1
Watson School of Biological Sciences, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA
2
Departments of Cell Biology and Chemical Physiology, The Scripps Research Institute, La Jolla, CA, USA
RNA interference (RNAi) is an evolutionarily conserved mechanism for sequence-specific gene silencing. Recent advances in our understanding of RNAi machinery make it possible to reduce protein expression by introducing short hairpin RNA (shRNA) into cells of many systems, however, the efficacy of RNAi-mediated protein knockdown can be quite variable, especially in intact animals, and this limits its application. We built adaptable molecular tools, pSilencer (pSi) and pReporter (pRe) constructs, to evaluate the impact of different promoters, shRNA structures and overexpression of Ago2, the key enzyme in the RNA-induced silencing complex, on the efficiency of RNAi. The magnitude of RNAi knockdown was evaluated in cultured cells and intact animals by comparing fluorescence intensity levels of GFP, the RNAi target, relative to mCherry, which was not targeted. Co-expression of human Ago2 with shRNA significantly enhanced efficiency of GFP knockdown in cell lines and in neurons of intact Xenopus tadpoles. Human H1- and U6-promotors alone or the U6-promotor with an enhancer element were equally effective at driving GFP knockdown. shRNA derived from the microRNA-30 design (shRNAmir30) enhanced the efficiency of GFP knockdown. Expressing pSi containing Ago2 with shRNA increased knockdown efficiency of an endogenous neuronal protein, the GluR2 subunit of the AMPA receptor, functionally accessed by recording AMPA receptor-mediated spontaneous synaptic currents in Xenopus CNS neurons. Our data suggest that co-expression of Ago2 and shRNA is a simple method to enhance RNAi in intact animals. While morpholino antisense knockdown is effective in Xenopus and Zebrafish, a principle advantage of the RNAi method is the possibility of spatial and temporal control of protein knockdown by use of cell type specific and regulatable pol II promoters to drive shRNA and Ago2. This should extend the application of RNAi to study gene function of intact brain circuits.
RNA interference (RNAi) is a natural biological process of sequence-specific gene silencing triggered by double-stranded RNA (Fire et al., 1998
). The capability of RNAi to induce loss-of-function phenotypes has been developed into a powerful tool to study gene function in cell cultures and a variety of organisms (Davidson and Boudreau, 2007
; Hannon, 2002
). Expressing exogenous short hairpin RNA (shRNA) in cells can take advantage of endogenous RNAi machinery and result in the degradation of target mRNA (Paddison et al., 2002
). Besides the requirement for Drosha, which cleaves microRNA, the endogenous source of double-stranded RNA, into a stem-loop precursor of ∼70 nucleotides (Lee et al., 2003
; Zeng et al., 2005
), exogenous shRNAs and microRNA share an overlapping molecular pathway which involves nuclear export by Exportin 5 (Lund et al., 2004
; Yi et al., 2003
) as well as cleavage by Dicer, a cytoplasmic RNase, into active double-stranded small interfering RNAs (siRNA) of ∼ 22 nucleotides in size (Hammond et al., 2000
; Zamore et al., 2000
). The antisense strand from the siRNA becomes incorporated into a protein complex called RNA-induced silencing complex (RISC) and targets the homologous mRNA for degradation (Hammond et al., 2000
; Zamore et al., 2000
). Recent studies showed that Argonaute2 (Ago2), a component of RISC, is the key enzyme for degradation of target mRNA for gene silencing based on its RNase activity (Hammond et al., 2001
; Hutvagner and Simard, 2008
; Liu et al., 2004
).
When used as a biological tool, however, the inconsistent efficacy of RNAi in protein knockdown has limited its application. One source of variable efficacy apparently comes from the choice of shRNA sequences. As a result, it is recommended that at least three to six shRNAs for each gene of interest be screened to identify those with highest knockdown efficiency (Paddison et al., 2004a
). Another source of variability is that shRNAs which show effective knockdown in cell lines often have lower knockdown efficiency in intact animals. Consequently, in Zebrafish and Xenopus, many studies have used morpholino antisense oligonucleotides as an alternative method for loss-of-function experiments (Anichtchik et al., 2008
; Bestman and Cline, 2008
; Chiu et al., 2008
; Kenwrick et al., 2004
; Nakaya et al., 2008
; Wang and Poo, 2005
), although there are some reports of successful application of shRNAs for protein knockdown in Xenopus (Li and Rohrer, 2006
; Miskevich et al., 2006
). Therefore, the application of RNAi technology would be greatly increased by enhancing RNAi efficiency, particularly in intact animals. Several attempts have been made to improve RNAi efficacy in cultured cells, for example by testing different type of promoters (Makinen et al., 2006
; Paddison et al., 2004b
), structure of shRNAs (Paddison et al., 2004b
; Silva et al., 2005
) and co-expression of some key components with shRNAs in the RNAi machinery (Diederichs et al., 2008
; Mikuma et al., 2004
; Yi et al., 2005
), however, application of these strategies to study physiological gene function in intact animals have not yet been explored.
We have developed an effective strategy to enhance RNAi in intact Xenopus. We first built molecular tools to evaluate RNAi efficiency in cultured cell lines and tested promoters driving shRNA expression and shRNA structures. We also compared the RNAi efficiency with co-expression of Ago2, the key enzyme for degradation of target mRNA. Our data demonstrate that co-expression of Ago2 with shRNA targeting GFP significantly enhances the level of GFP knockdown compared to expression of the shRNA alone in both cultured cell lines and in CNS neurons of intact Xenopus tadpoles. We further applied this strategy to test shRNA knockdown of the endogenous GluR2 subunit of the AMPA receptors in optic tectal neurons of Xenopus tadpoles. While neurons transfected with shRNAs against GluR2 have spontaneous AMPA receptor-mediated synaptic currents comparable to controls, co-expression of GluR2 shRNAs and Ago2 significantly reduced spontaneous AMPA receptor-mediated synaptic currents. Moreover, expression of ectopic Ago2 does not affect gross dendritic arbor morphology. Together, our results suggest that co-expression of Ago2 and shRNA increases the efficacy of RNAi in intact Xenopus CNS. This simple strategy should increase the utility of RNAi in testing gene function in neuronal or circuit development.
Silencer Constructs
The silencer vector (pSilencer, pSi) was modified from the Clontech pEGFP-N1 vector, in which the EGFP and multiple cloning sites were removed by BglII and NotI. A SacI site was inserted at position 3771 for the promoter cassette to drive shRNAs. Human U6 (U6) or human H1 (H1) promoters were amplified with 5′ primer with SacI and KpnI overhang and 3′ primer with XhoI, EcoRI and SacI overhang (Figure 1
A) for subsequent cloning into the SacI site in the silencer vector to make pSiU6 and pSiH1 constructs. To increase the potency of the U6 promoter, the enhancer (En) of the cytomegalovirus (CMV) promoter from pEGFP-N1 position 59–465 was amplified and inserted with the KpnI site in front of the promoter cassette of pSiU6 to make the pSiEnU6 construct. For constructs with Ago protein expression, multiple cloning sites from pEGFP-N1 were removed by BglII and BamHI and EGFP was replaced with human Ago1 or Ago2 (gifts from Dr Gregory Hannon at Cold Spring Harbor Laboratory) by AgeI and NotI. A silent mutation was introduced into Ago2 at the XhoI site in its coding region for the subsequent cloning of the shRNA cassette. To generate the silencer construct with GFP expression to identify transfected neurons, the CMV:GFP with the poly A sequence from pEGFP-N1 was PCR amplified and inserted into the AflII site in pSiEnU6.
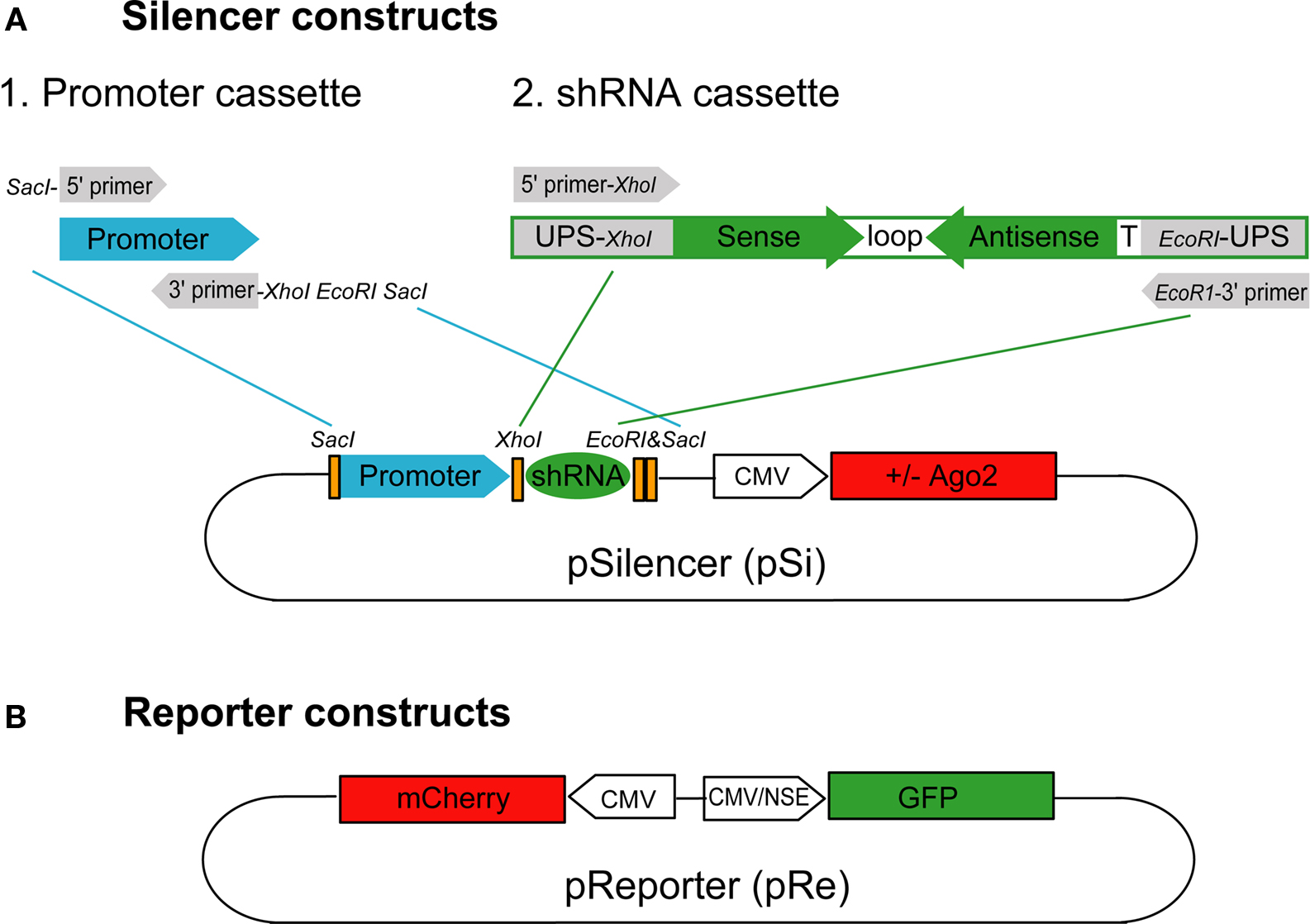
Figure 1. Silencer and reporter constructs used to test RNAi efficacy. (A) The silencer construct, pSi, includes two cassettes for easy exchange of promoters and shRNAs with or without Ago2 co-expression. The shRNA promoter fragments can be inserted into pSi through the SacI site on the vector and the shRNA fragments can then be inserted into pSi at XhoI and EcoRI sites. (B) The reporter construct, pRe, contains one CMV or NSE promoter for GFP expression and another CMV promoter for mCherry expression. The GFP silencing effect by shRNAs can be directly evaluated by calculating the green to red fluorescence intensity ratio in individual cells.
To construct the shRNA cassette, a template oligonucleotide including a 5′ universal primer sequence containing XhoI site (5′ UPS-XhoI; GACAGTGAGCGctcgag), sense sequence, loop sequence (CTTCCTGTCA), antisense sequence, termination signal (TTTTT) and 3′ UPS with EcoRI site (gaattcTGCCTACTGCCTC) in order was synthesized (Figure 1
A). For the GFP knockdown experiments, the sense sequences we used were: shGFP-1: CAGCCACAACGTCTATATCATG, shGFP-2: CGGCATCAAGGTGAACTTCAAG and shGFP-3: GGCAAGCTGACCCTGAAGTTCA. For the GluR2 knockdown experiment, the sense sequences we used were: shGluR2-1: CCGAATGAAGGTGGCAAAGAAT, shGluR2-2: CGCAACCTATAAGGAAGGTTAC and shScrambled: AGAG CTAAGCGAAATCTCTAGC, which was composed of scrambled sequence from shGluR2-2. The oligonucleotides were amplified by 5′ and 3′ universal primers containing restriction enzyme sites as shown in Figure 1
A. PCR products were pooled together for restriction enzyme digestion and subsequent cloning into the silencer vectors. All positive clones were sequenced. For shRNA derived from the artificial microRNA-30 structure (shRNAmir30), a fragment from pSM2c, a mir30 sequence containing plasmid, including the U6 promoter region and the microRNA region, was inserted into pSi vector with SacI. The pSM2c was a generous gift from Dr Hannon at Cold Spring Harbor Laboratory. GFP targeting sequences were incorporated into pSi as described (Paddison et al., 2004a
; Silva et al., 2005
).
Reporter Constructs
The reporter construct (pReporter, pRe) was modified from the pBiCS2 vector, a bidirectional plasmid containing two CMV promoters in opposite directions. GFP was inserted with BglII/NheI whereas mCherry was inserted with EcoRI/XbaI after each promoter to make pReCMV:mCherry CMV:GFP construct. To evaluate RNAi in neurons in intact Xenopus, we made another reporter construct with the neuron-specific enolase (NSE) promoter: pReCMV:mCherry NSE:GFP. One CMV promoter from pBiCS2 was replaced with the NSE promoter with AscI and HindIII sites. In addition, GFP was inserted with EcoRI/XbaI after the NSE promoter whereas mCherry was inserted with BglII/NheI after the CMV promoter.
Cell Culture and Transfection
Human embryonic kidney 293 (HEK293), COS7, NIH3T3 or HeLa cells were seeded on cover slips in six-well plates and cultivated in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA, USA) with 10% FBS (Gibco/Invitrogen) until they reached ∼70% confluence. Cells were transfected with pSi and pRe constructs in a 6:1 molar ratio with Fugene 6 (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer’s manual. Forty-eight and seventy-two hours after transfection, cells were fixed at room temperature for 15 min with 2% paraformaldehyde, mounted in ProLong Gold (Invitrogen) and imaged with a Zeiss LSM 510 Meta confocal microscope (Carl Zeiss, Jena, Germany).
Animals and Neuronal Transfection
Albino Xenopus laevis tadpoles obtained from our lab colony or commercial sources (Nasco, Fort Atkinson, WI, USA) were raised in an incubator with a 12-h light/12-h dark cycle. Tadpoles were staged according to standard criteria (Nieuwkoop et al., 1994
). Stage 47/48 tadpoles were used for all experiments. For RNAi evaluation or electrophysiology in intact tadpoles, bulk tectal cell transfections were accomplished by whole brain electroporation (Haas et al., 2002
) of DNA plasmids. For dendrite morphometric experiments, individual optic tectal neurons were transfected by single-cell electroporation (Bestman et al., 2006
) and only tadpoles with single well-labeled tectal neurons were selected for time-lapse imaging.
Image Quantification
For both cell cultures and intact animals, cells for the same set of experiments were imaged with the same settings on the confocal microscope. Single optical images with the highest mCherry signals were chosen for each transfected cell and the cell outlines were marked according to mCherry fluorescence. For each transfected cell we determined the maximal and averaged green and red fluorescence intensities from GFP and mCherry with NIH Image J software. Cells with any saturated pixels determined by maximal fluorescence intensities in either channel were excluded. The averaged green to red (G/R) fluorescence ratio was used as an index for RNAi efficiency of GFP knockdown. The G/R ratio from shGFPs-transfected cells was normalized to cells transfected with the same pSi construct without shRNA {G/R ratio = [(GreenshGFP/RedshGFP)/(Greenno shRNA/Redno shRNA)] × 100%}. One-way ANOVA and post hoc Scheffe’s test (ANOVA-Scheffe’s test) were used to test for statistical differences among groups.
Morphometric Analysis
One day after single-cell electroporation, tectal neurons were imaged in vivo using a custom designed laser scanning two-photon microscope. Images were collected with Olympus Fluoview software (Tokyo, Japan) at 2× zoom and 1.5 μm step-size in the z-axis to capture the entire extent of the dendritic arbor. Three-dimensional (3D) reconstructions of dendritic arbors were done manually by using Object Image software with custom macros (Ruthazer and Cline, 2002
). The Mann–Whitney test was used to test statistical significance between Ago2- and GFP-expressing neuron groups.
Electrophysiology
Electroporated tadpoles were anesthetized and their brains were rapidly dissected out and cut along the dorsal midline to expose the optic tectal cell bodies. Brains were constantly perfused with HEPES-buffered extracellular solution (115 mM NaCl, 4 mM KCl, 3 mM CaCl2, 3 mM MgCl2, 5 mM HEPES, 10 mM glucose, 10 μM glycine, pH 7.2 with NaOH, 255 mOsm) containing 1 μM tetrodotoxin (Alomone Labs, Jerusalem, Israel) and 100 μM picrotoxin (Tocris Biosciences, Ellisville, MO, USA). Whole-cell recording at −70 mV holding potential was performed at room temperature (21–23°C) using glass micropipettes (6–10 MΩ) filled with internal solution (80 mM cesium methanesulfonate, 5 mM MgCl2, 20 mM tetraethylammonium chloride, 10 mM EGTA, 20 mM HEPES, 2 mM ATP, 0.3 mM GTP, pH 7.2 with CsOH, 255 mOsm). Three to five minutes of recording were analyzed from each cell and mEPSCs were detected with a template matching algorithm using Clampfit 10.0 (Axon Instruments/Molecular Devices, Sunnyvale, CA, USA). Data were normalized by a Box–Cox transformation and subjected to a one-way ANOVA with subsequent pair-wise planned comparisons.
Building Tools for RNAi
To test RNAi efficacy in vivo, we first constructed a series of tools including a “silencer” construct, pSi, for gene silencing and a “reporter” construct, pRe, to evaluate knockdown efficiency. The pSi contains two easily exchangeable cassettes, the promoter cassette, for testing the effects of different promoters, and the shRNA cassette, for testing RNAi efficacy of different shRNAs (Figure 1
A). The design of the 5′ and 3′ universal primer sequences in our shRNA cassette provides a convenient and economical way to amplify candidate shRNA sequences together for subsequent cloning in one reaction.
We also made a dual-promoter reporter construct expressing GFP and mCherry under the control of CMV or NSE promoters (Figure 1
B) to demonstrate the principle of using cell-type specific promoters to drive expression. In addition, we designed three different shRNAs against different regions of GFP, termed shGFP-1, shGFP-2, shGFP-3. Since our shRNAs are designed specifically to target GFP but not mCherry, the green fluorescence intensity from GFP reflects the efficacy for RNAi knockdown whereas the red fluorescence intensity from mCherry controls for reporter expression level.
Ago2 Co-Expression Enhances RNAi Efficacy in HEK293 Cells
To optimize RNAi efficiency, we first tested the effect of different promoters in driving shRNAs in HEK293 cells. Human U6 (U6) and human H1 (H1) were used to drive shRNAs against GFP. A previous study has shown that an enhancer from the CMV promoter can enhance the U6 promoter activity and increase the synthesis of shRNAs (Xia et al., 2003
). We therefore made another construct adding this enhancer in front of U6 (EnU6) to test whether the increased U6 promoter potency can facilitate RNAi efficiency in our system.
We co-transfected HEK293 cells with the pSi transcribing shGFPs driven by different promoters and the pRe expressing GFP and mCherry driven by CMV promoters in the same plasmid (Figure 2
A). Forty-eight or seventy-two hours after transfection, HEK293 cells were fixed and imaged by confocal microscopy to calculate the green to red fluorescence intensity ratio (G/R ratio). The G/R ratio in shGFP-transfected cells was normalized to that in cells transfected with the corresponding pSi, which contains the same promoter but no shRNA.
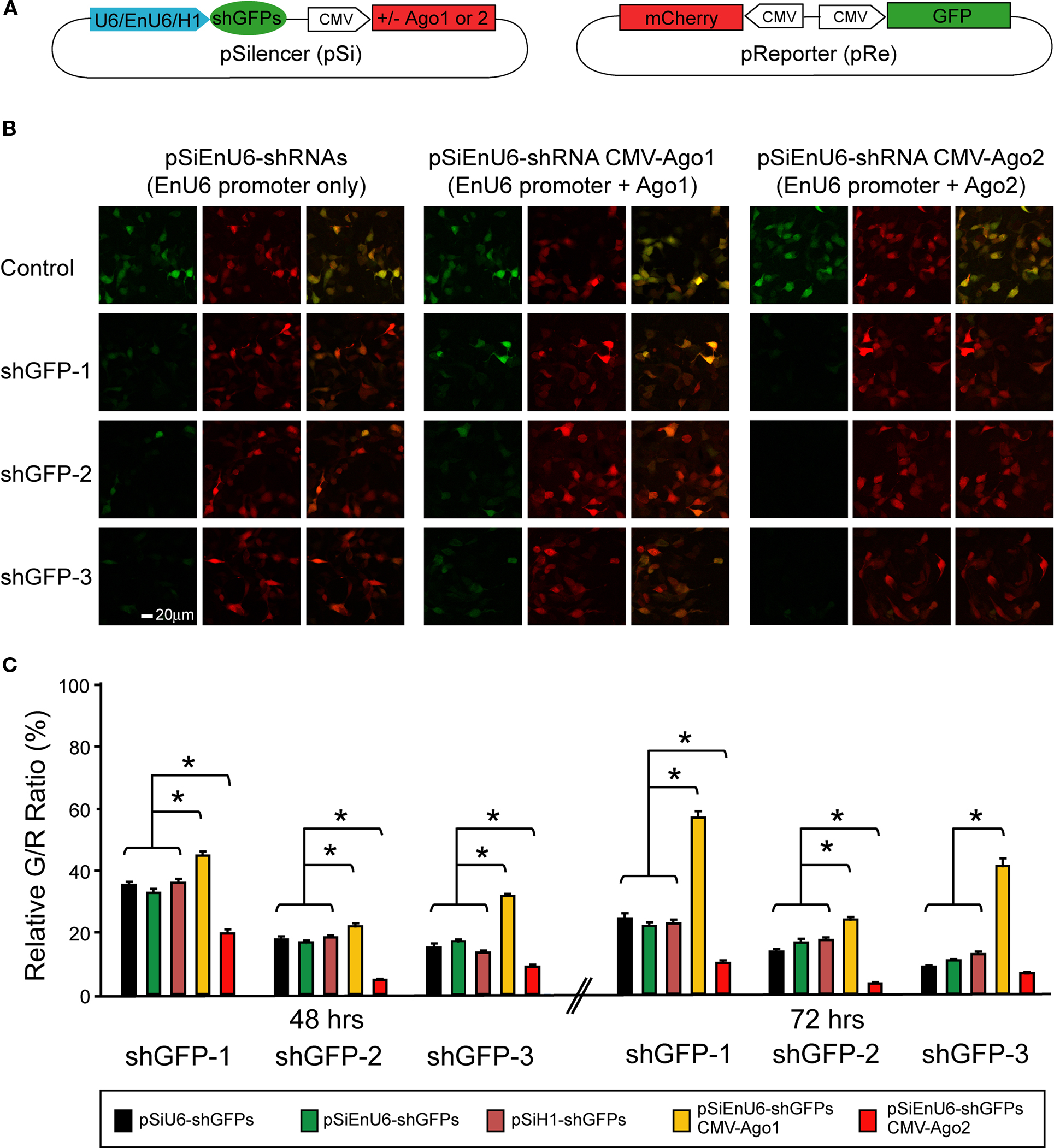
Figure 2. Effect of different promoters and Ago2 co-expression in RNAi efficacy. HEK293 cells, transfected with pSi and pRe constructs were imaged after 48 or 72 h to evaluate GFP knockdown efficiency by the green to red (G/R) fluorescence intensity ratio. (A) Diagram of the pSi constructs (left) containing U6, enhancer added U6 (EnU6) or H1 promoter driven shGFPs, and the pRe construct (right) for simultaneous GFP and mCherry expression. (B) Representative images from cells 2 days after transfection of pSi constructs containing EnU6 driven shGFPs with or without Ago protein co-expression. (C) G/R fluorescence intensity ratio measured from cells transfected with pSi constructs with or without Ago protein co-expression. The ratio in shGFP-transfected cells was normalized to the averaged G/R ratio from controls with no shRNA expression. shGFPs driven by U6, EnU6 and H1 promoters have comparable degrees of knockdown. However, EnU6-shGFPs with Ago2 co-expression show significant increases in GFP knockdown compared to EnU6-shGFPs alone or EnU6-shGFPs with Ago1 co-expression. Asterisks represent statistical significance (p < 0.05, ANOVA-Scheffe’s test).
HEK293 cells transfected for 48 h with shGFPs driven by U6, EnU6 and H1 promoters show similar degrees of GFP knockdown efficiency compared to corresponding controls (G/R ratios of 13.4–35.9%; Figure 2
C and Table 1
). Knockdown efficiency was only modestly improved by 72 h after transfection (Figure 2
C and Table 1
), suggesting that the knockdown approaches saturation by 2 days after transfection. These data demonstrate that the type of promoter and its potency are not the major determinants controlling the RNAi efficiency since U6, with or without enhancer, and H1 function comparably.
Overexpression of some key enzymes in the RNAi machinery has been used to enhance RNAi efficacy in cultured cells (Diederichs et al., 2008
; Mikuma et al., 2004
; Yi et al., 2005
). We tested whether co-expression of human Ago2 and shRNA can increase RNAi potency using our pRe and pSi constructs. We co-transfected HEK293 cells with the pRe expressing GFP/mCherry and a dual-promoter pSi expressing shGFPs under the control of the EnU6 promoter and either Ago1 or Ago2 under the control of the CMV promoter (Figure 2
A). Ago2 co-expression with shGFPs produced a dramatic decrease in G/R ratios of 4.6–19.8 and 3.4–9.9% in cells transfected for 48 and 72 h, respectively (Figures 2
B,C red bars and Table 1
). These effects are specific to Ago2 since HEK293 cells co-expressing Ago1, an Ago2 homolog without RNase activity (Liu et al., 2004
), showed less knockdown (Figures 2
B,C yellow bars and Table 1
). In fact, co-transfection of Ago1 appears to inhibit GFP knockdown in HEK293 cells compared to cells without co-transfection of Ago protein (Figures 2
B,C green bars and Table 1
). These results indicate that co-expression of Ago2 increases RNAi efficiency, consistent with a previous report in cell lines (Diederichs et al., 2008
). The results further indicate that Ago1 might interfere or compete with endogenous Ago2 function, suggesting that the endogenous levels of Ago1 and Ago2 may affect microRNA-mediated protein levels.
Co-Expression of Ago2 Broadly Facilitates RNAi Efficacy
Previous studies showed that shRNA derived from microRNA-30 structure (shRNAmir30) increased siRNA production which improves RNAi efficiency (Silva et al., 2005
). We tested whether shGFPsmir30 can facilitate GFP knockdown and how effective it is compared to Ago2 co-expression in our system. We incorporated the U6-mir30 fragment from pSM2c with the GFP shRNAs into our pSi constructs to generate shGFPsmir30 with or without Ago2 co-expression (Paddison et al., 2004a
; Silva et al., 2005
) (Figure 3
A; see Section “Materials and Methods” for detail).
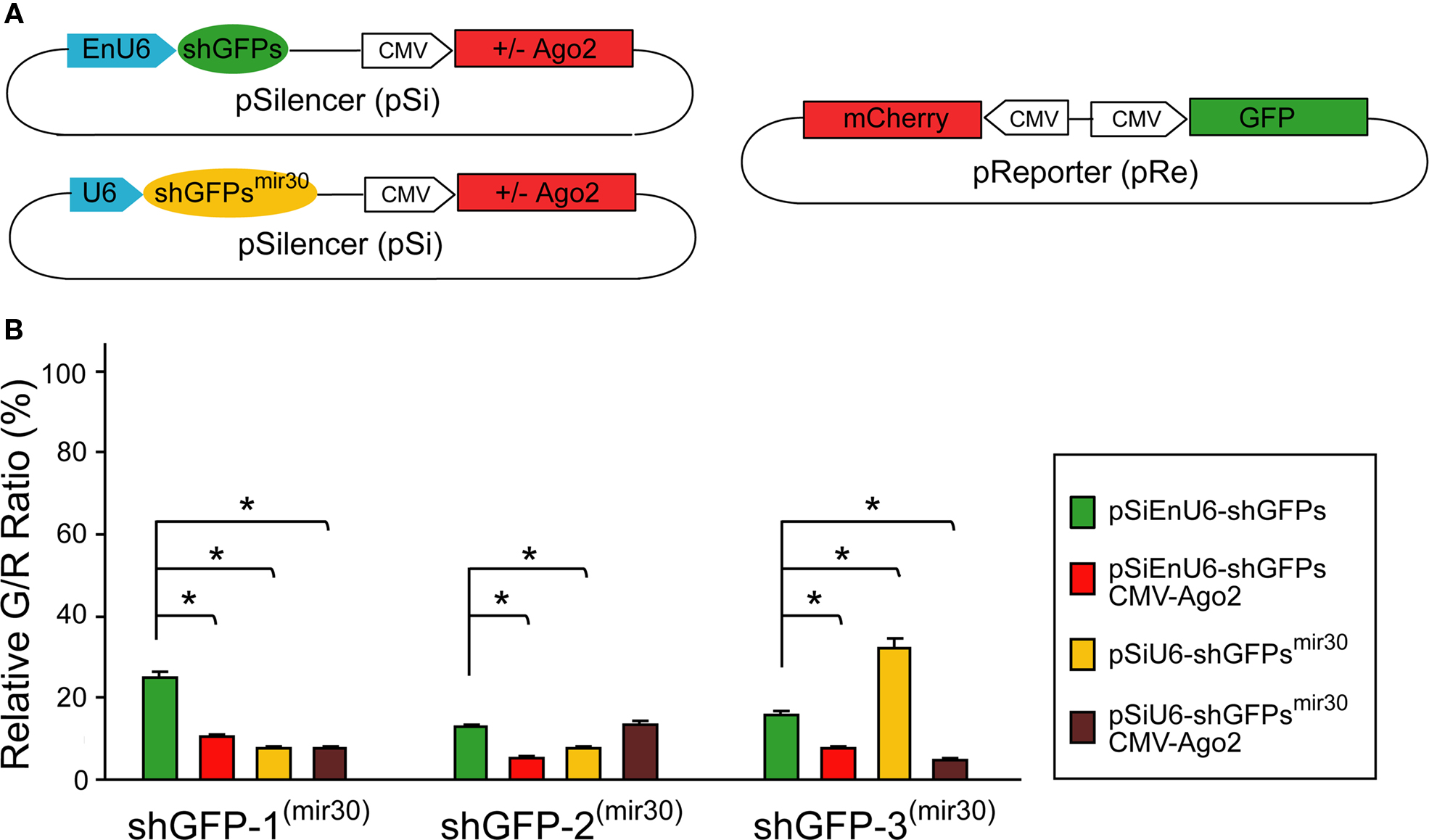
Figure 3. Effect of shRNA structure on RNAi efficacy. (A) Diagram of the pSi constructs containing shGFP or shGFPmir30 with or without Ago2 co-expression (left), and the pRe construct (right) for simultaneous GFP and mCherry expression. (B) G/R fluorescence intensity ratio measured from cells transfected with pSi constructs containing shGFP or shGFPmir30 with or without Ago2 co-expression. For shGFPs, Ago2 co-expression broadly enhances GFP knockdown (red bars). For shGFPsmir30, the RNAi efficiency increases for shGFP-1mir30 and shGFP-2mir30 but not shGFP-3mir30 (yellow bars). Additionally, Ago2 co-expression enhances knockdown with all shGFPsmir30 to a level comparable to Ago2 co-expression with shGFPs. Asterisks represent statistical significance (p < 0.05, ANOVA-Scheffe’s test).
HEK293 cells transfected with EnU6-shGFPs for 72 h showed GFP knockdown efficiency of 12.8–24.9% (Figure 3
B green bars and Table 2
), however, cells co-transfected with EnU6-shGFPs along with Ago2 co-expression showed consistent improvement in GFP knockdown to give G/R ratios of 5.4–10.6% for all shGFPs tested (Figure 3
B red bars and Table 2
). By contrast, when cells were transfected with U6-shGFPsmir30, GFP knockdown efficiency varied depending on the shGFPs tested (Figure 3
B yellow bars and Table 2
). shGFP-1mir30 and shGFP-2mir30 showed improved GFP knockdown with G/R ratios of 7.5 and 7.8%, respectively. Expression of shGFP-3mir30 did not improve knockdown and these cells had a higher G/R ratio of 32.0%. Furthermore, when we co-transfected cells with Ago2 and shGFPsmir30, the G/R ratios were 5.0–13.2% for all shGFPsmir30 tested (Figure 3
B brown bars and Table 2
), comparable to cells co-transfected with Ago2 and shGFPs (Figure 3
B red bars and Table 2
). Of note, we also co-expressed Ago2 with shGFPs in different cell lines. Although NIH3T3 cells did not show improved knockdown efficiency with Ago2 co-expression, COS7 and HeLa cell lines increased GFP knockdown efficiency with Ago2 co-expression (Table 2
), implying that the Ago2 enhancement in RNAi can be applied to various experimental systems, but may have cell-type specific effects. It is not clear why murine NIH3T3 cells show lower knockdown efficiency. Ago2 is evolutionarily well conserved, with human Ago2 amino acid sequence sharing 99 and 96% identity to mouse and Xenopus Ago2, suggesting that differences in Ago2 function in the different cell lines does not account for the difference.
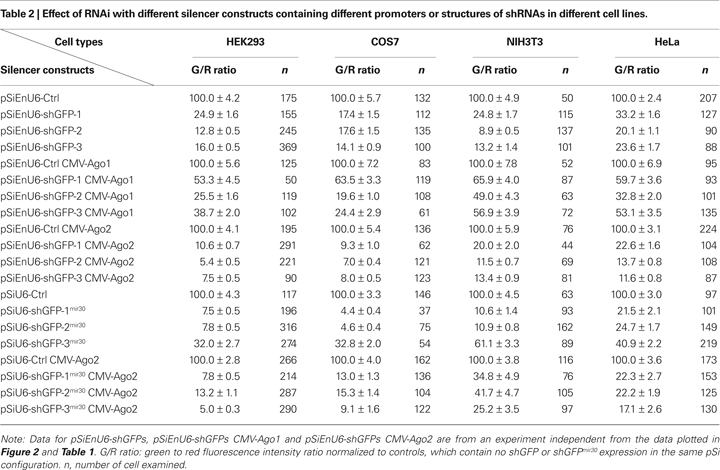
Ago2 Co-Expression Enhances RNAi in Xenopus Tadpoles In vivo
To test whether co-expression of Ago2 enhances RNAi in neurons in intact animals, we used live Xenopus laevis tadpoles as a model system. Although RNAi has been reported in Xenopus (Li and Rohrer, 2006
; Miskevich et al., 2006
), our own attempts and other reports suggest it is not efficient (Kenwrick et al., 2004
).
We transfected tadpole neurons by electroporation of the pSi containing EnU6-shGFPs with or without Ago2 co-expression, and the pRe expressing GFP and mCherry under the control of NSE and CMV promoters, respectively (Figure 4
A). Three or five days after electroporation, tadpoles were anesthetized and imaged by confocal microscopy. We evaluated the GFP knockdown efficiency by calculating the intensity ratio of green to red fluorescence.
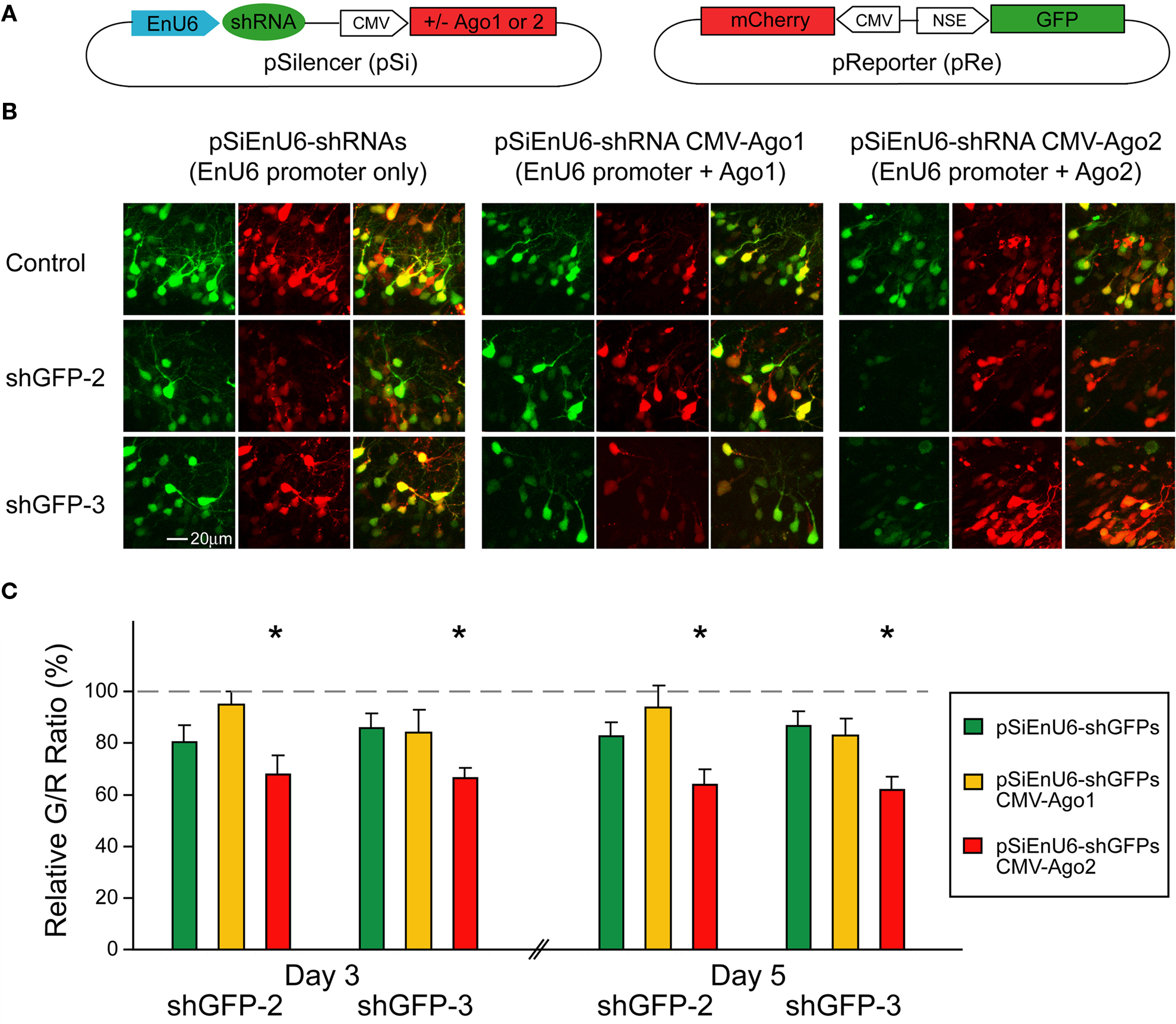
Figure 4. Ago2 expression enhances RNAi in neurons of living Xenopus tadpoles. Neurons, transfected with pSi and pRe constructs, were imaged after 3 or 5 days to evaluate GFP knockdown by G/R fluorescence intensity ratio. (A) Diagram of the pSi (left) containing EnU6-shGFPs with or without Ago protein co-expression, and the pRe (right) for simultaneous GFP and mCherry expression. (B) Representative images from CNS neurons 3 days after transfection of the pSi constructs as indicated. (C) G/R ratios measured from CNS neurons transfected with different pSi constructs driving shGFPs, normalized to control cells. Note that the pSi construct containing the EnU6-shGFPs and Ago2 (red bars) has greater knockdown than the EnU6 promoter alone (green bars) or EnU6-shGFPs with Ago1 (red bars). Asterisks represent statistical significance between neurons transfected with no shRNA and shGFPs with the same silencer configuration (p < 0.05, ANOVA-Scheffe’s test).
Three days after electroporation, neurons transfected with EnU6-shGFP2 or EnU6-shGFP3 did not show significant GFP knockdown compared to controls, even though these shRNAs had the highest GFP knockdown efficiency in cell lines (Figures 4
B,C green bars and Table 3
), Neurons transfected with pSi containing Ago2 and EnU6-shGFPs, had green to red fluorescence ratios that were significantly lower than controls (Figures 4
B,C red bars and Table 3
). A minor increase in GFP knockdown was observed 5 days after electroporation in neurons of living Xenopus tadpoles (Figures 4
B,C red bars and Table 3
), indicating that the RNAi effect was close to saturation 3 days after transfection. The enhanced RNAi efficiency was Ago2-specific because the G/R ratios in neurons transfected with pSi containing Ago1 and EnU6-shGFPs were comparable to controls (Figures 4
B,C yellow bars and Table 3
).

Neurons Expressing Ectopic Ago2 Acquire Normal Dendritic Morphology
The Xenopus optic tectal system is a well established system to study neuronal development in vivo. Numerous studies have demonstrated that the exquisite temporal and spatial regulation of multiple genes are required to control the dendritic arborization, synaptic connections and proper circuit function (Cline, 2001
; Wu et al., 1999
). To evaluate whether ectopic Ago2 expression affects neuron development, we monitored dendritic arbor structure of Ago2-transfected neurons in living Xenopus tadpoles using in vivo time-lapse imaging. Stage 46/47 tadpoles were electroporated with a dual promoter plasmid expressing Ago2 and GFP or GFP alone by single cell electroporation. One day after electroporation, animals with single labeled neurons were imaged daily with a laser-scanning two-photon microscope for 3 days. Dendritic branch lengths and branch tip numbers were quantified from 3D reconstructions of the dendritic arbor structures from in vivo images. For total dendritic branch length, neurons with ectopic Ago2 expression had similar branch length to GFP control neurons at each day of imaging (Figures 5
A,B; GFP: day1/2/3: 371.04 ± 76.67/772.72 ± 111.78/1001.02 ± 86.71; Ago2: day1/2/3: 385.81 ± 51.00/793.29 ± 52.87/986.10 ± 66.34). For the total branch tip number, neurons with ectopic Ago2 expression were also comparable to GFP controls (Figures 5
A,C; GFP: day1/2/3: 59.89 ± 14.59/121.67 ± 22.14/153.44 ± 19.07; Ago2: day1/2/3: 63.88 ± 8.18/133.88 ± 9.76/167.00 ± 11.78). These data indicate that ectopic Ago2 expression alone does not interfere with normal dendritic arbor elaboration during neuronal development.
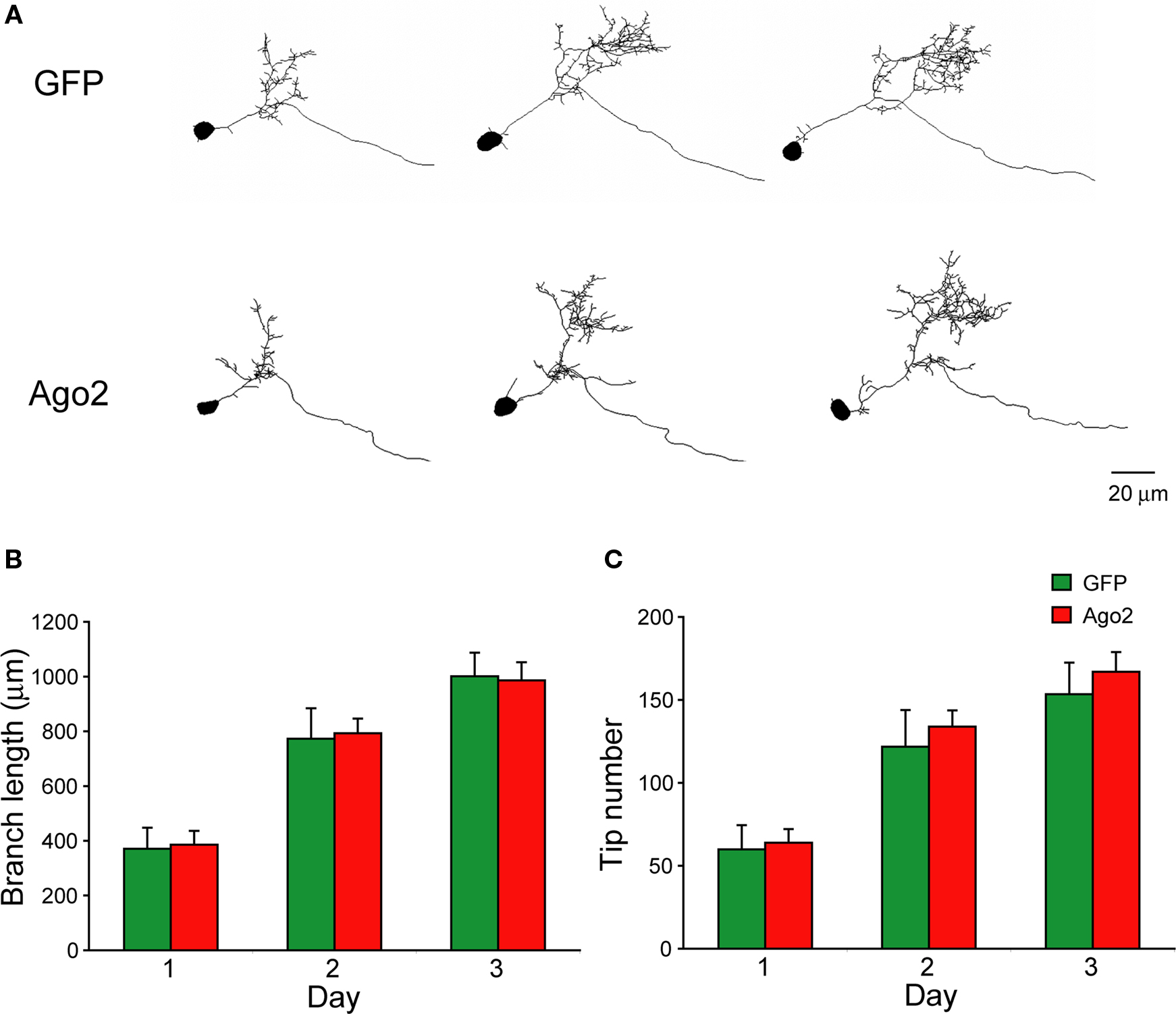
Figure 5. Neurons expressing ectopic Ago2 acquire normal dendritic arbor structure. Individual optic tectal neurons expressing GFP or GFP + Ago2 were imaged daily over 3 days in living animals (A). Total dendritic branch length (B) and branch tip number (C) were determined on 3D reconstructions of tectal neurons. Total branch length and branch tip number are comparable between GFP and Ago2-expressing neurons (p > 0.05, Mann–Whitney test).
Ago2 Co-Expression Enhances RNAi Efficiency In vivo
To test whether Ago2 co-expression enhances silencing of endogenous genes in Xenopus in vivo, we designed two shRNAs against the Xenopus GluR2 subunit of the AMPA receptor, shGluR2-1 and shGluR2-2, and a control shRNA, shScrambled, which encoded a scrambled sequence from shGluR2-2. AMPA receptors are heterotetramers, typically composed of GluR2 subunits combined with GluR1 or GluR3 (Hollmann and Heinemann, 1994
; Rosenmund et al., 1998
; Wenthold et al., 1996
). We reasoned that GluR2 knockdown would have a significant effect on AMPA-receptor mediated synaptic transmission. Tadpoles were electroporated with pSi containing EnU6-shRNAs for GluR2 knockdown and CMV driven GFP for cell identification with or without Ago2 expression under the control of another CMV promoter (Figure 6
A). Conventional western blot evaluation of the protein knockdown cannot be used in these experiments because of the low and variable transfection efficiency (∼3–10%) with electroporation (Chiu et al., 2008
). Isolation and enrichment of transfected neurons by FACS for further analysis is also not practical due to limited numbers of transfected cells in the optic tectum of Xenopus tadpoles. Furthermore, use of light microscope immunohistochemistry to evaluate knockdown of membrane proteins is not possible in the developing optic tectum because the cell bodies are extremely densely packed and it is difficult to distinguish quantitative changes in immuno-detection in membranes of neighboring cells. Therefore, we assessed AMPA receptor knockdown functionally by recording the frequency and amplitude of AMPA receptor-mediated spontaneous miniature excitatory postsynaptic current (AMPA mEPSC) 3 days after transfection.
Figure 6. Ago2 expression enhances RNAi reduction of AMPA receptor-mediated synaptic responses in vivo. Whole-cell recordings were made from CNS neurons of Xenopus tadpoles transfected with pSi construct expressing shRNAs targeting the GluR2 subunit of AMPA receptor with or without Ago2 co-expression. (A) Diagram of the pSi constructs containing EnU6-shGluR2s for GluR2 knockdown, Ago2 for RNAi enhancement and GFP for cell identification. (B) Representative traces of spontaneous AMPA mEPSCs, superimposed from 30 consecutive traces. Cells co-expressing Ago2 with shGluR2-1 or shGluR2-2 (red) appear to have fewer mEPSCs than cells expressing shGluR2-1 or shGluR2-2 alone (black) and other control (green) traces. (C) Frequency of AMPA mEPSC in neurons transfected with pSi construct expressing shGluR2s with Ago2 (red) is significantly less than cells transfected with the pSi construct expressing shScrambled + Ago2 or GFP alone ((green) and neurons expressing shGluRs only (black) (p < 0.05, ANOVA). Neurons expressing shGluR2s without Ago2 co-expression (black) have mEPSC frequency comparable to controls transfected with shScrambled + Ago2 and GFP alone (green). Neurons expressing GFP alone, Ago2 alone and shScrambled + Ago2 have comparable AMPA mEPSC frequencies. Dashed gray line indicates the averaged frequency of shScrambled control. (D) Amplitudes of AMPA mEPSC are not different between neurons transfected with GFP alone, Ago2 alone, and shScrambled or shGluR2s with or without Ago2 co-expression. Dashed gray line indicates the averaged amplitude of shScrambled control.
AMPA mEPSC frequency was significantly lower in neurons expressing shGluR2s with Ago2 than in neurons expressing shScrambled with Ago2 (Figures 6
B,C; Ago2 + shScrambled/Ago2 + shGluR2-1/Ago2 + shGluR2-2: 1.05 ± 0.23/0.38 ± 0.09/0.39 ± 0.10; n = 23, 24, 20) and neurons expressing shGluR2s alone (shGluR2-1/shGluR2-2: 0.75 ± 0.14/0.87 ± 0.21; n = 25, 21). Furthermore, 2 out of 24 or 3 out of 20 cells transfected with Ago2 and shGluR2-1 or shGluR2-2 did not have any detectable mEPSC throughout the 3–5 min recording period (although they have normal input and access resistances) suggesting a strong knockdown effect of endogenous GluR2. On the other hand, neurons expressing GFP alone (Figures 6
B,C; GFP: 1.22 ± 0.28; n = 18), shGluR2s without Ago2 co-expression (Figure 6
B,C, black traces and circles) and neurons expressing scrambled hairpins with Ago2 co-expression had similar mEPSC frequencies (Figures 6
B,C). These data indicate that co-expression of Ago2 facilitates the reduction of endogenous GluR2 function and can be applied to intact Xenopus system to enhance the RNAi efficiency.
We also compared the frequency of AMPA mEPSC in tectal neurons transfected with GFP alone, Ago2 alone or Ago2 + shScrambled co-expression. AMPA mEPSC frequency was comparable between these three groups of neurons (Figures 6
B,C; GFP/Ago2/Ago2 + shScrambled: 1.22 ± 0.28/0.74 ± 0.16/1.0 ± 0.23; n = 18, 21, 23). Although neurons with Ago2 expression seem to have lower AMPA mEPSC frequency, we did not detect a statistical difference between this group and either GFP and Ago2 + shScrambled controls. We did not find significant differences in AMPA mEPSC amplitudes between any groups of neurons tested (Figure 6
D; GFP/Ago2/Ago2 + shScrambled/shGluR2-1/Ago2 + shGluR2-1/shGluR2-2/Ago2 + shGluR2-2: −8.79 ± 0.56/−7.97 ± 0.55/−9.15 ± 0.59/−10.52 ± 0.89/−9.14 ± 0.96/−10.26 ± 1.10/−7.77 ± 0.96, n = 18, 21, 23, 25, 24, 21, 20).
RNAi is an evolutionarily conserved mechanism that has been developed into one of the most powerful genetic tools to inhibit gene expression in vivo. Compared to traditional knockout technology which is normally expensive, time consuming and only available for a limited number of species, RNAi methodology provides a rapid route to gene silencing, is relatively inexpensive, is applicable to a wide range of animal and plant species and requires only short sequence information for any gene of interest. However, the inconsistent efficacy of RNAi in gene knockdown remains an obstacle that limits its application. Here, we have evaluated RNAi efficacy and showed that co-expression of shRNA with Ago2 enhances knockdown of both exogenous GFP expressed in cultured cell lines and intact animals and endogenous AMPA receptors in CNS neurons of intact animals. Ago2 expression does not affect gross dendritic arbor morphology in CNS neurons of Xenopus tadpoles. These data together suggest that co-expression of Ago2 with shRNAs can be a simple method to enhance RNAi efficiency in intact animals.
Rate-Limiting Components of Endogenous RNAi Machinery
We hypothesized that the endogenous RNAi machinery may be expressed at low levels or occupied by endogenous microRNA processing, thereby limiting RNAi performance in intact animals. Once introduced into cells, the exogenous shRNAs would not be able to compete with endogenous microRNA for these shared molecules required for efficient RNAi, resulting in low protein knockdown efficiency in neurons of Xenopus tadpole. Increasing the amount of these limiting factors would then provide better capacity for a cell to execute targeted mRNA degradation and gene silencing.
Previous studies have tested whether key components of the RNAi machinery, including Drosha, Exportin, Dicer and Ago proteins, limit RNAi function in cultured cells (Diederichs et al., 2008
; Mikuma et al., 2004
; Yi et al., 2005
), and suggested that Ago2 can increase RNAi efficiency without induction of off-target effects (Diederichs et al., 2008
). Consistent with this, we found that a synthesized siRNA against GFP, which mimics the ∼22-mer Dicer products, does not show significant RNAi knockdown in Xenopus tadpole neurons (data not shown). These data suggest that the major limiting factor for knockdown efficiency in this system is downstream of Dicer. Indeed, we found that Ago2 co-expression with shRNA enhances knockdown of exogenous GFP and endogenous GluR2 subunit-containing AMPA receptors in Xenopus brain cells (Figures 4 and 6
). These data demonstrate that exogenous expression of Ago2 can increase RNAi efficiency and suggest that it is a rate-limiting factor in shRNA-induced gene knockdown in CNS neurons of intact Xenopus.
It is interesting to note that the three shGFPs that we know work efficiently in HEK293, HeLa, COS7 and NIH3T3 cell lines (Figure 2
; Tables 1 and 2
), function relatively poorly in intact animals (Figure 4
). In fact, without Ago2 co-expression, shGFPs only reduce exogenous GFP levels 15–20% compared to control neurons in Xenopus tadpoles. With Ago2 expression, we revealed moderate yet significant knockdown (∼34–38%) for exogenous GFP in Xenopus (Figure 4
). When we examined the endogenous neuronal protein, GluR2, we only detected a significant reduction of AMPA mEPSC frequency when Ago2 was co-expressed with shGluR2s (Figure 6
). AMPA mEPSC frequency is reduced to 0 in some neurons, although other neurons still have mEPSCs frequencies comparable to control neurons. This may reflect cell to cell variation in RNAi efficiency or variation in receptor subunit composition, and therefore sensitivity to loss of GluR2, at excitatory synapses. The differential efficiency of RNAi in intact animals compared to cells lines suggests that there might be multiple components of the RNAi machinery that are expressed at low levels or are occupied by endogenous microRNA processing in intact animals.
RNAi Enhancement by Artificial microRNA-Based Hairpins
Other strategies to improve RNAi efficiency are based on the presentation and structure of the shRNA. Based on the identification and understanding of natural microRNAs, it is possible to introduce artificial microRNA that not only preserves the overall structure of the primary microRNA but also incorporates engineered sequences for specific gene silencing (Zeng et al., 2002
). It is thought that the conserved structure from the primary microRNA transcript allows the artificial microRNA to enter the RNAi pathway more efficiently and can be subsequently processed into siRNA more effectively to yield higher RNAi efficacy. Indeed, shRNA derived from the primary microRNA-30 design yields more than a 10-fold increase in siRNA production and results in increased RNAi efficiency compared to shRNA in cultured cells (Silva et al., 2005
).
We find that two out of three shRNAmir30 constructs tested improve GFP knockdown. One possible explanation for lack of knockdown with shGFP-3mir30 is that some target sequences might interfere with the endogenous microRNA processing, such as Drosha cleavage, which affects the yield of siRNAs. Alternatively, shRNA and shRNAmir30 might be handled differently in the RNAi machinery even though they have identical shRNA sequences.
In addition, although shRNAmir30 and Ago2 co-expression with shRNA each enhanced RNAi efficiency individually, we did not see a synergistic effect on GFP knockdown when we expressed shGFPmir30 and Ago2 together in HEK293 cells. Interestingly, the poor GFP knockdown efficiency previously seen with shGFP-3mir30 was dramatically improved by ectopic Ago2 expression (Figure 3
). These data suggest that co-expression of Ago2 is more effective than shRNAmir30 at enhancing GFP knockdown and that Ago2 co-expression can be applied more consistently without concern for shRNA sequence selectivity.
Functional Assessment of Neuronal RNAi
AMPA receptors, the major excitatory neurotransmitter receptors in the CNS, are tetramers composed of four subunits, GluR1-4. The GluR2 subunit is thought to be present in the majority of synaptic AMPA receptors which are predominately made of GluR2 and GluR1 or GlurR2 and GluR3 (Hollmann and Heinemann, 1994
; Rosenmund et al., 1998
; Wenthold et al., 1996
). In Xenopus and rodents, blocking trafficking of GluR2-subunit containing AMPA receptors into synapses decreases AMPA receptor-mediated synaptic inputs and interferes with neuronal plasticity and circuit function (Haas et al., 2006
; Rumpel et al., 2005
; Shi et al., 2001
; Takahashi et al., 2003
). In addition, GluR2 knockdown decreases spine number whereas GluR2 overexpression increases spine number in cultured hippocampal neurons (Passafaro et al., 2003
). Therefore, reduced synaptic GluR2-containing receptors results in a reduction in excitatory synapse number and AMPA receptor mediated currents. We chose to test the efficacy of RNAi against an endogenous neuronal protein using GluR2, based on the aforementioned data in this system and others, and based on the highly quantitative nature of functional recording of mEPSCs.
To test whether co-expression of Ago2 enhances RNAi in intact animals, we designed shRNAs against GluR2 and recorded spontaneous AMPA receptor mediated mEPSC as a functional indicator for GluR2 knockdown. Indeed, when we introduced shGluR2 together with Ago2 in tectal neurons, AMPA receptor mediated mEPSC frequency was significantly reduced compared to control neurons transfected with GFP alone, or shScrambled + Ago2 and importantly, neurons transfected with shGluR2 alone. The data provide strong evidence that ectopically expressed Ago2 enhances RNAi efficacy in CNS neuron in vivo. The amplitude of AMPA mEPSCs was not different from controls, suggesting that there might be a threshold number of AMPA receptors which is required to maintain a synapse. RNAi-mediated decreases in synaptic receptor numbers may cause synapses to fall below a requisite level of synaptic transmission so that the functional synapse, detected here by spontaneous release of transmitter, is lost.
Promoter Selection and Future Perspectives
shRNAs introduced into cells by vector-based expression are transcribed by either RNA polymerase III (pol III) or polymerase II (pol II) promoters (Huang et al., 2006
; Paddison et al., 2004b
). The pol III promoters usually direct higher levels of shRNA expression for effective gene silencing and their smaller size facilitates cloning and packaging into viral vectors. We tested the two most commonly used pol III promoters, U6 and H1 promoters in driving shGFP expression for gene silencing. Consistent with previous findings (Paddison et al., 2004b
), we found that U6 or H1 promoters result in comparable GFP knockdown in cell lines. Adding an enhancer from the CMV promoter in front of the U6 promoter, which was previously shown to increase the U6 promoter activity and shRNA synthesis (Xia et al., 2003
), did not yield higher GFP knockdown compared to U6 or H1 promoters alone. These results suggest that the promoter activities of U6 and H1 are not rate-limiting for RNAi efficacy in the systems we tested.
Compared to pol III promoters, the application of pol II promoters for RNAi-mediated gene silencing is limited due to its lower shRNA expression activities. Even the strong CMV promoter has lower RNAi efficacy compared to pol III promoters, such as the H1 or U6 promoters (Paddison et al., 2004b
), however, the competence of pol II promoter to drive shRNA in a tissue-specific manner can be beneficial in studying gene function (Rao et al., 2006
) and gene therapy (Giering et al., 2008
) in specific types of cells or tissues. Several promoters, for example the calcium/calmodulin-dependent protein kinase II, glutamic acid decarboxylase 67 and tyrosine hydroxylase promoters are well characterized to have distinct expressions in pyramidal neurons, GABAergic neurons and dopaminergic neurons, respectively (Banerjee et al., 1992
; Makinae et al., 2000
; Mima et al., 2001
). Our strategy to enhance RNAi efficacy by co-expression of Ago2 with shRNAs can be applied to facilitate RNAi when these relative weak pol II promoters are used for cell-type specific expression. In addition, since natural microRNAs are transcribed predominantly by pol II promoters (Cai et al., 2004
; Lee et al., 2004
), and pol II promoters have been shown to drive artificial shRNAmir30 efficiently (Silva et al., 2005
), an alternative way to achieve cell-type specific gene silencing is to utilize shRNAmir30 under the control of pol II promoters.
Antisense morpholino oligonucleotides provide high levels of protein knockdown in frogs and fish (Anichtchik et al., 2008
; Bestman and Cline, 2008
; Chiu et al., 2008
; Kenwrick et al., 2004
; Nakaya et al., 2008
; Wang and Poo, 2005
), and this method on the whole is still more efficient than RNAi for protein knockdown in Xenopus. Nevertheless, a principle advantage of the RNAi method is the possibility of having spatial and temporal control of protein knockdown by use of cell type specific and regulatable pol II promoters to drive shRNA and Ago2. This provides significant opportunity to probe protein function that is not possible with morpholinos. Application of RNAi and further improvement on its use in intact animals is a valuable molecular strategy for exploring physiological gene functions in specific types of neurons within brain circuits with the potential for a therapeutic application in the treatment of neurological diseases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr Gregory Hannon for insightful discussions and the gift of the pSM2c plasmid and Ago1/Ago2 cDNAs. We also thank Dr Ed Ruthazer (McGill) and members of the Cline laboratory for helpful discussions. This research was support by the National Eye Institute (EY011261) and Dart Neuroscience LCC (H.T.C) and Elisabeth Sloan Livingston Foundation Fellowship (S.-L.C).
Makinae, K., Kobayashi, T., Kobayashi, T., Shinkawa, H., Sakagami, H., Kondo, H., Tashiro, F., Miyazaki, J., Obata, K., Tamura, S., and Yanagawa, Y. (2000). Structure of the mouse glutamate decarboxylase 65 gene and its promoter: preferential expression of its promoter in the GABAergic neurons of transgenic mice. J. Neurochem. 75, 1429–1437.