1
Max Delbrück Center for Molecular Medicine Berlin-Buch, Berlin, Germany
2
VolkswagenStiftung Research Group, Department of Experimental Neurology, Charité University Medicine Berlin, Berlin, Germany
3
CRTD-DFG Research Center for Regenerative Therapies Dresden, Dresden, Germany
4
Physiological Institute, University of Freiburg, Freiburg, Germany
Adult hippocampal neurogenesis is regulated by activity. But how do neural precursor cells in the hippocampus respond to surrounding network activity and translate increased neural activity into a developmental program? Here we show that long-term potentiation (LTP)-like synaptic activity within a cellular network of mature hippocampal neurons promotes neuronal differentiation of newly generated cells. In co-cultures of precursor cells with primary hippocampal neurons, LTP-like synaptic plasticity induced by addition of glycine in Mg2+-free media for 5 min, produced synchronous network activity and subsequently increased synaptic strength between neurons. Furthermore, this synchronous network activity led to a significant increase in neuronal differentiation from the co-cultured neural precursor cells. When applied directly to precursor cells, glycine- and Mg2+-free solution did not induce neuronal differentiation. Synaptic plasticity-induced neuronal differentiation of precursor cells was observed in the presence of GABAergic neurotransmission blockers but was dependent on NMDA-mediated Ca2+ influx. Most importantly, neuronal differentiation required the release of brain-derived neurotrophic factor (BDNF) from the underlying substrate hippocampal neurons as well as TrkB receptor phosphorylation in precursor cells. This suggests that activity-dependent stem cell differentiation within the hippocampal network is mediated via synaptically evoked BDNF signaling.
Adult hippocampal neurogenesis originating from neural precursor cells in the adult hippocampus has been linked to memory formation by providing structural network optimization ( Wiskott et al., 2006
; Aimone et al., 2009
; Garthe et al., 2009
). New neurons integrate into the existing circuitry and survive lifelong (Kempermann et al., 2003
). Neuronal activity regulates neuronal differentiation in the adult hippocampus via synaptic GABA and glutamate release (Tozuka et al., 2005
; Ge et al., 2007
). Kainic acid, an agonist at the non-NMDA receptor subtype of glutamate receptors, induces an increase in the number of newborn granule cells ( Bengzon et al., 1997
; Parent et al., 1997
). Although the proportion of active granule cells is sparse (Chawla et al., 2005
), single neurons fire brief high frequency bursts (Axmacher et al., 2006
) which might be important not only for induction of synaptic plasticity but – as we reasoned – also for the modulation of adult neurogenesis.
Network oscillations are believed to be important for temporal coding, information processing and storage of new memories (Buzsaki, 2002
). Theta rhythms (∼5–10 Hz) promote induction of synaptic plasticity in mature and newborn granule cells (Schmidt-Hieber et al., 2004
). Adult-born hippocampal neurons are highly excitable and show transiently enhanced LTP as compared to older granule cells at about 2–6 weeks after cell division (Wang et al., 2000a
; Snyder et al., 2001
; Schmidt-Hieber et al., 2004
; Saxe et al., 2006
; Garthe et al., 2009
), which might be important for preferential integration of young neurons in new memory circuits (Kee et al., 2007
). The general idea, supported by several studies demonstrating the increased survival of newborn neurons in response to learning stimuli, is that the new cells undergo a “critical period” after they were born (Kempermann et al., 1997
; Gould et al., 1999
; Greenough et al., 1999
; Van Praag et al., 1999b
; Kee et al., 2007
; Tashiro et al., 2007
). Much less is known about the early time periods, before GABAergic and glutamatergic synaptic transmission develop (van Praag et al., 2002
; Esposito et al., 2005
). This raised the important question, how neuronal network activity might influence the earliest stages of adult neurogenesis. We hypothesize that neural precursor cells can sense surrounding network activity and translate this information into a pro-neurogenic signal. Could therefore coherent synchronous network activity induce neuronal differentiation?
Behavioral activity such as voluntary physical exercise and environmental enrichment generate synchronized activity in the hippocampus and modulate learning and LTP (Van Praag et al., 1999a
; Duffy et al., 2001
). Similarly, environmental enrichment, physical exercise, and learning paradigms regulate neurogenesis in the dentate gyrus (DG) (Van Praag et al., 1999a
; Kempermann et al., 1997
; Gould et al., 1999
; Leuner et al., 2004
). Although both adult neurogenesis and synaptic plasticity are considered relevant for hippocampal function, the question whether there is a link between these two has hardly been addressed. It has been reported, though, that systems level LTP might increase adult hippocampal neurogenesis (Bruel-Jungerman et al., 2006
). However, it remained unknown whether synaptic plasticity in neurons could induce adult neurogenesis on the level of the precursor cells.
To determine the relationship between synaptic network plasticity and precursor cell behavior we developed a co-culture model consisting of hippocampal neurons and adult hippocampal precursor cells. Hippocampal neurons are an excellent model to study network activity and its synaptic relevance. In culture, they show relatively low synaptic release probability, which increases after brief calcium (Ca2+) rises mediated by NMDA receptor activation (Malenka, 2003
; Malenka and Bear, 2004
). Although the short-term cellular effects of such stimuli on mature functional neurons are well documented, the effects of such changes on neural precursor cells are not known.
Using this model we addressed the following questions: (1) Can neural precursor cells detect neuronal network activity? (2) How do precursor cells respond to changes in synaptic activity levels? (3) What are the signaling mechanisms by which synaptically active neurons influence precursor cells?
Adult Hippocampal Precursor Cells Culture
All institutional guidelines regarding animal handling and killing were followed. Precursor cells were cultured as described previously with slight modifications (Babu et al., 2007
). C57Bl6 mice were killed by an overdose of ketamine. The brains were removed and placed in Hepes buffer Hanks balanced salt saline solution (pH 7.3). The DG was isolated and digested in an enzyme cocktail containing 10 U/ml Papain (Worthington), 2 mg/ml dispase (Roche), and 1 U/ml DNase (Worthington) at 37°C for 40 min. Cell suspensions were washed with DPBS and enriched for precursor cells on a 22% percoll gradient (Amersham). The cells were plated on polystyrene surfaces coated with Poly-lysine (Sigma; 10 μg/ml) and Laminin (Roche; 5 μg/ml). The medium was serum free with added Neurobasal (Invitrogen), 2% B27 (Invitrogen) and 2 mM Glutamax (Invitrogen) and 20 ng/ml EGF (Peprotech) and 20 ng/ml bFGF (Peprotech). For differentiation, growth factors were withdrawn and the cells were allowed to differentiate for 4–6 days. In some of the differentiating cultures we also added 5 ng/ml of FGF-2.
For green fluorescent protein (EGFP) viral labeling, precursor cells were incubated with viral supernatant containing retrovirus with EGFP vector overnight for transducing and expressing the virus. The cells were washed subsequently and purified by fluorescence activated cell sorting (FACS) for EGFP expression and subsequent propagation. Purification was tested by immunostaining for EGFP expression.
Primary Postnatal Hippocampal Neuronal Culture
For cultures of primary hippocampal neurons, postnatal mice (P 0–1) were killed by decapitation and the hippocampus was dissected and subjected to digestion by Papain (20 U/ml) for 30–40 min at 37°C followed by Trypsin inhibitor (1 mg/ml). Cells were plated on to poly-lysine/laminin-coated coverslips at 5 × 104 cells/cm2 with medium containing Neurobasal, B27 (2%) and Glutamax (2 mM).
Under these conditions about 80% of the cells were MAP2ab positive neurons. The co-culture was established by trypsinizing the EGFP-labeled precursor cells and overlaying the primary hippocampal neurons with the precursor cell suspension at a density of 5 × 105 cells/cm2, replacing 75% of the medium. To sequester endogenously secreted BDNF recombinant Fc-trkB (R&D) and bioneutralizing antibody against BDNF (Santa-Cruz) were added at a concentration of 5–10 μg/ml. Rabbit anti-NT3 (Santa-Cruz) was used at a concentration of 5–10 μg/ml for neutralizing endogenously secreted neurotrophin-3 (NT3).
Immunocytochemisty and Immunohistochemistry
Cells were fixed in 4% paraformaldehyde for 20 min, washed with PBS, and blocked and permeabilized by incubating in PBS containing 0.25% Triton X-100 and 10% donkey serum. The primary antibodies were applied in 3% donkey serum in PBS overnight at 4°C. As primary antibodies we used mouse anti-βIII-tubulin (1:1000; Promega), rabbit anti-βIII-tubulin (1:2000; Covance), mouse anti-Synaptophysin (1:50; Abcam) mouse anti-MAP2ab (1:500; Sigma) and goat anti-GFP (1:1000; Axis).
Primary antibodies were detected by fluorophore-conjugated secondary antibodies (Dianova) incubated for 2 h at room temperature. The nuclei were stained with Hoechst 33258 (Sigma). For imaging we used a confocal microscope (TCS-SP2 Leica, Bensheim) in sequential scanning mode or an epifluorescence microscope (Zeiss Zeiss Axioplan2 epifluorescence microscope) with appropriate filters to prevent cross talk.
Identification of precursor cells in tissue sections derived from Nestin-GFP mice were performed as previously was reported (Kempermann et al., 2003
). Expression of neuronal markers was revealed with specific antibodies that recognized Calretinin and NeuN cells. Antibodies used were: rabbit anti-Calretinin antibody 1:250 (Swant), monoclonal mouse anti-NeuN antibody 1:100 (Chemicon) monoclonal mouse anti-MAP2ab (1:500; Sigma). All secondary antibodies were raised in donkey and diluted 1:250 (Jackson Immunoresearch, Dianova). Sections were mounted in polyvinyl alcohol with diazabicyclo-octane as anti-fading agent (DABCO).
LTP in Cultured Hippocampal Cells
On the day following the initiation of the co-culture the medium was replaced by Hepes-buffered extracellular saline (ECS) solutions consisting of: NaCl, 140 mM; KCl, 5 mM; CaCl2, 1 mM; Hepes, 25 mM; Glucose, 33 mM; adjusted to pH 7.4 with NaOH. To block GABAergic receptors Bicuculline (20 μM; Sigma) or SR95531 (10 μM; Gabazine; Tocris) was added to the extracellular solution. To stimulate opening of synaptic glutamate (NMDA) receptors and induce synaptic plasticity in the neuronal network glycine was added at 100 μM. Strychnine (Sigma) was added at 1 μM to prevent the stimulation of glycinergic receptors. In the control solution, MgCl2 was added instead of glycine at 1 mM to maintain the voltage-dependent block of NMDA receptors. The stimulations were done at room temperature. The NMDA receptor inhibitor APV (50 μM) and the CaMK inhibitor KN-93 (1 μM) were added directly to the glycine containing solution. After stimulation the cultures were replaced with fresh medium and returned to the CO2 incubator.
Electrophysiology
Whole-cell recordings were performed on cultured neurons at similar time points to that of cell biological experiments (10-14 days in vitro). The cell cultures were transferred to the recording chamber and continuously superfused with extracellular saline at a flow rate of ∼5 ml/min. The intracellular solution contained K-Gluconate, 110 mM; KCl, 25 mM; EGTA, 0.1 mM; Hepes, 10 mM; K2ATP, 4 mM and GTP, 0.3 mM; adjusted to pH 7.3 with KOH; osmolarity was 290–300 mOsm. Patch pipettes were pulled from borosilicate glass tubing (2.0 mm outer diameter, 0.5 mm wall thickness; Hilgenberg, Germany). An Axopatch 200A amplifier (Axon Instruments) was used for voltage-clamp recordings. Recorded currents were filtered at 5 kHz with a four-pole low pass Bessel filter and digitized at 10 kHz with a 1401plus interface (CED). Patch pipette resistance was 3–5 MΩ. The cells were voltage clamped at −80 mV to detect mEPSCs. Tetrodotoxin (TTX) was added at a concentration of 1 μM to select for mEPSCs. Data were analyzed using C-Stimfit (Christoph Schmidt-Hieber, University College London) based on template matching. Series resistance was not compensated and checked every 5 min. Experiments with more than 10% change in series resistance were discarded. Voltage-clamp recordings were made at 22–24°C.
Calcium Imaging
Measurements of the intracellular Ca2+ signals were performed as described previously (Normann et al., 2000
). Fura2-AM (Molecular Probes, Eugene, OR, USA) was dissolved in DMSO to generate a stock solution of 5 mM. The cells were incubated for ∼30 min in a solution with a final concentration of 10 μM Fura2-AM. Afterwards the cells were washed and the dye was allowed to de-esterify for ∼10 min before fluorescence was recorded in the presence of the extracellular solution as described before. The excitation light source (Polychrome II, TILL Photonics) was coupled to the epifluorescent port of the microscope (Axioskop FS2, Zeiss; 60× water immersion objective, Olympus) via a light guide. Light intensity was reduced to 10%. The filter combination for excitation and emission comprised a beam splitter (BSP410) and emission filters (LWP420, KP600) from Delta Light & Optics (Lyngby). The fluorescence was measured with a backilluminated frame-transfer CCD camera (EBFT 512, Princeton Instruments). Images with full spatial resolution were taken with exposure times of 5 s. For high-speed Ca2+ measurements (100 Hz repetition rate) we usually defined two rectangular regions of interest (ROIs) of about 5 × 5 μm at the soma of cultured neurons. The pixels included in the ROIs were binned on-chip and digitized subsequently by the controller (Micromax, 1 MHz, Princeton Instruments). The fluorescence signals were corrected for background, which was obtained simultaneously from ROIs shifted by 10–15 μm with respect to the original ROIs (Neher and Augustine, 1992
).
To convert the fluorescence signals into Ca2+ concentrations, we used the isosbestic ratioing method (Grynkiewicz et al., 1985
). The activity-induced fluorescence change was recorded at an excitation wavelength of 380 nm. The Ca2+-insensitive isosbestic fluorescence was measured immediately before and after this sweep, using an excitation wavelength of 356 nm. The ratio of the background corrected fluorescence signals R=F356/F380 was calculated, and converted into the Ca2+ concentration using the equation (Schiller et al., 1995
).
[Ca2+] = Keff (R − Rmin)/(Rmax − R),
where Rmin is the ratio in Ca2+-free solution and Rmax the ratio when Fura-2 is completely saturated with Ca2+. These values were determined by recording from hippocampal pyramidal cells with internal solutions containing either 30 mM EGTA (Rmin = 0.70 ± 0.01, n = 5) or 50 mM CaCl2 (Rmax = 6.97 ± 0.06, n = 5). Keff was calculated according to Neher and Augustine (1992)
as Keff = Kd (Rmax/Rmin), with the dissociation constant Kd = 250 nM (Balkowiec and Katz, 2002
).
All values are given as mean ± SEM, error bars in the figures also represent SEM. Derivatives of Ca2+ signals and cross-correlations were calculated using IGOR Pro (WaveMetrics, Lake Oswego, OR, USA). Statistical significance was assessed by a two-tailed Wilcoxon test or Students t-test at the significance level (P) indicated.
Western Blotting
Cells were lysed in lysis buffer consisting of 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100 and protease and phosphatase inhibitors. The proteins were quantified using Bradford reagent (Bio-Rad). The proteins were separated with 10% SDS–polyacrylamide gel electrophoresis after boiling the samples in Laemmli’s buffer. After electrophoresis the proteins were blotted onto a nitrocellulose or PVDF membrane and then probed with primary and horseradish peroxidase (HRP) conjugated secondary antibodies.
Determination of BDNF Release into the Medium
BDNF protein was assessed with a direct ELISA method as described previously but with slight modifications (Balkowiec and Katz, 2002
). Sterile 96-well plates were treated with poly-D-lysine (50 μg/ml) for 1 h at room temperature, rinsed with sterile double distilled water and air-dried. Equal amounts of the supernatant from the various time points after stimulations were adsorbed onto the plates by incubating the medium for 1 h at 37°C. The plates were then washed with tris buffer saline (TBS) with 0.05% Tween-20 and blocked for 45 min with 5% bovine serum albumin (BSA). Subsequently, anti-BDNF antibody (Santa Cruz) was added for 1 h at room temperature. After washing, the HRP coupled secondary antibody was added for 1 h at room temperature in blocking solution. After several washings the substrate orthophenylenediamine was added in Phosphate Citrate buffer. The plates were read at 490 nm with the reference wavelength of 405 nm in a multi-plate spectrophotometer (Wallac).
Adult Hippocampal Precursor Cells Undergo Neuronal Differentiation in Co-Cultures with Primary Neurons
In the DG, neural precursor cells are located in close proximity to the granule cell neurons (Figures 1
A,B). The precursor cells have complex morphologies and a subpopulation of them (type-1 cells) shows highly arborized processes that fan into the molecular layer (Filippov et al., 2003
), where the primary input fibers to the DG arrive from the entorhinal cortex (Figures 1
C,D). In the figure, the inner molecular layer is visualized with the help of a calretinin staining. The cell bodies of type-1 cells and the highly proliferative type-2 cells are found in the subgranular zone, a fiber-rich border zone between the granule cell layer and the hilus. For more detailed descriptions of this spatial relationship see for example (Filippov et al., 2003
; Kronenberg et al., 2003
; Plumpe et al., 2006
; Steiner et al., 2006
).
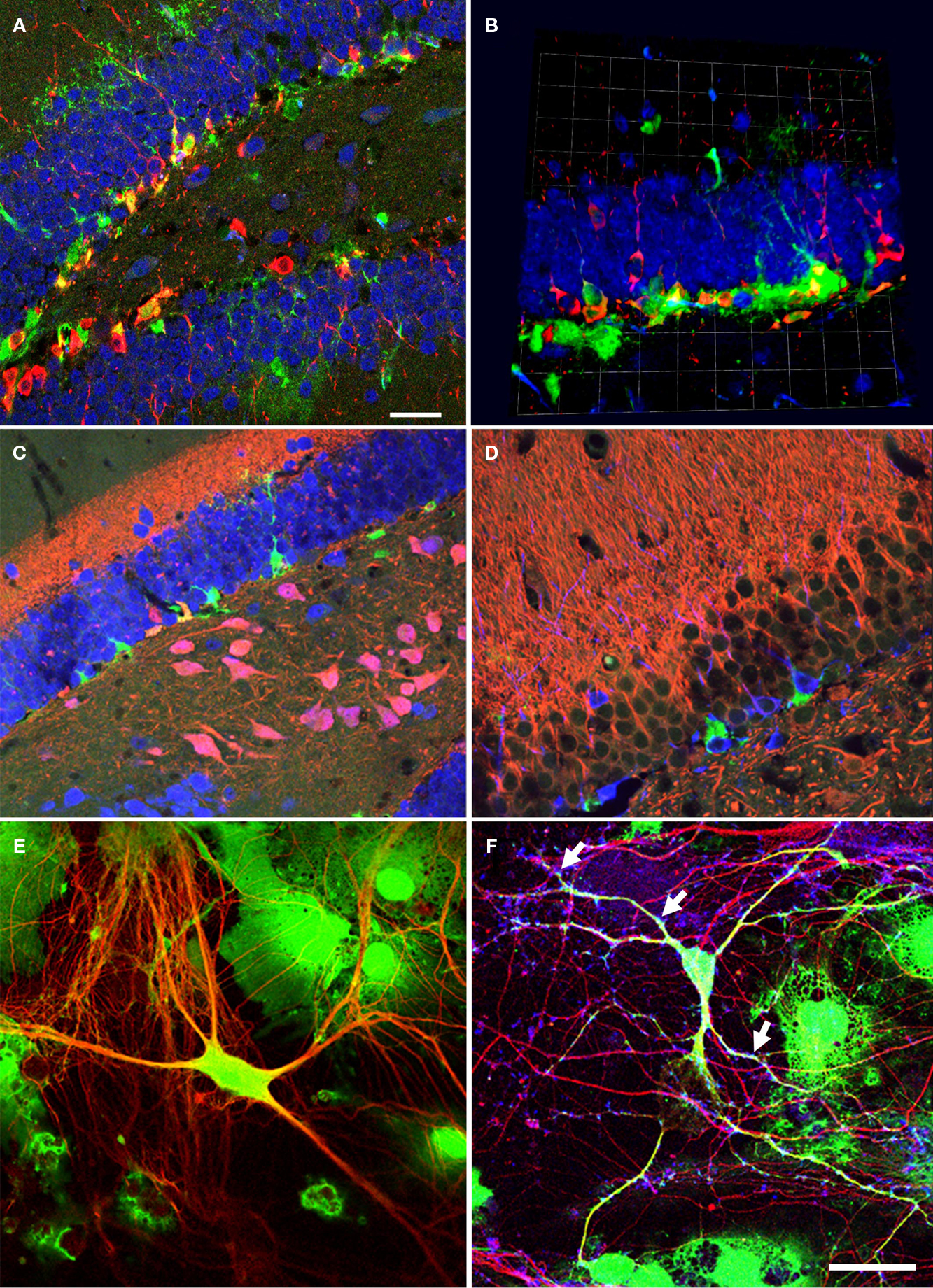
Figure 1. Neural precursor cells in their local microenvironment in vivo and in vitro. (A) Immunohistochemical analysis of a hippocampal section from mouse showing the close association of neural precursor cells with neurons. Confocal image (optical section of 1 μm thickness). Green: Nestin-GFP; Red: Dcx, Blue: NeuN; Scale bar, 25 μm. (B) Confocal projections (56×) of the dentate gyrus (z-stack of 11 optical sections with 2 μm thickness); Green: Nestin-GFP; Red: Dcx; Blue: NeuN; one square in the underlying grid equals 16.4 μm. (C) Calretinin-immunohistochemistry highlights the inner molecular layer. Radial glia-like type-1 cells (Green) reach into this area, which receives the input from the entorhinal cortex. Green: NestinEGFP; Red: Calretinin; Blue: NeuN. (D) Newborn neurons in the adult dentate gyrus are in close neighborhood with neurites in the mouse hippocampus; Green: NestinEGFP, Red: MAP2ab, Blue: Dcx. (E) Representative image of precursor cells differentiated into neurons in co-culture of precursor cells with primary mouse hippocampal neurons. EGFP cells differentiated into neurons and also non-neuronal cells types, presumably glial cell, in co-culture. Green: EGFP (precursor cells); Red: βIII-tubulin (neurons). (F) The neurons generated from precursor cells in co-culture integrate into the underlying neuronal circuit as suggested by the presence of synapses (arrows) onto it indicated by the synaptophysin staining. Green: EGFP; Red: βIII-tubulin, Blue: synaptophysin. Scale bar [in (F) for (C–F)]: C, 250 μm; D, 200 μm; E, 25 μm; F, 50 μm.
To mimic the intimate in vivo-relationship between neurons and precursor cells we co-cultured precursor cells on a layer of neurons and astrocytes derived from the hippocampus. This co-culture allowed us to subject the precursor cells to defined external changes in the neuronal layer.
Precursor cells from the adult dentate gyrus grow under adherent conditions and show stem cell-like properties (Palmer et al., 1997
; Babu et al., 2007
). We infected precursor cells with a retroviral vector constitutively expressing EGFP and purified them by FACS. The labeled precursor cells were then layered onto primary neuronal cultures from postnatal mice cultured for 10–14 days. This time period was chosen because after this interval most neurons have matured, synapse formation is almost complete, and the neurons elicit synaptic currents. The co-culture of the non-labeled neurons with GFP-labeled precursor cells enabled us to clearly distinguish the precursor cells and their progeny from the substrate culture.
After 6 (or in some instances more) days in culture, the cells were fixed and evaluated for neuronal differentiation with antibodies against pan-neuronal marker, βIII-tubulin, and for relatively more mature neurons in vitro MAP2ab (Dehmelt and Halpain, 2005
), together with EGFP (Figure 1
E). Co-cultured cells positive for EGFP and MAP2ab simultaneously have been shown to exhibit mature neuronal electrophysiological characteristics including synapse formation and network integration (Song et al., 2002
). After 6 days in culture about 12% of the precursor cells had differentiated into neurons. These cells showed characteristic neuronal morphologies with more than two fine processes and stained for βIII-tubulin and MAP2ab. Some cells that differentiated into neurons consistently showed a lower intensity of EGFP labeling than non-neuronal cells. The example in Figure 1
E strongly expresses EGFP. Generally, transgene downregulation has been reported previously and depends on the site of viral integration into the genome host cell (Xu et al., 1989
).
When the cells were cultured for more than a week the EGFP-labeled βIII-tubulin-positive neurons had formed synapses, which were positive for synaptophysin (Figure 1
F), a synaptic vesicle protein that identifies presynaptic compartments adjacent to dendritic spines and expressed in maturing neurons at the site of synapses (Calakos and Scheller, 1994
; Becher et al., 1999a
,b
; Hannah et al., 1999
). The characteristic immunoreaction was found along the EGFP-labeled dendrites. Since spines represent the major domain for excitatory synaptic input, its distribution throughout the dendritic tree suggests integration of the neuron into the existing neural network.
Stimuli Inducing Neuronal Synaptic Plasticity Increase Neuronal Differentiation in Precursor Cells
Neuronal activity can induce neuronal differentiation from neural precursor cells in co-culture with hippocampal neurons (Deisseroth et al., 2004
). However, it remains unknown, which kind of physiological neuronal activity patterns would be responsible for regulating adult neurogenesis. We addressed this question in our co-culture system, where complex neuronal activity such as synchronization of neuronal activity and synaptic plasticity can be imposed on neuronal networks and studied in vitro. In the hippocampus in vivo, synchronized bursts co-exist with population oscillation and are involved in information coding (Lisman, 1997
; Bacci et al., 1999
; Opitz et al., 2002
; Izhikevich et al., 2003
; Arnold et al., 2005
). NMDA-dependent synaptic activity has been observed in hippocampal neurons and leads to changes in synaptic strengths resulting in the induction of LTP (Malenka, 2003
).
We evoked enhanced synaptic activity in the co-culture model with Mg2+-free solution containing glycine to allow for maximal NMDA receptor activation. Extracellular Ca2+ enters via the NMDA receptors and activates the downstream signaling cascades within the hippocampal neurons. This model is considered to represent LTP and has been used with preparations of hippocampal neurons similar to ours and to study the physiological basis of learning and memory on the cellular level (Lu et al., 2001
; Malenka, 2003
).
We first performed intracellular calcium imaging with the Ca2+ sensitive fluorescent dye Fura-2. We found a synchronous rise in the intracellular Ca2+ concentration within the substrate neurons when the Mg2+-free solution with added glycine was applied to the bath (Figures 2
A,B). This increase was not a bulk rise in Ca2+ but rather appeared as periodic network activity that was present as long as glycine was applied. This was investigated by simultaneously recording the Ca2+ concentration in pairs of cells as shown in [Figures 2
B,C (red and blue traces)]. The change in Ca2+ as calculated by the first derivative over time [Figure 2
C (middle traces)] shows that the Ca2+ influx in neighboring neurons occurs in a synchronous manner. To quantify coherence of synchronous activity we calculated the cross-correlation coefficient between the Ca2+ changes in cell pairs and found a mean peak value of 0.74 ± 0.05 at a time interval of 6 ± 28 ms (n = 11 pairs; Figure 2
D). These data suggest that the neurons perform relative synchronous AP firing during the stimulation period leading to a periodic Ca2+ influx followed by subsequent extrusion similar to what was reported previously (Watt et al., 2004
). On average, the intracellular Ca2+ concentration increased from a basal level of 87 ± 11 nM to a mean concentration of 456 ± 52 nM (n = 22 cells) during the stimulation period (3–5 min Figure 2
E).
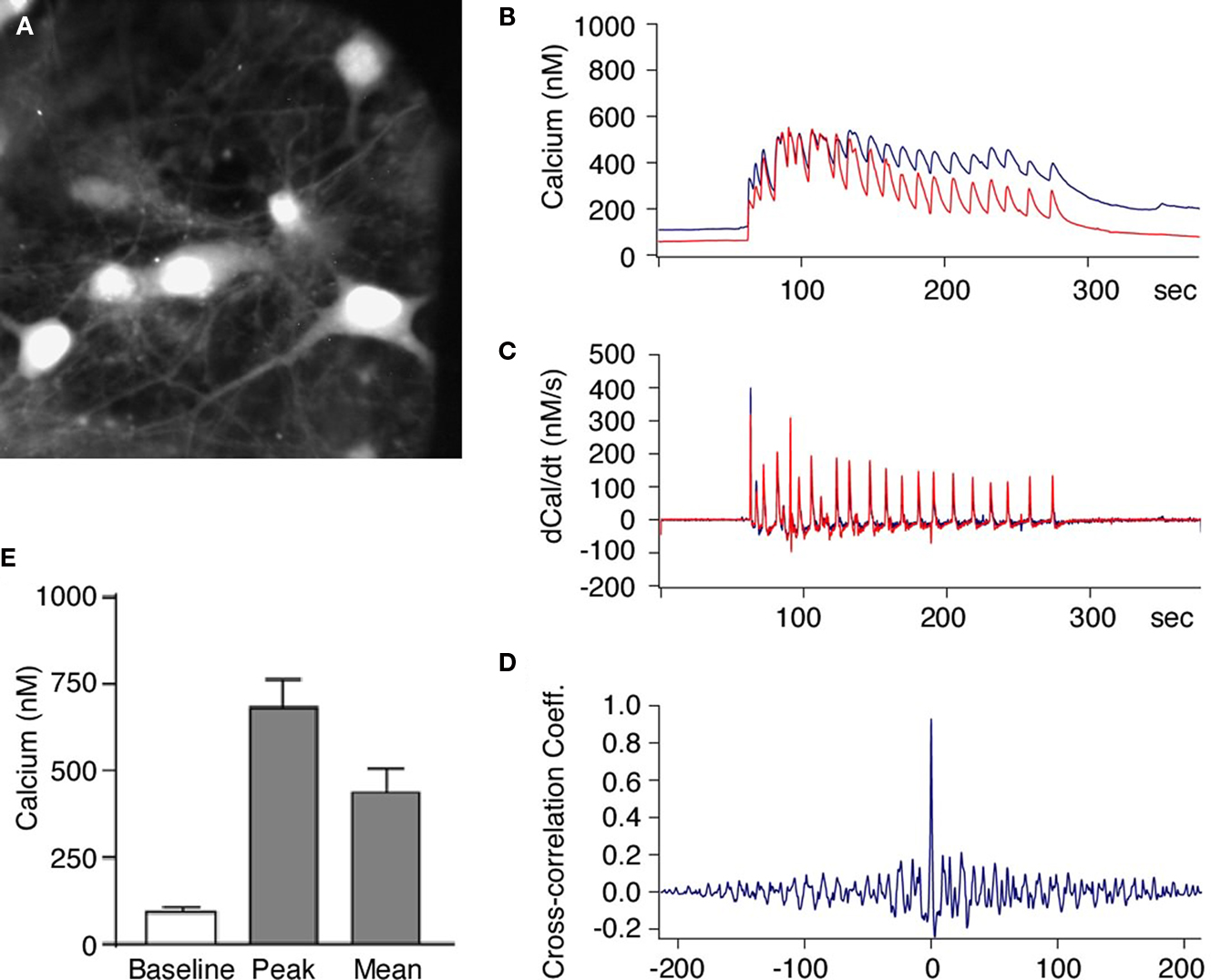
Figure 2. Activation of NMDA receptors induces synchronized increase of intracellular calcium in neurons within hippocampal neuronal network. (A) Primary hippocampal neurons loaded with the Ca2+ indicator dye Fura-2. The neurons were then stimulated with Mg2+-free solution and the intracellular Ca2+ concentration was measured in pairs of neurons. (B–D) Glycine induced increase in intracellular Ca2+ was synchronous in all neurons and showed simultaneous rise in fluorescence in different neurons [blue and red traces (B)]. The first derivative of the Ca2+ signals (C) and the cross-correlation coefficient of the first derivative calculated from cell pairs (D) indicates highly synchronous network activity. (E) When the stimulus solution was applied there was always an increase in the intracellular Ca2+ concentration all recorded neurons (n = 22). The intracellular Ca2+ concentration increased from a basal level of 87 ± 11 nM to a mean concentration of 456 ± 52 nM.
To study the effect of glycine on synaptic transmission between neurons, we performed whole-cell voltage-clamp recordings in the substrate neurons (Figure 3
A). We applied glycine to cultured neurons and simultaneously recorded the postsynaptic currents at a holding potential of −80 mV. The mEPSC were recorded in the presence of 1 μM TTX and 20 μM Bicuculline to block spontaneous APs and inhibitory GABAergic responses (Figure 3
B), before and after application of the glycine solution. We detected a robust increase in mEPSC frequency in the cultured neurons after stimulation as compared to the control period (Figure 3
C). This increase was observed as early as 5 min after glycine withdrawal and synaptic currents remained potentiated throughout the rest of the recording period with an average increase of the mEPSC frequency to 158 ± 24% (n = 4) of control. In some cases we recorded enhanced mEPSC frequencies as long as 40 min after stimulation. By contrast, the amplitude of mEPSCs was not significantly different from control (95 ± 14% of control; P > 0.5; n = 4). These data are consistent with an enhanced synaptic release probability and recruitment of silent synapses as typically reported for synaptic long-term potentiation (Malenka, 2003
).
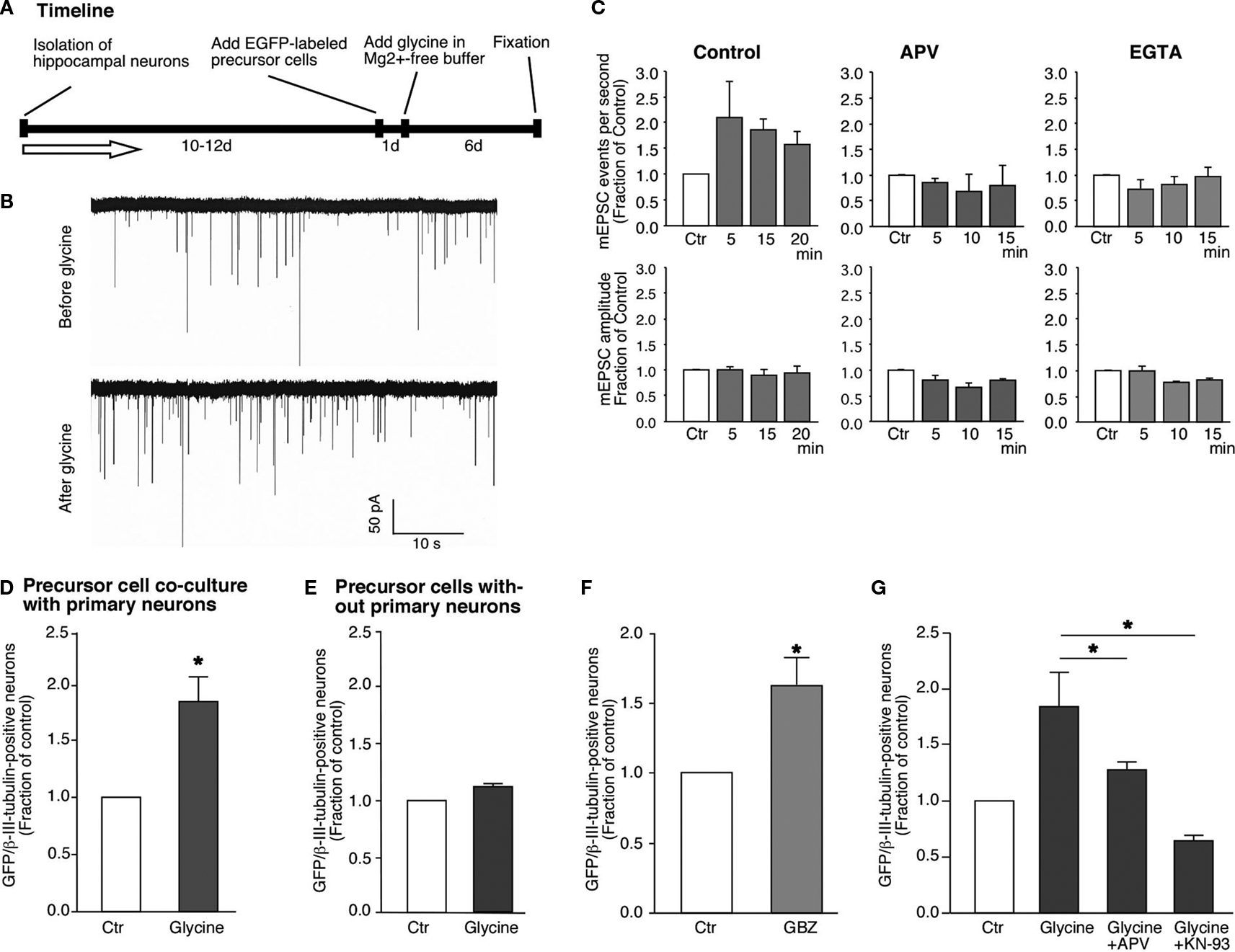
Figure 3. Glycine-induced synaptic plasticity changes are associated with neuronal differentiation from neural precursor cells. (A) Experimental time line for glycine induced synaptic plasticity changes and associated neuronal differentiation. (B) Glycine induced an increase in the frequency of mEPSCs in hippocampal neurons [158 ± 24% (n = 4) compared to the controls]. The recordings were performed before and after the addition of glycine. There was no change in the amplitude of the mEPSCs. The experiments were performed on neurons that were 12 days old. (C) This increase in the frequency of mEPSCs was blocked by the inhibition of NMDA receptors by APV and also by the chelating intracellular Ca2+ with EGTA. (D,E) Glycine induced increased neuronal differentiation from GFP-labeled neural precursor cells in co-culture with primary hippocampal neurons (Ctr; 1.0, LTP; 1.79 ± 0.22, P < 0.01). The co-cultures were stimulated with Mg2+-free glycine solution the day after the co-cultures were started as shown in (A). The number of cells per field of view remained unchanged in between the experimental conditions. There was no change in neuronal differentiation when precursor cells were stimulated with glycine in Mg2+-free media in the absence of hippocampal neurons. Experiments were performed at least three times in duplicates. (F) The increase in neuronal differentiation is not due to activation of GABAergic receptors on precursor cells by released GABA from neurons. Inhibiting GABA receptors with SR95531 paradoxically increases neuronal differentiation compared to control possibly by blocking synaptic GABA receptors (Ctr, 1.0; SR95531, 1.63 ± 0.20; P < 0.03). Experiments were done at least three times in duplicates. (G) The increase in neuronal differentiation is blocked by NMDA inhibitor APV and CaMK inhibitor KN-93. Each experiment was done at least three times.
If the intracellular calcium increase within neurons were the cause for the observed synaptic potentiation, sequestering calcium in the cells should decrease LTP. We thus performed patch-clamp recordings similar to those described above while adding the Ca2+ chelator EGTA (0.1 mM) to the patch pipette. The EGTA within the patch pipette would diffuse intracellularly and sequester Ca2+ within the neurons. As expected, addition of EGTA completely blocked LTP (Figure 3
C). Taken together, the data indicate that application of glycine added Mg2+-free solution induce change in synaptic network and a robust potentiation of synaptic currents in cultured neurons similar to what was reported previously (Liao et al., 2001
; Lu et al., 2001
).
To investigate how synaptic plasticity induced by glycine in a neuronal network would affect neuronal differentiation, the co-cultures were fixed 4–6 days after the stimulus and immunostained for MAP2 and βIII-tubulin expression. This analysis revealed a robust increase (∼79%) of EGFP-labeled cells that had acquired neuronal phenotype (Figure 3
D). To rule out a direct activation of the precursor cells by glycine- and Mg2+-free conditions we exposed precursor cells without substrate neurons and scored for the cells that underwent neural differentiation. In the isolated precursor cells we did not find an increase in the fraction of neurons generated, consistent with notion that network activity in the mature neurons is required (Figure 3
E). This observation indicated that neuronal differentiation was a result of the interaction between the neurons and the precursor cells. It is important to note that glycine was applied for only 5 min but produced an increase in neuronal differentiation from EGFP-labeled neural precursor cells that could be measured several days later. Our finding suggests that neural precursor cells sense the transient change in synaptic activity surrounding them and translate this stimulus into the initiation of neuronal differentiation.
We next addressed, whether these effects might be caused by GABA-dependent mechanisms. However, two observations ruled out a likely role of GABA in our co-culture model. First, the stimulation was done in the presence of Bicuculline or SR95531, both potent inhibitors of the GABAA receptor. Therefore, GABAergic transmission to the neuronal precursor cells could not be the activator of neuronal differentiation programs. Second, when we incubated the co-cultures in the continued presence of Bicuculline or SR95531 without the glycine induced LTP-like synaptic activity we still found an increase in neuronal differentiation from precursor cells (Ctr, 1.0; SR95531, 1.63 ± 0.20; P < 0.03; Figure 3
F). The underlying mechanism of this effect was not subject of our study but it is likely that seizure-like activity due to the complete disinhibition of the neuronal network was the cause. Independent of this, the results confirm that neuronal differentiation can occur in the absence of GABAergic stimulation of the precursor cells.
Blocking Synaptic Plasticity with NMDA-Antagonists Leads to Suppression of Neurogenesis
Blocking NMDA receptors abolishes LTP-like synaptic plasticity in vitro (Figurov et al., 1996
; Lu et al., 2001
; Patterson et al., 2001
). If our culture conditions were indeed inducing neurogenesis because the network of neurons in the cultures underwent LTP-like synaptic activity, then blocking the induction of synaptic plasticity should also block the positive regulation of neurogenesis. To address this question we added APV (2-Amino-5-Phosphonopentanoic acid) within the same stimulation paradigm described above to block activated NMDA receptors. APV along with glycine reduced the rise in mEPSC frequency observed to prestimulation levels (Figure 3
B). As expected, addition of APV along with glycine to the co-cultures abolished the increase in neuronal differentiation (Figure 3
G; Ctr, 1.0; glycine, 1.84 ± 0.22; glycine+APV, 1.27 ± 0.05). NMDA receptor-mediated Ca2+ influx is known to activate CaMK-IIα, which is a key enzyme in regulating the induction of LTP (Silva et al., 1992
; Wu et al., 2006
). Thus, if LTP-inducing synaptic response in the neuronal network was responsible for the increase in neuronal differentiation, blocking CaMK-IIα during the induction of LTP should prevent the induction of neuronal differentiation and recruitment into the neural network. When KN-93, a potent blocker of CaMK-IIα activity, was added to the cultures during the induction of LTP-like synaptic activity the cultures showed a significant reduction in the number of new neurons (Ctr, 1.0; glycine, 1.84 ± 0.22; glycine + KN-93, 0.64 ± 0.03; Figure 3
F).
BDNF Regulates the Increase in Neuronal Differentiation from Neural Precursor Cells
As direct action of the same experimental stimuli to the precursor cells did not produce identical or even similar effects than in the co-culture situation, it seemed plausible that a mediating factor secreted by the active neurons might underlie the induction of neuronal differentiation in response to LTP-like synaptic activity. The most plausible candidates for such mediators are neurotrophins, in particular BDNF, that are secreted by neurons and have been linked to a wide variety of plastic responses (Figurov et al., 1996
; Patterson et al., 2001
).
Several in vivo paradigms that increase adult hippocampal neurogenesis in vivo also show an increase in the transcription of neurotrophins (Sairanen et al., 2005
; Rossi et al., 2006
). We thus wondered whether neurotrophins would regulate neurogenesis in our paradigm. We first investigated whether induction of synaptic plasticity in our culture model would also lead to changes in neurotrophin levels. Primary neurons were subjected to LTP-like synaptic activity and the culture medium was harvested at several time points thereafter (Figure 4
A). Release of BDNF and NT3 into the medium was assessed by ELISA. As hypothesized, LTP-induced cultures had significantly higher levels of BDNF at 1h after the glycine stimulus and remained elevated for at least 6 h (Figure 4
B). This increase was seen only in the case of BDNF, whereas NT3 (acting via a different receptor) did not show changes in response to LTP-like synaptic activity (Figure 4
C). This suggested that the substrate neurons in our culture are a source for BDNF, whose secretion is regulated by synaptic activity.
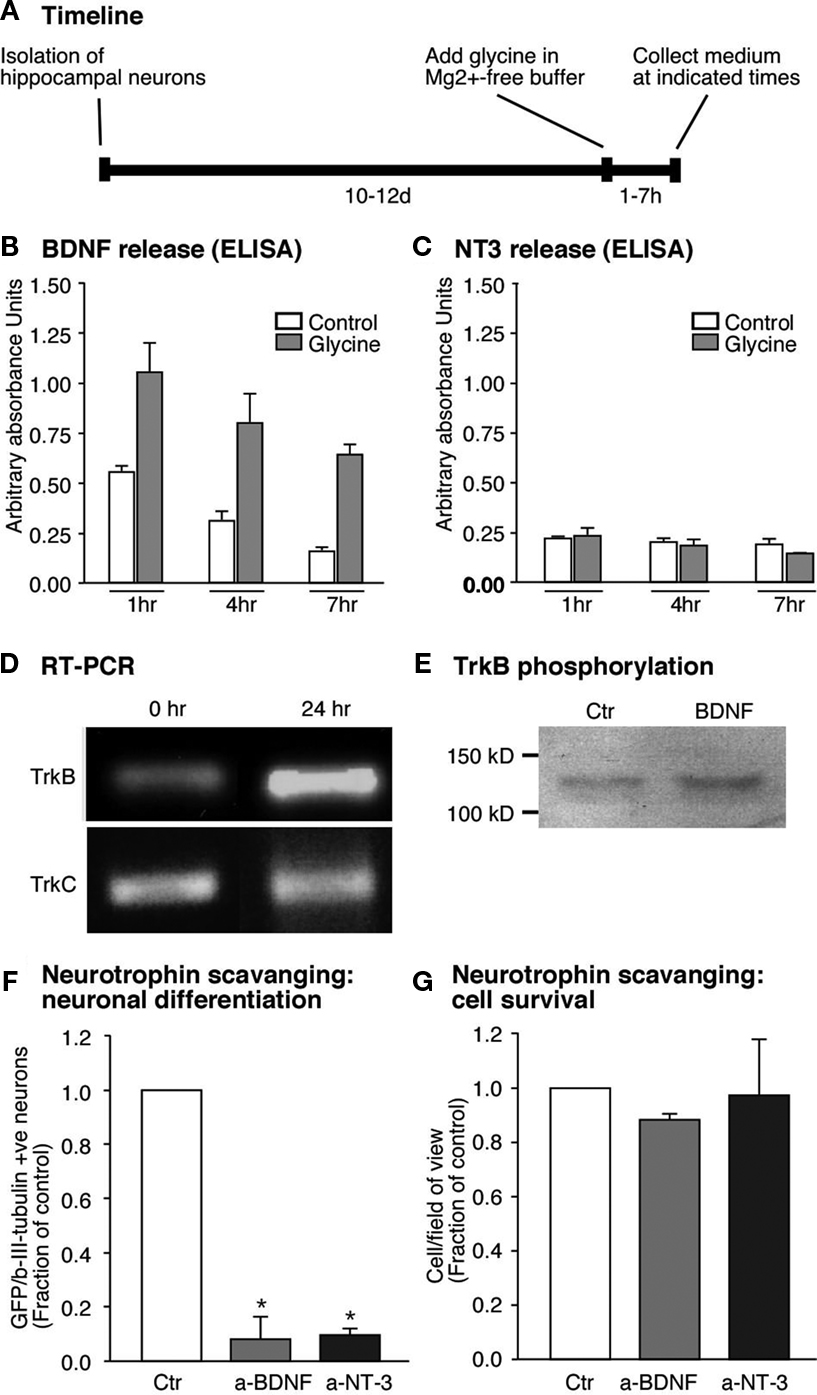
Figure 4. Glycine induced neuronal differentiation is mediated by the neurotrophin BDNF. (A) Time line for the experiments where glycine induced synaptic plasticity was induced and media collected from hippocampal neuronal cultures at set time points. (B,C) Glycine induced potentiation causes increased release of BDNF from the hippocampal neurons. But NT3 a closely related neurotrophin is not regulated by glycine (C). (D) RT-PCR analysis confirms expression of trkB- and trkC-receptors in neural precursor cells. The trkB receptor is upregulated early upon differentiation whereas the trkC receptor remains unchanged. (E) Precursor cells were cultured under differentiation conditions for 24 h, followed by stimulation with BDNF (50 ng/ml) for 15 min. Immunoblots of equal amounts of cell lysates were probed with anti phospho-TrkB antibody. (F,G) Neutralizing the neurotrophins released by the neurons results in a strong decrease in neuronal differentiation from the precursor cells. Each experiment was performed three times.
Detection of BDNF by the precursor cells would foremost require the presence of the appropriate trkB receptor. Mice lacking trkB receptor have been shown to have impaired adult hippocampal neurogenesis (Bergami et al., 2008
; Li et al., 2008
). RNA from the precursor cells was therefore assessed for the presence of trkB- and also trkC-transcripts. Both trkB- and trkC-transcripts were expressed in precursor cells, however it seems that only the trkB-transcript increased its expression 1 day after the start of the differentiation (Figure 4
D). We also detected trkB receptor protein as revealed by Western blot analysis (Figure 4
E). We stimulated precursor cells with BDNF and performed Western blot analyses on the cell lysates to test for trkB receptor phosphorylation. BDNF induced an increase in the phosphorylation of trkB receptors suggesting that functional trkB receptors are present on the precursor cells (Figure 4
E). If thus BDNF was indeed the key to the difference in the regulation of neurogenesis then blocking BDNF action should reduce neuronal differentiation after LTP-like synaptic activity induction. Indeed, scavenging secreted BDNF with bio-neutralizing antibodies led to a decrease in βIII-tubulin/MAP2ab-positive neurons (∼90% P < 0.005, Figure 4
F). In addition, BDNF scavenging slightly decreased cell survival (Figure 4
G). Incubating neutralizing antibody against NT3 also resulted in a decrease in neuronal differentiation from the precursor cells in co-culture even though its secretion was not regulated by the glycine-induced LTP-like synaptic activity (Figure 4
F). These results proved that BDNF released by neurons into the co-culture medium was responsible for the increase in neuronal differentiation from adult precursor cells in response to an LTP-like stimulus.
The present study indicates that structural cellular plasticity brought about by neurogenesis in the adult hippocampus could be the product of a complex and dynamically changing microenvironment, in which the neurons play a critical role. We have shown here ex vivo that adult hippocampal precursor cells respond to coherent neuronal network activity by initiating neuronal differentiation. This induction of neuronal differentiation from precursor cells was dependent on the presence of mature neurons in that it was the neuronal activity-dependent secretion of BDNF from the mature neurons that mediated the effect on the precursor cells. For this paracrine signaling mechanism, synaptic activation of NMDA receptors and CaMK activation was required, followed by Ca2+ influx into the neurons.
We thus propose that cellular plasticity originating from the precursor cells is linked to the network activity based on synaptic transmission between mature neurons. This does not principally rule out that additional effects based on direct neurotransmission to the precursor cells may play a role as previously suggested by others (Deisseroth et al., 2004
). In fact some effects of excitation on radial glia-like type-1 cells in vivo might depend on the presence of glutamate receptors on these cells. Type-1 cells respond to seizures induced by the application of kainic acid (Huttmann et al., 2003
; Steiner et al., 2008
). The largest population of precursor cells in vitro, however, apparently corresponds to the type-2a precursor cells in vivo, but no exact match of phenotypes in vivo and in vitro has yet been reached.
Mature neurons also undergo Ca2+ oscillations that may regulate synaptic strength (Berridge, 1998
; Wang et al., 2000b
). This is especially interesting in the adult DG where mature functional neurons exhibiting network activity coexist with precursor cells. Neuronally determined precursor cells represent a prospective pool of potentially recruitable neurons. Subsequently, more specific stimuli, presumably first GABAergic excitation then excitatory input from the entorhinal cortex recruit new neurons from this available pool for further integration and maturation (Tozuka et al., 2005
; Ge et al., 2007
). Thus different types of activity-dependent controls follow each other in the course of adult neurogenesis. Our present study, albeit in vitro, addressed the very first stage at which undetermined precursor cells become neuronally determined in response to neuronal network activity. In vivo, GABA would then drive the next step of maturation until the new neurons have made appropriate contacts, received correct glutamatergic input and have made axonal connection to CA3. Activity dependent synaptic plasticity in the new neurons themselves would control the terminal maturation and network integration.
We identified BDNF as key mediator between active neurons undergoing LTP-like synaptic plasticity and the trkB-expressing precursor cells. Several studies have shown that the release of BDNF is regulated by stimulus patterns with high frequency bursts being most effective (Balkowiec and Katz, 2002
) besides backpropagating action potentials (Kuczewski et al., 2008
). We used a stimulus that generated changes similar to the above-referred study, thus supporting the view that BDNF release from mature neurons was associated with patterned neuronal activity. A key question is whether BDNF is not only necessary but also sufficient to mediate neuronal differentiation evoked by synaptic network activity, in particular in the in vivo situation. Paracrine neurotrophin release might be a relatively non-specific means, only signaling the incidence of neuronal network activity. As one caveat, neurons might respond LTP-like stimuli with a paracrine release of BDNF even in the absence of synaptic plasticity. In such case, the release of BDNF would trigger neuronal differentiation in anticipation of neural network reorganization not the reorganization itself. BDNF might also be released in situations other than LTP in the network so that this mechanism is unlikely to be specific. In addition, the exact location within the DG where the release of BDNF can activate the precursor cells has not been identified. Immunohistochemical studies have shown that precursor cells are morphologically variable with several cells projecting processes well into the molecular layer (Filippov et al., 2003
). Precursor cells with processes could detect BDNF released from the neuronal dendrites far away from their cell bodies. Once the new neurons are more mature, specific synaptic input becomes dominant in determining the ultimate survival and terminal integration of the new cells. BDNF could also be involved in modulating electrophysiological properties of ion-channels during neuronal stem cells differentiation and development in our model (Leng et al., 2009
).
The reductionist setting of our model system might also obscure the relevance of additional mediators. Most notably, the serotonergic system has been suggested to be involved in the control of adult neurogenesis at this level. An accepted limitation of tissue culture preparation of neurons as used in this study is the alteration in the expression pattern of receptors and signaling molecules. Besides, hippocampal neuronal cultures besides dentate granule cells also include cells from CA1, CA3 and possibly other neighboring regions, which have different properties related to synaptic plasticity and may thus might influence the results. Hippocampal neuronal culture also contain non-neuronal cell, albeit in a minor proportion, which have the potential to interfere with in vivo extrapolation of tissue culture results. Irrespective of this, our present study clarifies the contribution of synchronous network activity to the initiation of neuronal development from adult hippocampal precursor cells. Therefore, stimulation of neuronal synaptic activity might offer new opportunities to modulate stem cell-based plasticity in the adult hippocampus.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Wolfgang Uckert for providing with the retrovirus used in this study. We thank Ruth Zarmstorff and Siegne Knespel for technical support. The study was financed with generous grants from Deutsche Forschungsgemeinschaft (DFG), Volkswagenstiftung, and Thyssenstiftung.
Chawla, M. K., Guzowski, J. F., Ramirez-Amaya, V., Lipa, P., Hoffman, K. L., Marriott, L. K., Worley, P. F., McNaughton, B. L., and Barnes, C. A. (2005). Sparse, environmentally selective expression of Arc RNA in the upper blade of the rodent fascia dentata by brief spatial experience. Hippocampus 15, 579–586.
Filippov, V., Kronenberg, G., Pivneva, T., Reuter, K., Steiner, B., Wang, L. P., Yamaguchi, M., Kettenmann, H., and Kempermann, G. (2003). Subpopulation of nestin-expressing progenitor cells in the adult murine hippocampus shows electrophysiological and morphological characteristics of astrocytes. Mol. Cell. Neurosci. 23, 373–382.
Plumpe, T., Ehninger, D., Steiner, B., Klempin, F., Jessberger, S., Brandt, M., Romer, B., Rodriguez, G. R., Kronenberg, G., and Kempermann, G. (2006). Variability of doublecortin-associated dendrite maturation in adult hippocampal neurogenesis is independent of the regulation of precursor cell proliferation. BMC Neurosci. 7, 77.
Rossi, C., Angelucci, A., Costantin, L., Braschi, C., Mazzantini, M., Babbini, F., Fabbri, M. E., Tessarollo, L., Maffei, L., Berardi, N., and Caleo, M. (2006). Brain-derived neurotrophic factor (BDNF) is required for the enhancement of hippocampal neurogenesis following environmental enrichment. Eur. J. Neurosci. 24, 1850–1856.
Saxe, M. D., Battaglia, F., Wang, J. W., Malleret, G., David, D. J., Monckton, J. E., Garcia, A. D., Sofroniew, M. V., Kandel, E. R., Santarelli, L., Hen, R., and Drew, M. R. (2006). Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc. Natl. Acad. Sci. U.S.A. 103, 17501–17506.