 Suzanne S. Bohlson
Suzanne S. Bohlson Sean D. O’Conner1
Sean D. O’Conner1 Holly Jo Hulsebus
Holly Jo Hulsebus Minh-Minh Ho
Minh-Minh Ho Deborah A. Fraser
Deborah A. Fraser- 1Department of Microbiology and Immunology, Des Moines University, Des Moines, IA, USA
- 2Department of Biologicial Sciences, California State University Long Beach, Long Beach, CA, USA
Complement is a critical system of enzymes, regulatory proteins, and receptors that regulates both innate and adaptive immune responses. Natural mutations in complement molecules highlight their requirement in regulation of a variety of human conditions including infectious disease and autoimmunity. As sentinels of the immune system, macrophages are specialized to respond to infectious microbes, as well as normal and altered self, and dictate appropriate immune responses. Complement components such as anaphylatoxins (C3a and C5a) and opsonins [C3b, C1q, mannan binding lectin (MBL)] influence macrophage responses. While anaphylatoxins C3a and C5a trigger inflammasome activation, opsonins such as C1q and related molecules (MBL and adiponectin) downregulate inflammasome activation and inflammation, and upregulate engulfment of apoptotic cells consistent with a pro-resolving or M2 macrophage phenotype. This review summarizes our current understanding of the influence of the complement system on macrophage polarization with an emphasis on C1q and related molecules.
Complement System
The complement system comprises over 35 cell associated and soluble molecules, which play a critical role in our innate immune response. Activation of complement begins with a recognition step. Recognition proteins of the complement system include C1q, mannan binding lectin (MBL), and ficolins (ficolin-1, -2, -3). These proteins are innate pattern recognition receptors (PRRs) and are capable of recognizing a wide range of structures including foreign organisms, either via binding directly to their pathogen associated molecular patterns (PAMPs) or when coated with antibody in an immune-complex (C1q) [reviewed in Ref. (1)]. In addition, these PRRs also recognize and bind to structures associated with cellular damage/debris such as apoptotic cell associated molecular patterns (ACAMPs) like phosphatidylserine, damage associated molecular patterns (DAMPs) like oxidation neo-epitopes, and fibrillar protein structures (2–8). Activation of complement via the classical pathway (C1q), lectin pathway (MBL/ficolins), or alternative pathway (C3 “tickover” autoactivation/properdin) begins a coordinated cascade of enzymatic cleavage events generating complement protein fragments that carry out effector functions. These include opsonization for enhanced phagocytosis, either directly or via production of C3b, triggering inflammation through production of anaphylatoxins C3a and C5a, and lysis of target cells through deposition of C5b-9, the membrane attack complex (MAC), in the complement terminal pathway (Figure 1).
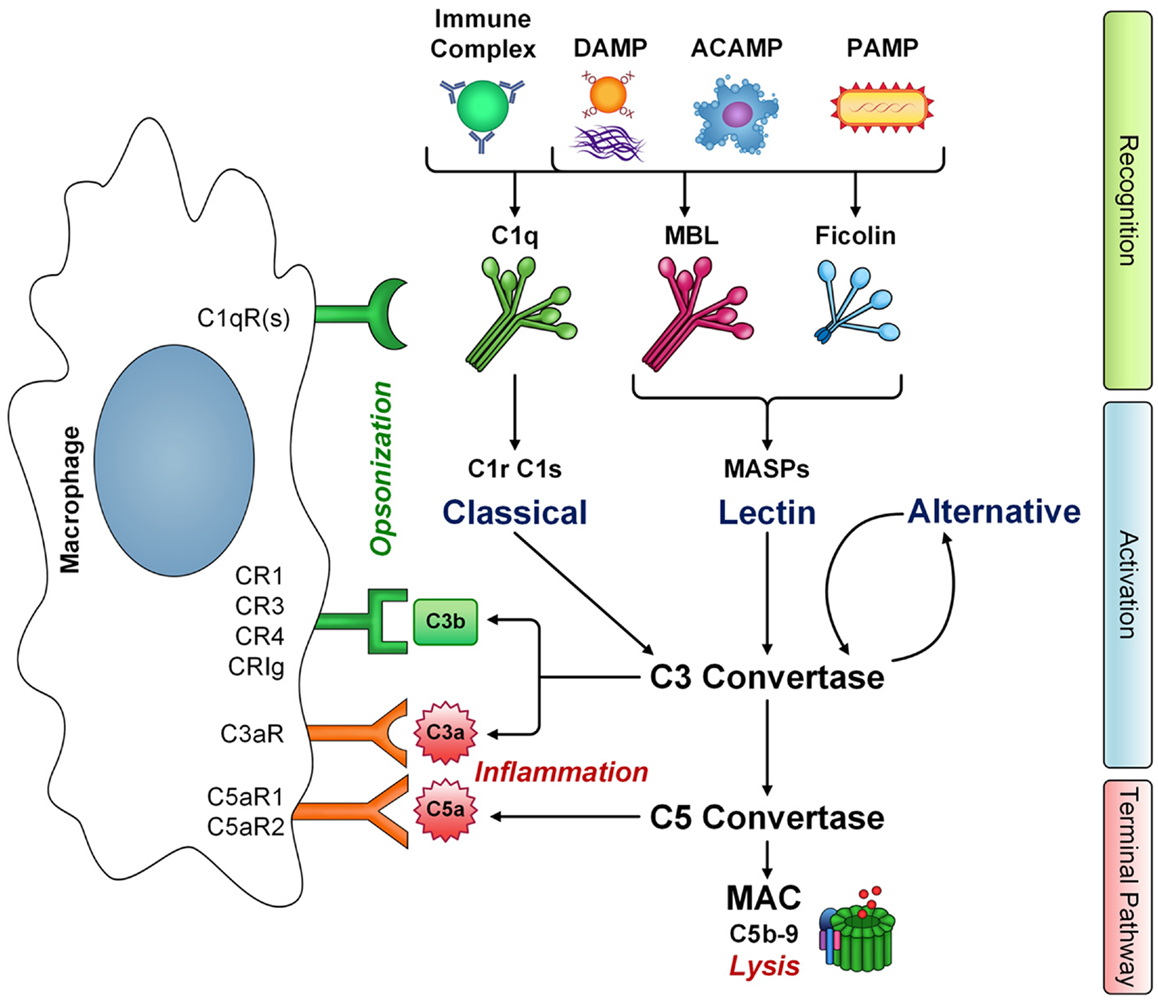
Figure 1. The interaction of macrophages with complement. Innate immune pattern recognition receptors C1q, MBL, and ficolins recognize a number of structures including immune complexes, damaged-self molecules expressing damage associated molecular patterns (DAMPS), apoptotic cells (ACAMPs), and pathogens (PAMPs), leading to activation of classical and lectin pathways, respectively, and amplification via the alternative pathway. Complement activation has three main effector functions: cytolysis via membrane attack complex (MAC) formation, inflammation mediated by anaphylatoxins C3a and C5a, and opsonization leading to phagocyte clearance via C3b deposition. However, C1q, MBL, and ficolins also have non-cascade activation related functions and are directly opsonic, leading to enhanced clearance of targets and phagocyte activation. Macrophages express a number of receptors that recognize complement components. The gC1qR and cC1qR bind the globular heads and collagen-like tail domains of C1q, respectively. However, additional C1q receptors likely exist (C1qR), some of which may also recognize the closely related collagen-like domains of other defense collagen family members, MBL and ficolins. Additional complement receptors on macrophages CR1, CR3, CR4, and CRIg recognize C3b-opsonized targets either as intact C3b or its degradation fragment iC3b. Macrophages also express receptors for complement activation fragments C3a (C3aR) and C5a (C5aR1 and C5aR2). Thus, macrophages are key players in carrying out the effector functions of complement activation.
Complement Deficiencies in Human Disease
Genetic deficiencies in complement components highlight its important role not only in clearance of pathogens but also in removal of dying cells/cellular debris and prevention of autoimmunity. For example, genetic deficiencies in lectin, alternative and terminal pathway components like MBL, factor D, properdin, C3, C5, C6, C7, C8, and C9, increase susceptibility to infections, in particular, by encapsulated bacteria [reviewed in Ref. (9)]. The strong link between late complement component deficiencies (C5–9) and recurrent neisserial infections indicates a critical role for C5b–9 MAC deposition in direct bactericidal defense. However, deficiency in early classical pathway component C1q is strongly associated with development of the autoimmune disease systemic lupus erythematosus (SLE), likely due to impaired opsonophagocytosis, and compromised removal of immune complexes and apoptotic cells [reviewed in Ref. (10)]. Weaker associations with SLE are also seen with deficiencies in other classical pathway components C1r/s, C2, C4, and C3. Thus, complement opsonization via C1q recognition plays a critical role in maintaining normal tissue homeostasis and prevention of autoimmunity. While excessive or inappropriate complement activation is associated with almost all inflammatory or inflammation-related diseases including cancer, Alzheimer’s disease (AD), and metabolic disease [reviewed in Ref. (11, 12)], associations of these diseases with (generally very rare) complement deficiencies in humans have not been reported. However, polymorphisms in MBL2 are common, and provide extensive evidence for links between MBL levels and cardiovascular disease [reviewed in Ref. (13)].
Complement Receptors on Macrophages
Macrophages are key players in carrying out complement effector functions. Complement components including C1q, MBL, C3b, C4b, C3a, and C5a interact with a variety of receptors on macrophages, leading to modulation of cytokine production/inflammatory responses and increased opsonophagocytic clearance of targets. For example, monocytes and macrophages express complement receptors CR1 (CD35), CR3 (CD11b/CD18), and CR4 (CD11c/CD18). CR1 binds complement opsonins C1q, C3b, and C4b, which are deposited on target cells/surfaces (14) and reviewed in Ref. (15). Binding to CR1 promotes phagocytosis of targets, along with degradation of C3b to its inactive fragment iC3b, preventing C5-convertase activity and thus inhibiting the complement terminal pathway. CR3 and CR4 bind iC3b and promote the phagocytosis of targets. Genetic deficiencies in these receptors are also a risk factor for the development of SLE [reviewed in Ref. (10, 16)]. A subset of tissue macrophages expresses the complement receptor CRIg. Gene expression of this receptor is associated with activated macrophages (17), and the protein is found in human liver Kupffer cells and in subsets of various resident tissue macrophages including alveolar and synovial macrophages (18). CRIg binds to C3b, and its degradation product, iC3b, and was shown to be important in clearance of C3b-opsonized pathogens from the circulation.
Beyond CR1, macrophages express other receptors capable of binding C1q. These include the ubiquitously co-expressed molecules gC1qR and cC1qR. gC1qR binds to the globular heads of C1q (19), whereas cC1qR (calreticulin) interacts with both the collagen-like tail and globular head domains of C1q (4). C1q opsonized targets are internalized more rapidly than in the absence of C1q via interaction of the collagen-like tail with a receptor on phagocytes (20). In addition, C1q bound to a variety of targets modulates macrophage inflammatory responses via its collagen-like domain (5, 21). The collagen-like domain of C1q is a feature shared with other so-called “defense collagens.” This family of molecules includes MBL and ficolins, which also trigger enhanced phagocytosis [reviewed in Ref. (1)] and modulate cytokine production (22). While the phagocytic receptor is not definitively identified for all targets, involvement of the cC1qR has been implicated in the C1q- and MBL-mediated removal of apoptotic cells by macrophages (6). An additional family of proteins, termed C1q/TNF-related proteins (CTRPs), contains C-terminal globular domains homologous to C1q (23, 24). Adiponectin is perhaps the best characterized member of the CTRP family, and interacts with macrophages, regulating inflammatory responses similar to C1q (25). However, the receptors for the metabolic actions of adiponectin were shown to be distinct from the C1qR, and include AdipoR1 and AdiopR2 (26).
Complement Anaphylatoxins
Complement anaphylatoxins C3a and C5a are soluble complement fragments produced from C3 or C5 by activation of the C3- or C5-convertase enzyme complex, respectively. They carry out their biological functions via interactions with three receptors. These include the C3a receptor (C3aR) (27), which binds C3a but not its degradation product C3a-desArg, the C5a receptor (C5aR1) (28), which binds C5a, and C5a receptor-like 2 (C5aR2, C5L2) (29), which binds C5a but has greater affinity for C5a-desArg. C5aR2 lacks signaling capabilities (30), and thus, is considered a decoy receptor, capable of sequestering the bioavailability of C5a/C5a-desArg and limiting their ability to activate via the C5aR1. C5aR2 may also bind C3a-desArg, but this is controversial (31). Cellular expression of the anaphylatoxin receptors is widespread but particularly includes immune cells like monocytes and macrophages (32, 33). Interestingly, LPS, associated with M1 macrophage polarization increases gene expression of C5aR1 in macrophages (34) while IL-4, associated with M2 macrophage polarization downregulates C5aR1 expression (35).
Macrophage Polarization
Macrophages are grouped as M1 and M2 in accordance with the Th1/Th2 nomenclature, and this terminology describes two macrophage phenotypes: the pro-inflammatory/classically activated macrophage (M1) and the pro-resolving/alternatively activated macrophage (M2). The expression of a variety of genes has been associated with macrophage polarization, most notably, the machinery required for enzymatic breakdown of arginine in pro-resolving and pro-inflammatory macrophages. By expressing arginase, pro-resolving macrophages generate ornithine, which promotes proliferation and repair, whereas pro-inflammatory (M1) macrophages express inducible nitric oxide synthase and generate nitric oxide (NO), an important molecule in host defense against invading pathogens, which also inhibits cell proliferation [reviewed in Ref. (36)]. Intermediates in the two enzymatic pathways act antagonistically, inhibiting the other when they are activated (37). Consistent with a role in promotion of inflammation, M1 macrophages are also often associated with an increased production of pro-inflammatory cytokines such as TNFα and IL-1β. In contrast, M2 macrophages, which are pro-resolving, are often associated with increased production of anti-inflammatory cytokine IL-10.
Although the idea of macrophages having separate phenotypes is helpful in defining their action, it is also somewhat misleading and over simplified. In vivo, macrophages are constantly encountering various external signals leading to a very fluid existence in terms of phenotypes (38). Even within the same site and population, macrophages can express different and constantly changing phenotypes, termed macrophage plasticity (39). Macrophage polarization is a result of a combination of external signals macrophages receive from their environment. Common signals that have been investigated include the Th1 cytokine interferon-γ (IFN-γ) and various PAMPs, which are associated with M1 polarization and the Th2 cytokines IL-4/IL-13, which are associated with M2 polarization. Here, we will review some recent work illuminating the role of the complement system in regulating macrophage activation and polarization.
Complement Anaphylatoxins Regulate Macrophage Activation
Complement anaphylatoxins, C3a and C5a, are pro-inflammatory and trigger monocyte and macrophage activation through various signaling mechanisms. For example, upon LPS stimulation in human monocytes, C3a induces NLRP3 inflammasome activation through an increase in ATP release mediated by extracellular signal-regulated kinase 1/2 (ERK1/2) (40). Samstad et al. found that C5a produced during complement activation by cholesterol crystals, influenced inflammation through NLRP3 inflammasome activation and IL-1β and TNFα release, and increased the production of reactive oxygen species (ROS) (41). C5a was also correlated with IL-6 induction and development of inflammatory T-helper 17 cells (42), as well as affecting IL-17 and IL-23 production (43). Interestingly, sublethal MAC (C5b-9) deposition rather than C3a or C5a was demonstrated to trigger inflammasome activation in murine dendritic cells following LPS stimulation (44). In the CNS, C5a provides a chemotactic and activation signal for microglia and astrocytes [reviewed in Ref. (45)]. It can also synergize with damage signals such as amyloid beta (Aβ) to trigger enhanced inflammatory cytokine production (46). Thus, activation of complement by extracellular Aβ plaques can exacerbate inflammation and may play a substantial role in the pathogenesis of AD. Indeed, treatment with a C5a receptor antagonist was shown to decrease deposition of fibrillar Aβ and inflammatory glia and improve cognitive performance in mice models of AD (47). Many other diseases are associated with anaphylatoxin signaling, including allergic, infectious, autoimmune diseases, and cancer (48). Clearly, the pro-inflammatory signaling provided by the anaphylatoxins contributes to both beneficial (pathogen clearing) and detrimental (inflammatory disease-related) inflammation.
Complement Opsonins Regulate Macrophage Activation
C3b-Mediated Opsonization
One of the major effector functions of the complement system is the tagging, or opsonization, of pathogens and/or cellular debris for clearance by phagocytes. Complement component C3 is the most abundant complement component in blood at about 1.2 mg/ml, and as such, permissible surfaces become readily coated in C3b and iC3b following cleavage of C3. CR3 is a major phagocytic receptor expressed on macrophages that is involved in clearance of iC3b opsonized particles (Figure 1). In contrast to other phagocytic receptors such as Fcγ receptors, engulfment of iC3b coated particles via CR3 has traditionally been considered anti-inflammatory. For example, CR3-dependent engulfment does not activate the arachidonic acid cascade (49) or the release of toxic oxygen products (50). In early experiments assessing macrophage heterogeneity, Stein and colleagues demonstrated increased secretion of TNFα from macrophages elicited into the peritoneal cavity by inflammatory mediators (e.g., thioglycollate) when compared to resident peritoneal macrophages. However, independent of the macrophage phenotype, all macrophages failed to secrete significant levels of TNFα following ligation of CR3 whereas ligation of Fcγ receptors led to TNFα release by all macrophage subsets (51). In studies with the macrophage intracellular pathogen Mycobacterium avium, C3-independent phagocytosis of M. avium resulted in enhanced TNFα production (52, 53). More recent reports assessing pathogenesis of Francisella tularensis support a role for CR3 in inducing immune suppression and facilitating infection with this intracellular pathogen (54). While CR3-dependent immune suppression is detrimental in the course of F. tularensis infection or other infectious disease processes, it is beneficial in the context of clearance of apoptotic cells and/or cellular debris.
Complement-Dependent Engulfment of Apoptotic Cells
Complement components readily coat the surface of apoptotic cells and facilitate ingestion by macrophages (55, 56). Ingestion of apoptotic cells is a silent process accompanied by the production of anti-inflammatory cytokines TGFβ and IL-10 (57). Complement mediated opsonization of apoptotic cells is largely dependent on the classical complement pathway, and deficiencies in early components of the classical pathway (C1q, C4, and C2) result in inefficient disposal of apoptotic cells and subsequent autoimmunity (58–60). While C1q deficiency results in development of lupus in virtually all cases, absence of C3, C2, and C4 results in lupus at lower frequency indicating a role for C1q beyond classical complement pathway activation in regulation of the immune response (61). Recent reports have indicated that C1q regulates the monocyte/macrophage/dendritic cell phenotype leading to development of a phagocyte that is specialized to resolve inflammation. Specifically, the C1q-stimulated phagocyte is pro-efferocytic and anti-inflammatory (Figure 2). As such, complement opsonins are mediating activity beyond the immediate stimulation of enhanced phagocytosis; they are inducing a macrophage phenotype, or polarizing macrophages toward a pro-resolving phenotype. This would be consistent with observation that C1q deficiency results in autoimmunity and chronic inflammation.
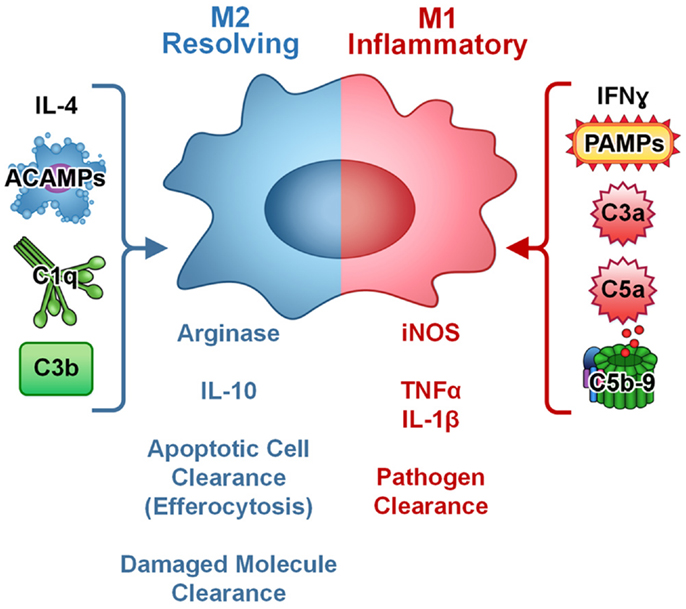
Figure 2. Complement regulates macrophage polarization. In culture, M1 macrophages (inflammatory, pathogen clearing) are induced by IFNγ or PAMPs and are characterized by production of iNOS and pro-inflammatory cytokines such as TNFα and IL-1β. M2 macrophages (resolving, apoptotic cell and damaged molecule clearing) are induced by IL-4 and characterized by arginase and anti-inflammatory cytokine IL-10 production. Complement components C3a, C5a, and C5b-9 modulate cytokine production in macrophages toward an inflammatory (M1-like) phenotype. Apoptotic cells and targets opsonized with complement components C1q or C3b increase clearance and modulate cytokine production in macrophages toward an anti-inflammatory, resolving (M2-like) phenotype and can block PAMP-mediated pro-inflammatory signaling. Thus, complement plays a dual role in macrophage activation and polarization depending on the target.
Korb and Ahearn were the first to describe a role for C1q in the clearance of apoptotic cells, and suggested that C1q-dependent engulfment of apoptotic cells was important in prevention of autoimmunity in lupus (62). There has been wide support for this hypothesis, and Bhatia et al. demonstrated that removal of apoptotic cells by C1q was also important in prevention of the inflammatory disease atherosclerosis (63). However, the receptors/signal transduction pathway leading to C1q-dependent efferocytosis has not been clearly delineated [reviewed in Ref. (56)]. C1q binds to apoptotic cells and serves as a bridging molecule linking the apoptotic cell to the phagocyte via calreticulin (cC1qR) and its binding partner, the phagocytic receptor LRP (CD91) (6). However, macrophages deficient in LRP still respond to C1q with enhanced phagocytosis and efferocytosis indicating that there are multiple mechanisms of C1q-dependent engulfment (64). We demonstrated that mouse bone marrow derived macrophages and peritoneal macrophages stimulated with C1q upregulated expression of engulfment machinery including Mer tyrosine kinase and the MerTK ligand Gas6, leading to development of a macrophage that is primed for efferocytosis (65). More recently, we showed that this pathway is shared with a C1q homolog, adiponectin (66) and not with MBL, a C1q-related collectin (65).
C1q and Adiponectin Mediate Efferocytosis via a Shared Pathway
Adiponectin is referred to as an adipokine; it is produced by adipocytes and is secreted into circulation where it modulates biological responses via several receptors including adiponectin receptor 1 (AdipoR1), adiponectin receptor 2 (AdipoR2), or T cadherin (T-cad) (67, 68). More recent studies have suggested there is an additional receptor on macrophages that mediates adiponectin signaling; however, this receptor has not been identified (69). Adiponectin signaling leads to activation of 5′ adenosine monophosphate-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor alpha (PPARα), and adiponectin-dependent regulation of metabolism is mediated via these key signaling nodes (70). Much information has been gathered regarding the role of AMPK in the regulation of metabolic activity; however, recent studies suggest that AMPK also influences the immune response, including macrophage cytokine expression and phagocytosis. For example, silencing of AMPK inhibits LPS- and fatty acid-mediated inflammation in macrophages (71). In addition, AMPK activation is associated with enhanced phagocytosis and efferocytosis, as well as macrophage polarization (72, 73). Similarly, C1q stimulates enhanced phagocytosis and diminution of pro-inflammatory cytokine production from myeloid cells, and we recently demonstrated that C1q and adiponectin-dependent Mer expression and efferocytosis require activation of AMPK (66). These studies have begun to define the mechanism by which C1q and related opsonins modulate macrophage activation.
C1q and MBL Inhibit Pro-Inflammatory and Promote Anti-Inflammatory Cytokine Production
In line with these observations, Fraser and colleagues demonstrated that both C1q-stimulated human monocytes and C1q-stimulated mouse microglia produce increased anti-inflammatory IL-10 and decreased pro-inflammatory cytokines following stimulation with the TLR4 ligand LPS (21, 22). Similar activity was shown for MBL indicating that this is a distinct mechanism for macrophage activation, independent of AMPK-mediated Mer expression since MBL failed to upregulate Mer-dependent efferocytosis. C1q/MBL-dependent activation of NFκB p50p50 homodimers were suggested to contribute to the anti-inflammatory phenotype via competitive inhibition of pro-inflammatory NFκB p50p65 heterodimer activation and/or via transcriptional activation of IL-10 (74). The same group demonstrated that C1q promoted M2 polarization and limited inflammasome activation in human monocyte derived macrophages (75). Interferon-alpha (IFN-α) is a pro-inflammatory cytokine that contributes to dendritic cell activation, a breakdown in peripheral tolerance and autoimmunity (76). C1q modulates IFN-α production from human phagocytes in response to a variety of stimuli (75, 77, 78). Santer and colleagues demonstrated that C1q deficiency in human lupus patients resulted in elevated IFN-α levels in serum and cerebrospinal fluid, and that C1q suppressed immune-complex stimulated IFN-α production from human monocytes (78–80). The anti-inflammatory effects of C1q are not limited to apoptotic cells. C1q has also been shown to enhance the uptake of atherogenic forms of lipoproteins such as oxidized or acetylated LDL (oxLDL, AcLDL) (81). During clearance of oxLDL by macrophages, C1q also modulates cytokine production toward an anti-inflammatory, resolving phenotype and dampens transcriptional activity by p50/p65 NFκB heterodimers, which may be important in limiting inflammation in the early atherosclerotic lesion (82). Combined, these data further support the hypothesis that C1q programs macrophages toward an anti-inflammatory, pro-efferocytic/phagocytic phenotype. Future studies should delineate the relative activity of C1q-dependent constitutive efferocytosis/phagocytosis versus programed polarization in the contribution toward protection from autoimmune and inflammatory disease.
Concluding Remarks
The complement system has traditionally been considered an arm of the innate immune response required for promotion of inflammation and pathogen clearance. While these functions of complement are essential to host defense, more recent advances demonstrate a novel role for components of the complement system in resolution of inflammation and protection from autoimmune and inflammatory diseases including SLE, neurodegenerative disease, and atherosclerosis. In particular, complement component C1q directs macrophage polarization leading to generation of pro-resolving macrophages that promote clearance of apoptotic cells with diminished pro-inflammatory cytokine production and increased anti-inflammatory cytokine production. C1q is synthesized by macrophages in response to tissue injury and is likely to be an important signal in resolution of inflammation independent of other complement components. Moreover, C1q-related molecules such as the complement component MBL, and the adipokine adiponectin, also downregulate macrophage-mediated inflammatory responses and upregulate efferocytosis. Identification of the molecular mechanisms by which these molecules govern macrophage activation, as well as their relative contribution to disease resolution, should reveal pathways to target for development of novel therapeutics in autoimmune and inflammatory disease.
Author Contributions
Suzanne Slater Bohlson outlined the review, invited co-authors, wrote the section on complement opsonins, extensively edited all sections, and prepared the manuscript for publication. Sean David O’Conner drafted the section on macrophage polarization. Holly Jo Hulsebus drafted the section on complement anaphylatoxins. Minh-Minh Ho designed and created the figures and figure legends. Deborah Ann Fraser wrote the section on complement and extensively edited the entire manuscript. All authors reviewed the final document prior to submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH NIAID R56AI099010-02 to Suzanne Slater Bohlson.
References
1. Fraser DA, Tenner AJ. Directing an appropriate immune response: the role of defense collagens and other soluble pattern recognition molecules. Curr Drug Targets (2008) 9(2):113–22. doi: 10.2174/138945008783502476
2. Biro A, Thielens NM, Cervenak L, Prohaszka Z, Fust G, Arlaud GJ. Modified low density lipoproteins differentially bind and activate the C1 complex of complement. Mol Immunol (2007) 44(6):1169–77. doi:10.1016/j.molimm.2006.06.013
3. Jiang H, Burdick D, Glabe CG, Cotman CW, Tenner AJ. Beta-amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J Immunol (1994) 152(10):5050–9.
4. Paidassi H, Tacnet-Delorme P, Verneret M, Gaboriaud C, Houen G, Duus K, et al. Investigations on the C1q-calreticulin-phosphatidylserine interactions yield new insights into apoptotic cell recognition. J Mol Biol (2011) 408(2):277–90. doi:10.1016/j.jmb.2011.02.029
5. Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem (2010) 112(3):733–43. doi:10.1111/j.1471-4159.2009.06494.x
6. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med (2001) 194(6):781–95. doi:10.1084/jem.194.6.781
7. Honore C, Hummelshoj T, Hansen BE, Madsen HO, Eggleton P, Garred P. The innate immune component ficolin 3 (Hakata antigen) mediates the clearance of late apoptotic cells. Arthritis Rheum (2007) 56(5):1598–607. doi:10.1002/art.22564
8. Matsushita M. Ficolins in complement activation. Mol Immunol (2013) 55(1):22–6. doi:10.1016/j.molimm.2012.08.017
9. Skattum L, van Deuren M, van der Poll T, Truedsson L. Complement deficiency states and associated infections. Mol Immunol (2011) 48(14):1643–55. doi:10.1016/j.molimm.2011.05.001
10. Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis (2014) 73(9):1601–6. doi:10.1136/annrheumdis-2014-205287
11. Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol (2011) 48(14):1592–603. doi:10.1016/j.molimm.2011.04.003
12. Hertle E, Stehouwer CD, van Greevenbroek MM. The complement system in human cardiometabolic disease. Mol Immunol (2014). doi:10.1016/j.molimm.2014.06.031
13. Pagowska-Klimek I, Cedzynski M. Mannan-binding lectin in cardiovascular disease. Biomed Res Int (2014) 2014:616817. doi:10.1155/2014/616817
14. Klickstein LB, Barbashov SF, Liu T, Jack RM, Nicholson-Weller A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity (1997) 7(3):345–55. doi:10.1016/S1074-7613(00)80356-8
15. Krych-Goldberg M, Atkinson JP. Structure-function relationships of complement receptor type 1. Immunol Rev (2001) 180:112–22. doi:10.1034/j.1600-065X.2001.1800110.x
16. Fagerholm SC, MacPherson M, James MJ, Sevier-Guy C, Lau CS. The CD11b-integrin (ITGAM) and systemic lupus erythematosus. Lupus (2013) 22(7):657–63. doi:10.1177/0961203313491851
17. Walker MG. Z39Ig is co-expressed with activated macrophage genes. Biochim Biophys Acta (2002) 1574(3):387–90. doi:10.1016/S0167-4781(01)00358-X
18. Helmy KY, Katschke KJ Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell (2006) 124(5):915–27. doi:10.1016/j.cell.2005.12.039
19. Ghebrehiwet B, Lim BL, Peerschke EI, Willis AC, Reid KB. Isolation, cDNA cloning, and overexpression of a 33-kD cell surface glycoprotein that binds to the globular “heads” of C1q. J Exp Med (1994) 179(6):1809–21. doi:10.1084/jem.179.6.1809
20. Bobak DA, Gaither TA, Frank MM, Tenner AJ. Modulation of FcR function by complement: subcomponent C1q enhances the phagocytosis of IgG-opsonized targets by human monocytes and culture-derived macrophages. J Immunol (1987) 138(4):1150–6.
21. Fraser DA, Laust AK, Nelson EL, Tenner AJ. C1q differentially modulates phagocytosis and cytokine responses during ingestion of apoptotic cells by human monocytes, macrophages, and dendritic cells. J Immunol (2009) 183(10):6175–85. doi:10.4049/jimmunol.0902232
22. Fraser DA, Bohlson SS, Jasinskiene N, Rawal N, Palmarini G, Ruiz S, et al. C1q and MBL, components of the innate immune system, influence monocyte cytokine expression. J Leukoc Biol (2006) 80(1):107–16. doi:10.1189/jlb.1105683
23. Ghai R, Waters P, Roumenina LT, Gadjeva M, Kojouharova MS, Reid KB, et al. C1q and its growing family. Immunobiology (2007) 212(4–5):253–66. doi:10.1016/j.imbio.2006.11.001
24. Schaffler A, Buechler C. CTRP family: linking immunity to metabolism. Trends Endocrinol Metab (2012) 23(4):194–204. doi:10.1016/j.tem.2011.12.003
25. Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood (2000) 96(5):1723–32.
26. Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med (2007) 13(3):332–9. doi:10.1038/nm1557
27. Ames RS, Li Y, Sarau HM, Nuthulaganti P, Foley JJ, Ellis C, et al. Molecular cloning and characterization of the human anaphylatoxin C3a receptor. J Biol Chem (1996) 271(34):20231–4. doi:10.1074/jbc.271.34.20231
28. Gerard NP, Gerard C. The chemotactic receptor for human C5a anaphylatoxin. Nature (1991) 349(6310):614–7. doi:10.1038/349614a0
29. Cain SA, Monk PN. The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-Arg(74). J Biol Chem (2002) 277(9):7165–9. doi:10.1074/jbc.C100714200
30. Okinaga S, Slattery D, Humbles A, Zsengeller Z, Morteau O, Kinrade MB, et al. C5L2, a nonsignaling C5A binding protein. Biochemistry (2003) 42(31):9406–15. doi:10.1021/bi034489v
31. Kalant D, Cain SA, Maslowska M, Sniderman AD, Cianflone K, Monk PN. The chemoattractant receptor-like protein C5L2 binds the C3a des-Arg77/acylation-stimulating protein. J Biol Chem (2003) 278(13):11123–9. doi:10.1074/jbc.M206169200
32. Monk PN, Scola AM, Madala P, Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol (2007) 152(4):429–48. doi:10.1038/sj.bjp.0707332
33. Zwirner J, Gotze O, Begemann G, Kapp A, Kirchhoff K, Werfel T. Evaluation of C3a receptor expression on human leucocytes by the use of novel monoclonal antibodies. Immunology (1999) 97(1):166–72. doi:10.1046/j.1365-2567.1999.00764.x
34. Hunt JR, Martin CB, Martin BK. Transcriptional regulation of the murine C5a receptor gene: NF-Y is required for basal and LPS induced expression in macrophages and endothelial cells. Mol Immunol (2005) 42(11):1405–15. doi:10.1016/j.molimm.2005.01.002
35. Soruri A, Kiafard Z, Dettmer C, Riggert J, Kohl J, Zwirner J. IL-4 down-regulates anaphylatoxin receptors in monocytes and dendritic cells and impairs anaphylatoxin-induced migration in vivo. J Immunol (2003) 170(6):3306–14. doi:10.4049/jimmunol.170.6.3306
36. Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol (2012) 32(6):463–88. doi:10.1615/CritRevImmunol.v32.i6.10
37. Morris SM Jr. Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol (2009) 157(6):922–30. doi:10.1111/j.1476-5381.2009.00278.x
38. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
39. Mills CD. Macrophage arginine metabolism to ornithine/urea or nitric oxide/citrulline: a life or death issue. Crit Rev Immunol (2001) 21(5):399–425. doi:10.1615/CritRevImmunol.v21.i5.10
40. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood (2013) 122(20):3473–81. doi:10.1182/blood-2013-05-502229
41. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol (2014) 192(6):2837–45. doi:10.4049/jimmunol.1302484
42. Fang C, Zhang X, Miwa T, Song WC. Complement promotes the development of inflammatory T-helper 17 cells through synergistic interaction with toll-like receptor signaling and interleukin-6 production. Blood (2009) 114(5):1005–15. doi:10.1182/blood-2009-01-198283
43. Grailer JJ, Bosmann M, Ward PA. Regulatory effects of C5a on IL-17A, IL-17F, and IL-23. Front Immunol (2013) 3:387. doi:10.3389/fimmu.2012.00387
44. Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, et al. Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1beta release. J Immunol (2013) 191(3):1006–10. doi:10.4049/jimmunol.1300489
45. Yao J, Harvath L, Gilbert DL, Colton CA. Chemotaxis by a CNS macrophage, the microglia. J Neurosci Res (1990) 27(1):36–42. doi:10.1002/jnr.490270106
46. O’Barr S, Cooper NR. The C5a complement activation peptide increases IL-1beta and IL-6 release from amyloid-beta primed human monocytes: implications for Alzheimer’s disease. J Neuroimmunol (2000) 109(2):87–94. doi:10.1016/S0165-5728(00)00291-5
47. Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol (2009) 183(2):1375–83. doi:10.4049/jimmunol.0901005
48. Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol (2009) 46(14):2753–66. doi:10.1016/j.molimm.2009.04.027
49. Aderem AA, Wright SD, Silverstein SC, Cohn ZA. Ligated complement receptors do not activate the arachidonic acid cascade in resident peritoneal macrophages. J Exp Med (1985) 161(3):617–22. doi:10.1084/jem.161.3.617
50. Wright SD, Silverstein SC. Receptors for C3b and C3bi promote phagocytosis but not the release of toxic oxygen from human phagocytes. J Exp Med (1983) 158(6):2016–23. doi:10.1084/jem.158.6.2016
51. Stein M, Gordon S. Regulation of tumor necrosis factor (TNF) release by murine peritoneal macrophages: role of cell stimulation and specific phagocytic plasma membrane receptors. Eur J Immunol (1991) 21(2):431–7. doi:10.1002/eji.1830210227
52. Bohlson SS, Strasser JA, Bower JJ, Schorey JS. Role of complement in Mycobacterium avium pathogenesis: in vivo and in vitro analyses of the host response to infection in the absence of complement component C3. Infect Immun (2001) 69(12):7729–35. doi:10.1128/IAI.69.12.7729-7735.2001
53. Irani VR, Maslow JN. Induction of murine macrophage TNF-alpha synthesis by Mycobacterium avium is modulated through complement-dependent interaction via complement receptors 3 and 4 in relation to M. avium glycopeptidolipid. FEMS Microbiol Lett (2005) 246(2):221–8. doi:10.1016/j.femsle.2005.04.008
54. Dai S, Rajaram MV, Curry HM, Leander R, Schlesinger LS. Fine tuning inflammation at the front door: macrophage complement receptor 3-mediates phagocytosis and immune suppression for Francisella tularensis. PLoS Pathog (2013) 9(1):e1003114. doi:10.1371/journal.ppat.1003114
55. Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med (1998) 188(12):2313–20. doi:10.1084/jem.188.12.2313
56. Galvan MD, Greenlee-Wacker MC, Bohlson SS. C1q and phagocytosis: the perfect complement to a good meal. J Leukoc Biol (2012) 92(3):489–97. doi:10.1189/jlb.0212099
57. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101(4):890–8. doi:10.1172/JCI1112
58. Gullstrand B, Martensson U, Sturfelt G, Bengtsson AA, Truedsson L. Complement classical pathway components are all important in clearance of apoptotic and secondary necrotic cells. Clin Exp Immunol (2009) 156(2):303–11. doi:10.1111/j.1365-2249.2009.03896.x
59. Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiology (2002) 205(4–5):395–406. doi:10.1078/0171-2985-00141
60. Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med (2000) 192(3):359–66. doi:10.1084/jem.192.3.359
61. Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology (1998) 199(2):265–85. doi:10.1016/S0171-2985(98)80032-6
62. Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol (1997) 158(10):4525–8.
63. Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol (2007) 170(1):416–26. doi:10.2353/ajpath.2007.060406
64. Lillis AP, Greenlee MC, Mikhailenko I, Pizzo SV, Tenner AJ, Strickland DK, et al. Murine low-density lipoprotein receptor-related protein 1 (LRP) is required for phagocytosis of targets bearing LRP ligands but is not required for C1q-triggered enhancement of phagocytosis. J Immunol (2008) 181(1):364–73. doi:10.4049/jimmunol.181.1.364
65. Galvan MD, Foreman DB, Zeng E, Tan JC, Bohlson SS. Complement component C1q regulates macrophage expression of Mer tyrosine kinase to promote clearance of apoptotic cells. J Immunol (2012) 188(8):3716–23. doi:10.4049/jimmunol.1102920
66. Galvan MD, Hulsebus H, Heitker T, Zeng E, Bohlson S. Complement protein C1q and adiponectin stimulate Mer tyrosine kinase dependent engulfment of apoptotic cells through a shared pathway. J Innate Immun (2014). doi:10.1159/000363295
67. Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev (2005) 26(3):439–51. doi:10.1210/er.2005-0005
68. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature (2003) 423(6941):762–9. doi:10.1038/nature01705
69. Awazawa M, Ueki K, Inabe K, Yamauchi T, Kubota N, Kaneko K, et al. Adiponectin enhances insulin sensitivity by increasing hepatic IRS-2 expression via a macrophage-derived IL-6-dependent pathway. Cell Metab (2011) 13(4):401–12. doi:10.1016/j.cmet.2011.02.010
70. Heiker JT, Kosel D, Beck-Sickinger AG. Molecular mechanisms of signal transduction via adiponectin and adiponectin receptors. Biol Chem (2010) 391(9):1005–18. doi:10.1515/bc.2010.104
71. Sag D, Carling D, Stout RD, Suttles J. Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol (2008) 181(12):8633–41. doi:10.4049/jimmunol.181.12.8633
72. Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, et al. AMPKalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab (2013) 18(2):251–64. doi:10.1016/j.cmet.2013.06.017
73. Jiang S, Park DW, Stigler WS, Creighton J, Ravi S, Darley-Usmar V, et al. Mitochondria and AMP-activated protein kinase-dependent mechanism of efferocytosis. J Biol Chem (2013) 288(36):26013–26. doi:10.1074/jbc.M113.489468
74. Fraser DA, Arora M, Bohlson SS, Lozano E, Tenner AJ. Generation of inhibitory NFkappaB complexes and phosphorylated cAMP response element-binding protein correlates with the anti-inflammatory activity of complement protein C1q in human monocytes. J Biol Chem (2007) 282(10):7360–7. doi:10.1074/jbc.M605741200
75. Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol (2012) 188(11):5682–93. doi:10.4049/jimmunol.1103760
76. Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev (2008) 19(1):41–52. doi:10.1016/j.cytogfr.2007.10.004
77. Lood C, Gullstrand B, Truedsson L, Olin AI, Alm GV, Ronnblom L, et al. C1q inhibits immune complex-induced interferon-alpha production in plasmacytoid dendritic cells: a novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum (2009) 60(10):3081–90. doi:10.1002/art.24852
78. Santer DM, Hall BE, George TC, Tangsombatvisit S, Liu CL, Arkwright PD, et al. C1q deficiency leads to the defective suppression of IFN-alpha in response to nucleoprotein containing immune complexes. J Immunol (2010) 185(8):4738–49. doi:10.4049/jimmunol.1001731
79. Santer DM, Yoshio T, Minota S, Moller T, Elkon KB. Potent induction of IFN-alpha and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J Immunol (2009) 182(2):1192–201. doi:10.4049/jimmunol.182.2.1192
80. Santer DM, Wiedeman AE, Teal TH, Ghosh P, Elkon KB. Plasmacytoid dendritic cells and C1q differentially regulate inflammatory gene induction by lupus immune complexes. J Immunol (2012) 188(2):902–15. doi:10.4049/jimmunol.1102797
81. Fraser DA, Tenner AJ. Innate immune proteins C1q and mannan-binding lectin enhance clearance of atherogenic lipoproteins by human monocytes and macrophages. J Immunol (2010) 185(7):3932–9. doi:10.4049/jimmunol.1002080
Keywords: macrophage, complement, C1q, adiponectin, inflammasome, cytokine, phagocytosis, efferocytosis
Citation: Bohlson SS, O’Conner SD, Hulsebus HJ, Ho M-M and Fraser DA (2014) Complement, C1q, and C1q-related molecules regulate macrophage polarization. Front. Immunol. 5:402. doi: 10.3389/fimmu.2014.00402
Received: 26 June 2014; Accepted: 07 August 2014;
Published online: 21 August 2014.
Edited by:
Laurel L. Lenz, National Jewish Health, USAReviewed by:
Cecilia Garlanda, Istituto Clinico Humanitas, ItalyDaisuke Kamimura, Hokkaido University, Japan
Copyright: © 2014 Bohlson, O’Conner, Hulsebus, Ho and Fraser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suzanne S. Bohlson, Department of Microbiology and Immunology, Des Moines University, 3200 Grand Avenue, Des Moines, IA 50312-4198, USA e-mail: suzanne.bohlson@dmu.edu