The Ca
2+ ion is a central signaling molecule in numerous cellular functions including apoptosis, energy production, gene regulation, cell proliferation, membrane excitability, synaptic transmission and plasticity. Due to the ubiquitous nature of Ca
2+ signaling, Ca
2+ is one of the most highly regulated ions with the concentration inside the cell maintained at a level 10,000 times lower than the concentration in the extracellular space (
Berridge et al., 2000
;
Rizzuto, 2001
;
Orrenius et al., 2003
). Accordingly, any change in Ca
2+ regulating mechanisms, unless compensated by another mechanism, will result in an alteration in cell function.
The Ca
2+ ‘dysregulation’ hypothesis of brain aging and Alzheimer’s disease formulated in the 1980s was based on discrete observations of alterations in processes that are regulated by Ca
2+ (
Landfield and Pitler, 1984
;
Gibson and Peterson, 1987
;
Khachaturian, 1989
). Over 20 years of research has accumulated substantial evidence for alterations in Ca
2+ homeostasis in contributing to cellular senescence. However, no single mechanism for Ca
2+ dysregulation has been found. Rather the causes and consequences of Ca
2+ dysregulation vary across the nervous system. As we increase our sophistication for identifying molecular and cellular processes, we are likely to find complex patterns of impaired/spared cellular function related to multiple Ca
2+ regulating mechanisms. Several recent reviews have detailed how different mechanisms for Ca
2+ dysregulation contribute to changes in cell excitability (
Disterhoft and Oh, 2006
) and synaptic plasticity (
Foster, 2007
) in the hippocampus. Conversely, in other regions of the nervous system, changes in Ca
2+ regulation may represent compensation to delay physiological aging (
Buchholz et al., 2007
;
Murchison and Griffith, 2007
), suggesting that cell specific differences in the expression of Ca
2+ regulating mechanisms may contribute to regional differences in the rate of brain aging. Similarly, cell specific differences in Ca
2+ regulating mechanisms may interact with neurodegenerative disorders to determine the pattern of cell death within the brain (
Morrison et al., 1998
;
LaFerla, 2002
;
Mattson, 2007
;
Chan et al., 2009
;
Naidoo, 2009
). Thus, mutations for genes involved in one aspect of Ca
2+ regulation may result is pathogenesis in regions which are more susceptible to other forms of Ca
2+ dysregulation. In this review, we focus on cell specificity in the operation of a subset of Ca
2+ regulatory mechanisms during aging and how this specificity might relate to the loss or preservation of cell function.
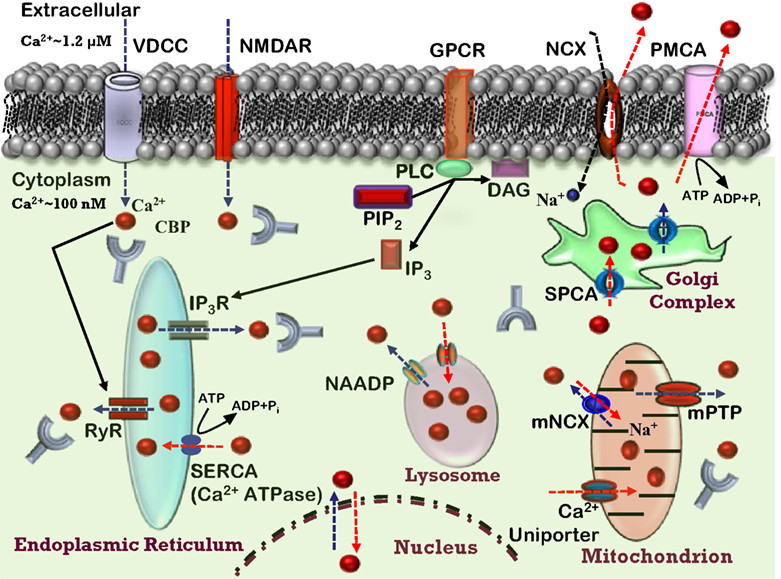
Ca
2+-signaling depends principally on a rapid and transient increase in intracellular Ca
2+ concentration through influx of Ca
2+ from several sources. In most cells, multiple mechanisms exist whereby elevation in intracellular Ca
2+ concentrations may occur. The major sources of intracellular Ca
2+ include Ca
2+ influx through ligand-gated glutamate receptors, such as
N-methyl-
D-aspartate (NMDA) receptor (NMDAR) or various voltage-dependent Ca
2+ channels (VDCCs), as well as the release of Ca
2+ from intracellular stores (
Ghosh et al., 1994
;
Geiger et al., 1995
;
Berridge, 1998
). The relative contribution of these sources will depend on the cell type: neuron, astrocyte, oligodendrocyte or microglia. In the case of neurons, Ca
2+ sources will vary depending on their size, transmitter system, and location in neural circuits (i.e., excitatory or inhibitory). Finally, we discuss age-related changes to the other aspect of Ca
2+ homeostasis, the Ca
2+ buffering and extrusion mechanisms (Figure
1
).
Figure 1. Ca2+ homeostasis in the neuron. Model depicting various Ca2+ sources, sequestrating, buffering mechanisms, and Ca2+ signaling events in a healthy neuron. Indicated are the voltage-dependent Ca2+ channels (VDCC), N-methyl-D-aspartate receptor (NMDAR), and G protein-coupled receptor (GPCR) involved in Ca2+ (red balls) influx into the cytosol (blue dashed arrows). The release of Ca2+ into the cytoplasm also occurs from the intracellular Ca2+ stores (ICS) through inositol (1,4,5)-trisphosphate receptor (IP3R) and ryanodine receptors (RyR). Organelles, including the endoplasmic reticulum (ER), mitochondria, and lysosomes act as a Ca2+ buffering system, releasing and sequestering Ca2+. Further, the model depicts Ca2+ buffering and extrusion pathways (red dashed arrows), involving Na+/Ca2+ exchanger (NCX) and plasma membrane Ca2+ ATPase (PMCA), sarcoplasmic reticulum Ca2+ ATPases (SERCA), nicotinic acid adenine dinucleotide phosphate (NAADP), various Ca2+ binding proteins (CBP). Mitochondrial permeability transition pore (mPTP) and mitochondrial Na+/Ca2+ exchanger (mNCX) and secretory pathway Ca2+-ATPases (SPCA) contribute to Ca2+ regulation.
NMDARs are ionotropic non-selective cationic glutamate receptors, which play a central role in the rapid regulation of synaptic plasticity. NMDARs are hetero-tetrameric protein complexes composed of two classes of subunits, the ubiquitously expressed and essential subunit (NR1) and a modulatory subunit (NR2A-NR2D) (
Moriyoshi et al., 1991
;
Kutsuwada et al., 1992
;
Meguro et al., 1992
;
Monyer et al., 1992
;
Cull-Candy et al., 2001
). The activation of NMDAR requires binding of a ligand (glutamate), membrane depolarization (to remove the Mg
2+ block of the channel), and binding of a co-agonist, glycine. Since NMDAR is a non-selective cation channel, its activation and opening leads to simultaneous influx of Na
+ and Ca
2+ ions (
Chen et al., 2005
). However, between the two predominant ionotropic glutamate receptors subtypes, the NMDARs are the most permeable to Ca
2+ ions (
Garaschuk et al., 1996
).
There is considerable evidence to indicate that aging is associated with a decline in NMDAR function within brain regions involved in higher brain function including learning and memory (
Gonzales et al., 1991
;
Pittaluga et al., 1993
;
Barnes et al., 1997
;
Magnusson, 1998
;
Eckles-Smith et al., 2000
;
Gore et al., 2002
;
Liu et al., 2008b
;
Zhao et al., 2009
). Perhaps the strongest evidence for a reduction in NMDAR function comes from physiological studies which indicate that the NMDAR mediated excitatory post synaptic potentials in the Schaeffer collateral pathway of the hippocampus are reduced by approximately 50% in aged animals (
Barnes et al., 1997
;
Eckles-Smith et al., 2000
;
Bodhinathan et al., 2007
). However, age-related changes in the amplitude of NMDA-evoked responses were not observed in dissociated cortical neurons suggesting the possibility of regional specificity in the loss of NMDAR function (
Kuehl-Kovarik et al., 2003
). Several studies indicate a decrease in the level of NMDAR protein expression in the hippocampus during aging (
Bonhaus et al., 1990
;
Kito et al., 1990
;
Miyoshi et al., 1991
;
Tamaru et al., 1991
;
Wenk et al., 1991
;
Magnusson, 1995
;
Magnusson et al., 2006
;
Billard and Rouaud, 2007
;
Das and Magnusson, 2008
;
Liu et al., 2008b
;
Zhao et al., 2009
); further, the decrease has primarily been localized to region CA1 (
Magnusson and Cotman, 1993
;
Gazzaley et al., 1996
;
Magnusson, 1998
;
Wenk and Barnes, 2000
). These studies report reduced binding of [
3H] glutamate (agonist site), [
3H] glycine (NR1 site), [
3H] CPP (a competitive antagonist to the
L-glutamate binding site), and [
3H] MK-801 (an open channel blocker) in the hippocampus and cerebral cortex of aged rats. However, others have reported no age-related change in antagonist binding (
Kito et al., 1990
;
Miyoshi et al., 1991
;
Araki et al., 1997
;
Shimada et al., 1997
) or an increased MK-801 binding in animals with learning and retention deficits (
Ingram et al., 1992
;
Topic et al., 2007
). It is interesting to note that MK-801 binds to the hydrophobic channel domain of NMDAR, exclusively labeling open channels. Thus, an apparent increase in NMDAR channel open time may act as a compensatory mechanism for the decrease in receptor number (
Serra et al., 1994
). However, the majority of reports, including our recent findings, indicate that the net function of the NMDARs decreases at CA3-CA1 hippocampal synaptic contacts during senescence (
Bodhinathan et al., 2007
).
One of the potential mechanisms for the observed decrease in the NMDAR function is related to altered expression of specific NMDAR subunits (
Magnusson, 2000
). Significant decreases have been observed in the expression of NR1 protein (
Eckles-Smith et al., 2000
;
Mesches et al., 2004
;
Liu et al., 2008a
) and NR1 mRNA (
Adams et al., 2001
) levels in the aged hippocampus. In contrast, other studies report no age-related decrease in NR1 protein expression in the whole hippocampus (
Sonntag et al., 2000
;
Zhao et al., 2009
). Despite the lack of congruent changes in the expression levels in the hippocampus, other brain regions exhibit a decline in NR1 mRNA expression during aging. Indeed, senescence-related decrease in the NR1 mRNA expression has been observed in the medial basal hypothalamus-median eminence (
Gore et al., 2002
), in the medial and lateral prefrontal cortices (
Magnusson et al., 2005
), and in the insular, orbital, and somatosensory cortices (
Das and Magnusson, 2008
).
Some studies indicate age-related changes in the modulatory NR2 subunits. A decrease in the NR2A protein expression has been observed in the hippocampus (
Sonntag et al., 2000
;
Liu et al., 2008b
), which is not observed in the frontal cortex (
Sonntag et al., 2000
). Furthermore, NR2A mRNA expression was reported to decline in the ventral hippocampus (
Adams et al., 2001
). In contrast, other studies report no significant change in the NR2A protein expression levels in the hippocampus and cortex (
Sonntag et al., 2000
;
Martinez Villayandre et al., 2004
). Age-related changes have also been reported for NR2B subunit of the NMDAR; in particular the expression of NR2B protein (
Mesches et al., 2004
;
Zhao et al., 2009
) and NR2B mRNA (
Adams et al., 2001
;
Magnusson, 2001
) declines in the hippocampus. This effect may be region specific since a decline in NR2B protein is not observed in the frontal cortex (
Sonntag et al., 2000
). In contrast, NR2B mRNA decreases in the frontal cortices of aging macaque monkeys, but not in the hippocampus (
Bai et al., 2004
).
From a physiological standpoint, the changes in the expression of specific NR2 subunits could have dramatic influences on NMDAR function through the regulation of mean channel open time and conductance of the NMDARs. Studies on recombinant NMDAR expressed in
Xenopus oocytes demonstrate that NMDARs containing the NR2A subunit (NR2A-NMDARs) have faster deactivation kinetics relative to NR2B containing NMDARs (NR2B-NMDARs) (
Cull-Candy et al., 2001
), such that smaller ion flux is observed for the NR2A-NMDARs, relative to the NR2B-NMDARs. Thus, a shift in the level of NR2 subunit expression could modify the time course and magnitude of the Ca
2+ signal leading to reduced Ca
2+ influx associated with loss of NR2B. A shift in NR2A and NR2B expression is thought to contribute to developmental changes in cognition and synaptic function (
Dumas, 2005
).
Alternatively it is possible that alterations in the NMDAR localization, through the insertion of receptors into the membrane or recruitment of extra-synaptic receptors into the synapse, may have important effects on NMDAR function during aging. It has been suggested that NR2B containing receptors may be more prevalent at extra-synaptic sites (
Massey et al., 2004
), which could temporarily house the NMDARs, before being internalized into the cytoplasm (
Blanpied et al., 2002
;
Lau and Zukin, 2007
). In the frontal cortex, the expression of the NR2B subunit is reduced in the synaptic membrane fraction, but not in the whole homogenate of senescent mice suggesting that NR2B containing receptor sequestration at the extra-synaptic sites may be the mechanism by which the NR2B levels decline during aging (
Zhao et al., 2009
). Finally, recent work indicates that extra-synaptic NMDARs couple to different signaling cascades, and initiate mechanisms that oppose synaptic potentiation, by shutting off the activity of cAMP response element binding protein and decreasing expression of brain-derived neurotropic factor (
Hardingham et al., 2002
;
Vanhoutte and Bading, 2003
). However, it remains to be determined whether altered localization of the NMDARs (specifically extra-synaptic localization) is the mechanism by which the NMDAR function declines during senescence.
Another likely candidate mechanism for regulating NMDAR function during aging is posttranslational modification of the receptor. In particular, the function of the NMDAR is influenced by its phosphorylation state. Activation of the tyrosine kinase (
Wang et al., 1994
;
Heidinger et al., 2002
), protein kinase C (
Ben-Ari et al., 1992
;
Chen and Huang, 1992
) and protein kinase A (
Raman et al., 1996
) increases NMDAR mediated currents. In contrast, protein phosphatases, including calcineurin and protein phosphatase 1, decrease NMDAR currents (
Lieberman and Mody, 1994
;
Wang et al., 1994
;
Raman et al., 1996
). Phosphorylation state of NR2A and NR2B subunits can rapidly regulate surface expression and localization of these receptors (
Gardoni et al., 2001
;
Chung et al., 2004
;
Hallett et al., 2006
;
Lin et al., 2006
). For example, phosphorylation of serine residues within the alternatively spliced cassettes of the C-terminal tail of NR1 promotes receptor trafficking from the endoplasmic reticulum (ER) and insertion into the postsynaptic membrane (
Scott et al., 2001
;
Carroll and Zukin, 2002
). Finally, increased phosphatase activity has been linked to the internalization of NMDARs (
Snyder et al., 2005
). Thus, the kinases and phosphatases act like molecular switches which increase or decrease NMDAR function, respectively. Interestingly, aging is associated with a shift in the balance of kinase/phosphatase activity, favoring an increase in the phosphatase activity (
Norris et al., 1998a
;
Foster et al., 2001
;
Foster, 2007
). Thus alterations in the phosphorylation state of the NMDAR could underlie the decrease in the NMDAR function during aging (
Coultrap et al., 2008
).
NMDAR function can be altered by the oxidation and reduction of sulfhydryl moieties on their structure. Previous research demonstrates that oxidizing agents like 5,5′-dithiobis(2-nitrobenzoic acid) (
Aizenman et al., 1989
), hydroxyl radicals generated by xanthine/xanthine oxidase (
Aizenman, 1995
) and oxidized glutathione (
Sucher and Lipton, 1991
) decrease NMDAR function in the neuronal cell cultures. The decrease in NMDAR function under oxidizing conditions is thought to result from the formation of disulfide bonds on the sulfhydryl group containing amino acid residues in NMDARs (
Aizenman et al., 1990
;
Sullivan et al., 1994
;
Choi et al., 2001
). The aging brain is associated with an increase in the levels of oxidative stress and/or a decrease in redox buffering capacity (
Foster, 2006
;
Poon et al., 2006
;
Parihar et al., 2008
), conditions that should promote a decrease in NMDAR function.
Finally, NMDAR function in neurons is regulated by local supporting cells, astrocytes and microglia, thus acting as an additional possible mechanism for the age-related changes to NMDAR function. Astrocytes are a major source of
D-serine an endogenous co-agonist for the NMDAR, which binds to the glycine site (
Schell et al., 1995
). An age-related loss of
D-serine is observed in the hippocampus and cortex of rats (
Williams et al., 2006
). Furthermore, the age-related decline in the NMDAR function is rescued by
D-cycloserine (
Billard and Rouaud, 2007
). Microglia contribute to the brain’s immune system and activated microglia can release
D-serine (
Wu and Barger, 2004
;
Wu et al., 2004
). In accordance with this idea, recent reports suggest that microglia can potentiate the NMDAR-mediated synaptic responses in cortical neurons (
Moriguchi et al., 2003
;
Hayashi et al., 2006
). Markers of neuroinflammation increase with age and in certain neurodegenerative disorders. Finally, there is evidence for a feedback reduction in NMDARs due to excess synaptic glutamate activity during microglial activation (
Rosi et al., 2004
,
2006
).
In light of the interaction of NMDARs and microglia, it is important to consider the possibility that the decrease in NMDAR function might represent a compensatory neuroprotective mechanism associated with inappropriate receptor activity or increased Ca
2+ due to other mechanisms (see below). Thus, impaired NMDAR-dependent synaptic plasticity and memory decline may be epiphenomena due to processes for cell preservation (
Foster, 1999
). Indeed, over expression of NR2B subunits improves synaptic plasticity and memory in aged mice (
Cao et al., 2007
) indicating that increased NMDAR function can ameliorate physiological aging. However, cognition and synaptic plasticity are also improved by treating with the low-affinity voltage-dependent NMDAR channel blocker, memantine (
Barnes et al., 1996
;
Norris and Foster, 1999
;
Pieta Dias et al., 2007
), possibly by reducing inappropriate NMDAR activity (
Rosi et al., 2006
;
Matute, 2007
;
Chang and Gold, 2008
). In the case of neurodegenerative disease, decreased expression of NR1 mRNA has been observed in brain regions that are most at risk for cell death, including Huntington’s disease, wherein a decrease in NR1 mRNA expression is observed in the neostriatum (
Arzberger et al., 1997
). Furthermore, there is evidence for decreased NMDA receptors expression in the hippocampus during the early stages of Alzheimer’s disease (
Jacob et al., 2007
;
Mishizen-Eberz et al., 2004
). Thus, it will be important for future research to determine whether enhancing or inhibiting NMDAR function will be beneficial in preserving memory during normal aging and in the face of neurodegenerative disease.
Voltage-Dependent Ca2+ Channels
VDCCs are ion channels in the plasma membrane, which open in response to membrane depolarization and allow Ca
2+ influx into the cell from the extracellular space. VDCCs, which are heteromultimers and are composed of an α
1 subunit and three auxiliary subunits, α
2δ, β
1–4, and γ (
Jones, 1998
;
Catterall, 2000
;
Kang et al., 2001
;
Dolphin, 2006
), provide one of the most effective sources of Ca
2+ influx into the neuron (
Bertolino and Llinas, 1992
). The pore forming α
1 subunit (190 kDa) is the primary subunit necessary for channel functioning. Each α
1 subunit has four homologous domains (I–IV), which are composed of six transmembrane helices. The fourth transmembrane helix of each domain contains the voltage-sensing motif. Two classes of VDCCs have been described; high-voltage-gated and low-voltage-gated channels, which are activated by strong and weak depolarization, respectively. On the basis of differential biophysical properties and sensitivity to pharmacological agents, high-voltage-gated channels are further classified into the L (Ca
v1.1-3), P/Q (Ca
v2.1), and N (Ca
v2.2) type channels. The low-voltage-gated channels include the T (Ca
v3.1) type channels; in addition, an intermediate-voltage-gated channel, R (Ca
v2.3) type is expressed throughout the central nervous system (
Veselovskii and Fedulova, 1983
;
Carbone and Lux, 1984
;
Fedulova et al., 1985
;
Nilius et al., 1985
;
Nowycky et al., 1985
;
Bean, 1989
;
Soong et al., 1993
).
In hippocampal CA1 pyramidal neurons of the rat, the whole-cell L-type Ca
2+ currents are increased (
Campbell et al., 1996
;
Brewer et al., 2009
) and an increase in the density of functional L-type VDCCs have been reported for aged animals (
Thibault and Landfield, 1996
). The idea that L-channels are increased in the hippocampus during senescence is also supported by mRNA and protein expression studies indicating an increase in Ca
v1.3 (
Herman et al., 1998
;
Chen et al., 2000
;
Veng et al., 2003
). Furthermore, posttranslational changes including the phosphorylation state of the Ca
v1.2 channel could contribute to age-associated increase in activity (
Norris et al., 1998a
;
Norris et al., 2002
;
Davare and Hell, 2003
). However, L-channel associated intracellular Ca
2+ transients may show region specific variations within the hippocampus itself. For example, a recent report indicates that CA3 interneurons in aged hippocampus exhibit no alterations in intracellular Ca
2+ transients at resting state; however larger Ca
2+ transients are evident in the presence of external excitatory drive produced by kainate application (
Lu et al., 2009
). Several cellular biomarkers of senescent physiology in the hippocampus are dependent on VDCC function and L-type channel blockers can reverse age-related changes in the magnitude of the afterhyperpolarization and spike frequency adaptation (
Moyer Jr. et al., 1992
;
Disterhoft et al., 1996
;
Norris et al., 1998b
;
Kumar and Foster, 2002
). In turn, the reduction in the afterhyperpolarization permits increase activation of NMDAR, to shift the threshold for induction of synaptic plasticity (
Shankar et al., 1998
;
Norris et al., 1998b
). It should be noted that L-channel blockade does not completely ameliorate age-related differences. The afterhyperpolarization amplitude is reduced but not to the levels observed in young animals (
Power et al., 2002
). In aged rats, under L-channel blockade, the induction of long-term potentiation is facilitated for low level synaptic activation, which would not induce synaptic modification in young animals (
Norris et al., 1998b
). Regardless, L-channel blockers appear to improve hippocampal-dependent learning and memory in aged animals (
Deyo et al., 1989
;
Straube et al., 1990
;
Thompson et al., 1990
;
Levy et al., 1991
;
Levere and Walker, 1992
;
Moyer Jr. et al., 1992
;
Quartermain et al., 1993
;
Kowalska and Disterhoft, 1994
;
Solomon et al., 1995
;
Woodruff-Pak et al., 1997
;
Quevedo et al., 1998
;
Veng et al., 2003
;
Rose et al., 2007
), non-human primates (
Sandin et al., 1990
), and humans (
Ban et al., 1990
;
Lopez de Armentia and Sah, 2004
;
Trompet et al., 2008
).
It is unclear exactly why L-channels increase in the hippocampus with advanced age. The increase in L-channel function appears to be specific to hippocampal pyramidal cells. The expression of L-channels in the cortex does not change or is decreased by age (
Tanaka and Ando, 2001
;
Iwamoto et al., 2004
), and peak currents for high-voltage-gated channels are not increased in the basal forebrain (
Murchison and Griffith, 1996
). The expression of L-channels in the hippocampus is regulated by the sex steroid estrogen, such that an increased expression is associated with the decline of the hormone during aging (
Foster, 2005
) Finally, it is possible that the increased L-channel function, increased afterhyperpolarization, and reduction in cell excitability represent compensatory mechanisms associated with Ca
2+ dysregulation during senescence, which attempts to limit depolarization and further influx of Ca
2+ through NMDARs (
Foster, 1999
).
In direct contrast to the hippocampus, altered Ca
2+ homeostasis in basal forebrain neurons of rats appears to result from the inactivation of high-voltage-gated currents and an increase in Ca
2+ from T-type, low-voltage-gated channels (
Murchison and Griffith, 1995
;
Murchison and Griffith, 1996
;
Murchison and Griffith, 2007
). T-channels have been localized on dendrites (
Magee and Johnston, 1995a
,
b
;
Christie et al., 1996a
,
b
;
Yasuda et al., 2003
) where they likely influence synaptic function (
Komatsu and Iwakiri, 1992
;
Yoshimura et al., 2008
;
Uebele et al., 2009
). Interestingly, the age-related shift in T-type channels in the basal forebrain appears to be specific to cholinergic neurons (
Han et al., 2005
) and may influence the magnitude of the afterhyperpolarization (
Murchison et al., 2009
). The influence of these changes on cell function and cognition remain to be determined. Similarly, much less is known concerning other high-voltage-gated channels and aging. N-channels may be decreased in the cortex with age (
Tanaka and Ando, 2001
). Animal models suggest that altered P/Q-type channel expression could contribute to age-related changes in cognition and motor function (
Alonso et al., 2008
). There is evidence that the expression of P/Q splice variants changes with age in different brain regions (
Tanaka and Ando, 2001
;
Iwamoto et al., 2004
;
Chang et al., 2007
;
Martella et al., 2008
). Thus, more research is required in order to determine how Ca
2+ channels other than the L-channel contribute to Ca
2+ regulation during aging.
Mounting evidence indicates that Ca
2+ channels in the membrane contribute to the specificity of cell loss and the progression of Parkinson’s and Alzheimer’s disease. For example, in the substantia nigra, a high level of Ca
v1.3 L-type channel activity contributes to the discharge pattern of dopamine pacemaker neurons. This activity results in a large Ca
2+ influx which is buffered by intracellular stores. However, in Parkinson’s disease, disruption of this buffering process due to genetic or environmental stress results in toxic levels of Ca
2+ leading to cell death (
Chan et al., 2009
). Furthermore, with advanced age, these neurons increase their reliance on L-channel activity to regulate pacemaker activity, increasing their liability (
Chan et al., 2007
). Thus, the activity of the L-channel provides the specificity for cell loss in Parkinson’s disease.
Temporal lobe regions, including the hippocampus, exhibit marked cell loss associated with Alzheimer’s disease. The beta amyloid protein of Alzheimer’s disease, increases cytosolic Ca
2+, impairs synaptic plasticity, and increases cell death through an L-channel-dependent mechanism (
Freir and Herron, 2003
;
Fu et al., 2006
;
Lopez et al., 2008
). Polymorphism of a recently identified Ca
2+ channel has been linked to late-onset Alzheimer’s disease (
Dreses-Werringloer et al., 2008
). Interestingly, this gene is predominantly expressed in brain regions, such as the hippocampus, which exhibit early and profound cell loss. Together, the results indicate that Ca
2+ channels provide a point of cross talk between age-related Ca
2+ dysregulation and signaling in neurodegenerative diseases resulting in selectivity of cell loss.
Intracellular Ca2+ Stores
In addition to Ca
2+ influx from outside the cell, intracellular Ca
2+ stores (ICS) play a major role in regulating larger Ca
2+ signals (
Mattson et al., 2000
;
Ly and Verstreken, 2006
). Organelles, including the ER, mitochondria, and lysosomes act as Ca
2+ buffering systems – releasing and sequestering Ca
2+ (
Duchen, 2000
;
Nicholls and Budd, 2000
;
Toescu et al., 2000
;
Solovyova et al., 2002
;
Toescu and Verkhratsky, 2004
;
McGuinness et al., 2007
;
Murchison and Griffith, 2007
). Thus, there are at least two possible mechanisms by which ICS regulate Ca
2+ homeostasis: (1) release of stored Ca
2+ to enhance Ca
2+ signals and (2) removing cytosolic Ca
2+ following a large influx.
Two pathways control the release of Ca
2+ from the ER, Ca
2+-induced Ca
2+ release (CICR) and the inositol (1,4,5)-trisphosphate (IP
3) pathway activated by G protein-coupled receptors (GPCR). GPCRs activate phospholipase C (PLC) to form diacylglycerol and IP
3 which act on IP
3 receptors (IP
3Rs) to release Ca
2+ from ICS. While several studies indicate age-related changes in GPCRs or PLC (
Roth, 1995
;
Mizutani et al., 1998
;
Nicolle et al., 1999
), we will focus on Ca
2+ release induced by IP
3R activation. Previous reports have observed a decrease in IP
3R in several brain regions (
Burnett et al., 1990
;
Igwe and Ning, 1993
;
Martini et al., 1994
;
Simonyi et al., 1998
). Despite a general decrease in the receptor, the literature suggests that a decrease in IP
3 induced Ca
2+ release is either limited to cortical cells (
Burnett et al., 1990
) or no age-related change is observed (
Stutzmann et al., 2006
). The disconnect between a reduction in IP
3R expression and the apparent absence of an effect of age on IP
3-induced Ca
2+ release may be due to increased oxidation of the IP
3Rs which has been demonstrated to increase IP
3R function in brain cells (
Peuchen et al., 1996
;
Long et al., 2009
). As such, reduced expression may act as compensation for an altered redox state, in order to maintain proper IP
3 signaling.
CICR is a Ca
2+ amplification process that is initiated by influx of Ca
2+ through membrane channels or from ICS through the activation of IP
3Rs. The intracellular Ca
2+ binds ryanodine receptors (RyRs) to release additional Ca
2+ into the cytosol from the ER. Accumulating evidence supports a role of altered CICR in contributing to altered physiology of normal aging. The increased involvement of RyRs does not appear to be due to increased RyR expression (
Martini et al., 1994
). Rather, an age-related increase in oxidative stress and a shift in the intracellular redox state may enhance the responsiveness of RyRs to intracellular Ca
2+ (
Hidalgo et al., 2004
;
Bull et al., 2007
;
Gokulrangan et al., 2007
). Thus, like IP
3Rs, a shift in the redox state may provide a general aging mechanism to promote Ca
2+ release from RyRs. Again, the contribution of this source of Ca
2+ to Ca
2+ dysregulation is cell specific and will depend on other Ca
2+ regulating mechanisms. For example, CICR is decreased during senescence in peripheral synapses, due in part to decreased expression of RyRs (
Buchholz et al., 2007
) and a decrease in the amplitude of Ca
2+ release in basal forebrain neurons is linked to enhanced mitochondrial buffering (
Murchison and Griffith, 1999
). Increased CICR appears to contribute to altered physiology in hippocampal neurons (
Kumar and Foster, 2004
;
Kumar and Foster, 2005
;
Gant et al., 2006
). As noted above, hippocampal cells exhibit increase Ca
2+ from L-type Ca
2+ channels, which could provide a source of Ca
2+ to fill ICS and activate CICR from ICS. Thus, the contribution of CICR to aging physiology in hippocampal cells may be due to due to a summation of various mechanisms.
Similarly, RyRs and IP
3Rs are likely to interact with other Ca
2+ regulating mechanisms and neurodegenerative diseases to contribute to cell specific vulnerability. The protein presenilin interacts with several signaling pathways including Ca
2+ signaling, contributing to the neurotoxicity and the pathology of familial Alzheimer’s disease (
Chan et al., 2000
;
Smith et al., 2005
;
Stutzmann, 2007
). Presenilin and beta amyloid have been associated with increased RyR function (
Supnet et al., 2006
;
Stutzmann, 2007
;
Hayrapetyan et al., 2008
). The influence of increased RyR activity on cell function (e.g., synaptic transmission) in the hippocampus may initially be masked by compensatory mechanisms and emerge with age and the disruption of other Ca
2+ regulatory mechanisms (
Chakroborty et al., 2009
). Polyglutamine expansion of the protein huntingtin in medium spiny neurons of the striatum results in Huntington’s disease, and this protein enhances activation of IP
3Rs resulting in uncontrolled Ca
2+ release and death of medium spiny neurons (
Tang et al., 2003
;
Tang et al., 2005
).
Recent studies suggest the possibility that lysosomes may act as a Ca
2+ storage organelle (
Lee, 2004
;
McGuinness et al., 2007
;
Brailoiu et al., 2009b
;
Pandey et al., 2009
). Release of Ca
2+ from lysosomes involves the pyridine nucleotide, nicotinic acid adenine dinucleotide phosphate (NAADP) which can stimulate a rise in cytosolic Ca
2+ from bafilomycin-sensitive Ca
2+ stores (
Brailoiu et al., 2005
,
2009a
). There are a few studies, which have investigated role of NAADP-induce Ca
2+ release in the regulation of neuronal Ca
2+ homeostasis and Ca
2+-dependent processes. Findings from recent studies provide evidence for the presence of NAADP-sensitive Ca
2+ stores in neurons (
Brailoiu et al., 2009b
;
Pandey et al., 2009
) and NAADP mediated Ca
2+ release can act to increase neurotransmitter release (
Brailoiu et al., 2001
;
Chameau et al., 2001
;
McGuinness et al., 2007
). Furthermore, pharmacological manipulations indicate that NAADP is a likely candidate for regulating Ca
2+ signaling in astrocytes (
Singaravelu and Deitmer, 2006
). It is unclear whether this source of Ca
2+ regulation is altered by aging; however, the aging brain is characterized by increased lysosomal markers and decreased lysosomal function (
Lynch and Bi, 2003
;
Keller et al., 2004
;
Stolzing et al., 2005
). Thus, the role of this source of Ca
2+ in susceptibility to Ca
2+ dysregulation should be examined in the future.
Ca2+ Buffering, Extrusion, and Sequestration
In most cases, a modest rise in intracellular Ca
2+ level is handled by rapid Ca
2+ buffering involving Ca
2+ binding proteins (CBPs) in the cytosol and extrusion into the extracellular space by Ca
2+ pumps or exchangers. In addition, Ca
2+ can be pumped into the ER or mitochondria for sequestration. A decrease in Ca
2+ buffering or delayed removal could result in the larger or prolonged Ca
2+ responses, which is a characteristic of aged neurons (
Verkhratsky and Toescu, 1998
;
Brewer et al., 2006
).
Over 250 proteins can be described as CBPs and the brain is a particularly rich source of these proteins (
Celio et al., 1996
). CBPs are generally considered to be neuroprotective (
Scharfman and Schwartzkroin, 1989
;
Mattson et al., 1991
;
Lukas and Jones, 1994
;
Berger et al., 1998
). Age-related changes in the expression of cytosolic CBPs (parvalbumin, calbindin-D28K, calretinin, calmodulin, hippocalcin) are cell and region specific (
Furuta et al., 1999
;
Bu et al., 2003
;
Geula et al., 2003a
,
b
;
Wu et al., 2003
;
Han et al., 2006
;
Ouda et al., 2008
). In general, age-related changes involve a decline in expression, which may be associated with a loss of function, rather than a compensatory up regulation. However, in a series of studies, Murchison and Griffith and associates (
Murchison and Griffith, 2007
) demonstrate rapid Ca
2+ buffering in aged basal forebrain neurons, likely mediated by CBPs, which can act as compensation for increased Ca
2+ influx and impaired mitochondrial uptake.
Na
+/Ca
2+ exchangers and plasma membrane Ca
2+ ATPase are major transport system capable of rapidly extruding substantial amounts of Ca
2+ from the cell cytosol, across the plasma membrane, to extracellular space. The Sarco/ER Ca
2+ ATPases are Ca
2+ ion ATPases, which resides in the membrane of sarco/ER and transport Ca
2+ from the cytosol to the intracellular membranous compartment of the sarco/ER. An age-related decline in the function of these transports systems has been suggested to underlie altered Ca
2+ homeostasis during brain aging and contribute to age-related neurodegenerative diseases (
Michaelis et al., 1984
,
1996
;
Zaidi et al., 1998
;
Hanahisa and Yamaguchi, 2001
;
Pottorf et al., 2001
;
Gomez-Villafuertes et al., 2007
). The decrease in activity may be linked to oxidation of pump proteins or CBPs that regulate pump activity (
Zaidi and Michaelis, 1999
;
Squier and Bigelow, 2000
;
Bartlett et al., 2003
). As such, it is likely that altered pump activity will vary with regional differences in oxidative stress.
Reports of age-related changes in the buffering function of the ER and mitochondria are highly variable across different cell types and brain regions (
Brown et al., 2004
). For example, studies of the peripheral nervous system indicate that buffering of cytosolic Ca
2+ by the ER is decreased during aging (
Tsai et al., 1998
) with no change in mitochondrial function (
Buchholz et al., 2007
). In contrast, ER-mediated Ca
2+ buffering may be intact (
Burnett et al., 1990
;
Pottorf et al., 2001
) and mitochondrial buffering may be altered at synapses in the central nervous system (
Martinez-Serrano et al., 1992
;
Satrustegui et al., 1996
;
Toescu and Verkhratsky, 2000
,
2003
,
2004
;
Toescu et al., 2000
;
Murchison et al., 2004
).
Mitochondria from aged animals show structural alterations to mitochondrial DNA (
Cortopassi and Arnheim, 1990
;
Toescu et al., 2000
) and to the mitochondrial membrane (
Yan and Sohal, 1998
;
Kwong and Sohal, 2000
), which could contribute to a net decrease in the Ca
2+ buffering capacity during senescence. More important is decreased Ca
2+ uptake capacity of aged mitochondria, which arises as a direct consequence of the decreased electrochemical gradient across the mitochondrial membrane (
Xiong et al., 2004
). Mitochondrial depolarization may increase the threshold level of Ca
2+ needed to initiate mitochondrial uptake. As such, an age-dependent delay in Ca
2+ sequestration or recovery would become apparent under conditions of a large rise in intracellular Ca
2+ (
Xiong et al., 2002
,
2004
;
Murchison et al., 2004
). Finally, mitochondria provide a source for oxidative stress and regional variability in oxidative stress (
Dubey et al., 1996
;
Rebrin et al., 2007
) and mitochondrial damage has been reported (
Corral-Debrinski et al., 1992
;
Filburn et al., 1996
), which appear to correspond to regions that are vulnerable to neurodegenerative disease.
The Ca2+ ion is a central signaling molecule in numerous cellular functions including apoptosis, energy production, gene regulation, cell proliferation, membrane excitability, synaptic transmission and plasticity. Since the first observations in the 1980s that led to Ca2+ ‘dysregulation’ hypothesis of brain aging and Alzheimer’s disease, new findings have shaped ideas concerning the mechanisms and the pervasiveness of Ca2+ dysregulation. It is becoming clear that Ca2+ dysregulation is not ubiquitous and mechanisms of dysregulation are restricted to specific cell populations. For example, an age-related increase in L-type Ca2+ channels is relatively specific to hippocampal pyramidal cells. Furthermore, an age-related decrease in NMDA receptor function, specifically in the hippocampus or frontal cortex, suggests a possible compensatory mechanism to limit intracellular Ca2+ levels. However, such a mechanism may protect the cell at the expense of cell function. Thus, memory decline associated with a decrease in NMDA receptor function might be considered an epiphenomenon due to the activation of mechanisms for cell survival.
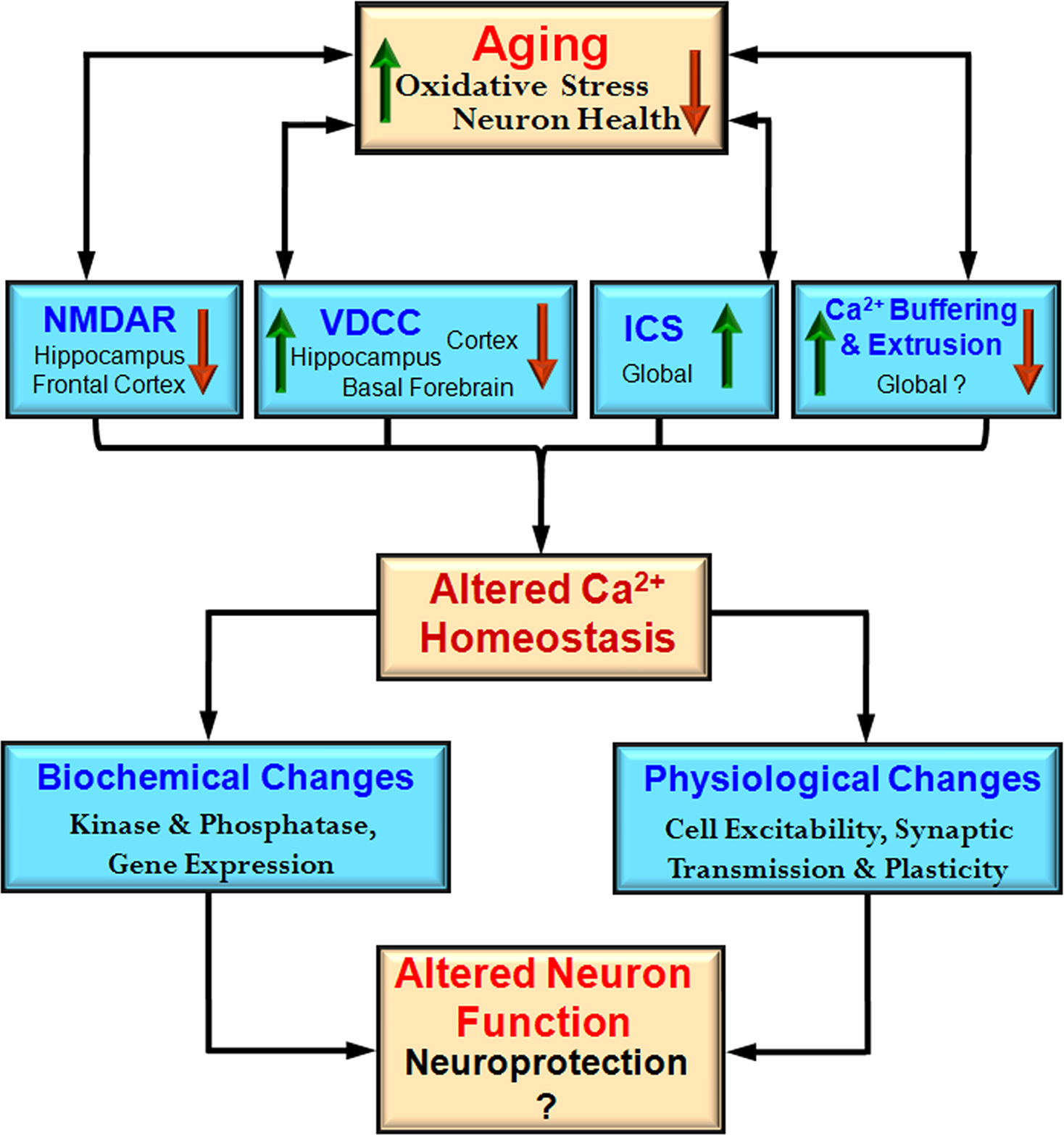
Cell specific susceptibility to Ca
2+ dysregulation depends on environmental and genomic factors in addition to the availability of mechanisms for handling Ca
2+. The level of neural activity may render some regions more susceptible to oxidative stress, resulting in multiple changes to increase intracellular Ca
2+ including increased release of Ca
2+ from ICS, impaired Ca
2+ pumps, and weakened Ca
2+ buffering (Figure
2
). In turn, gene mutations may interact with age and cell specific alterations in Ca
2+ regulation to produce the pattern of neuronal death which characterizes neurodegenerative diseases.
Figure 2. Integrative model of the impact of aging on the Ca2+ handling mechanisms and physiological processes. During aging there is an interaction between increased oxidative stress and decreased neuron health with mechanisms for Ca2+ regulation including NMDA receptors (NMDAR), voltage-dependent Ca2+ channels (VDCC), intracellular calcium stores (ICS), and Ca2+ buffering and extrusion mechanisms. These changes are region and cell specific rather than representing a global change. An indication of regional specificity (hippocampus, frontal cortex, cortex, basal forebrain) and the direction of change (increase – red arrow and decrease – green arrow) for each mechanism are also provided. The shift in Ca2+ homeostatic mechanisms may represent neuroprotective mechanisms to decrease further rise in intracellular Ca2+ by decreasing neuron activity. These changes also impair the function of the neuron.
Due to the importance of Ca2+ as a central signaling molecule, selective regulation of Ca2+ in a particular set of neurons may be a daunting task for treating age-related diseases. Clearly future research will need to delineate contribution of several mechanisms in optimizing Ca2+ homeostasis or mediating Ca2+ dysregulation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Financial support by National Institutes of Health Grants AG014979, MH 059891, and the Evelyn F. McKnight Brain Research Grant and a University of Florida Alumni Fellowship (Karthik Bodhinathan) is highly appreciated.
Adams, M. M., Morrison, J. H., and Gore, A. C. (2001).
N-methyl-
D-aspartate receptor mRNA levels change during reproductive senescence in the hippocampus of female rats.
Exp. Neurol. 170, 171–179.
Aizenman, E. (1995). Modulation of
N-methyl-
D-aspartate receptors by hydroxyl radicals in rat cortical neurons in vitro.
Neurosci. Lett. 189, 57–59.
Aizenman, E., Hartnett, K. A., and Reynolds, I. J. (1990). Oxygen free radicals regulate NMDA receptor function via a redox modulatory site.
Neuron 5, 841–846.
Aizenman, E., Lipton, S. A., and Loring, R. H. (1989). Selective modulation of NMDA responses by reduction and oxidation.
Neuron 2, 1257–1263.
Alonso, I., Marques, J. M., Sousa, N., Sequeiros, J., Olsson, I. A., and Silveira, I. (2008). Motor and cognitive deficits in the heterozygous leaner mouse, a Cav2.1
voltage-gated Ca2+ channel mutant.
Neurobiol. Aging 29, 1733–1743.
Araki, T., Kato, H., Nagaki, S., Shuto, K., Fujiwara, T., and Itoyama, Y. (1997). Effects of vinconate on age-related alterations in [3H]MK-801, [3H]glycine, sodium-dependent
D-[3H]aspartate, [3H]FK-506 and [3H]PN200-110 binding in rats.
Mech. Ageing Dev. 95, 13–29.
Arzberger, T., Krampfl, K., Leimgruber, S., and Weindl, A. (1997). Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease--an in situ hybridization study.
J. Neuropathol. Exp. Neurol. 56, 440–454.
Bai, L., Hof, P. R., Standaert, D. G., Xing, Y., Nelson, S. E., Young, A. B., and Magnusson, K. R. (2004). Changes in the expression of the NR2B subunit during aging in macaque monkeys.
Neurobiol. Aging 25, 201–208.
Ban, T. A., Morey, L., Aguglia, E., Azzarelli, O., Balsano, F., Marigliano, V., Caglieris, N., Sterlicchio, M., Capurso, A., Tomasi, N. A., Crepaldi, G., Volpe, D., Palmieri, G., Ambrosi, G., Polli, E., Cortellaro, M., Zanussi, C., and Froldi, M. (1990). Nimodipine in the treatment of old age dementias.
Prog. Neuropsychopharmacol. Biol. Psychiatry 14, 525–551.
Barnes, C. A., Rao, G., and McNaughton, B. L. (1996). Functional integrity of NMDA-dependent LTP induction mechanisms across the lifespan of F-344 rats.
Learn. Mem. 3, 124–137.
Barnes, C. A., Rao, G., and Shen, J. (1997). Age-related decrease in the
N-methyl-
D-aspartate R-mediated excitatory postsynaptic potential in hippocampal region CA1.
Neurobiol. Aging 18, 445–452.
Bartlett, R. K., Bieber Urbauer, R. J., Anbanandam, A., Smallwood, H. S., Urbauer, J. L., and Squier, T. C. (2003). Oxidation of Met144 and Met145 in calmodulin blocks calmodulin dependent activation of the plasma membrane Ca-ATPase.
Biochemistry 42, 3231–3238.
Bean, B. P. (1989). Classes of calcium channels in vertebrate cells.
Annu. Rev. Physiol. 51, 367–384.
Ben-Ari, Y., Aniksztejn, L., and Bregestovski, P. (1992). Protein kinase C modulation of NMDA currents: an important link for LTP induction.
Trends Neurosci. 15, 333–339.
Berger, F., Gage, F. H., and Vijayaraghavan, S. (1998). Nicotinic receptor-induced apoptotic cell death of hippocampal progenitor cells.
J. Neurosci. 18, 6871–6881.
Berridge, M. J. (1998). Neuronal calcium signaling.
Neuron 21, 13–26.
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling.
Nat. Rev. Mol. Cell Biol. 1, 11–21.
Bertolino, M., and Llinas, R. R. (1992). The central role of voltage-activated and receptor-operated calcium channels in neuronal cells.
Annu. Rev. Pharmacol. Toxicol. 32, 399–421.
Billard, J. M., and Rouaud, E. (2007). Deficit of NMDA receptor activation in CA1 hippocampal area of aged rats is rescued by
D-cycloserine.
Eur. J. Neurosci. 25, 2260–2268.
Blanpied, T. A., Scott, D. B., and Ehlers, M. D. (2002). Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines.
Neuron 36, 435–449.
Bodhinathan, K., Kumar, A., and Foster, T. C. (2007). Oxidative stress decreases NMDA receptor function in the hippocampus of aged animals.
Soc. Neurosci. Abstr. N18/256.8.
Bonhaus, D. W., Perry, W. B., and McNamara, J. O. (1990). Decreased density, but not number, of
N-methyl-
D-aspartate, glycine and phencyclidine binding sites in hippocampus of senescent rats.
Brain Res. 532, 82–86.
Brailoiu, E., Churamani, D., Cai, X., Schrlau, M. G., Brailoiu, G. C., Gao, X., Hooper, R., Boulware, M. J., Dun, N. J., Marchant, J. S., and Patel, S. (2009a). Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling.
J. Cell Biol. 186, 201–209.
Brailoiu, G. C., Brailoiu, E., Parkesh, R., Galione, A., Churchill, G. C., Patel, S., and Dun, N. J. (2009b). NAADP-mediated channel ‘chatter’ in neurons of the rat medulla oblongata.
Biochem. J. 419, 91–97, 2 p following 97.
Brailoiu, E., Hoard, J. L., Filipeanu, C. M., Brailoiu, G. C., Dun, S. L., Patel, S., and Dun, N. J. (2005). Nicotinic acid adenine dinucleotide phosphate potentiates neurite outgrowth.
J. Biol. Chem. 280, 5646–5650.
Brailoiu, E., Miyamoto, M. D., and Dun, N. J. (2001). Nicotinic acid adenine dinucleotide phosphate enhances quantal neurosecretion at the frog neuromuscular junction: possible action on synaptic vesicles in the releasable pool.
Mol. Pharmacol. 60, 718–724.
Brewer, L. D., Dowling, A. L., Curran-Rauhut, M. A., Landfield, P. W., Porter, N. M., and Blalock, E. M. (2009). Estradiol reverses a calcium-related biomarker of brain aging in female rats.
J. Neurosci. 29, 6058–6067.
Brewer, L. D., Porter, N. M., Kerr, D. S., Landfield, P. W., and Thibault, O. (2006). Chronic 1alpha,25-(OH)2 vitamin D3 treatment reduces Ca2+-mediated hippocampal biomarkers of aging.
Cell Calcium 40, 277–286.
Brown, M. R., Geddes, J. W., and Sullivan, P. G. (2004). Brain region-specific, age-related, alterations in mitochondrial responses to elevated calcium.
J. Bioenerg. Biomembr. 36, 401–406.
Bu, J., Sathyendra, V., Nagykery, N., and Geula, C. (2003). Age-related changes in calbindin-D28k, calretinin, and parvalbumin-immunoreactive neurons in the human cerebral cortex.
Exp. Neurol. 182, 220–231.
Buchholz, J. N., Behringer, E. J., Pottorf, W. J., Pearce, W. J., and Vanterpool, C. K. (2007). Age-dependent changes in Ca(2+) homeostasis in peripheral neurones: implications for changes in function.
Aging Cell 6, 285–296.
Bull, R., Finkelstein, J. P., Humeres, A., Behrens, M. I., and Hidalgo, C. (2007). Effects of ATP, Mg2+, and redox agents on the Ca2+ dependence of RyR channels from rat brain cortex.
Am. J. Physiol. Cell Physiol. 293, C162–C171.
Burnett, D. M., Daniell, L. C., and Zahniser, N. R. (1990). Decreased efficacy of inositol 1,4,5-trisphosphate to elicit calcium mobilization from cerebrocortical microsomes of aged rats.
Mol. Pharmacol. 37, 566–571.
Campbell, L. W., Hao, S. Y., Thibault, O., Blalock, E. M., and Landfield, P. W. (1996). Aging changes in voltage-gated calcium currents in hippocampal CA1 neurons.
J. Neurosci. 16, 6286–6295.
Cao, X., Cui, Z., Feng, R., Tang, Y. P., Qin, Z., Mei, B., and Tsien, J. Z. (2007). Maintenance of superior learning and memory function in NR2B transgenic mice during ageing.
Eur. J. Neurosci. 25, 1815–1822.
Carbone, E., and Lux, H. D. (1984). A low voltage-activated, fully inactivating Ca channel in vertebrate sensory neurones.
Nature 310, 501–502.
Carroll, R. C., and Zukin, R. S. (2002). NMDA-receptor trafficking and targeting: implications for synaptic transmission and plasticity.
Trends Neurosci. 25, 571–577.
Catterall, W. A. (2000). Structure and regulation of voltage-gated Ca2+ channels.
Annu. Rev. Cell Dev. Biol. 16, 521–555.
Celio, M. R., Pauls, T. L., and Schwaller, B. (1996). Introduction to EF-hand calcium binding proteins. In Guidebook to the Calcium-Binding Proteins, M. R. Celio, ed. (Oxford, Sambrook and Tooze Publication), pp. 15–20.
Chakroborty, S., Goussakov, I., Miller, M. B., and Stutzmann, G. E. (2009). Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice.
J. Neurosci. 29, 9458–9470.
Chameau, P., Van de Vrede, Y., Fossier, P., and Baux, G. (2001). Ryanodine-, IP3- and NAADP-dependent calcium stores control acetylcholine release.
Pflugers Arch. 443, 289–296.
Chan, C. S., Gertler, T. S., and Surmeier, D. J. (2009). Calcium homeostasis, selective vulnerability and Parkinson’s disease.
Trends Neurosci. 32, 249–256.
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., Meredith, G. E., and Surmeier, D. J. (2007). ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease.
Nature 447, 1081–1086.
Chan, S. L., Mayne, M., Holden, C. P., Geiger, J. D., and Mattson, M. P. (2000). Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons.
J. Biol. Chem. 275, 18195–18200.
Chang, Q., and Gold, P. E. (2008). Age-related changes in memory and in acetylcholine functions in the hippocampus in the Ts65Dn mouse, a model of Down syndrome.
Neurobiol. Learn. Mem. 89, 167–177.
Chang, S. Y., Yong, T. F., Yu, C. Y., Liang, M. C., Pletnikova, O., Troncoso, J., Burgunder, J. M., and Soong, T. W. (2007). Age and gender-dependent alternative splicing of P/Q-type calcium channel EF-hand.
Neuroscience 145, 1026–1036.
Chen, K. C., Blalock, E. M., Thibault, O., Kaminker, P., and Landfield, P. W. (2000). Expression of alpha 1D subunit mRNA is correlated with L-type Ca2+ channel activity in single neurons of hippocampal “zipper” slices.
Proc. Natl. Acad. Sci. U.S.A. 97, 4357–4362.
Chen, L., and Huang, L. Y. (1992). Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation.
Nature 356, 521–523.
Chen, P. E., Geballe, M. T., Stansfeld, P. J., Johnston, A. R., Yuan, H., Jacob, A. L., Snyder, J. P., Traynelis, S. F., and Wyllie, D. J. (2005). Structural features of the glutamate binding site in recombinant NR1/NR2A
N-methyl-
D-aspartate receptors determined by site-directed mutagenesis and molecular modeling.
Mol. Pharmacol. 67, 1470–1484.
Choi, Y., Chen, H. V., and Lipton, S. A. (2001). Three pairs of cysteine residues mediate both redox and zn2+ modulation of the nmda receptor.
J. Neurosci. 21, 392–400.
Christie, B. R., Magee, J. C., and Johnston, D. (1996a). Dendritic calcium channels and hippocampal long-term depression.
Hippocampus 6, 17–23.
Christie, B. R., Magee, J. C., and Johnston, D. (1996b). The role of dendritic action potentials and Ca2+ influx in the induction of homosynaptic long-term depression in hippocampal CA1 pyramidal neurons.
Learn. Mem. 3, 160–169.
Chung, H. J., Huang, Y. H., Lau, L. F., and Huganir, R. L. (2004). Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand.
J. Neurosci. 24, 10248–10259.
Corral-Debrinski, M., Horton, T., Lott, M. T., Shoffner, J. M., Beal, M. F., and Wallace, D. C. (1992). Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age.
Nat. Genet. 2, 324–329.
Cortopassi, G. A., and Arnheim, N. (1990). Detection of a specific mitochondrial DNA deletion in tissues of older humans.
Nucleic Acids Res. 18, 6927–6933.
Coultrap, S. J., Bickford, P. C., and Browning, M. D. (2008). Blueberry-enriched diet ameliorates age-related declines in NMDA receptor-dependent LTP.
Age (Dordr.) 30, 263–272.
Cull-Candy, S., Brickley, S., and Farrant, M. (2001). NMDA receptor subunits: diversity, development and disease.
Curr. Opin. Neurobiol. 11, 327–335.
Das, S. R., and Magnusson, K. R. (2008). Relationship between mRNA expression of splice forms of the zeta1 subunit of the
N-methyl-
D-aspartate receptor and spatial memory in aged mice.
Brain Res. 1207, 142–154.
Davare, M. A., and Hell, J. W. (2003). Increased phosphorylation of the neuronal L-type Ca(2+) channel Ca(v)1.2 during aging.
Proc. Natl. Acad. Sci. U.S.A. 100, 16018–16023.
Deyo, R. A., Straube, K. T., Moyer, J. R. Jr., and Disterhoft, J. F. (1989). Nimodipine ameliorates aging-related changes in open-field behaviors of the rabbit.
Exp. Aging Res. 15, 169–175.
Disterhoft, J. F., and Oh, M. M. (2006). Learning, aging and intrinsic neuronal plasticity.
Trends Neurosci. 29, 587–599.
Disterhoft, J. F., Thompson, L. T., Moyer, J. R., and Mogul, D. J. (1996). Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus.
Life Sci. 59, 413–420.
Dolphin, A. C. (2006). A short history of voltage-gated calcium channels.
Br. J. Pharmacol. 147(Suppl. 1), S56–S62.
Dreses-Werringloer, U., Lambert, J. C., Vingtdeux, V., Zhao, H., Vais, H., Siebert, A., Jain, A., Koppel, J., Rovelet-Lecrux, A., Hannequin, D., Pasquier, F., Galimberti, D., Scarpini, E., Mann, D., Lendon, C., Campion, D., Amouyel, P., Davies, P., Foskett, J. K., Campagne, F., and Marambaud, P. (2008). A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk.
Cell 133, 1149–1161.
Dubey, A., Forster, M. J., Lal, H., and Sohal, R. S. (1996). Effect of age and caloric intake on protein oxidation in different brain regions and on behavioral functions of the mouse.
Arch. Biochem. Biophys. 333, 189–197.
Duchen, M. R. (2000). Mitochondria and calcium: from cell signalling to cell death.
J. Physiol. (Lond.) 529(Pt. 1), 57–68.
Dumas, T. C. (2005). Developmental regulation of cognitive abilities: modified composition of a molecular switch turns on associative learning.
Prog. Neurobiol. 76, 189–211.
Eckles-Smith, K., Clayton, D., Bickford, P., and Browning, M. D. (2000). Caloric restriction prevents age-related deficits in LTP and in NMDA receptor expression.
Brain Res. Mol. Brain Res. 78, 154–162.
Fedulova, S. A., Kostyuk, P. G., and Veselovsky, N. S. (1985). Two types of calcium channels in the somatic membrane of new-born rat dorsal root ganglion neurones.
J. Physiol. (Lond.) 359, 431–446.
Filburn, C. R., Edris, W., Tamatani, M., Hogue, B., Kudryashova, I., and Hansford, R. G. (1996). Mitochondrial electron transport chain activities and DNA deletions in regions of the rat brain.
Mech. Ageing Dev. 87, 35–46.
Foster, T. C. (1999). Involvement of hippocampal synaptic plasticity in age-related memory decline.
Brain Res. Rev. 30, 236–249.
Foster, T. C. (2005). Interaction of rapid signal transduction cascades and gene expression in mediating estrogen effects on memory over the life span.
Front. Neuroendocrinol. 26, 51–64.
Foster, T. C. (2006). Biological markers of age-related memory deficits: treatment of senescent physiology.
CNS Drugs 20, 153–166.
Foster, T. C. (2007). Calcium homeostasis and modulation of synaptic plasticity in the aged brain.
Aging Cell 6, 319–325.
Foster, T. C., Sharrow, K. M., Masse, J. R., Norris, C. M., and Kumar, A. (2001). Calcineurin links Ca2+ dysregulation with brain aging.
J. Neurosci. 21, 4066–4073.
Freir, D. B., and Herron, C. E. (2003). Inhibition of L-type voltage dependent calcium channels causes impairment of long-term potentiation in the hippocampal CA1 region in vivo.
Brain Res. 967, 27–36.
Fu, H., Li, W., Lao, Y., Luo, J., Lee, N. T., Kan, K. K., Tsang, H. W., Tsim, K. W., Pang, Y., Li, Z., Chang, D. C., Li, M., and Han, Y. (2006). Bis(7)-tacrine attenuates beta amyloid-induced neuronal apoptosis by regulating L-type calcium channels.
J. Neurochem. 98, 1400–1410.
Furuta, Y., Kobayashi, M., Masaki, T., and Takamatsu, K. (1999). Age-related changes in expression of hippocalcin and NVP2 in rat brain.
Neurochem. Res. 24, 651–658.
Gant, J. C., Sama, M. M., Landfield, P. W., and Thibault, O. (2006). Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release.
J. Neurosci. 26, 3482–3490.
Garaschuk, O., Schneggenburger, R., Schirra, C., Tempia, F., and Konnerth, A. (1996). Fractional Ca2+ currents through somatic and dendritic glutamate receptor channels of rat hippocampal CA1 pyramidal neurones.
J. Physiol. (Lond.) 491(Pt. 3), 757–772.
Gardoni, F., Schrama, L. H., Kamal, A., Gispen, W. H., Cattabeni, F., and Di Luca, M. (2001). Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor.
J. Neurosci. 21, 1501–1509.
Gazzaley, A. H., Weiland, N. G., McEwen, B. S., and Morrison, J. H. (1996). Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus.
J. Neurosci. 16, 6830–6838.
Geiger, J. R., Melcher, T., Koh, D. S., Sakmann, B., Seeburg, P. H., Jonas, P., and Monyer, H. (1995). Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS.
Neuron 15, 193–204.
Geula, C., Bu, J., Nagykery, N., Scinto, L. F., Chan, J., Joseph, J., Parker, R., and Wu, C. K. (2003a). Loss of calbindin-D28k from aging human cholinergic basal forebrain: relation to neuronal loss.
J. Comp. Neurol. 455, 249–259.
Geula, C., Nagykery, N., Wu, C. K., and Bu, J. (2003b). Loss of calbindin-D28K from aging human cholinergic basal forebrain: relation to plaques and tangles.
J. Neuropathol. Exp. Neurol. 62, 605–616.
Ghosh, A., Ginty, D. D., Bading, H., and Greenberg, M. E. (1994). Calcium regulation of gene expression in neuronal cells.
J. Neurobiol. 25, 294–303.
Gibson, G. E., and Peterson, C. (1987). Calcium and the aging nervous system.
Neurobiol. Aging 8, 329–343.
Gokulrangan, G., Zaidi, A., Michaelis, M. L., and Schoneich, C. (2007). Proteomic analysis of protein nitration in rat cerebellum: effect of biological aging.
J. Neurochem. 100, 1494–1504.
Gomez-Villafuertes, R., Mellstrom, B., and Naranjo, J. R. (2007). Searching for a role of NCX/NCKX exchangers in neurodegeneration.
Mol. Neurobiol. 35, 195–202.
Gonzales, R. A., Brown, L. M., Jones, T. W., Trent, R. D., Westbrook, S. L., and Leslie, S. W. (1991).
N-methyl-
D-aspartate mediated responses decrease with age in Fischer 344 rat brain.
Neurobiol. Aging 12, 219–225.
Gore, A. C., Oung, T., and Woller, M. J. (2002). Age-related changes in hypothalamic gonadotropin-releasing hormone and
N-methyl-
D-aspartate receptor gene expression, and their regulation by oestrogen, in the female rat.
J. Neuroendocrinol. 14, 300–309.
Hallett, P. J., Spoelgen, R., Hyman, B. T., Standaert, D. G., and Dunah, A. W. (2006). Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking.
J. Neurosci. 26, 4690–4700.
Han, M., Schottler, F., Lei, D., Dong, E. Y., Bryan, A., and Bao, J. (2006). Bcl-2 over-expression fails to prevent age-related loss of calretinin positive neurons in the mouse dentate gyrus.
Mol. Neurodegener. 1, 9.
Han, S. H., Murchison, D., and Griffith, W. H. (2005). Low voltage-activated calcium and fast tetrodotoxin-resistant sodium currents define subtypes of cholinergic and noncholinergic neurons in rat basal forebrain.
Brain Res. Mol. Brain Res. 134, 226–238.
Hanahisa, Y., and Yamaguchi, M. (2001). Decrease in Ca2+-ATPase activity in the brain plasma membrane of rats with increasing age: involvement of brain calcium accumulation.
Int. J. Mol. Med. 7, 407–411.
Hardingham, G. E., Fukunaga, Y., and Bading, H. (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways.
Nat. Neurosci. 5, 405–414.
Hayashi, Y., Ishibashi, H., Hashimoto, K., and Nakanishi, H. (2006). Potentiation of the NMDA receptor-mediated responses through the activation of the glycine site by microglia secreting soluble factors.
Glia 53, 660–668.
Hayrapetyan, V., Rybalchenko, V., Rybalchenko, N., and Koulen, P. (2008). The N-terminus of presenilin-2 increases single channel activity of brain ryanodine receptors through direct protein-protein interaction.
Cell Calcium 44, 507–518.
Heidinger, V., Manzerra, P., Wang, X. Q., Strasser, U., Yu, S. P., Choi, D. W., and Behrens, M. M. (2002). Metabotropic glutamate receptor 1-induced upregulation of NMDA receptor current: mediation through the Pyk2/Src-family kinase pathway in cortical neurons.
J. Neurosci. 22, 5452–5461.
Herman, J. P., Chen, K. C., Booze, R., and Landfield, P. W. (1998). Up-regulation of alpha1D Ca2+ channel subunit mRNA expression in the hippocampus of aged F344 rats.
Neurobiol. Aging 19, 581–587.
Hidalgo, C., Bull, R., Behrens, M. I., and Donoso, P. (2004). Redox regulation of RyR-mediated Ca2+ release in muscle and neurons.
Biol. Res. 37, 539–552.
Igwe, O. J., and Ning, L. (1993). Inositol 1,4,5-trisphosphate arm of the phosphatidylinositide signal transduction pathway in the rat cerebellum during aging.
Neurosci. Lett. 164, 167–170.
Ingram, D. K., Garofalo, P., Spangler, E. L., Mantione, C. R., Odano, I., and London, E. D. (1992). Reduced density of NMDA receptors and increased sensitivity to dizocilpine-induced learning impairment in aged rats.
Brain Res. 580, 273–280.
Iwamoto, M., Hagishita, T., Shoji-Kasai, Y., Ando, S., and Tanaka, Y. (2004). Age-related changes in the levels of voltage-dependent calcium channels and other synaptic proteins in rat brain cortices.
Neurosci. Lett. 366, 277–281.
Jacob, C. P., Koutsilieri, E., Bartl, J., Neuen-Jacob, E., Arzberger, T., Zander, N., Ravid, R., Roggendorf, W., Riederer, P., and Grunblatt, E. (2007). Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease.
J. Alzheimers Dis. 11, 97–116.
Jones, S. W. (1998). Overview of voltage-dependent calcium channels.
J. Bioenerg. Biomembr. 30, 299–312.
Kang, M. G., Chen, C. C., Felix, R., Letts, V. A., Frankel, W. N., Mori, Y., and Campbell, K. P. (2001). Biochemical and biophysical evidence for gamma 2 subunit association with neuronal voltage-activated Ca2+ channels.
J. Biol. Chem. 276, 32917–32924.
Keller, J. N., Dimayuga, E., Chen, Q., Thorpe, J., Gee, J., and Ding, Q. (2004). Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain.
Int. J. Biochem. Cell Biol. 36, 2376–2391.
Khachaturian, Z. S. (1989). Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview.
Ann. N. Y. Acad. Sci. 568, 1–4.
Kito, S., Miyoshi, R., and Nomoto, T. (1990). Influence of age on NMDA receptor complex in rat brain studied by in vitro autoradiography.
J. Histochem. Cytochem. 38, 1725–1731.
Komatsu, Y., and Iwakiri, M. (1992). Low-threshold Ca2+ channels mediate induction of long-term potentiation in kitten visual cortex.
J. Neurophysiol. 67, 401–410.
Kowalska, M., and Disterhoft, J. F. (1994). Relation of nimodipine dose and serum concentration to learning enhancement in aging rabbits.
Exp. Neurol. 127, 159–166.
Kuehl-Kovarik, M. C., Partin, K. M., and Magnusson, K. R. (2003). Acute dissociation for analyses of NMDA receptor function in cortical neurons during aging.
J. Neurosci. Methods 129, 11–17.
Kumar, A., and Foster, T. C. (2002). 17beta-Estradiol benzoate decreases the AHP amplitude in CA1 pyramidal neurons.
J. Neurophysiol. 88, 621–626.
Kumar, A., and Foster, T. C. (2004). Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores.
J. Neurophysiol. 91, 2437–2444.
Kumar, A., and Foster, T. C. (2005). Intracellular calcium stores contribute to increased susceptibility to LTD induction during aging.
Brain Res. 1031, 125–128.
Kutsuwada, T., Kashiwabuchi, N., Mori, H., Sakimura, K., Kushiya, E., Araki, K., Meguro, H., Masaki, H., Kumanishi, T., Arakawa, M., and Mishina, M. (1992). Molecular diversity of the NMDA receptor channel.
Nature 358, 36–41.
Kwong, L. K., and Sohal, R. S. (2000). Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse.
Arch. Biochem. Biophys. 373, 16–22.
LaFerla, F. M. (2002). Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease.
Nat. Rev. Neurosci. 3, 862–872.
Landfield, P. W., and Pitler, T. A. (1984). Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats.
Science 226, 1089–1092.
Lau, C. G., and Zukin, R. S. (2007). NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders.
Nat. Rev. Neurosci. 8, 413–426.
Lee, H. C. (2004). Multiplicity of Ca2+ messengers and Ca2+ stores: a perspective from cyclic ADP-ribose and NAADP.
Curr. Mol. Med. 4, 227–237.
Levere, T. E., and Walker, A. (1992). Old age and cognition: enhancement of recent memory in aged rats by the calcium channel blocker nimodipine.
Neurobiol. Aging 13, 63–66.
Levy, A., Kong, R. M., Stillman, M. J., Shukitt-Hale, B., Kadar, T., Rauch, T. M., and Lieberman, H. R. (1991). Nimodipine improves spatial working memory and elevates hippocampal acetylcholine in young rats.
Pharmacol. Biochem. Behav. 39, 781–786.
Lieberman, D. N., and Mody, I. (1994). Regulation of NMDA channel function by endogenous Ca(2+)-dependent phosphatase.
Nature 369, 235–239.
Lin, Y., Jover-Mengual, T., Wong, J., Bennett, M. V., and Zukin, R. S. (2006). PSD-95 and PKC converge in regulating NMDA receptor trafficking and gating.
Proc. Natl. Acad. Sci. U.S.A. 103, 19902–19907.
Liu, F., Day, M., Muniz, L. C., Bitran, D., Arias, R., Revilla-Sanchez, R., Grauer, S., Zhang, G., Kelley, C., Pulito, V., Sung, A., Mervis, R. F., Navarra, R., Hirst, W. D., Reinhart, P. H., Marquis, K. L., Moss, S. J., Pangalos, M. N., and Brandon, N. J. (2008a). Activation of estrogen receptor-beta regulates hippocampal synaptic plasticity and improves memory.
Nat. Neurosci. 11, 334–343.
Liu, P., Smith, P. F., and Darlington, C. L. (2008b). Glutamate receptor subunits expression in memory-associated brain structures: regional variations and effects of aging.
Synapse 62, 834–841.
Long, L. H., Liu, J., Liu, R. L., Wang, F., Hu, Z. L., Xie, N., Fu, H., and Chen, J. G. (2009). Differential effects of methionine and cysteine oxidation on [Ca2+] i in cultured hippocampal neurons.
Cell. Mol. Neurobiol. 29, 7–15.
Lopez, J. R., Lyckman, A., Oddo, S., Laferla, F. M., Querfurth, H. W., and Shtifman, A. (2008). Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice.
J. Neurochem. 105, 262–271.
Lopez de Armentia, M., and Sah, P. (2004). Firing properties and connectivity of neurons in the rat lateral central nucleus of the amygdala.
J. Neurophysiol. 92, 1285–1294.
Lu, C. B., Hamilton, J. B., Powell, A. D., Toescu, E. C., and Vreugdenhil, M. (2009). Effect of ageing on CA3 interneuron sAHP and gamma oscillations is activity-dependent.
Neurobiol. Aging. [Epub ahead of print].
Lukas, W., and Jones, K. A. (1994). Cortical neurons containing calretinin are selectively resistant to calcium overload and excitotoxicity in vitro.
Neuroscience 61, 307–316.
Ly, C. V., and Verstreken, P. (2006). Mitochondria at the synapse.
Neuroscientist 12, 291–299.
Lynch, G., and Bi, X. (2003). Lysosomes and brain aging in mammals.
Neurochem. Res. 28, 1725–1734.
Magee, J. C., and Johnston, D. (1995a). Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons.
J. Physiol. (Lond.) 487(Pt. 1), 67–90.
Magee, J. C., and Johnston, D. (1995b). Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons.
Science 268, 301–304.
Magnusson, K. R. (1995). Differential effects of aging on binding sites of the activated NMDA receptor complex in mice.
Mech. Ageing Dev. 84, 227–243.
Magnusson, K. R. (1998). The aging of the NMDA receptor complex.
Front. Biosci. 3, e70–e80.
Magnusson, K. R. (2000). Declines in mRNA expression of different subunits may account for differential effects of aging on agonist and antagonist binding to the NMDA receptor.
J. Neurosci. 20, 1666–1674.
Magnusson, K. R. (2001). Influence of diet restriction on NMDA receptor subunits and learning during aging.
Neurobiol. Aging 22, 613–627.
Magnusson, K. R., Bai, L., and Zhao, X. (2005). The effects of aging on different C-terminal splice forms of the zeta1(NR1) subunit of the
N-methyl-
D-aspartate receptor in mice.
Brain Res. Mol. Brain Res. 135, 141–149.
Magnusson, K. R., and Cotman, C. W. (1993). Age-related changes in excitatory amino acid receptors in two mouse strains.
Neurobiol. Aging 14, 197–206.
Magnusson, K. R., Kresge, D., and Supon, J. (2006). Differential effects of aging on NMDA receptors in the intermediate versus the dorsal hippocampus.
Neurobiol. Aging 27, 324–333.
Martella, G., Spadoni, F., Sciamanna, G., Tassone, A., Bernardi, G., Pisani, A., and Bonsi, P. (2008). Age-related functional changes of high-voltage-activated calcium channels in different neuronal subtypes of mouse striatum.
Neuroscience 152, 469–476.
Martinez-Serrano, A., Blanco, P., and Satrustegui, J. (1992). Calcium binding to the cytosol and calcium extrusion mechanisms in intact synaptosomes and their alterations with aging.
J. Biol. Chem. 267, 4672–4679.
Martinez Villayandre, B., Paniagua, M. A., Fernandez-Lopez, A., Chinchetru, M. A., and Calvo, P. (2004). Effect of vitamin E treatment on
N-methyl-
D-aspartate receptor at different ages in the rat brain.
Brain Res. 1028, 148–155.
Martini, A., Battaini, F., Govoni, S., and Volpe, P. (1994). Inositol 1,4,5-trisphosphate receptor and ryanodine receptor in the aging brain of Wistar rats.
Neurobiol. Aging 15, 203–206.
Massey, P. V., Johnson, B. E., Moult, P. R., Auberson, Y. P., Brown, M. W., Molnar, E., Collingridge, G. L., and Bashir, Z. I. (2004). Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression.
J. Neurosci. 24, 7821–7828.
Mattson, M. P. (2007). Calcium and neurodegeneration.
Aging Cell 6, 337–350.
Mattson, M. P., LaFerla, F. M., Chan, S. L., Leissring, M. A., Shepel, P. N., and Geiger, J. D. (2000). Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders.
Trends Neurosci. 23, 222–229.
Mattson, M. P., Rychlik, B., Chu, C., and Christakos, S. (1991). Evidence for calcium-reducing and excito-protective roles for the calcium-binding protein calbindin-D28k in cultured hippocampal neurons.
Neuron 6, 41–51.
Matute, C. (2007). Interaction between glutamate signalling and immune attack in damaging oligodendrocytes.
Neuron Glia Biol. 3, 281–285.
McGuinness, L., Bardo, S. J., and Emptage, N. J. (2007). The lysosome or lysosome-related organelle may serve as a Ca2+ store in the boutons of hippocampal pyramidal cells.
Neuropharmacology 52, 126–135.
Meguro, H., Mori, H., Araki, K., Kushiya, E., Kutsuwada, T., Yamazaki, M., Kumanishi, T., Arakawa, M., Sakimura, K., and Mishina, M. (1992). Functional characterization of a heteromeric NMDA receptor channel expressed from cloned cDNAs.
Nature 357, 70–74.
Mesches, M. H., Gemma, C., Veng, L. M., Allgeier, C., Young, D. A., Browning, M. D., and Bickford, P. C. (2004). Sulindac improves memory and increases NMDA receptor subunits in aged Fischer 344 rats.
Neurobiol. Aging 25, 315–324.
Michaelis, M. L., Bigelow, D. J., Schoneich, C., Williams, T. D., Ramonda, L., Yin, D., Huhmer, A. F., Yao, Y., Gao, J., and Squier, T. C. (1996). Decreased plasma membrane calcium transport activity in aging brain.
Life Sci. 59, 405–412.
Michaelis, M. L., Johe, K., and Kitos, T. E. (1984). Age-dependent alterations in synaptic membrane systems for Ca2+ regulation.
Mech. Ageing Dev. 25, 215–225.
Mishizen-Eberz, A. J., Rissman, R. A., Carter, T. L., Ikonomovic, M. D., Wolfe, B. B., and Armstrong, D. M. (2004). Biochemical and molecular studies of NMDA receptor subunits NR1/2A/2B in hippocampal subregions throughout progression of Alzheimer’s disease pathology.
Neurobiol. Dis. 15, 80–92.
Miyoshi, R., Kito, S., Doudou, N., and Nomoto, T. (1991). Influence of age on
N-methyl-
D-aspartate antagonist binding sites in the rat brain studied by in vitro autoradiography.
Synapse 8, 212–217.
Mizutani, T., Nakashima, S., and Nozawa, Y. (1998). Changes in the expression of protein kinase C (PKC), phospholipases C (PLC) and D (PLD) isoforms in spleen, brain and kidney of the aged rat: RT-PCR and western blot analysis.
Mech. Ageing Dev. 105, 151–172.
Monyer, H., Sprengel, R., Schoepfer, R., Herb, A., Higuchi, M., Lomeli, H., Burnashev, N., Sakmann, B., and Seeburg, P. H. (1992). Heteromeric NMDA receptors: molecular and functional distinction of subtypes.
Science 256, 1217–1221.
Moriguchi, S., Mizoguchi, Y., Tomimatsu, Y., Hayashi, Y., Kadowaki, T., Kagamiishi, Y., Katsube, N., Yamamoto, K., Inoue, K., Watanabe, S., Nabekura, J., and Nakanishi, H. (2003). Potentiation of NMDA receptor-mediated synaptic responses by microglia.
Brain Res. Mol. Brain Res. 119, 160–169.
Moriyoshi, K., Masu, M., Ishii, T., Shigemoto, R., Mizuno, N., and Nakanishi, S. (1991). Molecular cloning and characterization of the rat NMDA receptor.
Nature 354, 31–37.
Morrison, B. M., Hof, P. R., and Morrison, J. H. (1998). Determinants of neuronal vulnerability in neurodegenerative diseases.
Ann. Neurol. 44, S32–S44.
Moyer, J. R. Jr., Thompson, L. T., Black, J. P., and Disterhoft, J. F. (1992). Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner.
J. Neurophysiol. 68, 2100–2109.
Murchison, D., and Griffith, W. H. (1995). Low-voltage activated calcium currents increase in basal forebrain neurons from aged rats.
J. Neurophysiol. 74, 876–887.
Murchison, D., and Griffith, W. H. (1996). High-voltage-activated calcium currents in basal forebrain neurons during aging.
J. Neurophysiol. 76, 158–174.
Murchison, D., and Griffith, W. H. (1999). Age-related alterations in caffeine-sensitive calcium stores and mitochondrial buffering in rat basal forebrain.
Cell Calcium 25, 439–452.
Murchison, D., and Griffith, W. H. (2007). Calcium buffering systems and calcium signaling in aged rat basal forebrain neurons.
Aging Cell 6, 297–305.
Murchison, D., McDermott, A. N., LaSarge, C. A., Peebles, K. A., Bizon, J. L., and Griffith, W. H. (2009). Enhanced calcium buffering in F344 rat cholinergic basal forebrain neurons is associated with age-related cognitive impairment.
J Neurophysiol. 102, 2194–2207.
Murchison, D., Zawieja, D. C., and Griffith, W. H. (2004). Reduced mitochondrial buffering of voltage-gated calcium influx in aged rat basal forebrain neurons.
Cell Calcium 36, 61–75.
Naidoo, N. (2009). ER and aging-protein folding and the ER stress response.
Ageing Res. Rev. 8, 150–159.
Nicholls, D. G., and Budd, S. L. (2000). Mitochondria and neuronal survival.
Physiol. Rev. 80, 315–360.
Nicolle, M. M., Colombo, P. J., Gallagher, M., and McKinney, M. (1999). Metabotropic glutamate receptor-mediated hippocampal phosphoinositide turnover is blunted in spatial learning-impaired aged rats.
J. Neurosci. 19, 9604–9610.
Nilius, B., Hess, P., Lansman, J. B., and Tsien, R. W. (1985). A novel type of cardiac calcium channel in ventricular cells.
Nature 316, 443–446.
Norris, C. M., Blalock, E. M., Chen, K. C., Porter, N. M., and Landfield, P. W. (2002). Calcineurin enhances L-type Ca(2+) channel activity in hippocampal neurons: increased effect with age in culture.
Neuroscience 110, 213–225.
Norris, C. M., and Foster, T. C. (1999). MK-801 improves retention in aged rats: implications for altered neural plasticity in age-related memory deficits.
Neurobiol. Learn. Mem. 71, 194–206.
Norris, C. M., Halpain, S., and Foster, T. C. (1998a). Alterations in the balance of protein kinase/phosphatase activities parallel reduced synaptic strength during aging.
J. Neurophysiol. 80, 1567–1570.
Norris, C. M., Halpain, S., and Foster, T. C. (1998b). Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels.
J. Neurosci. 18, 3171–3179.
Nowycky, M. C., Fox, A. P., and Tsien, R. W. (1985). Three types of neuronal calcium channel with different calcium agonist sensitivity.
Nature 316, 440–443.
Orrenius, S., Zhivotovsky, B., and Nicotera, P. (2003). Regulation of cell death: the calcium-apoptosis link.
Nat. Rev. Mol. Cell Biol. 4, 552–565.
Ouda, L., Druga, R., and Syka, J. (2008). Changes in parvalbumin immunoreactivity with aging in the central auditory system of the rat.
Exp. Gerontol. 43, 782–789.
Pandey, V., Chuang, C. C., Lewis, A. M., Aley, P., Brailoiu, E., Dun, N., Churchill, G. C., and Patel, S. (2009). Recruitment of NAADP-sensitive acidic Ca2+ stores by glutamate.
Biochem. J. 422, 503–512.
Parihar, M. S., Kunz, E. A., and Brewer, G. J. (2008). Age-related decreases in NAD(P)H and glutathione cause redox declines before ATP loss during glutamate treatment of hippocampal neurons.
J. Neurosci. Res. 86, 2339–2352.
Peuchen, S., Duchen, M. R., and Clark, J. B. (1996). Energy metabolism of adult astrocytes in vitro.
Neuroscience 71, 855–870.
Pieta Dias, C., Martins de Lima, M. N., Presti-Torres, J., Dornelles, A., Garcia, V. A., Siciliani Scalco, F., Rewsaat Guimaraes, M., Constantino, L., Budni, P., Dal-Pizzol, F., and Schroder, N. (2007). Memantine reduces oxidative damage and enhances long-term recognition memory in aged rats.
Neuroscience 146, 1719–1725.
Pittaluga, A., Fedele, E., Risiglione, C., and Raiteri, M. (1993). Age-related decrease of the NMDA receptor-mediated noradrenaline release in rat hippocampus and partial restoration by
D-cycloserine.
Eur. J. Pharmacol. 231, 129–134.
Poon, H. F., Calabrese, V., Calvani, M., and Butterfield, D. A. (2006). Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by
L-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress.
Antioxid. Redox Signal. 8, 381–394.
Pottorf, W. J., De Leon, D. D., Hessinger, D. A., and Buchholz, J. N. (2001). Function of SERCA mediated calcium uptake and expression of SERCA3 in cerebral cortex from young and old rats.
Brain Res. 914, 57–65.
Power, J. M., Wu, W. W., Sametsky, E., Oh, M. M., and Disterhoft, J. F. (2002). Age-related enhancement of the slow outward calcium-activated potassium current in hippocampal CA1 pyramidal neurons in vitro.
J. Neurosci. 22, 7234–7243.
Quartermain, D., Hawxhurst, A., Ermita, B., and Puente, J. (1993). Effect of the calcium channel blocker amlodipine on memory in mice.
Behav. Neural Biol. 60, 211–219.
Quevedo, J., Vianna, M., Daroit, D., Born, A. G., Kuyven, C. R., Roesler, R., and Quillfeldt, J. A. (1998). L-type voltage-dependent calcium channel blocker nifedipine enhances memory retention when infused into the hippocampus.
Neurobiol. Learn. Mem. 69, 320–325.
Raman, I. M., Tong, G., and Jahr, C. E. (1996). Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase.
Neuron 16, 415–421.
Rebrin, I., Forster, M. J., and Sohal, R. S. (2007). Effects of age and caloric intake on glutathione redox state in different brain regions of C57BL/6 and DBA/2 mice.
Brain Res. 1127, 10–18.
Rizzuto, R. (2001). Intracellular Ca(2+) pools in neuronal signalling.
Curr. Opin. Neurobiol. 11, 306–311.
Rose, G. M., Ong, V. S., and Woodruff-Pak, D. S. (2007). Efficacy of MEM 1003, a novel calcium channel blocker, in delay and trace eyeblink conditioning in older rabbits.
Neurobiol. Aging 28, 766–773.
Rosi, S., Ramirez-Amaya, V., Hauss-Wegrzyniak, B., and Wenk, G. L. (2004). Chronic brain inflammation leads to a decline in hippocampal NMDA-R1 receptors.
J. Neuroinflammation 1, 12.
Rosi, S., Vazdarjanova, A., Ramirez-Amaya, V., Worley, P. F., Barnes, C. A., and Wenk, G. L. (2006). Memantine protects against LPS-induced neuroinflammation, restores behaviorally-induced gene expression and spatial learning in the rat.
Neuroscience 142, 1303–1315.
Roth, G. S. (1995). Changes in tissue responsiveness to hormones and neurotransmitters during aging.
Exp. Gerontol. 30, 361–368.
Sandin, M., Jasmin, S., and Levere, T. E. (1990). Aging and cognition: facilitation of recent memory in aged nonhuman primates by nimodipine.
Neurobiol. Aging 11, 573–575.
Satrustegui, J., Villalba, M., Pereira, R., Bogonez, E., and Martinez-Serrano, A. (1996). Cytosolic and mitochondrial calcium in synaptosomes during aging.
Life Sci. 59, 429–434.
Scharfman, H. E., and Schwartzkroin, P. A. (1989). Protection of dentate hilar cells from prolonged stimulation by intracellular calcium chelation.
Science 246, 257–260.
Schell, M. J., Molliver, M. E., and Snyder, S. H. (1995).
D-Serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release.
Proc. Natl. Acad. Sci. U.S.A. 92, 3948–3952.
Scott, D. B., Blanpied, T. A., Swanson, G. T., Zhang, C., and Ehlers, M. D. (2001). An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing.
J. Neurosci. 21, 3063–3072.
Serra, M., Ghiani, C. A., Foddi, M. C., Motzo, C., and Biggio, G. (1994). NMDA receptor function is enhanced in the hippocampus of aged rats.
Neurochem. Res. 19, 483–487.
Shankar, S., Teyler, T. J., and Robbins, N. (1998). Aging differentially alters forms of long-term potentiation in rat hippocampal area CA1.
J. Neurophysiol. 79, 334–341.
Shimada, A., Mukhin, A., Ingram, D. K., and London, E. D. (1997).
N-methyl-
D-aspartate receptor binding in brains of rats at different ages.
Neurobiol. Aging 18, 329–333.
Simonyi, A., Xia, J., Igbavboa, U., Wood, W. G., and Sun, G. Y. (1998). Age differences in the expression of metabotropic glutamate receptor 1 and inositol 1,4,5-trisphosphate receptor in mouse cerebellum.
Neurosci. Lett. 244, 29–32.
Singaravelu, K., and Deitmer, J. W. (2006). Calcium mobilization by nicotinic acid adenine dinucleotide phosphate (NAADP) in rat astrocytes.
Cell Calcium 39, 143–153.
Smith, I. F., Hitt, B., Green, K. N., Oddo, S., and LaFerla, F. M. (2005). Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease.
J. Neurochem. 94, 1711–1718.
Snyder, E. M., Nong, Y., Almeida, C. G., Paul, S., Moran, T., Choi, E. Y., Nairn, A. C., Salter, M. W., Lombroso, P. J., Gouras, G. K., and Greengard, P. (2005). Regulation of NMDA receptor trafficking by amyloid-beta.
Nat. Neurosci. 8, 1051–1058.
Solomon, P. R., Wood, M. S., Groccia-Ellison, M. E., Yang, B. Y., Fanelli, R. J., and Mervis, R. F. (1995). Nimodipine facilitates retention of the classically conditioned nictitating membrane response in aged rabbits over long retention intervals.
Neurobiol. Aging 16, 791–796.
Solovyova, N., Veselovsky, N., Toescu, E. C., and Verkhratsky, A. (2002). Ca(2+) dynamics in the lumen of the endoplasmic reticulum in sensory neurons: direct visualization of Ca(2+)-induced Ca(2+) release triggered by physiological Ca(2+) entry.
EMBO J. 21, 622–630.
Sonntag, W. E., Bennett, S. A., Khan, A. S., Thornton, P. L., Xu, X., Ingram, R. L., and Brunso-Bechtold, J. K. (2000). Age and insulin-like growth factor-1 modulate
N-methyl-
D-aspartate receptor subtype expression in rats.
Brain Res. Bull. 51, 331–338.
Soong, T. W., Stea, A., Hodson, C. D., Dubel, S. J., Vincent, S. R., and Snutch, T. P. (1993). Structure and functional expression of a member of the low voltage-activated calcium channel family.
Science 260, 1133–1136.
Squier, T. C., and Bigelow, D. J. (2000). Protein oxidation and age-dependent alterations in calcium homeostasis.
Front. Biosci. 5, D504–D526.
Stolzing, A., Sethe, S., and Grune, T. (2005). Chronically active: activation of microglial proteolysis in ageing and neurodegeneration.
Redox Rep. 10, 207–213.
Straube, K. T., Deyo, R. A., Moyer, J. R. Jr., and Disterhoft, J. F. (1990). Dietary nimodipine improves associative learning in aging rabbits.
Neurobiol. Aging 11, 659–661.
Stutzmann, G. E. (2007). The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”?
Neuroscientist 13, 546–559.
Stutzmann, G. E., Smith, I., Caccamo, A., Oddo, S., Laferla, F. M., and Parker, I. (2006). Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice.
J. Neurosci. 26, 5180–5189.
Sucher, N. J., and Lipton, S. A. (1991). Redox modulatory site of the NMDA receptor-channel complex: regulation by oxidized glutathione.
J. Neurosci. Res. 30, 582–591.
Sullivan, J. M., Traynelis, S. F., Chen, H. S., Escobar, W., Heinemann, S. F., and Lipton, S. A. (1994). Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor.
Neuron 13, 929–936.
Supnet, C., Grant, J., Kong, H., Westaway, D., and Mayne, M. (2006). Amyloid-beta-(1-42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice.
J. Biol. Chem. 281, 38440–38447.
Tamaru, M., Yoneda, Y., Ogita, K., Shimizu, J., and Nagata, Y. (1991). Age-related decreases of the
N-methyl-
D-aspartate receptor complex in the rat cerebral cortex and hippocampus.
Brain Res. 542, 83–90.
Tanaka, Y., and Ando, S. (2001). Age-related changes in the subtypes of voltage-dependent calcium channels in rat brain cortical synapses.
Neurosci. Res. 39, 213–220.
Tang, T. S., Slow, E., Lupu, V., Stavrovskaya, I. G., Sugimori, M., Llinas, R., Kristal, B. S., Hayden, M. R., and Bezprozvanny, I. (2005). Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease.
Proc. Natl. Acad. Sci. U.S.A. 102, 2602–2607.
Tang, T. S., Tu, H., Chan, E. Y., Maximov, A., Wang, Z., Wellington, C. L., Hayden, M. R., and Bezprozvanny, I. (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1.
Neuron 39, 227–239.
Thibault, O., and Landfield, P. W. (1996). Increase in single L-type calcium channels in hippocampal neurons during aging.
Science 272, 1017–1020.
Thompson, L. T., Deyo, R. A., and Disterhoft, J. F. (1990). Nimodipine enhances spontaneous activity of hippocampal pyramidal neurons in aging rabbits at a dose that facilitates associative learning.
Brain Res. 535, 119–130.
Toescu, E. C., Myronova, N., and Verkhratsky, A. (2000). Age-related structural and functional changes of brain mitochondria.
Cell Calcium 28, 329–338.
Toescu, E. C., and Verkhratsky, A. (2000). Assessment of mitochondrial polarization status in living cells based on analysis of the spatial heterogeneity of rhodamine 123 fluorescence staining.
Pflugers Arch. 440, 941–947.
Toescu, E. C., and Verkhratsky, A. (2003). Neuronal ageing from an intraneuronal perspective: roles of endoplasmic reticulum and mitochondria.
Cell Calcium 34, 311–323.
Toescu, E. C., and Verkhratsky, A. (2004). Ca2+ and mitochondria as substrates for deficits in synaptic plasticity in normal brain ageing.
J. Cell. Mol. Med. 8, 181–190.
Topic, B., Willuhn, I., Palomero-Gallagher, N., Zilles, K., Huston, J. P., and Hasenohrl, R. U. (2007). Impaired maze performance in aged rats is accompanied by increased density of NMDA, 5-HT1A, and alpha-adrenoceptor binding in hippocampus.
Hippocampus 17, 68–77.
Trompet, S., Westendorp, R. G., Kamper, A. M., and de Craen, A. J. (2008). Use of calcium antagonists and cognitive decline in old age. The Leiden 85-plus study.
Neurobiol. Aging 29, 306–308.
Tsai, H., Pottorf, W. J., Buchholz, J. N., and Duckles, S. P. (1998). Adrenergic nerve smooth endoplasmic reticulum calcium buffering declines with age.
Neurobiol. Aging 19, 89–96.
Uebele, V. N., Nuss, C. E., Santarelli, V. P., Garson, S. L., Barrow, J. C., Stauffer, S. R., Koblan, K. S., Renger, J. J., Aton, S., Seibt, J., Dumoulin, M., Jha, S. K., Coleman, T., and Frank, M. G. (2009). T-type calcium channels regulate cortical plasticity in vivo NR-D-08-7049.
Neuroreport 20, 257–262.
Vanhoutte, P., and Bading, H. (2003). Opposing roles of synaptic and extrasynaptic NMDA receptors in neuronal calcium signalling and BDNF gene regulation.
Curr. Opin. Neurobiol. 13, 366–371.
Veng, L. M., Mesches, M. H., and Browning, M. D. (2003). Age-related working memory impairment is correlated with increases in the L-type calcium channel protein alpha1D (Cav1.3) in area CA1 of the hippocampus and both are ameliorated by chronic nimodipine treatment.
Brain Res. Mol. Brain Res. 110, 193–202.
Verkhratsky, A., and Toescu, E. C. (1998). Calcium and neuronal ageing.
Trends Neurosci. 21, 2–7.
Veselovskii, N. S., and Fedulova, S. A. (1983). [2 types of calcium channels in the somatic membrane of spinal ganglion neurons in the rat].
Dokl. Akad. Nauk SSSR 268, 747–750.
Wang, L. Y., Orser, B. A., Brautigan, D. L., and MacDonald, J. F. (1994). Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A.
Nature 369, 230–232.
Wenk, G. L., and Barnes, C. A. (2000). Regional changes in the hippocampal density of AMPA and NMDA receptors across the lifespan of the rat.
Brain Res. 885, 1–5.
Wenk, G. L., Walker, L. C., Price, D. L., and Cork, L. C. (1991). Loss of NMDA, but not GABA-A, binding in the brains of aged rats and monkeys.
Neurobiol. Aging 12, 93–98.
Williams, S. M., Diaz, C. M., Macnab, L. T., Sullivan, R. K., and Pow, D. V. (2006). Immunocytochemical analysis of
D-serine distribution in the mammalian brain reveals novel anatomical compartmentalizations in glia and neurons.
Glia 53, 401–411.
Woodruff-Pak, D. S., Chi, J., Li, Y. T., Pak, M. H., and Fanelli, R. J. (1997). Nimodipine ameliorates impaired eyeblink classical conditioning in older rabbits in the long-delay paradigm.
Neurobiol. Aging 18, 641–649.
Wu, C. K., Nagykery, N., Hersh, L. B., Scinto, L. F., and Geula, C. (2003). Selective age-related loss of calbindin-D28k from basal forebrain cholinergic neurons in the common marmoset (
Callithrix jacchus).
Neuroscience 120, 249–259.
Wu, S., and Barger, S. W. (2004). Induction of serine racemase by inflammatory stimuli is dependent on AP-1.
Ann. N. Y. Acad. Sci. 1035, 133–146.
Wu, S. Z., Bodles, A. M., Porter, M. M., Griffin, W. S., Basile, A. S., and Barger, S. W. (2004). Induction of serine racemase expression and
D-serine release from microglia by amyloid beta-peptide.
J. Neuroinflammation 1, 2.
Xiong, J., Camello, P. J., Verkhratsky, A., and Toescu, E. C. (2004). Mitochondrial polarisation status and [Ca2+]i signalling in rat cerebellar granule neurones aged in vitro.
Neurobiol. Aging 25, 349–359.
Xiong, J., Verkhratsky, A., and Toescu, E. C. (2002). Changes in mitochondrial status associated with altered Ca2+ homeostasis in aged cerebellar granule neurons in brain slices.
J. Neurosci. 22, 10761–10771.
Yan, L. J., and Sohal, R. S. (1998). Mitochondrial adenine nucleotide translocase is modified oxidatively during aging.
Proc. Natl. Acad. Sci. U.S.A. 95, 12896–12901.
Yasuda, R., Sabatini, B. L., and Svoboda, K. (2003). Plasticity of calcium channels in dendritic spines.
Nat. Neurosci. 6, 948–955.
Yoshimura, Y., Inaba, M., Yamada, K., Kurotani, T., Begum, T., Reza, F., Maruyama, T., and Komatsu, Y. (2008). Involvement of T-type Ca2+ channels in the potentiation of synaptic and visual responses during the critical period in rat visual cortex.
Eur. J. Neurosci. 28, 730–743.
Zaidi, A., Gao, J., Squier, T. C., and Michaelis, M. L. (1998). Age-related decrease in brain synaptic membrane Ca2+-ATPase in F344/BNF1 rats.
Neurobiol. Aging 19, 487–495.
Zaidi, A., and Michaelis, M. L. (1999). Effects of reactive oxygen species on brain synaptic plasma membrane Ca(2+)-ATPase.
Free Radic. Biol. Med. 27, 810–821.
Zhao, X., Rosenke, R., Kronemann, D., Brim, B., Das, S. R., Dunah, A. W., and Magnusson, K. R. (2009). The effects of aging on
N-methyl-
D-aspartate receptor subunits in the synaptic membrane and relationships to long-term spatial memory.
Neuroscience 162, 933–945
.