 Roman Sankowski
Roman Sankowski Simone Mader
Simone Mader Sergio Iván Valdés-Ferrer
Sergio Iván Valdés-Ferrer- 1Elmezzi Graduate School of Molecular Medicine, Manhasset, NY, USA
- 2Feinstein Institute for Medical Research, Manhasset, NY, USA
- 3Department of Neurology, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, México City, Mexico
The nervous and immune systems have evolved in parallel from the early bilaterians, in which innate immunity and a central nervous system (CNS) coexisted for the first time, to jawed vertebrates and the appearance of adaptive immunity. The CNS feeds from, and integrates efferent signals in response to, somatic and autonomic sensory information. The CNS receives input also from the periphery about inflammation and infection. Cytokines, chemokines, and damage-associated soluble mediators of systemic inflammation can also gain access to the CNS via blood flow. In response to systemic inflammation, those soluble mediators can access directly through the circumventricular organs, as well as open the blood–brain barrier. The resulting translocation of inflammatory mediators can interfere with neuronal and glial well-being, leading to a break of balance in brain homeostasis. This in turn results in cognitive and behavioral manifestations commonly present during acute infections – including anorexia, malaise, depression, and decreased physical activity – collectively known as the sickness behavior (SB). While SB manifestations are transient and self-limited, under states of persistent systemic inflammatory response the cognitive and behavioral changes can become permanent. For example, cognitive decline is almost universal in sepsis survivors, and a common finding in patients with systemic lupus erythematosus. Here, we review recent genetic evidence suggesting an association between neurodegenerative disorders and persistent immune activation; clinical and experimental evidence indicating previously unidentified immune-mediated pathways of neurodegeneration; and novel immunomodulatory targets and their potential relevance for neurodegenerative disorders.
Introduction: An Evolutionary Perspective
All multicellular organisms have germ-line encoded surveillance systems destined to detect potentially dangerous, perhaps lethal, invaders (Medzhitov and Janeway, 1997). This extremely effective response, the innate immunity, depends on detection of specific molecular patterns present in invaders, but not expressed by self-tissues. Innate immunity aroused approximately 600 million years ago, and is evolutionarily conserved across species and kingdoms. The main components of innate immunity – from toll-like receptors (TLR) to inflammasomes – are highly conserved from plants to mammals (Jones and Dangl, 2006). Adaptive immunity appeared much later in evolution, perhaps 500 million years ago, and is only present in vertebrates (Cooper and Alder, 2006; Moresco et al., 2011). Unlike innate immunity, adaptive immunity depends on one individual’s experience, in which adaptive responses form with high efficiency only against antigens that have been recognized and presented by the innate immunity. Although the origin of the nervous system remains controversial, the first centralized nervous system probably appeared with the first bilateral organisms (organisms with two relatively identical halves, called bilaterians) during the Ediacaran period around 550 million years ago (Knoll et al., 2004). Ever since, both nervous and immune systems have co-evolved and are in constant communication.
The nervous system of the evolutionarily ancient nematode Caenorhabditis elegans has the ability to regulate innate immune responses (Andersson and Tracey, 2012), and aid in decision-making regarding finding bacteria that can be used as food and avoiding pathogenic bacteria (Reddy et al., 2009). In mammals, the nervous system also has the ability to sense inflammatory stimuli directly, thus allowing to recognize a potential source of damage through generation of pain, and to modulate the response to infection (Mina-Osorio et al., 2012; Chiu et al., 2013a). Although the afferent pathways and integration of immune information in the brain are areas of active research, there is evidence that central muscarinic signaling modulates inflammation in experimental sepsis (Pavlov et al., 2009; Rosas-Ballina et al., 2015), obesity (Satapathy et al., 2011), and inflammatory colitis (Ji et al., 2014). The efferent axis of neuroimmune control is better understood after the cholinergic anti-inflammatory pathway (CAP) (Borovikova et al., 2000), a cholinergic reflex system that regulates inflammation via the vagus nerve that stimulates the splenic nerve to release noradrenaline. Noradrenaline in turn stimulates a subset of acetylcholine (ACh)-producing splenic T-cells (CD4+CD44hiCD62Llo) to release ACh, which binds to α7 nichotinic receptors on the surface of macrophages, resulting in down-regulation of TNF by blocking the nuclear translocation of nuclear factor kappa B (NF-κB) (Rosas-Ballina et al., 2011). Thus far, this is a unique scenario in which an immune cell acts as interneuron in a reflex system. Electrical as well as chemical stimulation of the CAP have been shown to decrease the inflammatory burden and increase survival of experimental sepsis (Borovikova et al., 2000; Bernik et al., 2002).
Neuroimmune modulation is a flexible phenomenon that relies on environmental cues from a continuously changing milieu. People undergoing persistent stress have been found to display abnormal immune responses. For instance, caregivers of Alzheimer disease (AD) patients have higher levels of anxiety and depression than age-matched controls; otherwise, these healthy caregivers also have reduced total T-cells and T helper cells in peripheral circulation, as well as higher titers of anti-Epstein–Barr virus antibodies (Kiecolt-Glaser et al., 1987). Moreover, the observed immune and behavioral changes are increased in proportion to disease progression. T-cell response to mitogenic stimuli progressively decreases, and while all subjects have the same number of infectious episodes, the number of days unable to perform activities of daily living, as well as the number of doctor visits are significantly increased in AD caregivers (Kiecolt-Glaser et al., 1991). One mechanism for the immune dysfunction in response to chronic stress is a reduction of telomeres and telomerase activity, potentially leading to early immune senescence (Epel et al., 2004). Mice with naturally elevated anxiety levels have increased activated microglia and perivascular macrophages in the brain, than less anxious strains (Li et al., 2014), suggesting that anxiety can increase the inflammatory background in the brain.
The acute effects of systemic inflammation upon cognition and behavior are not limited to the elderly or the critically ill. As we have witnessed in ourselves and those near us, even a minor and self-limited common cold induces a transient syndrome known as sickness behavior (SB) marked by fatigue, depression, lack of drive, malaise, sleep disturbances, decreased physical activity, and social interactions, as well as cognitive impairment (Capuron et al., 1999, 2001). Healthy volunteers develop anxiety, depression, and memory impairment in response to a low dose of lipopolysaccharide (LPS), and the development of such clinical scenario correlates with TNF secretion (Reichenberg et al., 2001). Some chronic infections may go unrecognized for long periods, as is the case in tuberculosis, human immunodeficiency virus (HIV), hepatitis B virus (HVB), or hepatitis C virus (HCV). Unlike septic patients, patients with chronic infections have an organized and targeted immune response (versus a massive and diffuse one in sepsis), even if the response is ineffective to clear the infection. Those patients, however, have increased cognitive and behavioral problems. For instance, patients with HCV infection have increased rates of fatigue and depression, as well as evidence of metabolic brain dysfunction in the absence of acute hepatitis (Forton et al., 2001, 2002; Hilsabeck et al., 2002). Patients with chronic HCV that have been treated with pegylated interferon (IFN)-α develop significantly higher incidence of depression when compared to the patients’ state before IFN-α administration (Reichenberg et al., 2005). This supports the role of large loads of inflammatory cytokines in inducing and sustaining brain dysfunction. Experimentally, NADPH oxidative activity and nitric oxide synthase (iNOS) are induced in the brain shortly after systemic inflammation (Wong et al., 1996; Yokoo et al., 2012), potentially leading to NMDA-dependent neurotoxicity (Dawson et al., 1993). Evidence derived from post-mortem studies indicates that iNOS is also induced in the brain of patients dying of severe sepsis, but the role of iNOS as inductor of neurotoxicity in response to systemic inflammation has not been assessed in patients surviving severe sepsis (Sharshar et al., 2003). Experimentally, preemptive administration of the free radical scavenger endarvone before sepsis induction resulted in reduced neuronal damage and blood–brain barrier (BBB) permeability (Yokoo et al., 2012). Administration of the antioxidants N-acetylcysteine and deferoxamine shortly after murine sepsis induction has shown long-term neuroprotective effects (Barichello et al., 2007).
Immunity in Sepsis
The immune system is in a constant state of surveillance against potential pathogens and self-generated molecules indicative of damage. Under normal conditions, inflammation is a well-orchestrated response with constant fine-tuning. Once microorganisms have breached the skin and mucosal barriers, innate immunity is critical in preventing further invasion by launching inflammation. After the infection source has been cleared, the inflammatory response also plays an important role in tissue repair and functional healing. When the source of damage has been controlled, the same mechanisms that initiated and regulated inflammation will dampen the response. Large loads of pathogens, or infection by highly virulent pathogens, can trigger an en-masse systemic response that leads to sepsis and multiple organ failure (Deutschman and Tracey, 2014). Moreover, during sepsis the inflammatory response does not resolve for several days or weeks (Valdés-Ferrer, 2014), leading to persistent release of high-mobility group box-1 (HMGB1) (Valdés-Ferrer et al., 2013b). HMGB1 is a highly conserved nuclear protein that can be passively released by stressed cells, or actively secreted by monocytes and other immune cells in response to inflammatory signals, playing a critical role in the innate immune response to inflammatory and sterile injury alike (Andersson and Tracey, 2011). HMGB1 in turn primes resident monocytes toward an inflammatory, cytokine producing phenotype (Valdés-Ferrer et al., 2013a). Severe trauma, as well as surgery can lead to large loads of endogenous pro-inflammatory molecules (damage-associated molecular patterns (DAMPs) being released. A few DAMPs have been shown to induce brain dysfunction in vivo. Of those, TNF and IL-1 can mediate long-standing cognitive and behavioral changes and, in experimental settings, interfering with the effect of TNF reduces the effect of trauma in the formation of contextual memory (Terrando et al., 2010b). A number of genes regulating immune responses are also closely related to neurodegenerative diseases (Table 1).
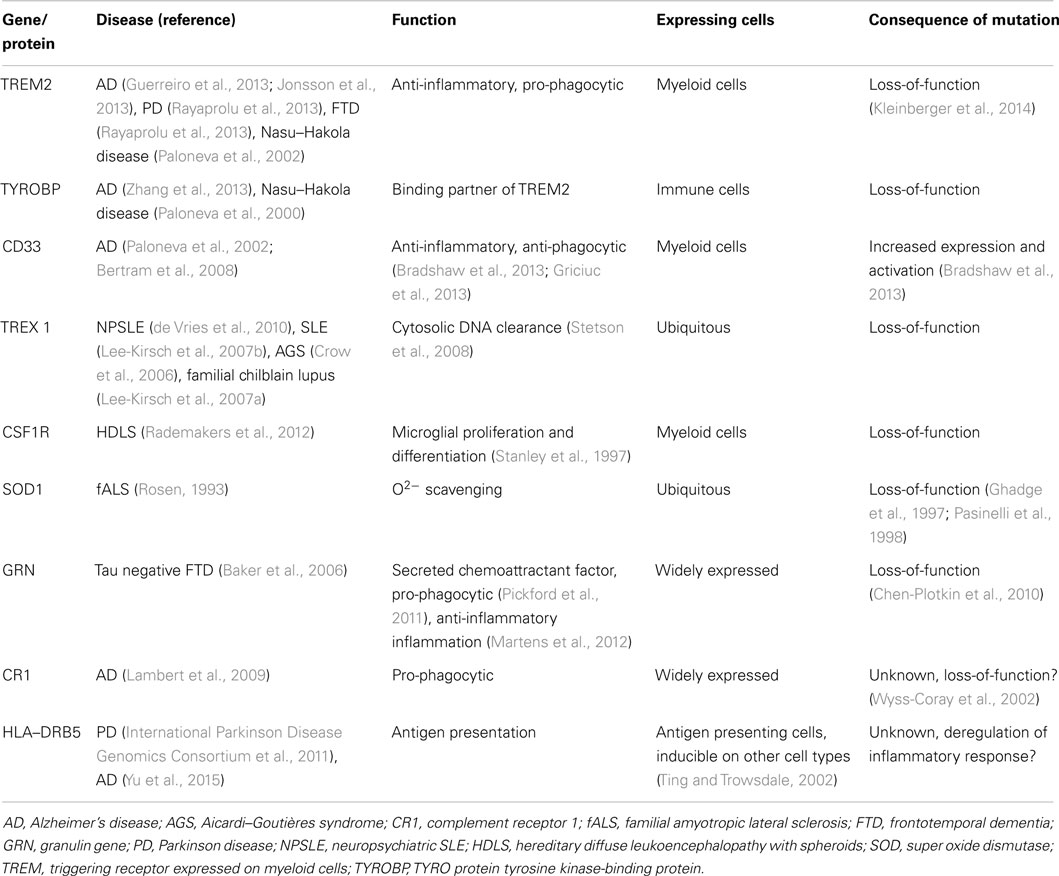
Table 1. Major immune genes associated with neurodegenerative diseases.
Cerebral Consequences of Systemic Inflammation: What We have Learned from Sepsis and Other Inflammatory Conditions
The nervous system is particularly vulnerable to damage in response to systemic inflammation. Inflammation-induced infiltration of immune cells and mediators into the brain leads to profound structural and functional changes (Figure 1). As a consequence, up to 81% of septic patients develop sepsis-associated delirium (SAD) (Ely et al., 2001, 2004), with elderly patients being at particularly high risk (McNicoll et al., 2003; Iwashyna et al., 2012). In the elderly, severe sepsis is sufficient to trigger new cognitive decline of sufficient importance as to profoundly interfere with quality of life (Iwashyna et al., 2010). SAD is not only common in septic patients but also is a reliable indicator of bad prognosis (Eidelman et al., 1996). Six-month mortality among septic patients is twice as high in patients who develop SAD at any point in time during hospitalization (Ely et al., 2004). Altered mental status is more common in septic patients with bacteremia than in those with negative blood cultures (Eidelman et al., 1996). Magnetic resonance imaging (MRI) in patients with septic shock has shown that new white matter lesions are common (Sharshar et al., 2007). Neonatal sepsis is also marked by abnormalities of the white matter (66% of infants in one cohort), and white matter lesions correlate to poorer mental and psychomotor development at 2 years (Shah et al., 2008). The burden of white matter damage worsens with duration of shock, indicating that sepsis interferes with brain connectivity (Figure 2). Gray matter damage in response to sepsis is far less clear. Imaging and post-mortem studies from patients, as well as data derived from experimental models, indicate that gray matter damage is the norm. In comparison to subjects dying of other causes, brain histology from severe sepsis patients shows a higher number of CD68+ and major histocompatibility complex (MHC)-class II microglia in the cerebral cortex and the white matter. Moreover, in septic brains ameboid-shaped activated microglia can be found in gray and white matter alike (Lemstra et al., 2007). An MRI study of premature infants showed reduced volume of deep gray matter structures, and although the findings were consistent in the small subset of septic infants, those were not different from premature non-septic infants (Boardman et al., 2006).
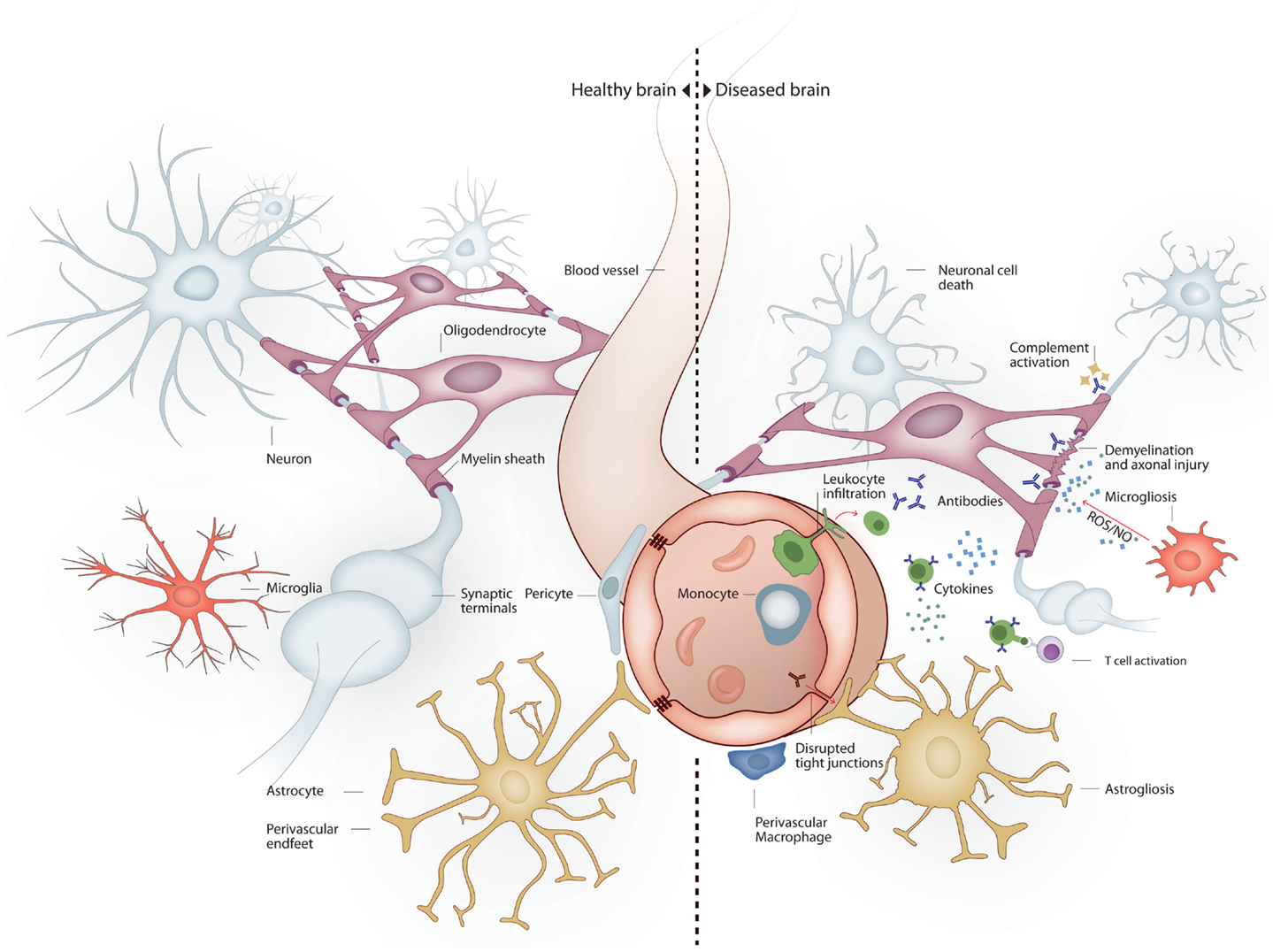
Figure 1. Brain milieu changes in response to systemic inflammation. Under healthy conditions the main cell types present in the brain are neurons, oligodendrocytes, astrocytes, and microglia. Neurons connect to each other through long axonal processes with synapses. Oligodendrocytes support axons with myelin sheaths. Astrocytes interact with blood vessels to form the blood–brain barrier and maintain neuronal synapses. Microglia form long processes that surveillance the brain and phagocytose apoptotic cells and prune inactive synapses without induction of inflammation. Under inflammatory conditions several mechanisms lead to neurodegeneration. Peripheral immune cells and inflammatory molecules traverse the blood–brain barrier exerting direct and indirect neuronal cytoxicity. Oligodendroglial myelin sheaths can be affected leading to axonal degeneration. Astrocytosis leads to reduced blood–brain barrier and synaptic maintenance. Microgliosis leads to a pro-inflammatory microglial phenotype with reduced phagocytic and tissue maintenance functions.
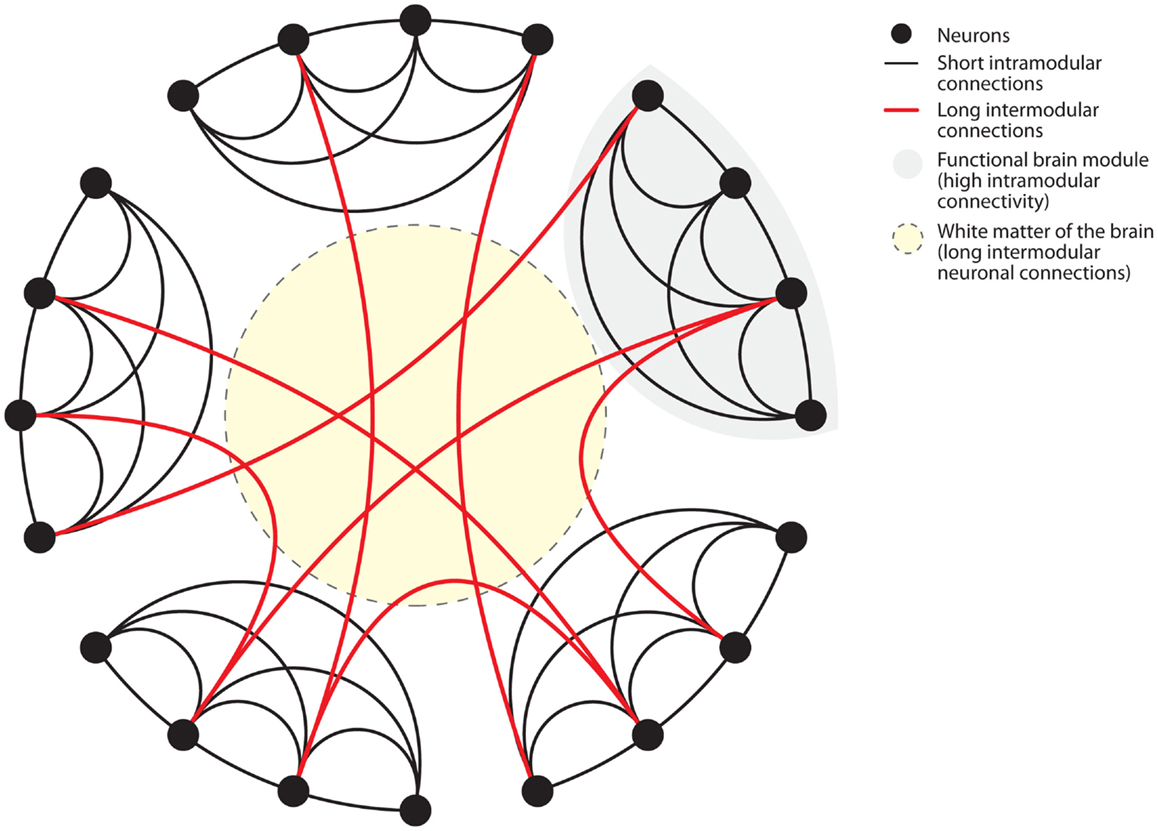
Figure 2. Connectome of the human brain. The human brain is organized as a small-world network. Neurons (black dots) form functional modules (gray shaded area). Within such modules, high connectivity is established by short intramodular connections (black lines). Additionally, long intermodular connections located in the white matter (red lines in yellow shaded area) connect different modules with each other. Small-world networks ensure parallel processing of different modes of information within specialized functional modules. Long intermodular connections (red lines) integrate different kinds of information to code a complex response by the brain. The “wiring cost” of neuronal connections is determined by the energetic requirements to maintain these connections. Short intramodular connections have low wiring costs (black line). Long intermodular connections ensure high network efficiency through parallel information processing at the expense of high wiring costs (red line). High wiring cost renders long intermodular connections (red lines) susceptible to energetic imbalance caused by systemic inflammation. Figure modified after Watts and Strogatz (1998).
In experimental sepsis, persistent cognitive impairment has been observed in rats completely recovered and with negative blood cultures. This indicates that clearing the trigger of sepsis does not prevent the appearance of persistent brain damage (Barichello et al., 2005). Brain mitochondria become dysfunctional during experimental sepsis, showing increased proton permeability, inadequate membrane potential recovery, and reduced oxidative phosphorylation (d’Avila et al., 2008). In mice, the BBB becomes leaky within 24 h following sepsis induction (Yokoo et al., 2012). While the number of hippocampal neurons is not reduced in mice surviving abdominal sepsis, there is neuronal degeneration (Yokoo et al., 2012), and the spine density of CA1 neurons is significantly diminished, and this finding correlates with circulating HMGB1 (Chavan et al., 2012). Anti-HMGB1 neutralizing monoclonal antibodies improve the memory deficit observed in experimental sepsis, although the mechanism of neuroprotection behind this protective effect is still under investigation. In response to systemic LPS, cortical mRNA expression of IL-1β, IL-6, TLR2, TLR4, Scavenger A, and glial fibrillary acidic protein (GFAP) are upregulated within 4 h, indicating that cortical inflammation and glial activation occur in parallel or shortly after systemic inflammation ensues (Noh et al., 2014; Silverman et al., 2014). LPS also increased brain mitochondrial complex II/III activity, and reduced brain glutathion levels within 1 h after systemic administration, supporting the role of systemic inflammation in cerebral mitochondrial dysfunction in sepsis (Noh et al., 2014). Interestingly, even a single and relatively low dose of LPS (that is sufficient to cause around 10% mortality) has been shown to have long-term behavioral and cognitive consequences. In one study, 30-day evaluation showed increased anhedonia and anxiety, altered working memory, as well as reduced exploratory behavior (Anderson et al., 2015). Consistent with that, in a model of endotoxemia in aged rats, a single systemic injection of LPS induced brain inflammation that lasted for at least 30 days. Interestingly, those rats had only transient increase of circulating TNF and IL-1 lasting <6 h in response to the injection (Fu et al., 2014). The hippocampus and dentate gyrus were particularly affected, showing astrogliosis and increased TNF, IL-1, and NF-κB mRNA and protein levels. This suggests that even transient bouts of systemic inflammation of only limited significance can cause sustained brain damage.
Delirium and brain dysfunction can be induced directly by soluble mediators such as TNF, IL-1, or HMGB1, as well as indirectly through activation of microglia and astrocytes. LPS has been shown to induce microglial activation and memory impairment in young mice through a mechanism that is at least partially dependent on interleukin (IL)-1, and HMGB1, both occurring within 24 h of LPS administration (Terrando et al., 2010a). However, while the rapid effect of LPS upon brain homeostasis is clear, a single sublethal injection of LPS (5 mg/kg) is sufficient to impair behavior and memory, mediated by a reduction in neural stem cell proliferation in the dentate gyrus, as well as inducing microglia invasion and activation to the hippocampus, all lasting for at least 30 days (Anderson et al., 2015). Minocycline, a tetracycline derivative used commonly for acne and other infections, inhibits activation and proliferation of microglia (Tikka et al., 2001). Intracerebroventricular (ICV) administration of minocycline immediately after sepsis induction by CLP reduced cerebral inflammation, decreased BBB permeability, and protected against memory impairment observed in untreated septic mice (Michels et al., 2015).
Susceptible Brain: Predisposing Factors Drive Neurodegeneration
While the nervous system is susceptible to peripheral challenges, one question under active research concerns to the role of a susceptible nervous system. Genes and molecular factors rendering rodents and humans prone to neurodegeneration have been identified recently. Genes predisposing to neurodegeneration are summarized in Table 1. The mouse strain DBA/2J has a natural anxious behavior. In response to a systemic challenge with low-dose LPS (1 mg/kg), DBA/2J mice develop increased anxiety, and increased expression of hypothalamic mRNA expression of the inflammatory genes Il1b, Il6, tnf, and Nos2 in comparison to behaviorally normal strains (Li et al., 2014), suggesting that emotional stress has a role in magnifying a systemic inflammatory stimulus.
Sepsis-induced cognitive decline can be exacerbated in individuals with a susceptible brain (Iwashyna et al., 2010). Interestingly, in community-based patients with AD, even mild inflammatory conditions can lead to cognitive decline and disease progression (Holmes et al., 2009). Similarly, in a mouse model of AD, within 24 h after the systemic administration of LPS, cerebral IL-1β and IL-6 were induced, followed by changes in β-amyloid precursor peptide (APP) isoforms similar to those observed in AD patients (Brugg et al., 1995). In the past few years, possible mechanisms of damage have been proposed based on rodent models. In contrast to healthy control mice, scrapie-infected mice – a model of prion disease – show markedly enhanced susceptibility to LPS, leading to altered working memory, synaptic loss, enhanced expression of inflammatory receptors, and microglial activation (Murray et al., 2012). Cognitive and motor coordination are more severely impaired in mice that have been scrapie-infected for longer time periods, and one-time inflammation seems to accelerate disease features of neurodegeneration (Cunningham et al., 2009). Moreover, in prion disease neurodegeneration occurring in response to systemic inflammation is proportional to the burden of preexisting neurodegeneration. In a rat model of toxin-induced Parkinson disease (PD) induced by 6-hydroxydopamine, persistent systemic inflammation induced by IL-1β resulted in a reduction in the number of neurons, as well as an increase in activated microglia in the substantia nigra (SN) (Pott Godoy et al., 2008). The damage to SN in response to inflammation is not limited to susceptible animals. In response to systemic LPS, 8-month-old wild type mice showed a rapid reduction of tyrosine hydroxylase (dopaminergic) neurons, with a reciprocal expansion in activated microglia in the SN occurring shortly thereafter (Reinert et al., 2014). Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by progressive, irreversible, and highly lethal motor neuron damage. In transgenic mice expressing a mutant form of superoxide dismutase 1 (SOD1), an experimental model of ALS, inflammation in the anterior horns of the spinal cord is a hallmark. Inflammation is driven by tissue-resident microglia displaying a neurodegeneration-specific pattern of gradual increase in insulin-like growth factor 1 (Igf1) and the tyrosine kinase receptor AXL (Axl) transcription that closely correlate with disease progression (Chiu et al., 2013b). The activated microglia induce motor neuron death through induction of NF-κB (Frakes et al., 2014). The peripheral nervous system of SOD1 mutant mice also displays inflammation that is proportional to disease progression; in this case, there is invasion of the anterior (motor) roots by circulation-derived active macrophages (Chiu et al., 2009). While spinal cord inflammation may be common in SOD1 mutant mice, those mice display an increased susceptibility to chronic systemic inflammation, leading to disease progression, induction of TLR2, early axonal damage, and accelerated mortality (Nguyen et al., 2004). Although the mechanism for increased susceptibility to systemic inflammation in SOD1 mutant mice is unknown, it is possible that persistent systemic inflammation can further activate microglia and induce the translocation of NF-κB and activation of downstream inflammatory mediators.
Altogether, current evidence indicates that cognitive impairment is common in sepsis; that a susceptible brain is fertile ground for systemic inflammatory-induced dysfunction; and that cognition and behavior are persistently challenged, even after resolution of acute sepsis.
Systemic Lupus Erythematosus: Systemic Autoimmune Inflammation as Driver of Neurodegeneration
Systemic lupus erythematosus (SLE) is a chronic relapsing–remitting autoimmune disease that affects multiple organs and often involves the central nervous system (CNS). It has a high female preponderance and occurs mainly in women of childbearing age. Immunologically, it is characterized by a loss of tolerance to self-antigens and abnormal B- and T-cell responses. Immunoglobulin complexes can deposit in tissue and can cause systemic inflammation. Anti-nuclear antibodies can be found in up to 98% of patients, yet can also be detected in other autoimmune conditions. Neuropsychiatric SLE (NPSLE) is an incompletely understood medical problem, and its clinical picture diverse. Depending on the study, the percentage of patients with neurological and psychiatric involvement can vary from 12 to 95% (Ainiala et al., 2001). CNS involvement indicates a more severe clinical presentation of SLE (Mak et al., 2012). Due to the wide range of motor, sensory, cognitive, and behavioral symptoms, diagnosis of neuropsychiatric symptoms is challenging (Jeltsch-David and Muller, 2014). Memory impairment is linked to neuronal cell death in the hippocampus, and might be caused by antibody mediated cytotoxicity following binding to neuronal cell surface receptors, such as the NMDA receptor (Faust et al., 2010) or neuronal surface P antigen (NSPA) (Bravo-Zehnder et al., 2015). Cognitive impairment in NPSLE has been associated with a higher frequency of hippocampal atrophy (Appenzeller et al., 2006). There is a great set of data demonstrating that neuropsychiatric symptoms could be caused by autoantibodies, cytokines or microvasculopathy, and thrombosis. The most common brain abnormalities in patients include microvasculopathy, which can be caused by antiphospholipid antibodies that bind to clotting factors and endothelial cells (Belmont et al., 1996). In addition to vasculopathy, post-mortem brain studies of patients show infarcts and hemorrhages, cortical atrophy, ischemic demyelination, as well as CNS demyelination (Hanly et al., 1992). Currently, it is not known whether CNS involvement develops independently or occurs as a consequence of systemic organ dysfunction or both. Neurological involvement might be related to treatment, infection, or metabolic disorders or may be part of a coexisting disease. Increased BBB permeability leads to CNS penetration of immunoglobulin, pro-inflammatory cytokines, and albumin. A limited number of studies addressed the genetics of NPSLE, and found an increased susceptibility in patients with brain involvement compared to SLE sparing CNS involvement. Particularly TREX 1 (DNase III) gene mutations have been reported in NPSLE (Table 1; Lee-Kirsch et al., 2007b; de Vries et al., 2010), a mutation that is also found in other brain diseases and is involved in apoptosis and oxidative stress. TREX 1 knockout mice develop severe inflammatory myocarditis, resulting in reduced survival rates (Morita et al., 2004) due to accumulation of single stranded DNA fragments, which facilitates the production of type 1 IFN (Miner and Diamond, 2014). Larger genome wide association studies (GWAS) comparing patients with and without neuropsychiatric involvement are needed to further understand NPSLE. Despite improved imaging and the availability of potential biomarkers (autoantibodies, cytokines, chemokines), NPSLE remains a diagnostic dilemma. So far, no specific treatment is available for NPSLE; however, the CD20 B-cell targeted treatment rituximab was shown to be promising in a small cohort of refractory NPSLE patients (Tokunaga et al., 2007). In addition, peptide mimotopes in patients with anti-brain reactive antibody responses may hold promise (Bloom et al., 2011).
Multiple Sclerosis: Brain Inflammation as Driver of Neurodegeneration
Multiple sclerosis (MS) is the most common inflammatory demyelinating diseases in young adults with a high risk of long-term disability affecting over 2.5 million people worldwide (Compston and Coles, 2008). Despite advances in diagnosis and treatment, the cause of MS remains unknown. While effective treatment for the relapsing–remitting form has improved significantly, treatment for the progressive disease course is still of limited utility (Lassmann et al., 2007). MS is presumably caused by an exogenous trigger in genetically predisposed individuals (Sospedra and Martin, 2005b), resulting in inflammation, demyelination, and neurodegeneration. MS is initiated by autoreactive T-cells against a yet unknown CNS antigen (Sospedra and Martin, 2005a). In addition to T-cells, in the last years, an important role of B-cells and antibodies has re-emerged as major contributors to the disease (Hauser et al., 2008). Experimental autoimmune encephalitis (EAE), the most widely used animal model for studying MS (Friese et al., 2006) is either induced by injection of myelin derived proteins together with adjuvant or by adoptive transfer of CD4+ encephalitogenic T-cells. It has been increasingly recognized that this model does not accurately represent the full spectrum of the human disease. MS is a heterogeneous disease, with patients experiencing a broad range of motor, cognitive, and neuropsychiatric impairment (Chiaravalloti and DeLuca, 2008). Approximately 80% of patients develop relapsing–remitting MS (RRMS), where patients experience relapses that are followed by partial or complete remission (Sospedra and Martin, 2005a). In an advanced disease stage, the majority of RRMS patients convert to secondary progressive MS (SPMS), with a steady disease progression in the absence of relapses and remissions (Sospedra and Martin, 2005b). A minority of patients (15%) suffer from an early onset progressive neurological decline, defined as primary progressive MS (PPMS) (Sospedra and Martin, 2005a,b; Miller and Leary, 2007). PPMS presentation is similar to that of SPMS patients (Confavreux and Vukusic, 2006). Since the progressive disease is not associated with the number of relapses or the extend of inflammation (Friese et al., 2014), it remains unknown what drives neurodegeneration in MS.
So far, the interplay, timing, and localization of inflammation, breakdown of the BBB, demyelination, axonal dysfunction, neurodegeneration, gliosis, atrophy, and repair mechanisms are incompletely understood (Figure 1) (Noseworthy et al., 2000; Bruck, 2005). The pathological hallmark of MS is CNS plaques, which are areas of focal demyelination in the white matter (Popescu and Lucchinetti, 2012). Whereas glial scarring was described as a characteristic feature of demyelinating plaques, most studies used to emphasize an initial preservation of axons. During RRMS, focal lesions, which are disseminated in space and time (McFarland and Martin, 2007), are associated with a BBB breakdown and an infiltration and activation of immune cells (Sospedra and Martin, 2005b). Lesions can be completely or partly resolved due to remyelination and resolution of inflammation. In contrast, the progressive disease stage results in irreversible deficits and is pathologically characterized by chronic axonal degeneration and gliosis. Although demyelination is described as primary event in MS, and neurodegeneration is believed to occur as a secondary event, it is now widely accepted that axonal loss occurs already in acute white matter plaques (Bruck and Stadelmann, 2003). MS was believed to be primarily a white matter disease, yet cortical gray matter lesions and atrophy in the brain and spinal cord can be observed at different time points of the disease, even as early as at the time of diagnosis (Peterson et al., 2001; Bø et al., 2003; Kutzelnigg et al., 2005, 2007; Geurts et al., 2007; Barkhof et al., 2009). In addition, the marker for neuronal integrity N-actetylaspartate (NAA) is decreased throughout the CNS at an early disease stage (De Stefano et al., 2001). NAA levels are also decreased in the normal appearing white matter and in the cortical gray matter. Increased concentrations of the neurofilament protein in the cerebrospinal fluid (CSF) can be observed at all stages of the disease, which additionally reflects early ongoing neurodegeneration (Kuhle et al., 2011).
While there has been major progress in treating RRMS with anti-inflammatory and immunomodulatory treatment, there are currently no effective treatment options for the progressive disease. The inefficiency of anti-inflammatory treatment in PPMS and SPMS was explained by the lack of inflammation during neurodegeneration. However, this could be due to impaired drug delivery to the brain due to a widely restored BBB integrity in the progressive disease stage (Lassmann et al., 2012). In contrast to the traditional view that neurodegeneration in MS occurs in the presence of limited or almost absent inflammation, it has been shown that inflammation is present at all stages of the disease (Frischer et al., 2009). In fact, there is a massive infiltration of microglia and immune cells into gray matter lesions, including the deep gray matter (Lucchinetti et al., 2011). However, chronic lesions in the gray matter show less extend of immune cell infiltration compared to chronic white matter lesions (Popescu and Lucchinetti, 2012). Cortical lesions can be observed even before detection of white matter lesions. Most patients have the highest prevalence of cortical lesions in the progressive disease stage, where diffuse meningeal inflammation correlates with the disease severity, suggesting that cytokines, chemokines, reactive oxygen species (ROS), and glutamate released by meningeal infiltrating immune cells contribute to neurodegeneration by disturbing the neuronal metabolic pathway (Friese et al., 2014). A redistribution of ion channels could be a compensatory effect of the inflamed axon but finally accelerates neurodegeneration.
Overall, the cause and mechanism of neurodegeneration in MS is unresolved, yet it is highly likely that there is a large variability within individual subgroups of MS patients. In some patients, inflammation can be caused by primary neurodegeneration, whereas demyelination can be the primary cause in others, causing axonal transection and thereby initiating Wallerian and retrograde neurodegeneration (Friese et al., 2014). It has been shown that demyelination can lead to axonal degeneration and neuronal loss by limited trophic support of oligodendrocytes (Fünfschilling et al., 2012; Lee et al., 2012). Neurodegeneration in MS can also occur, at least partly, in the absence of demyelination since axonal and neuronal injury in the normal appearing white matter is rather associated with global CNS inflammation but not with white matter demyelination (Friese et al., 2014). The continuous worsening of the disease can be explained by progressive neuronal loss, which cannot be compensated over time by protective repair mechanisms. However, the total lesion load does not correlate with the extent of neurodegeneration, suggesting that neurodegeneration is the result of inflammatory processes in non-lesioned white matter. Alternatively, MS can be a primary neurodegenerative disease rather than an autoimmune disease (Trapp and Nave, 2008). However, genetic findings show no correlation of genetic risk alleles in MS with other neurodegenerative diseases. A role of genetic heritability in MS was supported by early findings, which observed aggregations of MS cases in some families as well as an increased prevalence of MS in monozygotic twins compared to dizygotic twins (Ebers et al., 1995). Genetic susceptibility genes have been described but the disease does not follow a Mendelian inheritance pattern but a rather complex pattern of interaction. The most striking findings regarding genetic susceptibility in MS comes from studies obtained in the 1970s showing an association of MS with alleles of the MHC, particularly HLA-DRB1*15, of the class II gene HLA-DRB1, which is the most important risk allele in MS. GWAS and meta analysis enabled the discovery of many single nucleotide polymorphisms (SNIP) in MS. To date, around 350 MHC and non-MHC loci have been identified (Wang et al., 2011), which are mainly involved in immunological processes (Wang et al., 2011). Some of those risk genes overlap with other autoimmune diseases, whereas other risk genes are unique to MS. Based on the current studies, the genetic risk for MS is not linked to a single gene mutation but rather caused by a complex interplay of many SNIP, which amplify and result in small or moderate risk effects. MS is probably caused by a multifactorial pattern of inheritance, which needs further investigation in individual larger patients groups. Once GWAS studies identify SNIPs in MS patients, there is a great interest to correlate these findings with functional data. Prime examples are the findings of SNIPS in the TNFRSF1A gene that have been associated with a worse clinical outcome in MS patients. This mutation results in a novel soluble splice form of the TNF receptor (TNFR1) that, in contrast to the membrane bound form, lacks NF-κB activity and apoptotic activity but can block the function of TNF and thus mimics anti TNF therapies, which exacerbate MS (Gregory et al., 2012).
So far, most genetic studies focus on genes responsible for the initiation of the disease rather than on genes influencing the severity of the disease (Friese et al., 2014). Thus, future studies are needed to identify genes that trigger the progression of the disease in the presence of inflammation. One study showed that meningeal inflammation is associated with small fiber axonal loss in the spinal cord of patients that were HLA-DRB1*15 positive, correlating neurodegeneration with increased genetic susceptibility (DeLuca et al., 2013). Another study demonstrated higher glutamate levels in the brain of MS patients harboring certain risk alleles for genes involved in the glutamate pathway (Baranzini et al., 2010). A polymorphism of the inositol polyphosphate-4-phosphatase, type II (Inpp4b), which was described for MS results in decreased nerve conduction velocity, which could aggravate the disease (Lemcke et al., 2014). More GWAS studies are needed to confirm these results and there should be a particular focus on polymorphisms associated with the progressive disease stage. In addition to genetic susceptibility genes, several environmental factors have been suggested to trigger the disease, such as vitamin D, smoking, Epstein–Barr virus infection, and geographical location in relation to latitude gradient (Ascherio and Marrie, 2012).
Mechanisms of Neurodegeneration: Systemic Inflammation Drives Disruption of Brain Networks
Biological systems, such as the neuronal network of the human brain (Sporns et al., 2005; Achard et al., 2006; Bullmore and Sporns, 2009) have “small-world” properties (Watts and Strogatz, 1998). Small-world networks have two levels of organization (Figure 2). On the local level, groups of neurons specialized in a specific task form functional modules with high short intramodular connectivity. On the global level, different modules are connected through long intermodular connections. The advantage of the latter type of connections is enhanced computational efficiency through parallel processing of information (Watts and Strogatz, 1998; Latora and Marchiori, 2001; Bullmore and Sporns, 2009). Anatomically, long intermodular connections are formed by axonal fiber tracts in the white matter (Bullmore and Sporns, 2009; Toga et al., 2012). Long fibers are characterized by high energetic “wiring costs” (Bullmore and Sporns, 2012). To provide the energy for the maintenance of these long fibers the brain is relying on a constant energy supply. Recent findings have elegantly identified oligodendrocyte-derived lactate as the main energetic substrates for axonal maintenance (Fünfschilling et al., 2012). Consistently, disruption of this oligodendrocyte-neuronal metabolic coupling triggered neurodegeneration (Lee et al., 2012). Systemic inflammation poses dramatic challenges to the energetic supply of the brain.
To cover its wiring costs the brain is highly reliant on a constant nutrient supply. Nutrient supply through blood vessels can be compromised through vascular pathologies associated with systemic inflammation. During severe sepsis, disseminated intravascular coagulation leads to diffuse intravascular formation of thrombi and hemorrhages due to depletion of coagulation factors. The consequences are diffuse ischemic foci throughout the body and dysfunction of affected organs. Infarctions or hemorrhages occurring in the course of long connecting tracts in the brain lead to disconnection of distant brain regions and reduced efficiency of neuronal networks. These changes might contribute to white matter lesions observed in MRI studies of acute sepsis cases (Sharshar et al., 2007) and sepsis survivors (Morandi et al., 2012; Semmler et al., 2013). Mitochondrial dysfunction is another complication of sepsis affecting brain function. Despite normal or even increased tissue oxygen availability (Boekstegers et al., 1991), oxygen utilization is drastically reduced in critically ill patients leading to multi-organ failure and mortality (Brealey et al., 2002). Brain mitochondrial dysfunction was shown in rodent models of sepsis (d’Avila et al., 2008). This hibernation-like metabolic state was proposed as a physiological protective tissue reaction (Singer et al., 2004). However, mitochondrial dysfunction might persist in sepsis survivors (Comim et al., 2011). The cause for mitochondrial dysfunction might be a combination between induction of ROS during sepsis (Wong et al., 1996) and impaired mitochondrial turnover (Singer, 2014). Taken together, the complicated interrelation between focal hypoperfusion and reduced oxygen utilization lead to a complex acute and chronic phenotype referred to as sepsis-associated encephalopathy (Gofton and Young, 2012; Sonneville et al., 2013).
Autoimmune disorders have a chronic course of vascular pathology with acute flares. The most common vascular pathology is the autoantibody-associated antiphospholipid syndrome (Ben Salem, 2013; Giannakopoulos and Krilis, 2013; Million and Raoult, 2013). Patients with antiphospholipid syndrome display cognitive deficits (Gómez-Puerta et al., 2005; Tektonidou et al., 2006). MRI studies found diffuse infarctions and white matter lesions in these patients (Gómez-Puerta et al., 2005; Tektonidou et al., 2006; Valdés-Ferrer et al., 2008). The role of mitochondrial dysfunction in autoimmune diseases is not clear. In SLE, mitochondrial dysfunction and ATP depletion were shown as triggers of lymphocytic cell death (Gergely et al., 2002). An interesting view of mitochondrial dysfunction in the context of MS was recently proposed (Lassmann, 2011). In line with the concept of high “wiring costs” imposed on the brain by long intermodular connections (Bullmore and Sporns, 2012), Hans Lassmann argues that inflammation in MS causes mitochondrial damage and inability of the brain to maintain neuronal processes. The source of mitochondrial damage is radicals formed as a consequence of inflammation in MS (Lassmann, 2011; Fischer et al., 2012). Disruption of neuron–glia metabolic coupling might be another potential mechanism causing neurodegeneration and network disruption (Allaman et al., 2011; Lee et al., 2012).
Taken together these findings indicate that systemic inflammation leads to an energy crisis of the brain that reduces its connectivity. Oxidative stress might be the main mediator of this pathology. Thus, inflammation-induced changes in the brain resemble hallmarks of the aged brain where oxidative damage leads to decreased expression of genes associated with synaptic plasticity and increased expression of stress-response genes (Lu et al., 2004). Likewise, the brain during systemic inflammation shows hallmarks of neurodegenerative diseases where oxidative stress and mitochondrial damage have consistently been found (Lin and Beal, 2006). Studies with bigger sample sizes are needed to identify common mechanisms between systemic inflammation, brain aging, and neurodegeneration.
Systemic Inflammation-Associated Immunopathology Irreparably Damages the Architecture of the Brain
With approximately 90 billion neuronal and non-neuronal cells, respectively (Azevedo et al., 2009), the human brain is characterized by high architectural complexity (Braitenberg and Schüz, 1991). Due to this complexity, the repair capacity is limited rendering the human brain highly susceptible to tissue damage. As primary preventive measures the brain is protected from different modes of tissue damage; for example, by the skull bone from mechanical damage or by the BBB from blood-borne pathogens. The BBB is mainly formed by endothelial cells and astrocytes (Abbott et al., 2006; Weiss et al., 2009); the formation of tight junctions between endothelial cells forms a highly selective barrier that becomes more permeable during systemic inflammation (McColl et al., 2008; Weiss et al., 2009). A third kind potential source of brain tissue damage is the immune system itself. As pathogen defense is invariably associated with host tissue damage (Graham et al., 2005) an anti-inflammatory milieu preserves the brain from aberrant immune activation. Under physiological conditions, astrocytes and neurons actively modulate the activation of brain immune cells (Neumann, 2001; Tian et al., 2012). Through this cross-talk brain cells can actively recruit immune cells for purposes of brain homeostasis such as synaptic plasticity, induction of inflammation, clearance of debris, and resolution of inflammation.
Brain-resident microglia and peripheral immune cells maintain immune surveillance of brain parenchyma, CSF, and perivascular space for infectious agents or damage-associated milieu changes (Ousman and Kubes, 2012; Ransohoff and Engelhardt, 2012). In the case of brain infection, complete eradication of some invading pathogens can only be achieved at the cost of irreparable damage to brain tissue. To prevent such damage, the immune system has established active mechanisms of pathogen tolerance (Medzhitov et al., 2012). Examples for coexistence-prone pathogens are herpes simplex virus type I (Khanna et al., 2004) or Cryptococcus gattii (Cheng et al., 2009). A growing body of evidence indicates that not only immune tolerance but also resolution of neuroinflammation is a tightly regulated active immunological process (Schwartz and Baruch, 2014). Taken together, anti-inflammatory brain milieu, pathogen tolerance, and resolution of neuroinflammation require a balanced action between different branches of the immune system. An imbalance within the immune system leading to systemic inflammation may be a driver of neurodegeneration through the mechanisms discussed below.
Caspases as Mediators of Inflammation and Neurodegeneration
Apoptosis is one of the main drivers of neurodegeneration. Apoptosis and cell death constantly occur under physiological conditions throughout the human body and cell debris is cleared by immune cells mostly without induction of chronic inflammation (Green et al., 2009). However, during systemic inflammation, apoptosis of stressed cells might further exacerbate the underlying pathology (Zitvogel et al., 2010). Activators of apoptosis lead to direct or indirect activation of caspases. Interestingly, caspases are not only classical executors of apoptosis (Friedlander, 2003) but also inflammatory caspases are crucial for the activation of the innate immune system through the inflammasome (Martinon and Tschopp, 2004). Caspase-1, as the cleaving enzyme for IL-1β and constituent of the inflammasome, is the prototypic representative of the latter class of caspases (Martinon et al., 2002).
Activation of the innate immune system through the inflammasome is a driver of pathology in age-associated and autoimmune neurodegenerative disorders. In AD, the NLRP3 inflammasome was described a sensor of β-amyloid (Heneka et al., 2013). Of interest, deletion of Caspase-1 or NLRP3 rescued the phenotype in APP/PS1 AD transgenic mice (Heneka et al., 2013). Consistently with that the deletion of the inflammasome scavenger receptor CD36 (Sheedy et al., 2013) ameliorated pathology in the Tg2576 AD mouse model (Park et al., 2013). MS-like lesions were found in humans with mutations of proteins associated with the inflammasome (Compeyrot-Lacassagne et al., 2009). Additionally, NLRP3 and IL-1 knockout mice had decreased pathology following EAE (Matsuki et al., 2006; Gris et al., 2010). Approved clinical treatment options of MS such as IFN β (Guarda et al., 2011) or glatiramer acetate (Burger et al., 2009) have been shown to decrease IL-1β levels, the main cytokine processed by the inflammasome. Additionally, novel evidence extends the functions of the classical apoptotic caspases linking neuroinflammation to neurodegeneration. Activation of microglial caspase-8, -3, and -7 can drive neurodegeneration (Burguillos et al., 2011). Finally, recent evidence from C. elegans has described protective effects through mediators of the intrinsic apoptosis pathway against ROS (Yee et al., 2014). Taken together, these finding show an intricate relationship between inflammation and activation of apoptosis. However, the definite role of these mediators in the brain remains to be characterized.
Microglia- and Macrophage-Derived Microvesicles as Inducers of Neurodegeneration
Cellular components of innate immunity can pack and secrete inflammatory messengers in microvesicles (MVs). Peripheral macrophages, as well as brain microglia can secrete inflammasome components (caspase-1, IL-1β, and IL-18) in MVs, and the presence of extravesicular inflammatory inducers (e.g., astrocitic ATP) is sufficient to induce the neurotoxicity by the inflammatory load of MVs (Bianco et al., 2005; Sarkar et al., 2009; Gulinelli et al., 2012). Although MVs are visible in the CSF in healthy controls, the load of MVs in RRMS, neuromyelitis optica (NMO), brain infections, and brain tumors is significantly increased and, in MS, correlates with disease activity (Verderio et al., 2012). Recent evidence suggest that MVs play a critical role in the spectrum of AD as well. MVs released by activated microglia participate in the neurodegenerative process of AD by promoting the generation of highly neurotoxic soluble forms of β-amyloid (Joshi et al., 2014). Based on this collective evidence, it is now clear that EVs produced by peripheral myeloid cells, as well as immune brain cells, are novel and potentially critical biomarkers for neuroinflammatory conditions by providing a link between inflammation and neurodegeneration.
Immune Cells and Immune Mediators as Drivers of Neurodegeneration
Various triggers of apoptosis have been described with respect to the brain. Neuronal apoptosis can be directly induced by ROS, pro-inflammatory cytokines or activated immune cells. The consequences of ROS-induced mitochondrial damage on brain metabolism have been discussed above. Additionally, damaged mitochondria are a major source of ROS (Rego and Oliveira, 2003) and mediators of apoptosis (Green and Kroemer, 2004). Conversely, inactivation of ROS has anti-apoptotic effects (Hockenbery et al., 1993; Greenlund et al., 1995). The inflammatory cytokine TNFα (Tamatani et al., 1999; Kaur et al., 2014) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (Aktas et al., 2005) directly induce neuronal apoptosis (Figure 3). Additionally, intracerebroventricularly injected TNFα was shown to induce depression-like symptoms (Kaster et al., 2012). Cytokine mediated induction of apoptosis was also observed by IL-1β (Wang et al., 2005; Kaur et al., 2014). Sources of cytokines under systemic inflammation are brain resident, paravascular or peripheral immune cells (Dantzer et al., 2008). Furthermore, activated immune cells can directly induce neuronal cell death (Figures 1 and 3). Brain-resident microglia convey neuronal toxicity through various mechanisms including secretion of neurotoxic factors (Liu and Hong, 2003; Block et al., 2007), as well as through activation of cyclooxygenase/prostaglandin E2 (COX/PGE2) pathways (Montine et al., 1999; Liang et al., 2005). In fact, blocking the COX/PGE2 pathway by experimentally deleting the prostaglandine receptor EP2 increases mitochondrial degradation of β-amyloid, potentially opening a new therapeutic avenue for AD (Johansson et al., 2015).
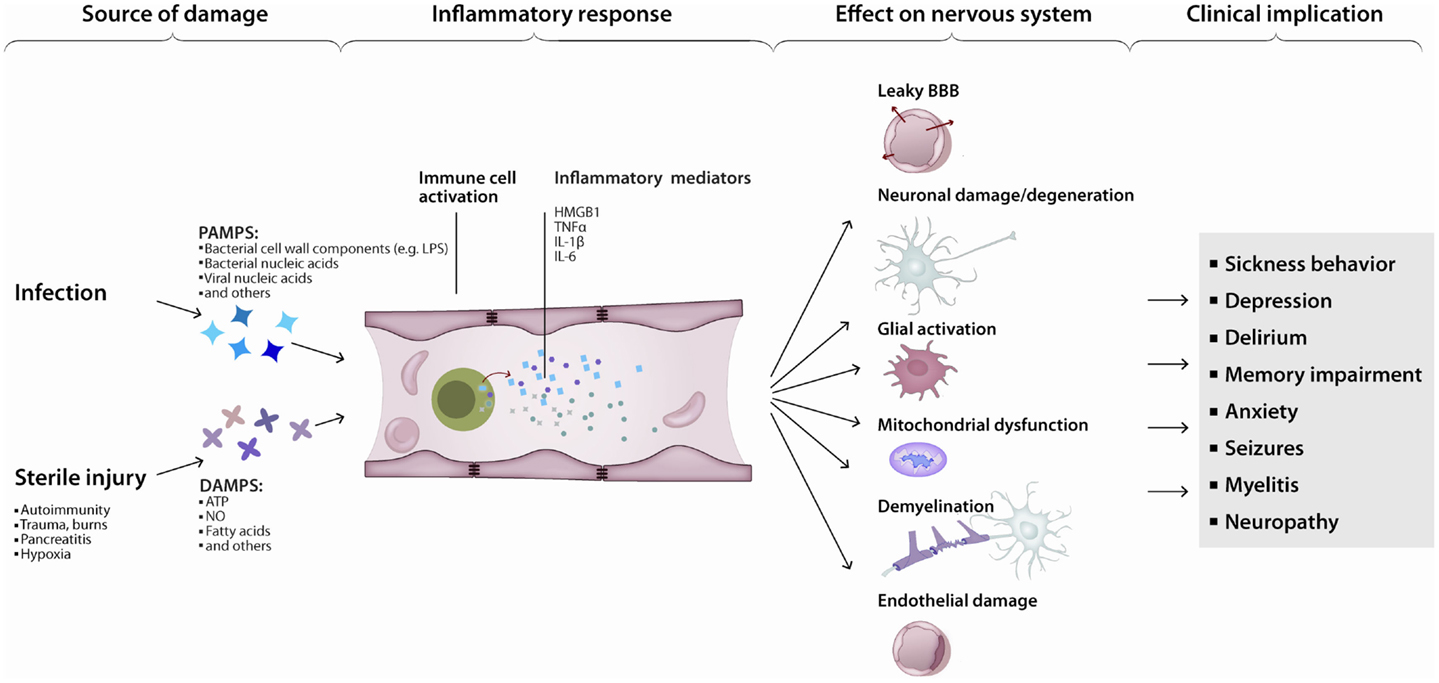
Figure 3. Inflammation leads to neurodegeneration: a simplified model. Pathogen- or damage-derived antigens released in sufficient quantity activate systemic inflammation. In turn, peripheral (e.g., monocytes) as well as central (e.g., microglia) immune cells activate, increasing the production and release of inflammatory cytokines, chemokines, and other immunologically active peptides. Those mediators can induce neuronal dysfunction directly or indirectly, by interfering with neuronal homeostasis or disrupting the neuronal milieu. The end-result is a continuum of clinical manifestations from local and transient, to diffuse and persistent.
Peripheral immune cells can penetrate the BBB under conditions of systemic inflammation (Schmitt et al., 2012) and contribute to brain pathology (Rezai-Zadeh et al., 2009) (Figure 3). Cytotoxic T-cells were shown to be directly neurotoxic in autoimmune and aging-associated neurodegenerative disorders of the CNS (Neumann et al., 2002). Co-localization of T-cells with neurons and neuron-specific cytotoxicity of T-cells was shown in vivo and in vitro (Giuliani et al., 2003; Nitsch et al., 2004). The pivotal role of pathogenic T-cells in MS is suggested by the fact that adoptive transfer of myelin reactive T-cells is sufficient to induce EAE in rodents. Finally, B-cells have multiple roles in MS (Krumbholz et al., 2012). On the one hand, B-cells can serve as antigen presenting cells for T-cells and B-cell derived cytokines can activate pathogenic T-cells (Schneider et al., 2011). On the other hand, intrathecal IgG synthesis and persistent oligoclonal bands in the CSF are a hallmark finding in MS. Furthermore, antibodies and complement deposition is present in acute MS lesions (Lucchinetti et al., 2000). B-cells were shown to mature in the draining cervical lymph nodes and migrate to the brain (Stern et al., 2014). With advancing pathology, B-cells can be found in serum, brain parenchyma, and meninges of patients (Serafini et al., 2004; Kuerten et al., 2014); and B-cell depletion was shown to be beneficial in a subgroup of MS patients (Hauser et al., 2008).
Anti-Brain Antibodies as Drivers of Neurodegeneration
B-cell-derived anti-brain antibodies have been identified as drivers of brain pathology in various diseases. In the last decade, an increasing number of anti-brain antibodies has been detected that can affect cognition and behavior (Diamond et al., 2013). For many of these newly discovered antibodies their frequency in disease and involvement in pathogenesis has not yet been determined. Although anti-brain antibodies can be present in around 5% of healthy individuals (Diamond et al., 2013), an intact BBB restricts entry of antibodies into the brain. Under pathological conditions, antibodies may penetrate the BBB through different mechanisms including local and systemic inflammation, or antigen mediated endocytosis (Diamond et al., 2013). In addition, fenestrated endothelial cells of the circumventricular organs, which lack the tight junction of the BBB, may facilitate entry of antibodies into the brain parenchyma (Popescu et al., 2011); or antibodies may be produced intrathecally by B-cells, which migrate into the CNS.
So far, few antibodies have been confirmed to be directly neurotoxic. The most striking evidence is provided by tumor-associated autoantibodies causing paraneoplastic neurological disorders. These antibodies are cross-reactive to neuronal antigens expressed on cancer cells; neuronal pathology is induced through neuronal cell death after antibody binding to neuronal antigens. Clinically, patients present with severe neuropsychiatric symptoms. Rapid removal of paraneoplastic antibodies and surgical removal of the tumor can prevent further neuronal cell death und may reverse neurological pathology. Patients that harbor an antibody response to intracellular antigens have a worse response to treatment compared to patients with antibodies to extracellular neuronal autoantigens. This could be due to an involvement of pathogenic T-cells, which cause irreversible neuronal cell death (Lancaster and Dalmau, 2012).
Anti-NMDA receptor encephalitis is the most common paraneoplastic disorder, with ovarian teratomas as the underlying malignancy in approximately 50% of the patients (Dalmau et al., 2011). These antibodies bind the extracellular domain of the NMDA receptor subunit 1 (GluN1) and cause psychiatric symptoms such as anxiety, memory loss, and psychosis (Dalmau et al., 2007). Binding of antibodies results in reduced levels of NMDA receptor in vitro and in vivo through antibody mediated capping, crosslinking, and internalization of the receptor (Hughes et al., 2010), which can be reversed upon removal of the antibodies (Seki et al., 2008). Interestingly, patients with high titers of NMDA receptor antibodies have lower levels of the NMDA receptors in postsynaptic dendrites (Dalmau et al., 2008). Direct injection of the antibodies in the cortex of rodents results in excitotoxicity by increased glutamate release (Hughes et al., 2010). A recent study found NMDA receptor antibodies in patients with demyelinating disorders indicating that these antibodies might cause neuropsychiatric symptoms in these patients (Titulaer et al., 2014).
Furthermore, NMDA-receptor-specific antibodies to the subunit 2 (GluN2) have been found in a subset of SLE patients with neuropsychiatric symptoms. These antibodies are cross-reactive to DNA and bind the consensus sequence D/E W D/E Y S/G (DWEYS) present in the extracellular domains of GluN2A and GluN2B subunits of the NMDA receptor (Gaynor et al., 1997). DNA–NMDA receptor antibodies preferentially bind the open configuration of the NMDA receptor and augment NMDA receptor-mediated excitatory postsynaptic potentials (Faust et al., 2010). DNA–NMDA receptor antibodies have been found in serum, CSF, and brain tissue of SLE patients (DeGiorgio et al., 2001; Kowal et al., 2006). Notably, patient derived DNA–NMDA receptor antibodies cause hippocampal neuronal loss and persistent memory impairment in rodents (Kowal et al., 2006). These findings indicate that DNA–NMDA receptor antibodies are directly involved in neurodegenerative processes in SLE. Hippocampal brain abnormalities in NPSLE patients further support this notion (Appenzeller et al., 2006). Depending on the antibody concentrations, DNA–NMDA receptor antibodies can cause either neuronal dysfunction by transiently enhancing excitatory postsynaptic potentials or can result in neuronal cell death (Kowal et al., 2006; Faust et al., 2010). This evidence could be of high relevance in terms of reversibility of symptoms. Furthermore, anti-brain antibodies were also shown to induce neuropsychiatric symptoms in patients with other autoimmune disorders such as celiac disease (Alaedini et al., 2007; Hadjivassiliou et al., 2013) or inflammatory bowel diseases (Häuser et al., 2011; Papathanasiou et al., 2014). Taken together, anti-brain antibodies were shown to cause neuropsychiatric pathology in different diseases presenting novel therapeutic options.
These promising findings of anti-brain antibodies in systemic inflammatory disorders encouraged the search for anti-brain antibodies in inflammatory brain disorders. MS is characterized by oligoclonal bands in the CSF and antibodies in acute MS lesions (Lucchinetti et al., 2000). Histopathological studies confirm antibody mediated demyelination (Storch et al., 1998). Despite these findings all attempts to identify pathogenic antibodies to CNS antigens and infectious agents in the context of MS were rather unsatisfactory. In contrast to MS, highly specific anti-brain antibodies were discovered in NMO (Lennon et al., 2005), a disease which can closely resemble MS but requires different treatment. Antibodies to the water channel protein aquaporin-4 (AQP4) are detectable in around 80–90% of patients with NMO (Mader et al., 2010; Waters et al., 2012). AQP4 is localized on astrocytic endfeet forming the BBB. NMO is characterized by optic neuritis and longitudinal extensive transverse myelitis over three or more vertebral segments, which can lead to blindness and paralysis of patients (Wingerchuk et al., 1999; Cree, 2008). AQP4 antibody seropositivity is highly predictive for the disease (Matiello et al., 2008) and enables early treatment of patients. This is particularly important since IFN β, commonly used for treating MS, can worsen the disease outcome of NMO patients (Palace et al., 2010). Pathological findings show immunoglobulin and complement deposition around blood vessels with AQP4 specific loss in brain and spinal cord lesions (Lucchinetti et al., 2002). Rodents develop NMO-like lesions in the brain and spinal cord upon injection of patient derived AQP4 antibodies in an EAE animal model (Bennett et al., 2009; Bradl et al., 2009; Kinoshita et al., 2009). In addition, one study showed loss of astrocytes and demyelinating lesions after injection of AQP4 IgG into the brain together with human complement (Saadoun et al., 2010). AQP4 antibodies might require the help of encephalitogenic T-cells to breach the BBB (Saadoun et al., 2010; Pohl et al., 2013) and in addition CNS specific T-cells may require a local inflammatory environment for the antibodies at the lesion site (Pohl et al., 2013). Antibodies to AQP4 bind to astrocytes and lead to complement depend cytotoxicity as well as antibody dependent cytotoxicity, resulting in astrocytosis (Papadopoulos et al., 2014). Demyelination and neuronal cell death could occur as a secondary inflammatory response, due to secretion of toxic compounds such as nitrogen species, reactive oxygen or glutamate by activated astrocytes (Brosnan and Raine, 2013), which could lead to limited trophic support to myelin (Levy, 2014). Recently, cognitive impairment has been reported in NMO (Saji et al., 2013), yet this finding needs to be replicated in larger cohorts. The pathological role of AQP4 antibodies in cortical neuronal loss at areas of high AQP4 expression is not well understood. Although NMO is a rather rare disease, the findings obtained from AQP4 IgG and its contribution in glial injury, demyelination and neurodegeneration could be of direct relevance for MS and related inflammatory diseases, and will help to better understand the role of astrocytes in inflammation and neurodegeneration.
Immune-Mediated Disruption of the Neurogenic Niche may Contribute to Neurodegeneration
A final mechanism potentially connecting systemic inflammation and neurodegeneration is impairment of neurogenesis. Neurogenesis is a central mechanism required for neuronal maintenance and adaptive plasticity in the healthy and diseased brain (Jin et al., 2006a). Inflammatory mediators have various effects on neurogenesis (Whitney et al., 2009). Impairment of neurogenesis was shown in neurodegenerative diseases such as AD (Lazarov and Marr, 2010) and neuropsychiatric disorders such as depression (Sahay and Hen, 2007). Interestingly, approved AD drugs (Jin et al., 2006b; Kotani et al., 2008) and chronic antidepressant treatment (Malberg et al., 2000; Santarelli et al., 2003) induce neurogenesis. Inflammation and microglial activation is detrimental for neurogenesis that can be restored by anti-inflammatory treatment (Ekdahl et al., 2003; Monje et al., 2003). Moreover, microglia are not only involved in the maintenance of the neurogenic niche (Sierra et al., 2010) but also in synaptic maintenance (Stevens et al., 2007; Parkhurst et al., 2013). Of interest, systemic immune cells were shown to be involved in regulation of neurogenesis. CD4+ T-cells were shown to promote (Wolf et al., 2009) while CD8+ T-cells impair proliferation of neural progenitor cells (Hu et al., 2014). An effect of B-cells on neurogenesis was not observed (Wolf et al., 2009). These findings need to be independently replicated. However, one may speculate that neuropsychiatric symptoms elicited by chronic inflammation may be driven by detrimental changes of neuronal homeostasis. Thus, specific immune modulatory treatment might be beneficial.
Concluding Remarks
The immune and nervous systems have co-evolved from early invertebrates to higher mammals, creating an intricate bi-directional modulating dialog. The CAP is a good example of the influence of the nervous system upon the immune response. In the opposite direction, the activation of innate immunity in response to serious, as well as non-life threatening, infections induces the maturation and release of TNF, IL-1β, and other inflammatory cytokines that in turn cause transient anorexia, malaise, depression, and other features of the sickness syndrome (Figure 3). This is not surprising if we take into consideration that glia constitutes no less than half of the cells in a mammalian brain.
Sustained systemic inflammation is a common feature of many autoimmune disorders, and is present in most sepsis survivors. Cognitive impairment is common in sepsis survivors, as well as patients suffering from chronic inflammatory conditions. Cognition and behavior are persistently challenged, even after apparent resolution of acute sepsis. Moreover, systemic inflammation occurring in a susceptible brain (e.g., patients with AD) may lead to even further disruption in quality of life and activities of daily living. Up to 95% of patients with SLE develop neuropsychiatric dysfunction. In SLE, part of the repertoire of DNA-binding autoantibodies cross-react with hippocampal NMDA receptors, and – through a leaky BBB – gain access to the brain, inducing cognitive decline and other neuropsychiatric manifestations. In patients with rheumatoid arthritis, the baseline vagal tone of is persistently low, suggesting a possible mechanism for persistent inflammation. Those examples indicate that the normal neuroimmune cross-talk in health can become deleterious during disease, particularly in a primed brain – one with preexistent damage. Recently, cellular, molecular, environmental, and genetic components have been linked to the persistent brain disfunction of systemic inflammation. Here, we have discussed mechanistic evidence for the intricate interrelation between inflammation and neurodegeneration. Identification of druggable targets derived from these mechanisms holds the promise to prevent long-term disability and improve the quality of life in patients with chronic inflammatory conditions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Simone Mader work was supported by S.L.E. Lupus Foundation. The authors want to thank Benjamin Obholzer for the image design.
References
Abbott, N. J., Rönnbäck, L., and Hansson, E. (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53. doi:10.1038/nrn1824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Achard, S., Salvador, R., Whitcher, B., Suckling, J., and Bullmore, E. (2006). A resilient, low-frequency, small-world human brain functional network with highly connected association cortical hubs. J. Neurosci. 26, 63–72. doi:10.1523/JNEUROSCI.3874-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ainiala, H., Loukkola, J., Peltola, J., Korpela, M., and Hietaharju, A. (2001). The prevalence of neuropsychiatric syndromes in systemic lupus erythematosus. Neurology 57, 496–500. doi:10.1212/WNL.57.3.496
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aktas, O., Smorodchenko, A., Brocke, S., Infante-Duarte, C., Schulze Topphoff, U., Vogt, J., et al. (2005). Neuronal damage in autoimmune neuroinflammation mediated by the death ligand TRAIL. Neuron 46, 421–432. doi:10.1016/j.neuron.2005.03.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alaedini, A., Okamoto, H., Briani, C., Wollenberg, K., Shill, H. A., Bushara, K. O., et al. (2007). Immune cross-reactivity in celiac disease: anti-gliadin antibodies bind to neuronal synapsin I. J. Immunol. 178, 6590–6595. doi:10.4049/jimmunol.178.10.6590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Allaman, I., Bélanger, M., and Magistretti, P. J. (2011). Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 34, 76–87. doi:10.1016/j.tins.2010.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anderson, S. T., Commins, S., Moynagh, P. N., and Coogan, A. N. (2015). Lipopolysaccharide-induced sepsis induces long-lasting affective changes in the mouse. Brain Behav. Immun. 43, 98–109. doi:10.1016/j.bbi.2014.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andersson, U., and Tracey, K. J. (2011). HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29, 139–162. doi:10.1146/annurev-immunol-030409-101323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andersson, U., and Tracey, K. J. (2012). Neural reflexes in inflammation and immunity. J. Exp. Med. 209, 1057–1068. doi:10.1084/jem.20120571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Appenzeller, S., Carnevalle, A. D., Li, L. M., Costallat, L. T., and Cendes, F. (2006). Hippocampal atrophy in systemic lupus erythematosus. Ann. Rheum. Dis. 65, 1585–1589. doi:10.1136/ard.2005.049486
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ascherio, A., and Marrie, R. A. (2012). Vitamin D in MS: a vitamin for 4 seasons. Neurology 79, 208–210. doi:10.1212/WNL.0b013e31825fe131
Azevedo, F. A., Carvalho, L. R., Grinberg, L. T., Farfel, J. M., Ferretti, R. E., Leite, R. E., et al. (2009). Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 513, 532–541. doi:10.1002/cne.21974
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baker, M., Mackenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. doi:10.1038/nj7102-596a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baranzini, S. E., Srinivasan, R., Khankhanian, P., Okuda, D. T., Nelson, S. J., Matthews, P. M., et al. (2010). Genetic variation influences glutamate concentrations in brains of patients with multiple sclerosis. Brain 133, 2603–2611. doi:10.1093/brain/awq192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barichello, T., Machado, R. A., Constantino, L., Valvassori, S. S., Réus, G. Z., Martins, M. R., et al. (2007). Antioxidant treatment prevented late memory impairment in an animal model of sepsis. Crit. Care Med. 35, 2186–2190. doi:10.1097/01.CCM.0000281452.60683.96
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barichello, T., Martins, M. R., Reinke, A., Feier, G., Ritter, C., Quevedo, J., et al. (2005). Cognitive impairment in sepsis survivors from cecal ligation and perforation. Crit. Care Med. 33, 221–223. doi:10.1097/01.CCM.0000150741.12906.BD
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barkhof, F., Calabresi, P. A., Miller, D. H., and Reingold, S. C. (2009). Imaging outcomes for neuroprotection and repair in multiple sclerosis trials. Nat. Rev. Neurol. 5, 256–266. doi:10.1038/nrneurol.2009.41
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belmont, H. M., Abramson, S. B., and Lie, J. T. (1996). Pathology and pathogenesis of vascular injury in systemic lupus erythematosus. Interactions of inflammatory cells and activated endothelium. Arthritis Rheum. 39, 9–22. doi:10.1002/art.1780390103
Ben Salem, C. (2013). The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med. 368, 2334. doi:10.1056/NEJMc1304515
Bennett, J. L., Lam, C., Kalluri, S. R., Saikali, P., Bautista, K., Dupree, C., et al. (2009). Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 66, 617–629. doi:10.1002/ana.21802
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bernik, T. R., Friedman, S. G., Ochani, M., DiRaimo, R., Ulloa, L., Yang, H., et al. (2002). Pharmacological stimulation of the cholinergic antiinflammatory pathway. J. Exp. Med. 195, 781–788. doi:10.1084/jem.20011714
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bertram, L., Lange, C., Mullin, K., Parkinson, M., Hsiao, M., Hogan, M. F., et al. (2008). Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83, 623–632. doi:10.1016/j.ajhg.2008.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bianco, F., Pravettoni, E., Colombo, A., Schenk, U., Möller, T., Matteoli, M., et al. (2005). Astrocyte-derived ATP induces vesicle shedding and IL-1β release from microglia. J. Immunol. 174, 7268–7277. doi:10.4049/jimmunol.174.11.7268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi:10.1038/nrn2038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bloom, O., Cheng, K. F., He, M., Papatheodorou, A., Volpe, B. T., Diamond, B., et al. (2011). Generation of a unique small molecule peptidomimetic that neutralizes lupus autoantibody activity. Proc. Natl. Acad. Sci. U.S.A. 108, 10255–10259. doi:10.1073/pnas.1103555108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bø, L., Vedeler, C. A., Nyland, H. I., Trapp, B. D., and Mørk, S. J. (2003). Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J. Neuropathol. Exp. Neurol. 62, 723–732.
Boardman, J. P., Counsell, S. J., Rueckert, D., Kapellou, O., Bhatia, K. K., Aljabar, P., et al. (2006). Abnormal deep grey matter development following preterm birth detected using deformation-based morphometry. Neuroimage 32, 70–78. doi:10.1016/j.neuroimage.2006.03.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boekstegers, P., Weidenhöfer, S., Pilz, G., and Werdan, K. (1991). Peripheral oxygen availability within skeletal muscle in sepsis and septic shock: comparison to limited infection and cardiogenic shock. Infection 19, 317–323. doi:10.1007/BF01645355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Borovikova, L. V., Ivanova, S., Zhang, M., Yang, H., Botchkina, G. I., Watkins, L. R., et al. (2000). Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458–462. doi:10.1038/35013070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bradl, M., Misu, T., Takahashi, T., Watanabe, M., Mader, S., Reindl, M., et al. (2009). Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann. Neurol. 66, 630–643. doi:10.1002/ana.21837
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bradshaw, E. M., Chibnik, L. B., Keenan, B. T., Ottoboni, L., Raj, T., Tang, A., et al. (2013). CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850. doi:10.1038/nn.3435
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Braitenberg, V., and Schüz, A. (1991). Anatomy of the Cortex: Statistics and Geometry. Berlin: Springer-Verlag.
Bravo-Zehnder, M., Toledo, E. M., Segovia-Miranda, F., Serrano, F. G., Benito, M. J., Metz, C., et al. (2015). Anti-ribosomal P protein autoantibodies from neuropsychiatric lupus impair memory. Arthritis Rheumatol. 67, 204–214. doi:10.1002/art.38900
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brealey, D., Brand, M., Hargreaves, I., Heales, S., Land, J., Smolenski, R., et al. (2002). Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360, 219–223. doi:10.1016/S0140-6736(02)09459-X
Brosnan, C. F., and Raine, C. S. (2013). The astrocyte in multiple sclerosis revisited. Glia 61, 453–465. doi:10.1002/glia.22443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bruck, W. (2005). The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J. Neurol. 252(Suppl. 5), v3–v9. doi:10.1007/s00415-005-5002-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bruck, W., and Stadelmann, C. (2003). Inflammation and degeneration in multiple sclerosis. Neurol. Sci. 24(Suppl. 5), S265–S267. doi:10.1007/s10072-003-0170-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brugg, B., Dubreuil, Y. L., Huber, G., Wollman, E. E., Delhaye-Bouchaud, N., and Mariani, J. (1995). Inflammatory processes induce beta-amyloid precursor protein changes in mouse brain. Proc. Natl. Acad. Sci. U.S.A. 92, 3032–3035. doi:10.1073/pnas.92.7.3032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bullmore, E., and Sporns, O. (2009). Complex brain networks: graph theoretical analysis of structural and functional systems. Nat. Rev. Neurosci. 10, 186–198. doi:10.1038/nrn2575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bullmore, E., and Sporns, O. (2012). The economy of brain network organization. Nat. Rev. Neurosci. 13, 336–349. doi:10.1038/nrn3214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burger, D., Molnarfi, N., Weber, M. S., Brandt, K. J., Benkhoucha, M., Gruaz, L., et al. (2009). Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1beta in human monocytes and multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 106, 4355–4359. doi:10.1073/pnas.0812183106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burguillos, M. A., Deierborg, T., Kavanagh, E., Persson, A., Hajji, N., Garcia-Quintanilla, A., et al. (2011). Caspase signalling controls microglia activation and neurotoxicity. Nature 472, 319–324. doi:10.1038/nature09788
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Capuron, L., Lamarque, D., Dantzer, R., and Goodall, G. (1999). Attentional and mnemonic deficits associated with infectious disease in humans. Psychol. Med. 29, 291–297. doi:10.1017/S0033291798007740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Capuron, L., Ravaud, A., and Dantzer, R. (2001). Timing and specificity of the cognitive changes induced by interleukin-2 and interferon-α treatments in cancer patients. Psychosom. Med. 63, 376–386. doi:10.1097/00006842-200105000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chavan, S. S., Huerta, P. T., Robbiati, S., Valdes-Ferrer, S. I., Ochani, M., Dancho, M., et al. (2012). HMGB1 mediates cognitive impairment in sepsis survivors. Mol. Med. 18, 930–937. doi:10.2119/molmed.2012.00195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheng, P. Y., Sham, A., and Kronstad, J. W. (2009). Cryptococcus gattii isolates from the British Columbia cryptococcosis outbreak induce less protective inflammation in a murine model of infection than Cryptococcus neoformans. Infect. Immun. 77, 4284–4294. doi:10.1128/IAI.00628-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen-Plotkin, A. S., Xiao, J., Geser, F., Martinez-Lage, M., Grossman, M., Unger, T., et al. (2010). Brain progranulin expression in GRN-associated frontotemporal lobar degeneration. Acta Neuropathol. 119, 111–122. doi:10.1007/s00401-009-0576-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiaravalloti, N. D., and DeLuca, J. (2008). Cognitive impairment in multiple sclerosis. Lancet Neurol. 7, 1139–1151. doi:10.1016/S1474-4422(08)70259-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiu, I. M., Heesters, B. A., Ghasemlou, N., Von Hehn, C. A., Zhao, F., Tran, J., et al. (2013a). Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501, 52–57. doi:10.1038/nature12479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiu, I. M., Morimoto, E. T., Goodarzi, H., Liao, J. T., O’Keeffe, S., Phatnani, H. P., et al. (2013b). A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 4, 385–401. doi:10.1016/j.celrep.2013.06.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiu, I. M., Phatnani, H., Kuligowski, M., Tapia, J. C., Carrasco, M. A., Zhang, M., et al. (2009). Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 106, 20960–20965. doi:10.1073/pnas.0911405106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Comim, C. M., Cassol-Jr, O. J., Constantino, L. S., Felisberto, F., Petronilho, F., Rezin, G. T., et al. (2011). Alterations in inflammatory mediators, oxidative stress parameters and energetic metabolism in the brain of sepsis survivor rats. Neurochem. Res. 36, 304–311. doi:10.1007/s11064-010-0320-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Compeyrot-Lacassagne, S., Tran, T. A., Guillaume-Czitrom, S., Marie, I., and Koné-Paut, I. (2009). Brain multiple sclerosis-like lesions in a patient with Muckle-Wells syndrome. Rheumatology (Oxford) 48, 1618–1619. doi:10.1093/rheumatology/kep321
Compston, A., and Coles, A. (2008). Multiple sclerosis. Lancet 372, 1502–1517. doi:10.1016/S0140-6736(08)61620-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Confavreux, C., and Vukusic, S. (2006). Natural history of multiple sclerosis: a unifying concept. Brain 129(Pt 3), 606–616. doi:10.1093/brain/awh714
Cooper, M. D., and Alder, M. N. (2006). The evolution of adaptive immune systems. Cell 124, 815–822. doi:10.1016/j.cell.2006.02.001
Cree, B. (2008). Neuromyelitis optica: diagnosis, pathogenesis, and treatment. Curr. Neurol. Neurosci. Rep. 8, 427–433. doi:10.1007/s11910-008-0066-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crow, Y. J., Hayward, B. E., Parmar, R., Robins, P., Leitch, A., Ali, M., et al. (2006). Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 38, 917–920. doi:10.1038/ng1845
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cunningham, C., Campion, S., Lunnon, K., Murray, C. L., Woods, J. F., Deacon, R. M., et al. (2009). Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol. Psychiatry 65, 304–312. doi:10.1016/j.biopsych.2008.07.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dalmau, J., Gleichman, A. J., Hughes, E. G., Rossi, J. E., Peng, X., Lai, M., et al. (2008). Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 7, 1091–1098. doi:10.1016/S1474-4422(08)70224-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dalmau, J., Lancaster, E., Martinez-Hernandez, E., Rosenfeld, M. R., and Balice-Gordon, R. (2011). Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 10, 63–74. doi:10.1016/S1474-4422(10)70253-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dalmau, J., Tüzün, E., Wu, H. Y., Masjuan, J., Rossi, J. E., Voloschin, A., et al. (2007). Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 61, 25–36. doi:10.1002/ana.21050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dantzer, R., O’Connor, J. C., Freund, G. G., Johnson, R. W., and Kelley, K. W. (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. doi:10.1038/nrn2297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
d’Avila, J. C., Santiago, A. P., Amâncio, R. T., Galina, A., Oliveira, M. F., and Bozza, F. A. (2008). Sepsis induces brain mitochondrial dysfunction. Crit. Care Med. 36, 1925–1932. doi:10.1097/CCM.0b013e3181760c4b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dawson, V. L., Dawson, T. M., Bartley, D. A., Uhl, G. R., and Snyder, S. H. (1993). Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J. Neurosci 13, 2651–2661.
De Stefano, N., Narayanan, S., Francis, G. S., Arnaoutelis, R., Tartaglia, M. C., Antel, J. P., et al. (2001). Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch. Neurol. 58, 65–70. doi:10.1001/archneur.58.1.65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Vries, B., Steup-Beekman, G. M., Haan, J., Bollen, E. L., Luyendijk, J., Frants, R. R., et al. (2010). TREX1 gene variant in neuropsychiatric systemic lupus erythematosus. Ann. Rheum. Dis. 69, 1886–1887. doi:10.1136/ard.2009.114157
DeGiorgio, L. A., Konstantinov, K. N., Lee, S. C., Hardin, J. A., Volpe, B. T., and Diamond, B. (2001). A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat. Med. 7, 1189–1193. doi:10.1038/nm1101-1189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DeLuca, G. C., Alterman, R., Martin, J. L., Mittal, A., Blundell, S., Bird, S., et al. (2013). Casting light on multiple sclerosis heterogeneity: the role of HLA-DRB1 on spinal cord pathology. Brain 136(Pt 4), 1025–1034. doi:10.1093/brain/awt031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deutschman, C. S., and Tracey, K. J. (2014). Sepsis: current dogma and new perspectives. Immunity 40, 463–475. doi:10.1016/j.immuni.2014.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Diamond, B., Honig, G., Mader, S., Brimberg, L., and Volpe, B. T. (2013). Brain-reactive antibodies and disease. Annu. Rev. Immunol. 31, 345–385. doi:10.1146/annurev-immunol-020711-075041
Ebers, G. C., Sadovnick, A. D., and Risch, N. J. (1995). A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature 377, 150–151. doi:10.1038/377150a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eidelman, L. A., Putterman, D., Putterman, C., and Sprung, C. L. (1996). The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA 275, 470–473. doi:10.1001/jama.1996.03530300054040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ekdahl, C. T., Claasen, J. H., Bonde, S., Kokaia, Z., and Lindvall, O. (2003). Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. U.S.A. 100, 13632–13637. doi:10.1073/pnas.2234031100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ely, E. W., Gautam, S., Margolin, R., Francis, J., May, L., Speroff, T., et al. (2001). The impact of delirium in the intensive care unit on hospital length of stay. Intensive Care Med. 27, 1892–1900. doi:10.1007/s00134-001-1132-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ely, E. W., Shintani, A., Truman, B., Speroff, T., Gordon, S. M., Harrell, F. E. Jr., et al. (2004). Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA 291, 1753–1762. doi:10.1001/jama.291.14.1753
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Epel, E. S., Blackburn, E. H., Lin, J., Dhabhar, F. S., Adler, N. E., Morrow, J. D., et al. (2004). Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. U.S.A. 101, 17312–17315. doi:10.1073/pnas.0407162101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Faust, T. W., Chang, E. H., Kowal, C., Berlin, R., Gazaryan, I. G., Bertini, E., et al. (2010). Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc. Natl. Acad. Sci. U.S.A. 107, 18569–18574. doi:10.1073/pnas.1006980107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fischer, M. T., Sharma, R., Lim, J. L., Haider, L., Frischer, J. M., Drexhage, J., et al. (2012). NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135(Pt 3), 886–899. doi:10.1093/brain/aws012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Forton, D. M., Allsop, J. M., Main, J., Foster, G. R., Thomas, H. C., and Taylor-Robinson, S. D. (2001). Evidence for a cerebral effect of the hepatitis C virus. Lancet 358, 38–39. doi:10.1016/S0140-6736(01)07051-9
Forton, D. M., Thomas, H. C., Murphy, C. A., Allsop, J. M., Foster, G. R., Main, J., et al. (2002). Hepatitis C and cognitive impairment in a cohort of patients with mild liver disease. Hepatology 35, 433–439. doi:10.1053/jhep.2002.30688
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frakes, A. E., Ferraiuolo, L., Haidet-Phillips, A. M., Schmelzer, L., Braun, L., Miranda, C. J., et al. (2014). Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 81, 1009–1023. doi:10.1016/j.neuron.2014.01.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Friedlander, R. M. (2003). Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 348, 1365–1375. doi:10.1056/NEJMra022366
Friese, M. A., Montalban, X., Willcox, N., Bell, J. I., Martin, R., and Fugger, L. (2006). The value of animal models for drug development in multiple sclerosis. Brain 129(Pt 8), 1940–1952. doi:10.1093/brain/awl083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Friese, M. A., Schattling, B., and Fugger, L. (2014). Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 10, 225–238. doi:10.1038/nrneurol.2014.37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frischer, J. M., Bramow, S., Dal-Bianco, A., Lucchinetti, C. F., Rauschka, H., Schmidbauer, M., et al. (2009). The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132(Pt 5), 1175–1189. doi:10.1093/brain/awp070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fu, H. Q., Yang, T., Xiao, W., Fan, L., Wu, Y., Terrando, N., et al. (2014). Prolonged neuroinflammation after lipopolysaccharide exposure in aged rats. PLoS ONE 9:e106331. doi:10.1371/journal.pone.0106331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fünfschilling, U., Supplie, L. M., Mahad, D., Boretius, S., Saab, A. S., Edgar, J., et al. (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. doi:10.1038/nature11007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gaynor, B., Putterman, C., Valadon, P., Spatz, L., Scharff, M. D., and Diamond, B. (1997). Peptide inhibition of glomerular deposition of an anti-DNA antibody. Proc. Natl. Acad. Sci. U.S.A. 94, 1955–1960. doi:10.1073/pnas.94.5.1955
Gergely, P. Jr., Grossman, C., Niland, B., Puskas, F., Neupane, H., Allam, F., et al. (2002). Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 46, 175–190. doi:10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geurts, J. J., Bö, L., Roosendaal, S. D., Hazes, T., Daniëls, R., Barkhof, F., et al. (2007). Extensive hippocampal demyelination in multiple sclerosis. J. Neuropathol. Exp. Neurol. 66, 819–827. doi:10.1097/nen.0b013e3181461f54
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghadge, G. D., Lee, J. P., Bindokas, V. P., Jordan, J., Ma, L., Miller, R. J., et al. (1997). Mutant superoxide dismutase-1-linked familial amyotrophic lateral sclerosis: molecular mechanisms of neuronal death and protection. J. Neurosci. 17, 8756–8766.
Giannakopoulos, B., and Krilis, S. A. (2013). The pathogenesis of the anti- phospholipid syndrome. N. Engl. J. Med. 368, 1033–1044. doi:10.1056/NEJMra1112830
Giuliani, F., Goodyer, C. G., Antel, J. P., and Yong, V. W. (2003). Vulnerability of human neurons to T cell-mediated cytotoxicity. J. Immunol. 171, 368–379. doi:10.4049/jimmunol.171.1.368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gofton, T. E., and Young, G. B. (2012). Sepsis-associated encephalopathy. Nat. Rev. Neurol. 8, 557–566. doi:10.1038/nrneurol.2012.183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gómez-Puerta, J. A., Cervera, R., Calvo, L. M., Gómez-Ansón, B., Espinosa, G., Claver, G., et al. (2005). Dementia associated with the antiphospholipid syndrome: clinical and radiological characteristics of 30 patients. Rheumatology (Oxford) 44, 95–99. doi:10.1093/rheumatology/keh408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Graham, A. L., Allen, J. E., and Read, A. F. (2005). Evolutionary causes and consequences of immunopathology. Annu. Rev. Ecol. Evol. Syst. 36, 373–397. doi:10.1146/annurev.ecolsys.36.102003.152622
Green, D. R., Ferguson, T., Zitvogel, L., and Kroemer, G. (2009). Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363. doi:10.1038/nri2545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Green, D. R., and Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science 305, 626–629. doi:10.1126/science.1099320
Greenlund, L. J., Deckwerth, T. L., and Johnson, E. M. Jr. (1995). Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron 14, 303–315. doi:10.1016/0896-6273(95)90287-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gregory, A. P., Dendrou, C. A., Attfield, K. E., Haghikia, A., Xifara, D. K., Butter, F., et al. (2012). TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 488, 508–511. doi:10.1038/nature11307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. doi:10.1016/j.neuron.2013.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gris, D., Ye, Z., Iocca, H. A., Wen, H., Craven, R. R., Gris, P., et al. (2010). NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 185, 974–981. doi:10.4049/jimmunol.0904145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guarda, G., Braun, M., Staehli, F., Tardivel, A., Mattmann, C., Förster, I., et al. (2011). Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223. doi:10.1016/j.immuni.2011.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi:10.1056/NEJMoa1211851
Gulinelli, S., Salaro, E., Vuerich, M., Bozzato, D., Pizzirani, C., Bolognesi, G., et al. (2012). IL-18 associates to microvesicles shed from human macrophages by a LPS/TLR-4 independent mechanism in response to P2X receptor stimulation. Eur. J. Immunol. 42, 3334–3345. doi:10.1002/eji.201142268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hadjivassiliou, M., Aeschlimann, P., Sanders, D. S., Mäki, M., Kaukinen, K., Grünewald, R. A., et al. (2013). Transglutaminase 6 antibodies in the diagnosis of gluten ataxia. Neurology 80, 1740–1745. doi:10.1212/WNL.0b013e3182919070
Hanly, J. G., Walsh, N. M., and Sangalang, V. (1992). Brain pathology in systemic lupus erythematosus. J. Rheumatol. 19, 732–741.
Hauser, S. L., Waubant, E., Arnold, D. L., Vollmer, T., Antel, J., Fox, R. J., et al. (2008). B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 358, 676–688. doi:10.1056/NEJMoa0706383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Häuser, W., Janke, K. H., Klump, B., and Hinz, A. (2011). Anxiety and depression in patients with inflammatory bowel disease: comparisons with chronic liver disease patients and the general population. Inflamm. Bowel Dis. 17, 621–632. doi:10.1002/ibd.21346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi:10.1038/nature11729
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hilsabeck, R. C., Perry, W., and Hassanein, T. I. (2002). Neuropsychological impairment in patients with chronic hepatitis C. Hepatology 35, 440–446. doi:10.1053/jhep.2002.31257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hockenbery, D. M., Oltvai, Z. N., Yin, X. M., Milliman, C. L., and Korsmeyer, S. J. (1993). Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75, 241–251. doi:10.1016/0092-8674(93)80066-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holmes, C., Cunningham, C., Zotova, E., Woolford, J., Dean, C., Kerr, S., et al. (2009). Systemic inflammation and disease progression in Alzheimer disease. Neurology 73, 768–774. doi:10.1212/WNL.0b013e3181b6bb95
Hu, S., Rotschafer, J. H., Lokensgard, J. R., and Cheeran, M. C. (2014). Activated CD8+ T lymphocytes inhibit neural stem/progenitor cell proliferation: role of interferon-gamma. PLoS ONE 9:e105219. doi:10.1371/journal.pone.0105219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hughes, E. G., Peng, X., Gleichman, A. J., Lai, M., Zhou, L., Tsou, R., et al. (2010). Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 30, 5866–5875. doi:10.1523/JNEUROSCI.0167-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
International Parkinson Disease Genomics Consortium, Nalls, M. A., Plagnol, V., Hernandez, D. G., Sharma, M., Sheerin, U. M., et al. (2011). Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 377, 641–649. doi:10.1016/S0140-6736(10)62345-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwashyna, T. J., Cooke, C. R., Wunsch, H., and Kahn, J. M. (2012). Population burden of long-term survivorship after severe sepsis in older Americans. J. Am. Geriatr. Soc. 60, 1070–1077. doi:10.1111/j.1532-5415.2012.03989.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwashyna, T. J., Ely, E. W., Smith, D. M., and Langa, K. M. (2010). Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304, 1787–1794. doi:10.1001/jama.2010.1553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeltsch-David, H., and Muller, S. (2014). Neuropsychiatric systemic lupus erythematosus: pathogenesis and biomarkers. Nat. Rev. Neurol. 10, 579–596. doi:10.1038/nrneurol.2014.148
Ji, H., Rabbi, M. F., Labis, B., Pavlov, V. A., Tracey, K. J., and Ghia, J. E. (2014). Central cholinergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal Immunol. 7, 335–347. doi:10.1038/mi.2013.52
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jin, K., Wang, X., Xie, L., Mao, X. O., Zhu, W., Wang, Y., et al. (2006a). Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl. Acad. Sci. U.S.A. 103, 13198–13202. doi:10.1073/pnas.0601164103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jin, K., Xie, L., Mao, X. O., and Greenberg, D. A. (2006b). Alzheimer’s disease drugs promote neurogenesis. Brain Res. 1085, 183–188. doi:10.1016/j.brainres.2006.02.081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johansson, J. U., Woodling, N. S., Wang, Q., Panchal, M., Liang, X., Trueba-Saiz, A., et al. (2015). Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J. Clin. Invest. 125, 350–364. doi:10.1172/JCI77487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, J. D. G., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi:10.1038/nature05286
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi:10.1056/NEJMoa1211103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joshi, P., Turola, E., Ruiz, A., Bergami, A., Libera, D. D., Benussi, L., et al. (2014). Microglia convert aggregated amyloid-[beta] into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 21, 582–593. doi:10.1038/cdd.2013.180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaster, M. P., Gadotti, V. M., Calixto, J. B., Santos, A. R., and Rodrigues, A. L. (2012). Depressive-like behavior induced by tumor necrosis factor-α in mice. Neuropharmacology 62, 419–426. doi:10.1016/j.neuropharm.2011.08.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaur, C., Sivakumar, V., Zou, Z., and Ling, E. A. (2014). Microglia-derived proinflammatory cytokines tumor necrosis factor-alpha and interleukin-1beta induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct. Funct. 219, 151–170. doi:10.1007/s00429-012-0491-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khanna, K. M., Lepisto, A. J., Decman, V., and Hendricks, R. L. (2004). Immune control of herpes simplex virus during latency. Curr. Opin. Immunol. 16, 463–469. doi:10.1016/j.coi.2004.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiecolt-Glaser, J. K., Dura, J. R., Speicher, C. E., Trask, O. J., and Glaser, R. (1991). Spousal caregivers of dementia victims: longitudinal changes in immunity and health. Psychosom. Med. 53, 345–362. doi:10.1097/00006842-199107000-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiecolt-Glaser, J. K., Glaser, R., Shuttleworth, E. C., Dyer, C. S., Ogrocki, P., and Speicher, C. E. (1987). Chronic stress and immunity in family caregivers of Alzheimer’s disease victims. Psychosom. Med. 49, 523–535. doi:10.1097/00006842-198709000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kinoshita, M., Nakatsuji, Y., Kimura, T., Moriya, M., Takata, K., Okuno, T., et al. (2009). Neuromyelitis optica: passive transfer to rats by human immunoglobulin. Biochem. Biophys. Res. Commun. 386, 623–627. doi:10.1016/j.bbrc.2009.06.085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kleinberger, G., Yamanishi, Y., Suárez-Calvet, M., Czirr, E., Lohmann, E., Cuyvers, E., et al. (2014). TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6, 243ra286. doi:10.1126/scitranslmed.3009093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Knoll, A. H., Walter, M. R., Narbonne, G. M., and Christie-Blick, N. (2004). A new period for the geologic time scale. Science 305, 621–622. doi:10.1126/science.1098803
Kotani, S., Yamauchi, T., Teramoto, T., and Ogura, H. (2008). Donepezil, an acetylcholinesterase inhibitor, enhances adult hippocampal neurogenesis. Chem. Biol. Interact. 175, 227–230. doi:10.1016/j.cbi.2008.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kowal, C., Degiorgio, L. A., Lee, J. Y., Edgar, M. A., Huerta, P. T., Volpe, B. T., et al. (2006). Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc. Natl. Acad. Sci. U.S.A. 103, 19854–19859. doi:10.1073/pnas.0608397104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krumbholz, M., Derfuss, T., Hohlfeld, R., and Meinl, E. (2012). B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat. Rev. Neurol. 8, 613–623. doi:10.1038/nrneurol.2012.203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuerten, S., Pommerschein, G., Barth, S. K., Hohmann, C., Milles, B., Sammer, F. W., et al. (2014). Identification of a B cell-dependent subpopulation of multiple sclerosis by measurements of brain-reactive B cells in the blood. Clin. Immunol. 152, 20–24. doi:10.1016/j.clim.2014.02.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuhle, J., Leppert, D., Petzold, A., Regeniter, A., Schindler, C., Mehling, M., et al. (2011). Neurofilament heavy chain in CSF correlates with relapses and disability in multiple sclerosis. Neurology 76, 1206–1213. doi:10.1212/WNL.0b013e31821432ff
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kutzelnigg, A., Faber-Rod, J. C., Bauer, J., Lucchinetti, C. F., Sorensen, P. S., Laursen, H., et al. (2007). Widespread demyelination in the cerebellar cortex in multiple sclerosis. Brain Pathol. 17, 38–44. doi:10.1111/j.1750-3639.2006.00041.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kutzelnigg, A., Lucchinetti, C. F., Stadelmann, C., Brück, W., Rauschka, H., Bergmann, M., et al. (2005). Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128(Pt 11), 2705–2712. doi:10.1093/brain/awh641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lambert, J. C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099. doi:10.1038/ng.439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lancaster, E., and Dalmau, J. (2012). Neuronal autoantigens – pathogenesis, associated disorders and antibody testing. Nat. Rev. Neurol. 8, 380–390. doi:10.1038/nrneurol.2012.99
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lassmann, H. (2011). Mechanisms of neurodegeneration shared between multiple sclerosis and Alzheimer’s disease. J. Neural Transm. 118, 747–752. doi:10.1007/s00702-011-0607-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lassmann, H., Bruck, W., and Lucchinetti, C. F. (2007). The immunopathology of multiple sclerosis: an overview. Brain Pathol. 17, 210–218. doi:10.1111/j.1750-3639.2007.00064.x
Lassmann, H., van Horssen, J., and Mahad, D. (2012). Progressive multiple sclerosis: pathology and pathogenesis. Nat. Rev. Neurol. 8, 647–656. doi:10.1038/nrneurol.2012.168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Latora, V., and Marchiori, M. (2001). Efficient behavior of small-world networks. Phys. Rev. Lett. 87, 198701. doi:10.1103/PhysRevLett.87.198701
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lazarov, O., and Marr, R. A. (2010). Neurogenesis and Alzheimer’s disease: at the crossroads. Exp. Neurol. 223, 267–281. doi:10.1016/j.expneurol.2009.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi:10.1038/nature11314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee-Kirsch, M. A., Chowdhury, D., Harvey, S., Gong, M., Senenko, L., Engel, K., et al. (2007a). A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J. Mol. Med. (Berl.) 85, 531–537. doi:10.1007/s00109-007-0199-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee-Kirsch, M. A., Gong, M., Chowdhury, D., Senenko, L., Engel, K., Lee, Y. A., et al. (2007b). Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 39, 1065–1067. doi:10.1038/ng2091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lemcke, S., Müller, S., Möller, S., Schillert, A., Ziegler, A., Cepok-Kauffeld, S., et al. (2014). Nerve conduction velocity is regulated by the inositol polyphosphate-4-phosphatase II gene. Am. J. Pathol. 184, 2420–2429. doi:10.1016/j.ajpath.2014.05.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lemstra, A. W., Groen in’tWoud, J. C., Hoozemans, J. J., van Haastert, E. S., Rozemuller, A. J., Eikelenboom, P., et al. (2007). Microglia activation in sepsis: a case-control study. J. Neuroinflammation 15, 4.
Lennon, V. A., Kryzer, T. J., Pittock, S. J., Verkman, A. S., and Hinson, S. R. (2005). IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 202, 473–477. doi:10.1084/jem.20050304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levy, M. (2014). Does aquaporin-4-seronegative neuromyelitis optica exist? JAMA Neurol. 71, 271–272. doi:10.1001/jamaneurol.2013.5865
Li, Z., Ma, L., Kulesskaya, N., Võikar, V., and Tian, L. (2014). Microglia are polarized to M1 type in high-anxiety inbred mice in response to lipopolysaccharide challenge. Brain Behav. Immun. 38, 237–248. doi:10.1016/j.bbi.2014.02.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liang, X., Wang, Q., Hand, T., Wu, L., Breyer, R. M., Montine, T. J., et al. (2005). Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J. Neurosci. 25, 10180–10187. doi:10.1523/JNEUROSCI.3591-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi:10.1038/nature05292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, B., and Hong, J. S. (2003). Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 304, 1–7. doi:10.1124/jpet.102.043406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, T., Pan, Y., Kao, S. Y., Li, C., Kohane, I., Chan, J., et al. (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891. doi:10.1038/nature02661
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lucchinetti, C., Brück, W., Parisi, J., Scheithauer, B., Rodriguez, M., and Lassmann, H. (2000). Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47, 707–717. doi:10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lucchinetti, C. F., Mandler, R. N., McGavern, D., Bruck, W., Gleich, G., Ransohoff, R. M., et al. (2002). A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 125(Pt 7), 1450–1461. doi:10.1093/brain/awf151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lucchinetti, C. F., Popescu, B. F., Bunyan, R. F., Moll, N. M., Roemer, S. F., Lassmann, H., et al. (2011). Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 365, 2188–2197. doi:10.1056/NEJMoa1100648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mader, S., Lutterotti, A., Di Pauli, F., Kuenz, B., Schanda, K., Aboul-Enein, F., et al. (2010). Patterns of antibody binding to aquaporin-4 isoforms in neuromyelitis optica. PLoS ONE 5:e10455. doi:10.1371/journal.pone.0010455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mak, A., Cheung, M. W., Chiew, H. J., Liu, Y., and Ho, R. C. (2012). Global trend of survival and damage of systemic lupus erythematosus: meta-analysis and meta-regression of observational studies from the 1950s to 2000s. Semin. Arthritis Rheum. 41, 830–839. doi:10.1016/j.semarthrit.2011.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malberg, J. E., Eisch, A. J., Nestler, E. J., and Duman, R. S. (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20, 9104–9110.
Martens, L. H., Zhang, J., Barmada, S. J., Zhou, P., Kamiya, S., Sun, B., et al. (2012). Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Invest. 122, 3955–3959. doi:10.1172/JCI63113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. doi:10.1016/S1097-2765(02)00599-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martinon, F., and Tschopp, J. (2004). Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 117, 561–574. doi:10.1016/j.cell.2004.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matiello, M., Lennon, V. A., Jacob, A., Pittock, S. J., Lucchinetti, C. F., Wingerchuk, D. M., et al. (2008). NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology 70, 2197–2200. doi:10.1212/01.wnl.0000303817.82134.da
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsuki, T., Nakae, S., Sudo, K., Horai, R., and Iwakura, Y. (2006). Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int. Immunol. 18, 399–407. doi:10.1093/intimm/dxh379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McColl, B. W., Rothwell, N. J., and Allan, S. M. (2008). Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J. Neurosci. 28, 9451–9462. doi:10.1523/JNEUROSCI.2674-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McFarland, H. F., and Martin, R. (2007). Multiple sclerosis: a complicated picture of autoimmunity. Nat. Immunol. 8, 913–919. doi:10.1038/ni1507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McNicoll, L., Pisani, M. A., Zhang, Y., Ely, E. W., Siegel, M. D., and Inouye, S. K. (2003). Delirium in the intensive care unit: occurrence and clinical course in older patients. J. Am. Geriatr. Soc. 51, 591–598. doi:10.1034/j.1600-0579.2003.00201.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Medzhitov, R., and Janeway, C. A. (1997). Innate immunity: the virtues of a nonclonal system of recognition. Cell 91, 295–298. doi:10.1016/S0092-8674(00)80412-2
Medzhitov, R., Schneider, D. S., and Soares, M. P. (2012). Disease tolerance as a defense strategy. Science 335, 936–941. doi:10.1126/science.1214935
Michels, M., Vieira, A. S., Vuolo, F., Zapelini, H. G., Mendonça, B., Mina, F., et al. (2015). The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav. Immun. 43, 54–59. doi:10.1016/j.bbi.2014.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, D. H., and Leary, S. M. (2007). Primary-progressive multiple sclerosis. Lancet Neurol. 6, 903–912. doi:10.1016/S1474-4422(07)70213-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Million, M., and Raoult, D. (2013). The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med. 368, 2335. doi:10.1056/NEJMc1300484
Mina-Osorio, P., Rosas-Ballina, M., Valdes-Ferrer, S. I., Al-Abed, Y., Tracey, K. J., and Diamond, B. (2012). Neural signaling in the spleen controls B-cell responses to blood-borne antigen. Mol. Med. 18, 618–627. doi:10.2119/molmed.2012.00027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miner, J. J., and Diamond, M. S. (2014). MDA5 and autoimmune disease. Nat. Genet. 46, 418–419. doi:10.1038/ng.2959
Monje, M. L., Toda, H., and Palmer, T. D. (2003). Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765. doi:10.1126/science.1088417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Montine, T. J., Sidell, K. R., Crews, B. C., Markesbery, W. R., Marnett, L. J., Roberts, L. J. II, et al. (1999). Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology 53, 1495–1495. doi:10.1212/WNL.53.7.1495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morandi, A., Rogers, B. P., Gunther, M. L., Merkle, K., Pandharipande, P., Girard, T. D., et al. (2012). The relationship between delirium duration, white matter integrity, and cognitive impairment in intensive care unit survivors as determined by diffusion tensor imaging: the VISIONS prospective cohort magnetic resonance imaging study. Crit. Care Med. 40, 2182–2189. doi:10.1097/CCM.0b013e318250acdc
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moresco, E. M. Y., LaVine, D., and Beutler, B. (2011). Toll-like receptors. Curr. Biol. 21, R488–R493. doi:10.1016/j.cub.2011.05.039
Morita, M., Stamp, G., Robins, P., Dulic, A., Rosewell, I., Hrivnak, G., et al. (2004). Gene-targeted mice lacking the Trex1 (DNase III) 3’ – >5’ DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol. 24, 6719–6727. doi:10.1128/MCB.24.22.9736-9743.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murray, C., Sanderson, D. J., Barkus, C., Deacon, R. M., Rawlins, J. N., Bannerman, D. M., et al. (2012). Systemic inflammation induces acute working memory deficits in the primed brain: relevance for delirium. Neurobiol. Aging 33, 603–616e603. doi:10.1016/j.neurobiolaging.2010.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neumann, H. (2001). Control of glial immune function by neurons. Glia 36, 191–199. doi:10.1002/glia.1108
Neumann, H., Medana, I. M., Bauer, J., and Lassmann, H. (2002). Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 25, 313–319. doi:10.1016/S0166-2236(02)02154-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nguyen, M. D., D’Aigle, T., Gowing, G., Julien, J. P., and Rivest, S. (2004). Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 24, 1340–1349. doi:10.1523/JNEUROSCI.4786-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nitsch, R., Pohl, E. E., Smorodchenko, A., Infante-Duarte, C., Aktas, O., and Zipp, F. (2004). Direct impact of T cells on neurons revealed by two-photon microscopy in living brain tissue. J. Neurosci. 24, 2458–2464. doi:10.1523/JNEUROSCI.4703-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Noh, H., Jeon, J., and Seo, H. (2014). Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem. Int. 69, 35–40. doi:10.1016/j.neuint.2014.02.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Noseworthy, J. H., Lucchinetti, C., Rodriguez, M., and Weinshenker, B. G. (2000). Multiple sclerosis. N. Engl. J. Med. 343, 938–952. doi:10.1056/NEJM200009283431307
Ousman, S. S., and Kubes, P. (2012). Immune surveillance in the central nervous system. Nat. Neurosci. 15, 1096–1101. doi:10.1038/nn.3161
Palace, J., Leite, M. I., Nairne, A., and Vincent, A. (2010). Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch. Neurol. 67, 1016–1017. doi:10.1001/archneurol.2010.188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paloneva, J., Kestilä, M., Wu, J., Salminen, A., Böhling, T., Ruotsalainen, V., et al. (2000). Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 25, 357–361. doi:10.1038/77153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paloneva, J., Manninen, T., Christman, G., Hovanes, K., Mandelin, J., Adolfsson, R., et al. (2002). Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662. doi:10.1086/342259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Papadopoulos, M. C., Bennett, J. L., and Verkman, A. S. (2014). Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat. Rev. Neurol. 10, 493–506. doi:10.1038/nrneurol.2014.141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Papathanasiou, A., Nikakis, P., Bonakis, A., Kilidireas, K., Dimitrakopoulos, A., Michopoulos, S., et al. (2014). Rapidly progressive dementia as presenting feature in inflammatory bowel disease. Alzheimer Dis. Assoc. Disord. 28, 294–295. doi:10.1097/WAD.0b013e31826a96b2
Park, L., Zhou, J., Zhou, P., Pistick, R., El Jamal, S., Younkin, L., et al. (2013). Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. U.S.A. 110, 3089–3094. doi:10.1073/pnas.1300021110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. III, Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi:10.1016/j.cell.2013.11.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pasinelli, P., Borchelt, D. R., Houseweart, M. K., Cleveland, D. W., and Brown, R. H. Jr. (1998). Caspase-1 is activated in neural cells and tissue with amyotrophic lateral sclerosis-associated mutations in copper-zinc superoxide dismutase. Proc. Natl. Acad. Sci. U.S.A. 95, 15763–15768. doi:10.1073/pnas.95.26.15763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pavlov, V. A., Parrish, W. R., Rosas-Ballina, M., Ochani, M., Puerta, M., Ochani, K., et al. (2009). Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav. Immun. 23, 41–45. doi:10.1016/j.bbi.2008.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peterson, J. W., Bö, L., Mörk, S., Chang, A., and Trapp, B. D. (2001). Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann. Neurol. 50, 389–400. doi:10.1002/ana.1123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pickford, F., Marcus, J., Camargo, L. M., Xiao, Q., Graham, D., Mo, J. R., et al. (2011). Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am. J. Pathol. 178, 284–295. doi:10.1016/j.ajpath.2010.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pohl, M., Kawakami, N., Kitic, M., Bauer, J., Martins, R., Fischer, M. T., et al. (2013). T cell-activation in neuromyelitis optica lesions plays a role in their formation. Acta Neuropathol. Commun. 1, 85. doi:10.1186/2051-5960-1-85
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Popescu, B. F., Lennon, V. A., Parisi, J. E., Howe, C. L., Weigand, S. D., Cabrera-Gómez, J. A., et al. (2011). Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology 76, 1229–1237. doi:10.1212/WNL.0b013e318214332c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Popescu, B. F., and Lucchinetti, C. F. (2012). Pathology of demyelinating diseases. Annu. Rev. Pathol. 7, 185–217. doi:10.1146/annurev-pathol-011811-132443
Pott Godoy, M. C., Tarelli, R., Ferrari, C. C., Sarchi, M. I., and Pitossi, F. J. (2008). Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain 131(Pt 7), 1880–1894. doi:10.1093/brain/awn101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rademakers, R., Baker, M., Nicholson, A. M., Rutherford, N. J., Finch, N., Soto-Ortolaza, A., et al. (2012). Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 44, 200–205. doi:10.1038/ng.1027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ransohoff, R. M., and Engelhardt, B. (2012). The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 12, 623–635. doi:10.1038/nri3265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rayaprolu, S., Mullen, B., Baker, M., Lynch, T., Finger, E., Seeley, W. W., et al. (2013). TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 8, 19. doi:10.1186/1750-1326-8-19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reddy, K. C., Andersen, E. C., Kruglyak, L., and Kim, D. H. (2009). A polymorphism in npr-1 is a behavioral determinant of pathogen susceptibility in C. elegans. Science 323, 382–384. doi:10.1126/science.1166527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rego, A. C., and Oliveira, C. R. (2003). Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochem. Res. 28, 1563–1574. doi:10.1023/A:1025682611389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reichenberg, A., Gorman, J. M., and Dieterich, D. T. (2005). Interferon-induced depression and cognitive impairment in hepatitis C virus patients: a 72 week prospective study. AIDS 19, S174–S178. doi:10.1097/01.aids.0000192087.64432.ae
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reichenberg, A., Yirmiya, R., Schuld, A., Kraus, T., Haack, M., Morag, A., et al. (2001). Cytokine-associated emotional and cognitive disturbances in humans. Arch. Gen. Psychiatry 58, 445–452. doi:10.1001/archpsyc.58.5.445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reinert, K. R., Umphlet, C. D., Quattlebaum, A., and Boger, H. A. (2014). Short-term effects of an endotoxin on substantia nigra dopamine neurons. Brain Res. 1557, 164–170. doi:10.1016/j.brainres.2014.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rezai-Zadeh, K., Gate, D., and Town, T. (2009). CNS infiltration of peripheral immune cells: D-day for neurodegenerative disease? J. Neuroimmune Pharmacol. 4, 462–475. doi:10.1007/s11481-009-9166-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rosas-Ballina, M., Olofsson, P. S., Ochani, M., Valdés-Ferrer, S. I., Levine, Y. A., Reardon, C., et al. (2011). Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101. doi:10.1126/science.1209985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rosas-Ballina, M., Valdés-Ferrer, S. I., Dancho, M. E., Ochani, M., Katz, D., Cheng, K. F., et al. (2015). Xanomeline suppresses excessive pro-inflammatory cytokine responses through neural signal-mediated pathways and improves survival in lethal inflammation. Brain Behav. Immun. 44, 19–27. doi:10.1016/j.bbi.2014.07.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rosen, D. R. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364, 362. doi:10.1038/364362d0
Saadoun, S., Waters, P., Bell, B. A., Vincent, A., Verkman, A. S., and Papadopoulos, M. C. (2010). Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 133(Pt 2), 349–361. doi:10.1093/brain/awp309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sahay, A., and Hen, R. (2007). Adult hippocampal neurogenesis in depression. Nat. Neurosci. 10, 1110–1115. doi:10.1038/nn1969
Saji, E., Arakawa, M., Yanagawa, K., Toyoshima, Y., Yokoseki, A., Okamoto, K., et al. (2013). Cognitive impairment and cortical degeneration in neuromyelitis optica. Ann. Neurol. 73, 65–76. doi:10.1002/ana.23721
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., et al. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805–809. doi:10.1126/science.1083328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sarkar, A., Mitra, S., Mehta, S., Raices, R., and Wewers, M. D. (2009). Monocyte derived microvesicles deliver a cell death message via encapsulated caspase-1. PLoS ONE 4:e7140. doi:10.1371/journal.pone.0007140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Satapathy, S. K., Ochani, M., Dancho, M., Hudson, L. K., Rosas-Ballina, M., Valdes-Ferrer, S. I., et al. (2011). Galantamine alleviates inflammation and other obesity-associated complications in high-fat diet-fed mice. Mol. Med. 17, 599–606. doi:10.2119/molmed.2011.00083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmitt, C., Strazielle, N., and Ghersi-Egea, J. F. (2012). Brain leukocyte infiltration initiated by peripheral inflammation or experimental autoimmune encephalomyelitis occurs through pathways connected to the CSF-filled compartments of the forebrain and midbrain. J. Neuroinflammation 9, 187. doi:10.1186/1742-2094-9-187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneider, R., Mohebiany, A. N., Ifergan, I., Beauseigle, D., Duquette, P., Prat, A., et al. (2011). B cell-derived IL-15 enhances CD8 T cell cytotoxicity and is increased in multiple sclerosis patients. J. Immunol. 187, 4119–4128. doi:10.4049/jimmunol.1100885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwartz, M., and Baruch, K. (2014). The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J. 33, 7–22. doi:10.1002/embj.201386609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seki, M., Suzuki, S., Iizuka, T., Shimizu, T., Nihei, Y., Suzuki, N., et al. (2008). Neurological response to early removal of ovarian teratoma in anti-NMDAR encephalitis. J. Neurol. Neurosurg. Psychiatr. 79, 324–326. doi:10.1136/jnnp.2007.136473
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Semmler, A., Widmann, C. N., Okulla, T., Urbach, H., Kaiser, M., Widman, G., et al. (2013). Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J. Neurol. Neurosurg. Psychiatr. 84, 62–69. doi:10.1136/jnnp-2012-302883
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Serafini, B., Rosicarelli, B., Magliozzi, R., Stigliano, E., and Aloisi, F. (2004). Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 14, 164–174. doi:10.1111/j.1750-3639.2004.tb00049.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shah, D. K., Doyle, L. W., Anderson, P. J., Bear, M., Daley, A. J., Hunt, R. W., et al. (2008). Adverse neurodevelopment in preterm infants with postnatal sepsis or necrotizing enterocolitis is mediated by white matter abnormalities on magnetic resonance imaging at term. J. Pediatr. 153, 170–5, 175.e1. doi:10.1016/j.jpeds.2008.02.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sharshar, T., Carlier, R., Bernard, F., Guidoux, C., Brouland, J. P., Nardi, O., et al. (2007). Brain lesions in septic shock: a magnetic resonance imaging study. Intensive Care Med. 33, 798–806. doi:10.1007/s00134-007-0598-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sharshar, T., Gray, F., Lorin de la Grandmaison, G., Hopkinson, N. S., Ross, E., Dorandeu, A., et al. (2003). Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet 362, 1799–1805. doi:10.1016/S0140-6736(03)14899-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sheedy, F. J., Grebe, A., Rayner, K. J., Kalantari, P., Ramkhelawon, B., Carpenter, S. B., et al. (2013). CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820. doi:10.1038/ni.2639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sierra, A., Encinas, J. M., Deudero, J. J., Chancey, J. H., Enikolopov, G., Overstreet-Wadiche, L. S., et al. (2010). Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495. doi:10.1016/j.stem.2010.08.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Silverman, H. A., Dancho, M., Regnier-Golanov, A., Nasim, M., Ochani, M., Olofsson, P. S., et al. (2014). Brain region-specific alterations in the gene expression of cytokines, immune cell markers and cholinergic system components during peripheral endotoxin-induced inflammation. Mol. Med. 20, doi:10.2119/molmed.2014.00147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singer, M. (2014). The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 5, 66–72. doi:10.4161/viru.26907
Singer, M., De Santis, V., Vitale, D., and Jeffcoate, W. (2004). Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet 364, 545–548. doi:10.1016/S0140-6736(04)16815-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sonneville, R., Verdonk, F., Rauturier, C., Klein, I. F., Wolff, M., Annane, D., et al. (2013). Understanding brain dysfunction in sepsis. Ann. Intensive Care 3, 15. doi:10.1186/2110-5820-3-15
Sospedra, M., and Martin, R. (2005a). Antigen-specific therapies in multiple sclerosis. Int. Rev. Immunol. 24, 393–413. doi:10.1080/08830180500371256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sospedra, M., and Martin, R. (2005b). Immunology of multiple sclerosis. Annu. Rev. Immunol. 23, 683–747. doi:10.1146/annurev.immunol.23.021704.115707
Sporns, O., Tononi, G., and Kötter, R. (2005). The human connectome: a structural description of the human brain. PLoS Comput. Biol. 1:e42. doi:10.1371/journal.pcbi.0010042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stanley, E. R., Berg, K. L., Einstein, D. B., Lee, P. S., Pixley, F. J., Wang, Y., et al. (1997). Biology and action of colony – stimulating factor-1. Mol. Reprod. Dev. 46, 4–10. doi:10.1002/(SICI)1098-2795(199701)46:1<4::AID-MRD2>3.0.CO;2-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stern, J. N., Yaari, G., Vander Heiden, J. A., Church, G., Donahue, W. F., Hintzen, R. Q., et al. (2014). B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 6, 248ra107. doi:10.1126/scitranslmed.3008879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stetson, D. B., Ko, J. S., Heidmann, T., and Medzhitov, R. (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598. doi:10.1016/j.cell.2008.06.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi:10.1016/j.cell.2007.10.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Storch, M. K., Piddlesden, S., Haltia, M., Iivanainen, M., Morgan, P., and Lassmann, H. (1998). Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann. Neurol. 43, 465–471. doi:10.1002/ana.410430409
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamatani, M., Che, Y. H., Matsuzaki, H., Ogawa, S., Okado, H., Miyake, S., et al. (1999). Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J. Biol. Chem. 274, 8531–8538. doi:10.1074/jbc.274.13.8531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tektonidou, M. G., Varsou, N., Kotoulas, G., Antoniou, A., and Moutsopoulos, H. M. (2006). Cognitive deficits in patients with antiphospholipid syndrome: association with clinical, laboratory, and brain magnetic resonance imaging findings. Arch. Intern. Med. 166, 2278–2284. doi:10.1001/archinte.166.20.2278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Terrando, N., Fidalgo, A. R., Vizcaychipi, M., Cibelli, M., Ma, D., Monaco, C., et al. (2010a). The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit. Care 14, 1–9. doi:10.1186/cc9019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Terrando, N., Monaco, C., Ma, D., Foxwell, B. M., Feldmann, M., and Maze, M. (2010b). Tumor necrosis factor-alpha triggers a cytokine cascade yielding postoperative cognitive decline. Proc. Natl. Acad. Sci. U.S.A. 107, 20518–20522. doi:10.1073/pnas.1014557107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tian, L., Ma, L., Kaarela, T., and Li, Z. (2012). Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflammation 9, 155. doi:10.1186/1742-2094-9-155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tikka, T., Fiebich, B. L., Goldsteins, G., Keinänen, R., and Koistinaho, J. (2001). Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J. Neurosci. 21, 2580–2588.
Ting, J. P., and Trowsdale, J. (2002). Genetic control of MHC class II expression. Cell 109(Suppl.), S21–S33. doi:10.1016/S0092-8674(02)00696-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Titulaer, M. J., Höftberger, R., Iizuka, T., Leypoldt, F., McCracken, L., Cellucci, T., et al. (2014). Overlapping demyelinating syndromes and anti-N-methyl-D-aspartate receptor encephalitis. Ann. Neurol. 75, 411–428. doi:10.1002/ana.24117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Toga, A. W., Clark, K. A., Thompson, P. M., Shattuck, D. W., and Van Horn, J. D. (2012). Mapping the human connectome. Neurosurgery 71, 1–5. doi:10.1227/NEU.0b013e318258e9ff
Tokunaga, M., Saito, K., Kawabata, D., Imura, Y., Fujii, T., Nakayamada, S., et al. (2007). Efficacy of rituximab (anti-CD20) for refractory systemic lupus erythematosus involving the central nervous system. Ann. Rheum. Dis. 66, 470–475. doi:10.1136/ard.2006.057885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trapp, B. D., and Nave, K. A. (2008). Multiple sclerosis: an immune or neurodegenerative disorder? Annu. Rev. Neurosci. 31, 247–269. doi:10.1146/annurev.neuro.30.051606.094313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Valdés-Ferrer, S. I. (2014). The challenges of long-term sepsis survivors: when surviving is just the beginning. Rev. Invest. Clin. 66, 439–449.
Valdés-Ferrer, S. I., Rosas-Ballina, M., Olofsson, P. S., Lu, B., Dancho, M. E., Li, J., et al. (2013a). HMGB1 mediates persistent splenocyte priming in sepsis survivors: evidence from a murine model. Shock 40, 492–495. doi:10.1097/SHK.0000000000000050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Valdés-Ferrer, S. I., Rosas-Ballina, M., Olofsson, P. S., Lu, B., Dancho, M. E., Ochani, M., et al. (2013b). HMGB1 mediates splenomegaly and expansion of splenic CD11b+ Ly-6C inflammatory monocytes in murine sepsis survivors. J. Intern. Med. 274, 381–390. doi:10.1111/joim.12104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Valdés-Ferrer, S. I., Vega, F., Cantú-Brito, C., Ceballos-Ceballos, J., Estañol, B., García-Ramos, G., et al. (2008). Cerebral changes in SLE with or without antiphospholipid syndrome. A case-control MRI study. J. Neuroimaging 18, 62–65. doi:10.1111/j.1552-6569.2007.00183.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verderio, C., Muzio, L., Turola, E., Bergami, A., Novellino, L., Ruffini, F., et al. (2012). Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann. Neurol. 72, 610–624. doi:10.1002/ana.23627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, J. H., Pappas, D., De Jager, PL., Pelletier, D., de Bakker, P. I., Kappos, L., et al. (2011). Modeling the cumulative genetic risk for multiple sclerosis from genome-wide association data. Genome Med. 3, 3. doi:10.1186/gm217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X. J., Kong, K. M., Qi, W. L., Ye, W. L., and Song, P. S. (2005). Interleukin-1 beta induction of neuron apoptosis depends on p38 mitogen-activated protein kinase activity after spinal cord injury. Acta Pharmacol. Sin. 26, 934–942. doi:10.1111/j.1745-7254.2005.00055.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waters, P. J., McKeon, A., Leite, M. I., Rajasekharan, S., Lennon, V. A., Villalobos, A., et al. (2012). Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 78, 665–671. doi:10.1212/WNL.0b013e318248dec1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watts, D. J., and Strogatz, S. H. (1998). Collective dynamics of ‘small-world’ networks. Nature 393, 440–442. doi:10.1038/30918
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weiss, N., Miller, F., Cazaubon, S., and Couraud, P. O. (2009). The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta 1788, 842–857. doi:10.1016/j.bbamem.2008.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Whitney, N. P., Eidem, T. M., Peng, H., Huang, Y., and Zheng, J. C. (2009). Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J. Neurochem. 108, 1343–1359. doi:10.1111/j.1471-4159.2009.05886.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wingerchuk, D. M., Hogancamp, W. F., O’Brien, P. C., and Weinshenker, B. G. (1999). The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 53, 1107–1114. doi:10.1212/WNL.53.5.1107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wolf, S. A., Steiner, B., Akpinarli, A., Kammertoens, T., Nassenstein, C., Braun, A., et al. (2009). CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J. Immunol. 182, 3979–3984. doi:10.4049/jimmunol.0801218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong, M. L., Rettori, V., al-Shekhlee, A., Bongiorno, P. B., Canteros, G., McCann, S. M., et al. (1996). Inducible nitric oxide synthase gene expression in the brain during systemic inflammation. Nat. Med. 2, 581–584. doi:10.1038/nm1196-1170a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wyss-Coray, T., Yan, F., Lin, A. H., Lambris, J. D., Alexander, J. J., Quigg, R. J., et al. (2002). Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc. Natl. Acad. Sci. U.S.A. 99, 10837–10842. doi:10.1073/pnas.162350199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yee, C., Yang, W., and Hekimi, S. (2014). The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 157, 897–909. doi:10.1016/j.cell.2014.02.055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yokoo, H., Chiba, S., Tomita, K., Takashina, M., Sagara, H., Yagisita, S., et al. (2012). Neurodegenerative evidence in mice brains with cecal ligation and puncture-induced sepsis: preventive effect of the free radical scavenger edaravone. PLoS ONE 7:e51539. doi:10.1371/journal.pone.0051539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, L., Chibnik, L. B., Srivastava, G. P., Pochet, N., Yang, J., Xu, J., et al. (2015). Association of brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 72, 15–24. doi:10.1001/jamaneurol.2014.3049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, B., Gaiteri, C., Bodea, L. G., Wang, Z., McElwee, J., Podtelezhnikov, A. A., et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. doi:10.1016/j.cell.2013.03.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zitvogel, L., Kepp, O., and Kroemer, G. (2010). Decoding cell death signals in inflammation and immunity. Cell 140, 798–804. doi:10.1016/j.cell.2010.02.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: neurodegeneration, systemic inflammation and sepsis, autoimmune disorders, anti-brain antibodies, TNF, HMGB1, connectome
Citation: Sankowski R, Mader S and Valdés-Ferrer SI (2015) Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front. Cell. Neurosci. 9:28. doi: 10.3389/fncel.2015.00028
Received: 29 November 2014; Accepted: 15 January 2015;
Published online: 02 February 2015.
Edited by:
Victoria Campos, Instituto Nacional de Neurologia y Neurocirugia, MexicoReviewed by:
Rafael Linden, Federal University of Rio de Janeiro, BrazilMuzamil Ahmad, Indian Institute of Integrative Medicine, India
Claudia Verderio, Istituto di Neuroscienze, Italy
Copyright: © 2015 Sankowski, Mader and Valdés-Ferrer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roman Sankowski, Simone Mader, and Sergio Iván Valdés-Ferrer, Feinstein Institute for Medical Research, 350 Community Dr., Manhasset, NY 11030, USA e-mail:cnNhbmtvd3NraUBuc2hzLmVkdQ==;c21hZGVyQG5zaHMuZWR1;c3ZhbGRlc2ZlckBuc2hzLmVkdSw=c2l2YWxkZXNAZ21haWwuY29t
†Roman Sankowski, Simone Mader and Sergio Iván Valdés-Ferrer have contributed equally to this work.