is crucial for the correct localization at plasma membrane and expression of DRD2. In GH-secreting tumors (right), SSTR2 does not require FLNA for membrane localization, but in the absence of FLNA, SSTR2 expression is downregulated after prolonged agonist stimulation. Moreover, FLNA is required for both DRD2 and SSTR2 intracellular signaling leading to inhibitory effects on hormone secretion and cell proliferation.
Experiments in rat MMQ cells, a cell model of prolactinoma endogenously expressing functional DRD2 and FLNA show that FLNA is not only required for DRD2 targeting to the cell membrane but also protects DRD2 against lysosomal degradation, suggesting a role for FLNA in the control of DRD2 fate toward recycling processes or lysosomal degradation (81). Since FLNA directly interacts with beta arrestins (82) that are involved in DRD2 trafficking (66), it is possible to hypothesize the formation of a complex receptor-FLNA–arrestin involved in the regulation of DRD2 stability. These data demonstrated a protective effect of FLNA from receptor instability, as also demonstrated for other receptors, including calcium-sensing receptor, calcitonin receptor, cystic fibrosis transmembrane conductance regulator, and the high-affinity IgG receptor FcgammaRI (83–86).
Beside a structural role in anchoring DRD2 to actin cytoskeleton and regulating receptor localization and stability, FLNA plays an additional functional role as scaffold for signaling molecules involved in DRD2 signal transduction. Indeed, since the pituitary has a substantial DRD2 reserve for PRL inhibition, and PRL response reaches the plateau at about 40% receptor occupancy in rat pituitary cells (87), the 60% reduction of DRD2 levels measured in prolactinomas and MMQ cells after FLNA knockdown might not entirely account for the loss of D2R effects on PRL release and cell proliferation (81).
Since FLNA is crucial for both DRD2 expression and signaling in lactotrophs, the loss of FLNA expression may be one of the mechanisms involved in loss of DA responsiveness previously documented in human prolactinomas.
Further studies are required to clarify the molecular events underlying FLNA reduced expression in resistant tumors. To date, no alterations in the FLNA gene CpG island with the highest probability to have regulatory functions have been found, excluding an epigenetic silencing (81).
In addition to DRD2, FLNA seems to have a critical role also in the regulation of SSTR2, with important implications for SS responsiveness in acromegalic patients.
A direct interaction of FLNA with SSTR2 has been first demonstrated by surface plasmon resonance (88). Specifically, this interaction involve SSTR2 first intracellular loop and FLNA repeats 19–20 and plays a role in SSTR2 stabilization at the cell membrane. Contrary to what observed for DRD2, SSTR2 was correctly targeted to the plasma membrane in M2 cells lacking FLNA, but after agonist stimulation SSTR2 internalization rate in M2 cells is increased with respect to A7 cells. The authors also found that FLNA is required for SSTR2 mediated inhibition of cell survival, and the molecular mechanism involves a competition of FLNA with p85, the regulatory subunit of PI3K, for direct binding to SSTR2. Ligand-stimulated FLNA binding results in the disruption of the SSTR2-p85 complex and the subsequent inhibition of PI3K (88).
The importance of FLNA in SSTR2 regulation in GH-secreting pituitary tumors has recently been examined (89) (Figure 3). In contrast to DRD2 in prolactinomas, the expression of FLNA in GH-secreting tumors did not correlate with SSTR2 levels, and FLNA silencing in human tumoral cells did not affect SSTR2 expression and membrane localization. However, by using different FLNA dominant negative mutants that selectively abolished the ability of FLNA to interact with SST2 (FLNA repeats 19–20) or to function as scaffold for partner proteins (FLNA 21–24), the authors demonstrated that FLNA plays both a structural role, by stabilizing SST2 expression after prolonged stimulation with a specific agonist, and a functional role, acting as scaffold protein for molecules involved in signal transduction.
Using as a model pituitary GH-secreting rat GH3 cells, the authors showed that disrupting the FLNA/SSTR2 interaction, receptor stability after prolonged agonist stimulation was strongly compromised. The mechanism probably involve an effect of FLNA in regulating lysosomal degradation of SSTR2, since SST2 ligand-induced down-regulation was abolished when lysosomal degradation was prevented. An effect of FLNA in regulating SSTR2 internalization rate was also reported (88).
Furthermore, in vitro experiments in tumoral somatotrophs demonstrated that FLNA is required for SSTR2-induced reduction of cyclin D1 and caspase3/7 activation, suggesting a crucial role for FLNA in mediating antiproliferative and proapoptotic effects of SSTR2. The observation that FLNA 21–24 which does not abolish the interaction of SSTR2 with endogenous FLNA, was able to block the apototic effects of SST2 and the SST2-mediated ERK1/2 inhibition suggests that the FLNA scaffold properties are required for the assembly of signal transduction complexes.
Overall, these data support a new role for FLNA in the responsiveness of patients with GH-secreting pituitary tumors to pharmacological treatment with SS analogs. Low levels of FLNA, causing loss of coupling of SSTR2 with downstream signal transduction molecules, might explain the resistance to SS analogs in GH-secreting pituitary tumors even if in the presence of SSTR2.
It is of interest to note that FLNA splice variant-1, that was found widely expressed at low levels, lacks 41 amino acids between repeats 19 and 20, the region involved in the interaction both with DRD2 and SST2. NMR spectroscopy analysis has revealed that in the splice variant the repeat 19 is intrinsically unfolded (90), with consequent biological effect. FLNA splice variant-1 showed an increased binding to integrins with respect to wild-type non-spliced FLNA, suggesting that this alternative splicing might function as a regulatory mechanism for the binding of partner proteins to FLNA, with possible implications in pituitary tumors responsiveness to SS/DA analogs.
Beside its role in GPCR regulation, FLNA is involved in the control of cell motility. Indeed, FLNA is recruited to membrane protrusions of migrating cells where it functions as scaffold for proteins involved in cell migration and adhesion.
The observation that in periventricular heterotopia FLNA null mutations caused impaired neuronal migration within the cerebral cortex (73), together with experimental evidences obtained in FLNA-deficient M2 cells that are not able to migrate (91), suggest that FLNA is crucial in promoting cell migration. In contrast, it has been shown that overexpression of FLNA inhibited cell motility (92) and FLNA knockdown promoted cell migration, invasion, and metastasis (93, 94), leading to the conclusion that FLNA positive or negative effect on cell motility depend on the cell type and on a balanced level of FLNA expression.
Pituitary tumors are generally benign, but frequently present local invasiveness that strongly reduces neurosurgery success. Since DA-resistance of prolactinomas has been associated with increased invasiveness and aggressiveness of these tumors (15, 20), and reduced FLNA levels correlate with resistance to dopaminergic drugs, it is possible to hypothesize that low levels of FLNA might be involved in pituitary tumors invasive behavior.
Up to now, no data are present in literature about the effects of FLNA expression levels on pituitary cell migration and invasion, and further studies are required to test a possible contribution of FLNA alterations in the invasiveness, aggressive behavior, and recurrences of pituitary tumors.
Molecular mechanisms underlying the resistance of pituitary tumor to pharmacological treatment with DA and SS analogs might involve different steps that from the receptor activation by agonist lead to the final biological responses in terms of hormone secretion inhibition as well as tumor shrinkage. In the absence of genetic defect in SSTR2, SSTR5, and DRD2 genes, alterations have been reported in molecules involved in the intracellular signal transduction pathway specific of these receptors, and in the regulation of receptors expression. Growing evidence revealed that DRD2 and SSTR2 expression, localization, and signaling are regulated by interaction with cytoskeleton protein FLNA, once again emphasizing the multiple roles of cytoskeleton in physiological cell functions. Future works searching for genetic and post-translational modifications affecting FLNA expression and function, as well as for a possible involvement of other cytoskeleton proteins, might help to clarify the complex mechanisms involved in pituitary tumors pharmacological resistance.
EP: preparing the manuscript, manuscript revision, final approval of the version to be published. DT, EG, EV, AL: preparing the manuscript, final approval of the version to be published. GM: manuscript revision, final approval of the version to be published.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by two AIRC (Associazione Italiana Ricerca Cancro – Milan) grants, one to GM (IG-2014) and the other to AL (IG-13051).
1. Saveanu A, Jaquet P. Somatostatin-dopamine ligands in the treatment of pituitary adenomas. Rev Endocr Metab Disord (2009) 10(2):83–90. doi:10.1007/s11154-008-9086-0
2. Beaulieu JM, Espinoza S, Gainetdinov RR. Dopamine receptors. IUPHAR Review 13. Br J Pharmacol (2015) 172(1):1–23. doi:10.1111/bph.12906
3. Colao A, Savastano S. Medical treatment of prolactinomas. Nat Rev Endocrinol (2011) 7:267–78. doi:10.1038/nrendo.2011.37
4. Petrossians P, Thonnard AS, Beckers A. Medical treatment in Cushing’s syndrome: dopamine agonists and cabergoline. Neuroendocrinology (2010) 92(Suppl 1):116–9. doi:10.1159/000317716
5. Colao A, Di Somma C, Pivonello R, Faggiano A, Lombardi G, Savastano S. Medical therapy for clinically non-functioning pituitary adenomas. Endocr Relat Cancer (2008) 15(4):905–15. doi:10.1677/ERC-08-0181
6. Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol (1999) 20:157–98. doi:10.1006/frne.1999.0183
7. Schulz S, Handel M, Schreff M, Schmidt H, Hollt V. Localization of five somatostatin receptors in the rat central nervous system using subtype-specific antibodies. J Physiol (2000) 94:259–64. doi:10.1016/S0928-4257(00)00212-6
8. Ben-Shlomo A, Melmed S. Pituitary somatostatin receptor signaling. Trends Endocrinol Metab (2010) 21(3):123–33. doi:10.1016/j.tem.2009.12.003
9. Cordelier P, Esteve JP, Bousquet C, Delesque N, O’Carroll AM, Schally AV, et al. Characterization of the antiproliferative signal mediated by somatostatin receptor subtype 5. Proc Natl Acad Sci U S A (1997) 94:9343–8. doi:10.1073/pnas.94.17.9343
10. Ferrante E, Pellegrini C, Bondioni S, Peverelli E, Locatelli M, Gelmini P, et al. Octreotide promotes apoptosis in human somatotroph tumor cells by activating somatostatin receptor type 2. Endocr Relat Cancer (2006) 13(3):955–62. doi:10.1677/erc.1.01191
11. Peverelli E, Lania AG, Mantovani G, Beck-Peccoz P, Spada A. Characterization of intracellular signaling mediated by human somatostatin receptor 5: role of the DRY motif and the third intracellular loop. Endocrinology (2009) 150(7):3169–76. doi:10.1210/en.2008-1785
12. Sharma K, Patel YC, Srikant CB. Subtype-selective induction of wild-type p53 and apoptosis, but not cell cycle arrest, by human somatostatin receptor 3. Mol Endocrinol (1996) 10(12):1688–96. doi:10.1210/mend.10.12.8961277
13. Lamberts SW, de Herder WW, Hofland LJ. Somatostatin analogs in the diagnosis and treatment of cancer. Trends Endocrinol Metab (2002) 13(10):451–7. doi:10.1016/S1043-2760(02)00667-7
14. Beck-Peccoz P, Persani L. Medical management of thyrotropin-secreting pituitary adenomas. Pituitary (2002) 5:83–8. doi:10.1023/A:1022360414062
15. Molitch ME. Pharmacologic resistance in prolactinoma patients. Pituitary (2005) 8:43–52. doi:10.1007/s11102-005-5085-2
16. Delgrange E, Daems T, Verhelst J, Abs R, Maiter D. Characterization of resistance to the prolactin-lowering effects of cabergoline in macroprolactinomas: a study in 122 patients. Eur J Endocrinol (2009) 160:747–52. doi:10.1530/EJE-09-0012
17. Colao A, Auriemma RS, Lombardi G, Pivonello R. Resistance to somatostatin analogs in acromegaly. Endocr Rev (2011) 32(2):247–71. doi:10.1210/er.2010-0002
18. Di Sarno A, Landi ML, Cappabianca P, Di Salle F, Rossi FW, Pivonello R, et al. Resistance to cabergoline as compared with bromocriptine inhyperprolactinemia: prevalence, clinical definition, and therapeuticstrategy. J Clin Endocrinol Metab (2001) 86:5256–61. doi:10.1210/jcem.86.11.8054
19. Giustina A, Chanson P, Bronstein MD, Klibanski A, Lamberts S, Casanueva FF, et al. Acromegaly consensus group. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab (2010) 95(7):3141–8. doi:10.1210/jc.2009-2670
20. Vroonen L, Jaffrain-Rea ML, Petrossians P, Tamagno G, Chanson P, Vilar L, et al. Prolactinomas resistant to standard doses of cabergoline: a multicenter study of 92 patients. Eur J Endocrinol (2012) 167(5):651–62. doi:10.1530/EJE-12-0236
21. Pellegrini I, Rasolonjanahary R, Gunz G, Bertrand P, Delivet S, Jedynak CP, et al. Resistance to bromocriptine in prolactinomas. J Clin Endocrinol Metab (1989) 69:500–9. doi:10.1210/jcem-69-3-500
22. Passos VQ, Fortes MAHZ, Giannella-Neto D, Bronstein MD. Genes differentially expressed in prolactinomas responsive and resistant to dopamine agonists. Neuroendocrinology (2009) 89:163–70. doi:10.1159/000156116
23. Caccavelli L, Feron F, Morange I, Rouer E, Benarous R, Dewailly D, et al. Decreased expression of the two D2 dopamine receptor isoforms in bromocriptine-resistant prolactinomas. Neuroendocrinology (1994) 60(3):314–22. doi:10.1159/000126764
24. Wu ZB, Zheng WM, Su ZP, Chen Y, Wu JS, Wang CD, et al. Expression of D2R mRNA isoforms and ER mRNA isoforms in prolactinomas: correlation with the response to bromocriptine and with tumor biological behavior. J Neurooncol (2010) 99:25–32. doi:10.1007/s11060-009-0107-y
25. Shimazu S, Shimatsu A, Yamada S, Inoshita N, Nagamura Y, Usui T, et al. Resistance to dopamine agonists in prolactinoma is correlated with reduction of dopamine D2 receptor long isoform mRNA levels. Eur J Endocrinol (2012) 166(3):383–90. doi:10.1530/EJE-11-0656
26. Reubi JC, Landolt AM. The growth hormone responses to octreotide in acromegaly correlate with tumor somatostatin receptor status. J Clin Endocrinol Metab (1989) 68:844–50. doi:10.1210/jcem-68-4-844
27. Plöckinger U, Albrecht S, Mawrin C, Saeger W, Buchfelder M, Petersenn S, et al. Selective loss of somatostatin receptor 2 in octreotide-resistant growth hormone-secreting tumors. J Clin Endocrinol Metab (2008) 93:1203–10. doi:10.1210/jc.2007-1986
28. Taboada GF, Luque RM, Neto LV, Machado Ede O, Sbaffi BC, Domingues RC, et al. Quantitative analysis of somatostatin receptor subtypes (1-5) gene expression levels in somatotropinomas and correlation to in vivo hormonal and tumor volume responses to treatment with octreotide LAR. Eur J Endocrinol (2008) 158:295–303. doi:10.1530/EJE-07-0562
29. Ferone D, de Herder WW, Pivonello R, Kros JM, van Koetsveld PM, de Jong T, et al. Correlation of in vitro and in vivo somatotropic tumor responsiveness to somatostatin analogs and dopamine agonists with immunohistochemical evaluation of somatostatin and dopamine receptors and electron microscopy. J Clin Endocrinol Metab (2008) 93:1412–7. doi:10.1210/jc.2007-1358
30. Wildemberg LE, Neto LV, Costa DF, Nasciuti LE, Takiya CM, Alves LM. Low somatostatin receptor subtype 2, but not dopamine receptor subtype 2 expression predicts the lack of biochemical response of somatotropinomas to treatment with somatostatin analogs. J Endocrinol Invest (2013) 36(1):38–43. doi:10.3275/8305
31. Bertherat J, Chanson P, Dewailly D, Enjalbert A, Jaquet P, Kordon C, et al. Resistance to somatostatin (SRIH) analog therapy in acromegaly. Re-evaluation of the correlation between the SRIH receptor status of the pituitary tumor and the in vivo inhibition of GH secretion in response to SRIH analog. Horm Res (1992) 38(1–2):94–9. doi:10.1159/000182500
32. Bertherat J, Chanson P, Dewailly D, Dupuy M, Jaquet P, Peillon F. Epelbaum J. Somatostatin receptors, adenylate cyclase activity and growth hormone response to octreotide in GH-secreting tumors. J Clin Endocrinol Metab (1993) 7:1577–83. doi:10.1210/jcem.77.6.7903312
33. Casarini AP, Jallad RS, Pinto EM, Soares IC, Nonogaki S, Giannella-Neto D, et al. Acromegaly: correlation between expression of somatostatin receptor subtypes and response to octreotide-lar treatment. Pituitary (2009) 12(4):297–303. doi:10.1007/s11102-009-0175-1
34. Brzana J, Yedinak CG, Gultekin SH, Delashaw JB, Fleseriu M. Growth hormone granulation pattern and somatostatin receptor subtype 2A correlate with postoperative somatostatin receptor ligand response in acromegaly: a large single center experience. Pituitary (2013) 16(4):490–8. doi:10.1007/s11102-012-0445-1
35. Friedman E, Adams EF, Höög A, Gejman PV, Carson E, Larsson C, et al. Normal structural dopamine type 2 receptor gene in prolactin-secreting and other pituitary tumors. J Clin Endocrinol Metab (1994) 78:568–74. doi:10.1210/jc.78.3.568
36. Wang F, Gao H, Li C, Bai J, Lu R, Cao L, et al. Low levels of PRB3 mRNA are associated with dopamine-agonist resistance and tumor recurrence in prolactinomas. J Neurooncol (2014) 116(1):83–8. doi:10.1007/s11060-013-1276-2
37. Gao H, Wang F, Lan X, Li C, Feng J, Bai J, et al. Lower PRDM2 expression is associated with dopamine-agonist resistance and tumor recurrence in prolactinomas. BMC Cancer (2015) 12(15):272. doi:10.1186/s12885-015-1267-0
38. Filopanti M, Barbieri AM, Angioni AR, Colao A, Gasco V, Grottoli S, et al. Dopamine D2 receptor gene polymorphisms and response to cabergoline therapy in patients with prolactin-secreting pituitary adenomas. Pharmacogenomics J (2008) 8:357–63. doi:10.1038/tpj.2008.1
39. Petersenn S, Heyens M, Lüdecke DK, Beil FU, Schulte HM. Absence of somatostatin receptor type 2A mutations and gip oncogene in pituitary somatotroph tumors. Clin Endocrinol (2000) 52:35–42. doi:10.1046/j.1365-2265.2000.00880.x
40. Corbetta S, Ballarè E, Mantovani G, Lania AG, Losa M, Di Blasio AM, et al. Somatostatin receptor subtype 2 and 5 in human GH-secreting pituitary tumors: analysis of gene sequence and mRNA expression. Eur J Clin Invest (2001) 31:208–14. doi:10.1046/j.1365-2362.2001.00786.x
41. Ballarè E, Persani L, Lania AG, Filopanti M, Giammona E, Corbetta S, et al. Mutation of somatostatin receptor type 5 in an acromegalic patient resistant to somatostatin analog treatment. J Clin Endocrinol Metab (2001) 86(8):3809–14. doi:10.1210/jcem.86.8.7787
42. Peverelli E, Busnelli M, Vitali E, Giardino E, Galés C, Lania AG, et al. Specific roles of Gi protein family members revealed by dissecting somatostatin receptor 5 (SST5) coupling in human pituitary cells. J Cell Sci (2013) 126(Pt 2):638–44. doi:10.1242/jcs.116434
43. Filopanti M, Ronchi C, Ballarè E, Bondioni S, Lania AG, Losa M, et al. Analysis of somatostatin receptors 2 and 5 polymorphisms in patients with acromegaly. J Clin Endocrinol Metab (2005) 90(8):4824–8. doi:10.1210/jc.2005-0132
44. Ciganoka D, Balcere I, Kapa I, Peculis R, Valtere A, Nikitina-Zake L, et al. Identification of somatostatin receptor type 5 gene polymorphisms associated with acromegaly. Eur J Endocrinol (2011) 165(4):517–25. doi:10.1530/EJE-11-0416
45. Filopanti M, Ballarè E, Lania AG, Bondioni S, Verga U, Locatelli M, et al. Loss of heterozygosity at the SS receptor type 5 locus in human GH- and TSH-secreting pituitary tumors. J Endocrinol Invest (2004) 27:937–42. doi:10.1007/BF03347536
46. Duran-Prado M, Gahete MD, Martinez-Fuentes AJ, Luque RM, Quintero A, Webb SM, et al. Identification and characterization of two novel truncated but functional isoforms of the somatostatin receptor subtype 5 differentially present in pituitary tumors. J Clin Endocrinol Metab (2009) 94:2634–43. doi:10.1210/jc.2008-2564
47. Duran-Prado M, Saveanu A, Luque M, Gahete MD, Gracia-Navarro F, Jaquet P, et al. A potential inhibitory role for the new truncated variant of somatostatin receptor 5, sst5TMD4, in pituitary adenomas poorly responsive to somatostatin analogs. J Clin Endocrinol Metab (2010) 95:2497–502. doi:10.1210/jc.2009-2247
48. Luque RM, Ibáñez-Costa A, Neto LV, Taboada GF, Hormaechea-Agulla D, Kasuki L, et al. Truncated somatostatin receptor variant sst5TMD4 confers aggressive features (proliferation, invasion and reduced octreotide response) to somatotropinomas. Cancer Lett (2015) 359(2):299–306. doi:10.1016/j.canlet.2015.01.037
49. Marina D, Burman P, Klose M, Casar-Borota O, Luque RM, Castaño JP, et al. Truncated somatostatin receptor 5 may modulate therapy response to somatostatin analogues – Observations in two patients with acromegaly and severe headache. Growth Horm IGF Res (2015) 25(5):262–7. doi:10.1016/j.ghir.2015.07.003
50. Resmini E, Dadati P, Ravetti JL, Zona G, Spaziante R, Saveanu A, et al. Rapid pituitary tumor shrinkage with dissociation between antiproliferative and antisecretory effects of a long-acting octreotide in an acromegalic patient. J Clin Endocrinol Metab (2007) 92:1592–9. doi:10.1210/jc.2006-2084
51. Danila DC, Haidar JNS, Zhang X, Katznelson L, Culler MD, Klibanski A. Somatostatin receptor-specific analogs: effects on cell proliferation and growth hormone secretion in human somatotroph tumors. Clin Endocrinol Metab (2001) 86:2976–81. doi:10.1210/jcem.86.7.7620
52. Murphy WA, Taylor JE, Moreau JP, Coy DH. Novel heptapeptide somatostatin analog displays anti-tumor activity independent of effects on growth hormone secretion. Pept Res (1989) 2:128–32.
53. Caccavelli L, Morange-Ramos I, Kordon C, Jaquet P, Enjalbert A. Alteration of G alpha subunits mRNA levels in bromocriptine resistant prolactinomas. J Neuroendocrinol (1996) 8(10):737–46. doi:10.1046/j.1365-2826.1996.04902.x
54. Ballarè E, Mantovani S, Bassetti M, Lania AG, Spada A. Immunodetection of G proteins in human pituitary adenomas: evidence for a low expression of proteins of the Gi subfamily. Eur J Endocrinol (1997) 137(5):482–9. doi:10.1530/eje.0.1370482
55. Spada A, Arosio M, Bochicchio D, Bazzoni N, Vallar L, Bassetti M, et al. Clinical, biochemical and morphological correlates in patients bearing growth hormone secreting tumors with or without constitutively active adenylyl cyclase. J Clin Endocrinol Metab (1990) 71:1421–6. doi:10.1210/jcem-71-6-1421
56. Barlier A, Gunz G, Zamora AJ, Morange-Ramos I, Figarella-Branger D, Dufour H, et al. Pronostic and therapeutic consequences of Gs alpha mutations in somatotroph adenomas. J Clin Endocrinol Metab (1998) 83:1604–10. doi:10.1210/jc.83.5.1604
57. Barlier A, Pellegrini-Bouiller I, Gunz G, Zamora AJ, Jaquet P, Enjalbert A. Impact of gsp oncogene on the expression of genes coding for Gsalpha, Pit-1, Gi2alpha, and somatostatin receptor 2 in human somatotroph adenomas: involvement in octreotide sensitivity. J Clin Endocrinol Metab (1999) 84:2759–65. doi:10.1210/jcem.84.8.5919
58. Daly AF, Tichomirowa MA, Petrossians P, Heliövaara E, Jaffrain-Rea ML, Barlier A, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab (2010) 95(11):E373–83. doi:10.1210/jc.2009-2556
59. Chahal HS, Trivellin G, Leontiou CA, Alband N, Fowkes RC, Tahir A, et al. Somatostatin analogs modulate AIP in somatotroph adenomas: the role of the ZAC1 pathway. J Clin Endocrinol Metab (2012) 97(8):E1411–20. doi:10.1210/jc.2012-1111
60. Tuominen I, Heliövaara E, Raitila A, Rautiainen M, Mehine M, Katainen R, et al. AIP inactivation leads to pituitary tumorigenesis through defective Gαi-cAMP signaling. Oncogene (2014) 34(9):1174–84. doi:10.1038/onc.2014.50
61. Iacovazzo D, Carlsen E, Lugli F, Chiloiro S, Piacentini S, Bianchi A, et al. Factors predicting pasireotide responsiveness in somatotroph pituitary adenomas resistant to first generation somatostatin analogues: an immunohistochemical study. Eur J Endocrinol (2015). doi:10.1530/EJE-15-0832
62. Oriola J, Lucas T, Halperin I, Mora M, Perales MJ, Alvarez-Escolá C, et al. Germline mutations of AIP gene in somatotropinomas resistant to somatostatin analogues. Eur J Endocrinol (2012) 168(1):9–13. doi:10.1530/EJE-12-0457
63. Fougner SL, Bollerslev J, Latif F, Hald JK, Lund T, Ramm-Pettersen J, et al. Low levels of raf kinase inhibitory protein in growth hormone-secreting pituitary adenomas correlate with poor response to octreotide treatment. J Clin Endocrinol Metab (2008) 93:1211–6. doi:10.1210/jc.2007-2272
64. Tulipano G, Stumm R, Pfeiffer M, Kreienkamp HJ, Höllt V, Schulz S. Differential beta-arrestin trafficking and endosomal sorting of somatostatin receptor subtypes. J Biol Chem (2004) 279(20):21374–82. doi:10.1074/jbc.M313522200
65. Peverelli E, Mantovani G, Calebiro D, Doni A, Bondioni S, Lania A, et al. The third intracellular loop of the human somatostatin receptor 5 is crucial for arrestin binding and receptor internalization after somatostatin stimulation. Mol Endocrinol (2008) 22(3):676–88. doi:10.1210/me.2007-0068
66. Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J Biol Chem (2001) 276(40):37409–14. doi:10.1074/jbc.M106728200
67. Gatto F, Feelders R, van der Pas R, Kros JM, Dogan F, van Koetsveld PM, et al. β-Arrestin 1 and 2 and G protein-coupled receptor kinase 2 expression in pituitary adenomas: role in the regulation of response to somatostatin analogue treatment in patients with acromegaly. Endocrinology (2013) 154(12):4715–25. doi:10.1210/en.2013-1672
68. Li Z, Liu Q, Li C, Zong X, Bai J, Wu Y, et al. The role of TGF-β/Smad signaling in dopamine agonist-resistant prolactinomas. Mol Cell Endocrinol (2015) 15(402):64–71. doi:10.1016/j.mce.2014.12.024
69. Wu ZB, Li WQ, Lin SJ, Wang CD, Cai L, Lu JL, et al. MicroRNA expression profile of bromocriptine-resistant prolactinomas. Mol Cell Endocrinol (2014) 395(1–2):10–8. doi:10.1016/j.mce.2014.07.014
70. van der Flier A, Sonnenberg A. Structural and functional aspects of filamins. Biochim Biophys Acta (2001) 1538(2–3):99–117. doi:10.1016/S0167-4889(01)00072-6
71. Hartwig JH, Stossel TP. Isolation and properties of actin, myosin, and a new actin binding protein in rabbit alveolar macrophages. J Biol Chem (1975) 250(14):5696–705.
72. Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol (2004) 6(11):1034–8. doi:10.1038/ncb1104-1034
73. Fox JW, Lamperti ED, Ekşioğlu YZ, Hong SE, Feng Y, Graham DA, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron (1998) 21(6):131525. doi:10.1016/S0896-6273(00)80651-0
74. Robertson SP, Twigg SR, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat Genet (2003) 33(4):487–91. doi:10.1038/ng1119
75. Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol (2001) 2:138–45. doi:10.1038/35052082
76. Nakamura F, Stossel TP, Hartwig JH. The filamins: organizers of cell structure and function. Cell Adh Migr (2011) 5(2):160–9. doi:10.4161/cam.5.2.14401
77. Pentikäinen U, Jiang P, Takala H, Ruskamo S, Campbell ID, Ylänne J. Assembly of a filamin four-domain fragment and the influence of splicing variant-1 on the structure. J Biol Chem (2011) 286(30):26921–30. doi:10.1074/jbc.M110.195958
78. Li M, Bermak JC, Wang ZW, Zhou QY. Modulation of dopamine D(2) receptor signaling by actin-binding protein (ABP-280). Mol Pharmacol (2000) 57:446–52. doi:10.1124/mol.57.3.446
79. Lin R, Karpa K, Kabbani N, Goldman-Rakic P, Levenson R. Dopamine D2 and D3 receptors are linked to the actin cytoskeleton via interaction with filamin A. Proc Natl Acad Sci (2001) 98:5258–63. doi:10.1073/pnas.011538198
80. Lin R, Canfield V, Levenson R. Dominant negative mutants of filamin A block cell surface expression of the D2 dopamine receptor. Pharmacology (2002) 66:173–81. doi:10.1159/000065531
81. Peverelli E, Mantovani G, Vitali E, Elli FM, Olgiati L, Ferrero S, et al. Filamin-A is essential for dopamine D2 receptor expression and signaling in tumorous lactotrophs. J Clin Endocrinol Metab (2012) 97(3):967–77. doi:10.1210/jc.2011-2902
82. Scott MG, Pierotti V, Storez H, Lindberg E, Thuret A, Muntaner O, et al. Cooperative regulation of extracellular signal-regulated kinase activation and cell shape change by filamin A and beta-arrestins. Mol Cell Biol (2006) 26(9):3432–45. doi:10.1128/MCB.26.9.3432-3445.2006
83. Zhang M, Breitwieser GE. High affinity interaction with filamin A protects against calcium-sensing receptor degradation. J Biol Chem (2005) 280:11140–6. doi:10.1074/jbc.M412242200
84. Seck T, Baron R, Horne WC. Binding of filamin to the C-terminal tail of the calcitonin receptor controls recycling. J Biol Chem (2003) 278:10408–16. doi:10.1074/jbc.M209655200
85. Thelin WR, Chen Y, Gentzsch M, Kreda SM, Sallee JL, Scarlett CO, et al. Direct interaction with filamins modulates the stability and plasma membrane expression of CFTR. J Clin Invest (2007) 117:364–74. doi:10.1172/JCI30376
86. Beekman JM, van der Poel CE, van der Linden JA, van den Berg DL, van den Berghe PV, van de Winkel JG, et al. Filamin A stabilizes Fc gamma RI surface expression and prevents its lysosomal routing. J Immunol (2008) 180:3938–45. doi:10.4049/jimmunol.180.6.3938
87. Meller E, Puza T, Miller JC, Friedhoff AJ, Schweitzer JW. Receptor reserve for D2 dopaminergic inhibition of prolactin release in vivo and in vitro. J Pharmacol Exp Ther (1991) 257:668–75.
88. Najib S, Saint-Laurent N, Estève JP, Schulz S, Boutet-Robinet E, Fourmy D, et al. A switch of G protein-coupled receptor binding preference from phosphoinositide 3-kinase (PI3K)-p85 to filamin A negatively controls the PI3K pathway. Mol Cell Biol (2012) 32(5):1004–16. doi:10.1128/MCB.06252-11
89. Peverelli E, Giardino E, Treppiedi D, Vitali E, Cambiaghi V, Locatelli M, et al. Filamin A (FLNA) plays an essential role in somatostatin receptor 2 (SST2) signaling and stabilization after agonist stimulation in human and rat somatotroph tumor cells. Endocrinology (2014) 155(8):2932–41. doi:10.1210/en.2014-1063
90. Travis MA, van der Flier A, Kammerer RA, Mould AP, Sonnenberg A, Humphries MJ. Interaction of filamin A with the integrin beta 7 cytoplasmic domain: role of alternative splicing and phosphorylation. FEBS Lett (2004) 569(1–3):185–90. doi:10.1016/j.febslet.2004.04.099
91. Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR, et al. Actin-binding protein requirement for cortical stability and efficient locomotion. Science (1992) 255(5042):325–7. doi:10.1126/science.1549777
92. Sarkisian MR, Bartley CM, Chi H, Nakamura F, Hashimoto-Torii K, Torii M, et al. MEKK4 signaling regulates filamin expression and neuronal migration. Neuron (2006) 52(5):789–801. doi:10.1016/j.neuron.2006.10.024
93. Xu Y, Bismar TA, Su J, Xu B, Kristiansen G, Varga Z, et al. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. J Exp Med (2010) 207(11):2421–37. doi:10.1084/jem.20100433
Keywords: pituitary tumors, dopamine, somatostatin, DRD2, SSTRs, filamin A
Citation: Peverelli E, Treppiedi D, Giardino E, Vitali E, Lania AG and Mantovani G (2015) Dopamine and Somatostatin Analogues Resistance of Pituitary Tumors: Focus on Cytoskeleton Involvement. Front. Endocrinol. 6:187. doi: 10.3389/fendo.2015.00187
Received: 05 November 2015; Accepted: 07 December 2015;
Published: 22 December 2015
Edited by:
Francisco Gracia-Navarro, University of Cordoba, SpainReviewed by:
Akira Shimatsu, National Hospital Organization Kyoto Medical Center, JapanCopyright: © 2015 Peverelli, Treppiedi, Giardino, Vitali, Lania and Mantovani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanna Mantovani, Z2lvdmFubmEubWFudG92YW5pQHVuaW1pLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.