Fear conditioning is a highly conserved form of emotional learning that occurs when stimuli in the environment predict aversive events. This type of learning allows previously neutral stimuli to elicit fear responses that prepare the animal for threat and aid in its avoidance (
LeDoux et al., 1988
;
Blanchard and Blanchard, 1989
;
Davis, 1992
;
Fendt and Fanselow, 1999
). The neural circuitry and cellular mechanisms that mediate fear conditioning have been extensively characterized. The amygdala is an essential component of this circuit and plasticity in this region has been tightly linked to fear learning (
Han et al., 2007
,
2009
;
Davis, 1997
;
Rodrigues et al., 2004
;
Maren, 2005
;
Rumpel et al., 2005
; ).
The amygdala is a heterogeneous structure consisting of multiple nuclei with unique afferent and efferent connections. The lateral nucleus (LA) of the amygdala, for example, receives auditory inputs from the thalamus and neocortex while spatial information is relayed from the hippocampus to the basolateral nucleus (BLA). The central nucleus serves as an output structure capable of activating a variety of fear responses via its connections with the hypothalamus and periaqueductal grey (
Fendt and Fanselow, 1999
;
LeDoux, 2000
;
Nader et al., 2001
). This compartmentalization is also observed in the genetic anatomy of the amygdala. For example, the α1 subunit of the GABA receptor is highly expressed in the lateral amygdala, moderately expressed in the BLA and completely absent from the central nucleus (
Fritschy and Mohler, 1995
;
Kaufmann et al., 2003
;
Marowsky et al., 2004
;
McDonald and Mascagni, 2004
). The function of this differential genetic expression pattern is currently unknown.
The cellular mechanisms that mediate fear learning have been thoroughly characterized in the lateral and basolateral amygdala. Blockade of NMDA receptors, CREB activation, protein synthesis and/or prevention of GluR1 insertion into the postsynaptic membrane interferes with fear learning in these regions (
Han et al., 2007
,
2009
;
Fanselow and Kim, 1994
;
Schafe and LeDoux, 2000
;
Rodrigues et al., 2001
;
Rumpel et al., 2005
; ). In contrast, much less is known about the role of inhibitory transmission in this process although recent work is beginning to shed light on this subject (
Davis et al., 1994
;
Pare et al., 2004
;
Heldt and Ressler, 2007
;
Cui et al., 2008
;
Ehrlich et al., 2009
). One technique that has emerged as a powerful tool for understanding the role of inhibition in learning and memory is the genetic deletion of specific GABA(A) receptor subunits (
Wiltgen et al., 2005
;
Mohler, 2007
). The region-specific location of these receptor subtypes and their distinct effects on inhibitory transmission provide an opportunity to gain functional insight into the role of specific neuronal circuits. The current experiments used targeted genetic deletion to study the role of the α1 subunit of the GABA(A) receptor in plasticity and fear learning in the amygdala of adult mice.
α1 function was examined using convergent cellular, molecular and behavioral strategies. Immunohistochemical analysis revealed high levels of α1 expression in the LA of the amygdala. Genetic deletion of this subunit enhanced activity-dependent plasticity in this region but not in the neighboring basolateral amygdala. We also observed that auditory fear learning was selectively enhanced in knockout animals while contextual learning remained unchanged. This phenotype appeared to be mediated by the loss of α1 in inhibitory neurons as mice lacking this subunit in excitatory cells showed no changes in fear learning. Lastly, pharmacological blockade of α1 receptors in the amygdala selectively impaired auditory fear learning while leaving context conditioning intact. Together, these results suggest that inhibitory transmission mediated by α1-containing GABA(A) receptors plays a critical role in amygdala plasticity and fear learning.
Animals
All mice were group housed with free access to food and tap water in the Herbert L. Washington Vivarium in the Department of Psychology at UCLA. They were maintained on a 12:12 h light:dark cycle and experiments were always performed during the light phase of the cycle. All animal protocols conformed to NIH guidelines and were approved by the UCLA Chancellor’s Animal Research Committee. Control mice (α1
+/+) harbored a GABA(A) receptor α1 subunit gene in which the exon encoding nucleotides 1307–1509 of the α1 cDNA (
Keir et al., 1991
) was flanked by loxP sites (i.e., floxed) as described (
Vicini et al., 2001
). Global α1 knockout mice (α1
−/−) were homozygous for a cre recombinase recombined locus in which the floxed exon was globally deleted (
Vicini et al., 2001
). Forebrain-selective conditional knockout mice (
Sonner et al., 2005
) were homozygous for the floxed α1 gene and hemizygous for a αCamKII-cre transgene (
Tsien et al., 1996
). Global α1
−/− mice, floxed α1
+/+ littermate controls and conditional knockouts were generated on a mixed C57BL/6J × Strain 129S1/X1 genetic background and genotyped as previously described (
Vicini et al., 2001
;
Sonner et al., 2005
). B6129F1 mice were purchased from Taconic.
Immunohistochemistry
Animals and tissue preparation
Seven α1−/− and five floxed age-matched adult male control mice (α1+/+) were used for immunohistochemical studies. They were deeply anesthetized with sodium pentobarbital (90 mg/kg) and perfused through the ascending aorta with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.3). After perfusion, the brains were maintained in situ at 4°C for 1 h and then removed and postfixed in the same fixative for 1 h. After thorough rinsing, the brains were cryoprotected in a 30% sucrose solution, blocked in the coronal plane, frozen on dry ice, and sectioned at 30 mm on a cryostat.
Antisera and immunohistochemistry
Prior to immunohistochemistry, free-floating sections were processed with a water bath heating antigen-retrieval method to reduce background staining and enhance specific labeling of the receptor subunits (
Peng et al., 2004
). Briefly, the sections were incubated in 1% H
2O
2 for 30 min to reduce endogenous peroxidase-like activity and then heated in a water bath in 0.05 M sodium citrate solution (pH 8.6) at 90°C for 70 min.
GABA
A receptor subunit-specific antisera that recognize the α1, α2, α3, α4, α5, α6, β2, β3, γ2 and δ subunits were used in this study. Guinea pig anti-α1, -α2, and -α5 subunits were kindly provided by J.-M. Fritschy; rabbit anti-α3, -γ2 and -δ subunits were kindly provided by W. Sieghart; and rabbit anti-α4, -β2 and -β3 subunits were obtained from Chemicon International (Temecula, CA, USA). The characterization and specificity of these antisera have been described previously (
Fritschy and Mohler, 1995
;
Sperk et al., 1997
;
Jechlinger et al., 1998
;
Peng et al., 2002
).
Free-floating sections that contained the lateral, basolateral and central amygdaloid nuclei at comparable levels were processed for immunohistochemistry with standard avidin-biotin-peroxidase methods (Vectastain Elite ABC; Vector Labo-ratories, Burlingame, CA, USA) as described previously (
Peng et al., 2004
). The concentrations of the primary antisera were: α1, 1:50,000; α2, 1:10,000; α3, 1:1500; α4, 1:1000; α5, 1:3000; α6, 1:10,000; β2, 1:1000; β3, 1:1000; δ, 1:4000; and γ2, 1:1:2000. After immunohistochemical labeling, sections were mounted on gelatin-coated slides, dehydrated, and coverslipped.
Immunolabeling analyses
Immunolabeling for all subunits was analyzed, and digital images were obtained with a Zeiss (Thornwood, NY, USA) AxioSkop 2 microscope equipped with an AxioCam digital camera system and AxioVision software (Version 4.4; Zeiss). Following qualitative analyses of GABAA receptor subunit changes in the α1−/− mice, differences in the intensity of immunolabeling for the α2, α3 and α4 subunits between the two genotypes were determined with densitometry. For each subunit, sections from all animals were processed identically in the same immunohistochemical experiment. Such experiments were repeated at least twice for each subunit to ensure the reliability of the results. Digital images of immunolabeling in the amygdala of both sides were obtained with the microscopic system described above, using a 5× objective. Images to be included in the same analysis were photographed under identical conditions on the same day with stabilized light levels. The densities of labeling (gray level values) were then determined with morphometric AxioVision software.
To ensure that comparable regions of each nucleus were analyzed in the two groups of animals, densitometric measurements were made in the dorsal part of the LA, the middle region of the BLA, and the complete region of the central nucleus. All values were corrected for background labeling by subtracting the gray level values of a rectangular area in the optic tract in the same section. The densitometry measurements were analyzed with Student’s t-test. For all analyses, p < 0.05 was considered significant. Graphs were prepared with Origin 7.5 software (OriginLab, Northampton, MA, USA).
Electrophysiology
Coronal amygdala slices, 400 μm thick, were obtained from halothane-anesthetized adult mice using standard techniques. Slices were maintained (at 30–31°C) in an interface recording chamber (Fine Science Tools, Inc.) and perfused (2–3 ml/min) with mouse artificial cerebral spinal fluid (ACSF) consisting of 124 mM NaCl, 4.4 mM KCl, 25 mM Na2HCO3, 1.0 mM Na2PO4, 1.2 mM MgSO4, 2 mM CaCl2, and 10 mM glucose, gassed with 95% O2, 5% CO2. In order to preserve GABAergic transmission in the amygdala slices, picrotoxin was not included in the perfusion solution. Field postsynaptic potentials (fPSPs) were recorded from either the LA or BLA of the amygdala. In some experiments, the stimulating electrode was positioned near presynaptic fibers passing from the thalamus to the LA (thalamo-LA pathway). In other experiments, the stimulating electrode was positioned in the LA and the recording electrode was positioned in the BLA (LA-BLA pathway). In all the experiments, fPSPs were evoked (at 0.05 Hz) using stimulation strengths sufficient to elicit fPSPs that were approximately 50% of the maximal fPSP amplitude. Five 1 s long trains of 100 Hz stimulation (intertrain interval = 1 min) elicited high-frequency stimulation-induced plasticity. Experiments from the knockout and floxed animals were interleaved and the experimenter was kept blind to the genotype of the subject during data acquisition. All values are reported as mean ± SEM. Planned comparisons using an unpaired two-tailed t-test were used to assess statistical significance of the plasticity data.
Behavioral Testing
Fear conditioning
Global α1 knockout mice. The apparatus (Med Associates, Inc., St Albans, VT, USA) and general procedures used in these experiments have been described previously (
Blaeser et al., 2006
). In experiment I, the mice were placed in the conditioning context for 2 min before receiving three tone (20 s, 2.8 kHz, 85 dB) shock (2 s, 0.6 mA) pairings spaced by 1-min intertrial intervals. The following day the mice received a 5-min tone test 2 min after placement into a novel environment. Twenty-four hours later the mice received a 5-min test in the training context. Freezing was scored via an automated system (
Anagnostaras et al., 2000
) during all sessions and used as an index of memory. In experiment II, mice in the signaled group were trained with five tone (30 s, 2.8 kHz, 85 dB) shock (2 s,75 mA) pairings spaced by 1-min intertrial intervals. Mice in the unsignaled group received five shock (2 s, 0.6 mA) presentations spaced by 1-min intertrial intervals. The following day the mice received a tone test 2 min after placement into a novel environment. During this test five 30 s tones were presented, each spaced by a 1-min intertrial interval. Twenty-four hours later the mice received a 5-min test in the training context. In experiment III, mice underwent discrimination training. In this procedure, the mice were placed into the training context on five consecutive days. Each day, two different auditory stimuli (20 s, whitenoise; clicker) were presented five times each. One of the stimuli was always paired with shock (2 s, 0.6 mA) (CS+) while the other (CS−) was never paired with shock. The stimulus used in each of these conditions was counterbalanced for α1
+/+ control mice and α1
−/− animals. Freezing behavior was scored during presentation of the auditory stimuli each day. After the last training day, mice received an extinction test in the training context where each auditory stimulus was presented five times in the absence of shock.
Conditional α1 knockout mice. The fear conditioning procedure used for these animals was identical to those used for global knockouts in experiment I.
3-PBC amygdala infusions. Mice received intra-amygdala infusions of 3-Propoxy-b-carboline hydrochloride (3-PBC) or saline and 20 min later were trained with 10 tone (30 s, 2.8 kHz, 85 dB) shock (2 s, 0.6 mA) pairings spaced by 1-min intertrial intervals. The following day the mice received a 10 min test in the training context. Twenty-four hours later, the mice received a tone test 2 min after placement into a novel environment. During this test ten 30 s tones were presented, each spaced by a 1-min intertrial interval.
Acoustic startle experiments
Threshold function. The apparatus (MED-ASR-310; Med Associates, Inc., St Albans, VT, USA) and procedures used in these experiments have been described previously (
Frankland et al., 2004
). Briefly, acoustic startle stimuli and prepulse stimuli were presented via a high-frequency speaker. The testing cylinder was positioned on a sensor platform and a piezoelectric accelerometer detected and transduced all cage movement, which were then digitized and stored by a computer. Following an acclimation period of 5 min, mice were presented with a total of 99 startle stimulus trials (at a fixed intertrial interval of 15 s) that varied between 75 and 120 dB at 5 dB increments. A block of 11 trials contained one stimulus of each intensity with a no stimulus catch trial (NS). Animals received nine such blocks. The startle stimuli were 40 ms noise bursts with a rise/fall time of less than 1 ms. Background noise levels were maintained at 65 dB throughout the test session. Startle threshold was defined as the minimal intensity at which responding was significantly greater than in the NS trials.
Pre-pulse inhibition. The apparatus (Med Associates, Inc., St Albans, VT, USA) and procedures used in these experiments have been described previously (
Frankland et al., 2004
). The mice were initially given a habituation session to acclimate them to the testing environment. In this session, mice were presented with 80 startle stimuli, delivered at a fixed intertrial interval of 15 s. The startle stimulus was a 40 ms, 120 dB noise burst with a rise/fall time of less than 1 ms. Background noise levels were maintained at 65 dB. The next day, prepulse inhibition was tested. Following an acclimation period of 5 min, mice were presented with a total of 90 trials. Three prepulse intensities were tested: 70, 75 and 80 dB. Prepulses were 20 ms in duration with a rise/fall time of less than 1 ms. For each prepulse intensity, there were three types of trials: prepulse alone, prepulse/startle stimulus and startle stimulus alone. In the prepulse/startle stimulus trial, the onset of the prepulse preceded the onset of the startle stimulus by 100 ms. Background noise levels were maintained at 68 dB throughout testing, and the trials were spaced 15 s apart.
Intra-amygdala infusions
Mice were were anesthetized with sodium pentobarbital (90 mg/kg) and mounted in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). The scalp of each animal was incised and retracted, and the skull was adjusted to place bregma and lambda in the same horizontal plane. Small burr holes were drilled at the appropriate injection sites. Plastic guide cannule (22 gauge; Plastics One, Roanoke, VA, USA) were inserted bilaterally at the following positions relative to bregma (mm): AP = −1.3, ML = ±3.3, DV = −4 and affixed with dental cement. Dummy cannule (28 gauge) were inserted into the guide cannule following the surgery. Mice were allowed to recover for 1 week prior to behavioral training. Twenty minutes prior to conditioning, the dummy cannule were removed and replaced with injection cannule (28 gauge) that projected an additional 0.8 mm from the tip of the guide cannule. 3-Propoxy-b-carboline hydrochloride (3-PBC; 10 μg/side) or ACSF was infused into the amygdala (0.25 μl/side; 0.1 μl/min). The injectors were left in place for 2 min after the end of the infusion to allow for diffusion. The mice were then returned to their homecage until training.
Histology. Histological verification of the cannula locations was performed at the end of behavioral testing. Mice were perfused transcardially with 0.9% saline, followed by 10% formalin. After extraction from the skull, the brains were postfixed in 10% formalin and then transferred to a 30% sucrose solution until sectioning. Coronal sections (40 μm thick, taken every 120 μm) were cut on a cryostat (−16°C) and mounted on glass microscope slides. After drying, the sections were stained with cresyl violet to identify neuronal cell bodies. Cannula tips were verified by visual inspection of the stained sections reconstructed on the mouse Allen Reference Atlas (
Dong, 2009
).
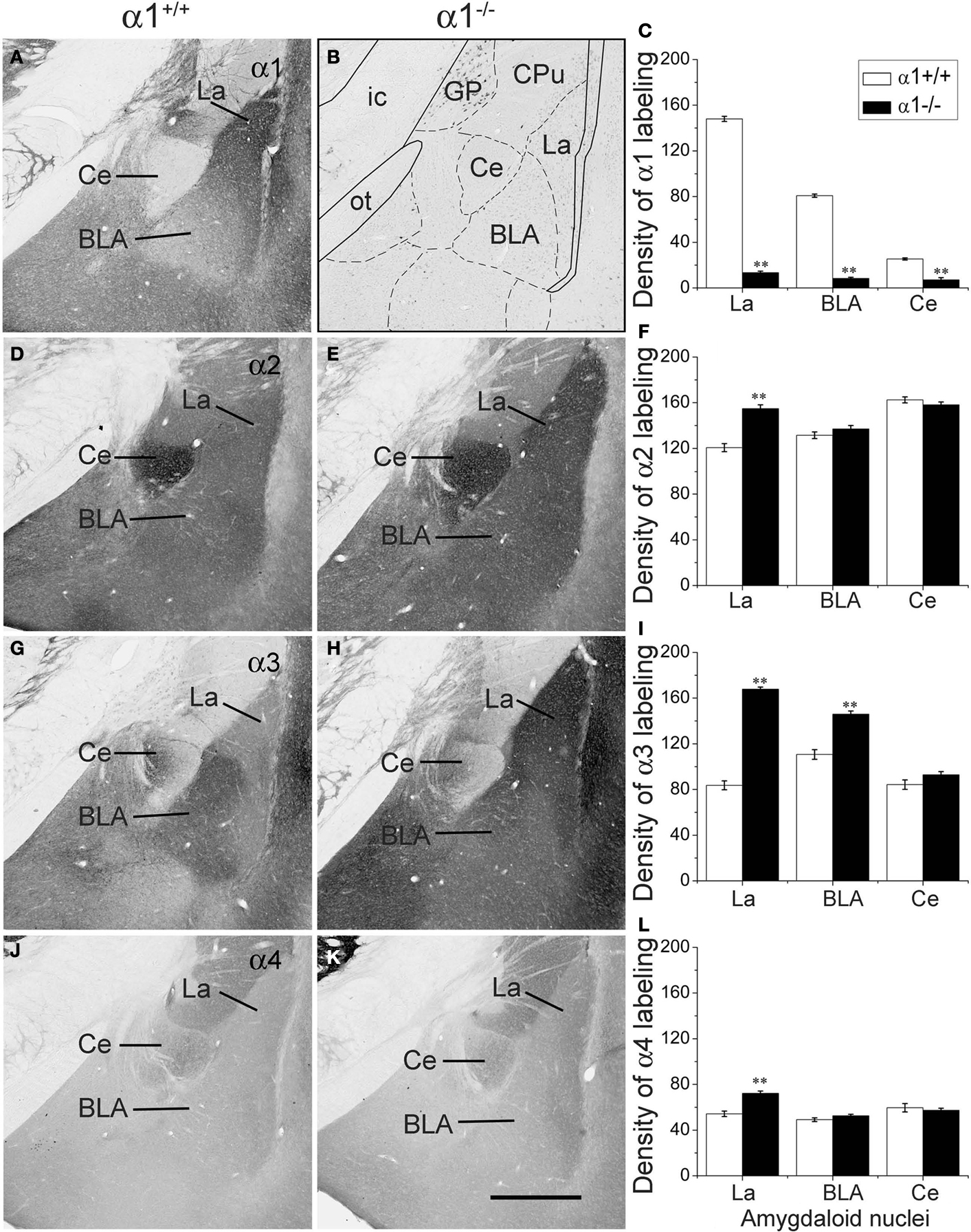
Distribution of the α1 Subunit in Amygdaloid Nuclei
Three amygdaloid nuclei, the lateral, basolateral and central nuclei, were selected for immunohistochemical analyses in this study, and each had different levels of α1 subunit expression in control mice (α1
+/+). Immunolabeling of α1 was highest in the LA, relatively low in the BLA, and virtually absent in the central nucleus, where the level of labeling was similar to the background labeling in the optic tract (Figures
1
A,C). This pattern of α1 subunit labeling was similar to that described previously in the mouse (
Marowsky et al., 2004
) but differs from that observed in rat, where α1 subunit labeling is also present at moderately high levels in the central nucleus (
Pirker et al., 2000
). In global α1
−/− mice, α1 labeling was absent in the amydaloid nuclei (Figures
1
B,C). Some isolated cell bodies were labeled by the α1 antiserum in a few brain regions, including the globus pallidus and hippocampus, as described previously by others (
Schneider Gasser et al., 2007
). However, such labeling was seldom observed in the amygdala.
Figure 1. Comparison of labeling for the α1 (A–C), α2 (D–F), α3 (G–I) and α4 (J–L) subunits in three amygdaloid nuclei in coronal sections of control and α1−/− mice. (A) In a control mouse, α1 labeling is high in the lateral amygdaloid nucleus (La) but relatively low in the basolateral nucleus (BLA). Virtually no labeling is evident in the central nucleus (Ce). (B) In a α1−/− mouse, no specific α1 labeling is present in the amygdaloid complex although a few cell bodies are labeled in the globus pallidus. A superimposed schematic drawing identifies the location of the amygdaloid nuclei and nearby structures, including the globus pallidus (GP), caudate putamen (CPu), optic tract (ot) and internal capsule (ic). (D) In a control mouse, moderate α2 subunit labeling is present in the lateral and basolateral nuclei, and strong α2 labeling is evident in the central nucleus. (E) In α1−/− mice, α2 subunit labeling is increased in the lateral nucleus, but no changes are evident in the basolateral and central nuclei. (G) In a control mouse, low levels of α3 subunit labeling are present in the lateral and central nuclei, but moderate α3 labeling is evident in the basolateral nucleus. (H) In a α1−/− mouse, α3 subunit labeling is substantially increased in the lateral nucleus and moderately increased in the basolateral nucleus. (J) In a control mouse, α4 subunit labeling is low in the three amygdaloid nuclei. (K) In a α1−/−mouse, α4 subunit labeling is slightly increased in the lateral nucleus, but remains low in this region, as in the other amygdaloid nuclei. (C, F, I, L) Bar graphs illustrate the virtual absence of α1 labeling in the α1−/− mouse (C), and a significant increase in α2, α3 and α4 labeling (F, I, L) in the lateral nucleus in α1−/− mice. The only other significant change is an increase in α3 labeling in the basolateral nucleus. Error bars represent SEM. **p < 0.01. Scale bar, 500 μm for all panels.
Multiple Changes in GABA(A) Receptor Subunits in the Amygdala of α1−/− Mice
The α2, α3 and α4 subunits had unique distribution patterns in the amygdaloid nuclei of α1
+/+ control mice, and the patterns were altered in the α1
−/− mice. In control mice, labeling for the α2 subunit was moderately strong in both the lateral and basolateral nuclei and was very strong in the central nucleus (Figure
1
D). In the α1
−/− mice, α2 subunit labeling was significantly increased in the LA (28.3% increase;
p < 0.01) (Figures
1
E,F). No changes were identified in the other two nuclei.
Labeling for the α3 subunit in control mice was moderately high in the BLA and relatively low in the lateral and central nuclei (Figure
1
G). In the α1
−/− mice, α3 subunit labeling increased substantially in the lateral and basolateral nuclei (100.1% and 31.9% respectively,
p < 0.01) (Figures
1
H,I). No changes were found in the central nucleus.
Labeling for the α4 subunit was low throughout the amygdaloid nuclei in control mice (Figure
1
J) but increased significantly (32.6%,
p < 0.01) in the LA in α1
−/− mice (Figures
1
K,L). Despite this increase, α4 expression remained low in the amygdaloid nuclei in the α1
−/− mice (Figure
1
K).
In summary, expression of the α2, α3 and α4 subunits increased in the LA of the amygdala in α1−/− mice, where α1 is normally present at high levels. Only α3 was increased significantly in the BLA, where the α1 subunit is normally present at relatively low levels. No changes in subunit expression were found in the central nucleus of α1−/− mice, consistent with the virtual absence of the α1 subunit in the central nucleus of control mice.
The α5 and α6 subunits are normally not detected in the lateral and basolateral nuclei and showed no increase in labeling in the a1−/− mice (data not shown).
Alterations in other GABA(A) receptor subunits were observed in the α1
−/− mice but were less striking (data not shown). Immunolabeling of the β2 subunit was decreased slightly in the LA in the α1
−/− mice, but the decrease was less marked than that in the cerebral cortex and several other brain regions. Labeling for β3 was normally quite low in the amygdaloid nuclei and remained low in α1
−/− mice. Immunolabeling of the γ2 subunit was also decreased in the LA, but, as for the β2 subunit, the decrease was not as great as that in the cerebral cortex. These findings are consistent with previous reports of decreased β2/β3 and γ2 subunit expression in other brain regions in α1 subunit-deficient mice (
Kralic et al., 2002a,
b
,
2006
;
Sur et al., 2001
; ). Despite such decreases, β2 and γ2 subunits remain in the amygdala of the α1
−/− mice, and thus could contribute to functional GABA(A) receptors in this region. δ subunit expression was very low or absent in the amygdaloid nuclei of control mice, and no change was observed in α1
−/− mice.
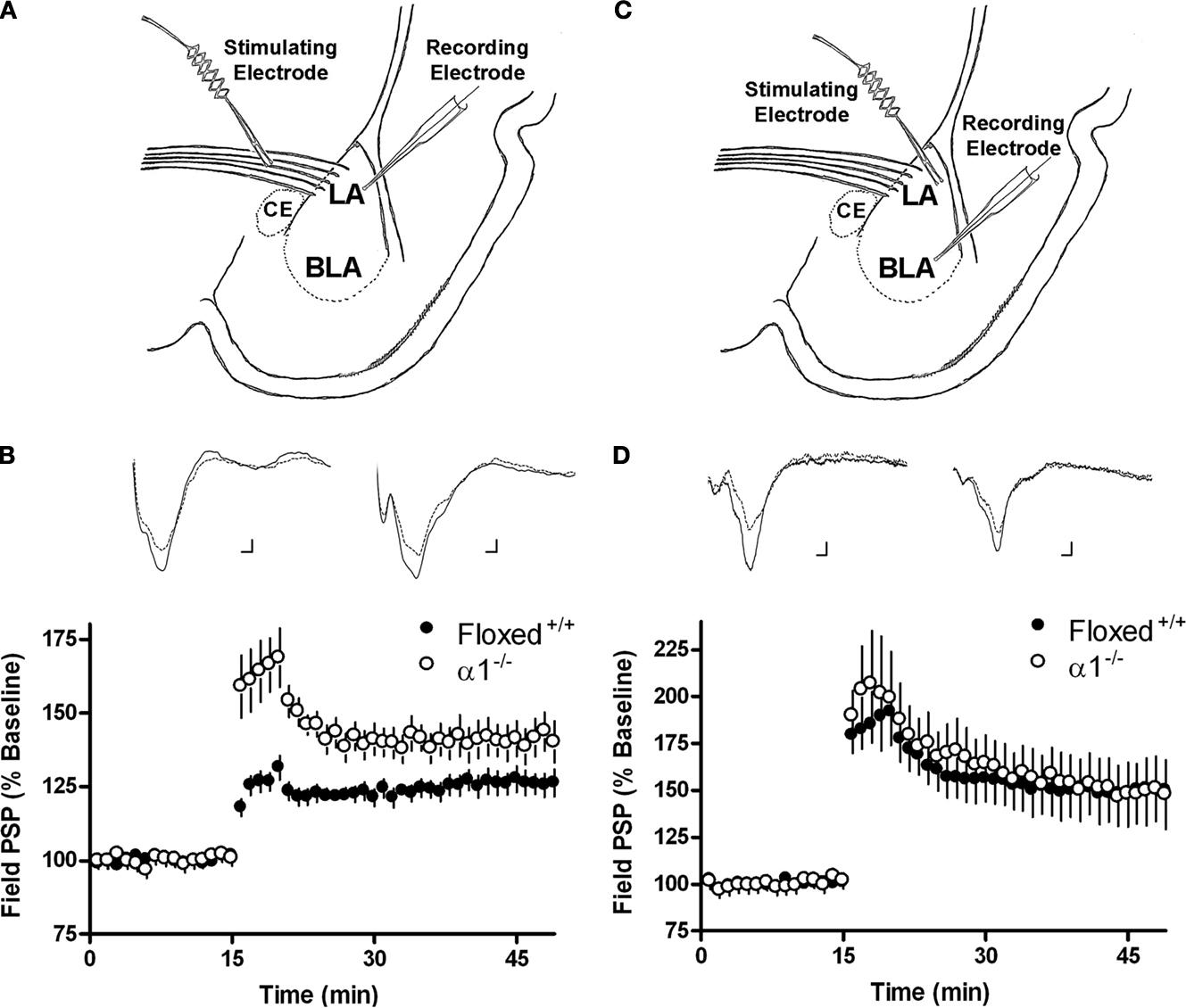
Plasticity in the Lateral Amygdala is Selectively Enhanced by α1 Deletion
Deletion of the α1 subunit dramatically reduces IPSC decay rate in the cerebellum and hippocampus and decreases GABAergic tone (
Vicini et al., 2001
;
Goldstein et al., 2002
;
Kralic et al., 2002a
). To examine the role of the α1 protein in activity-dependent synaptic plasticity, we compared the amygdala plasticity of floxed α1
+/+ controls and α1
−/− mice. In one set of experiments, population responses were recorded in the LA after activation of presynaptic fibers extending from the thalamus to the LA (Figure
2
A). The 100 Hz stimulation protocol produced a lasting increase in the amplitude of the LA field potential in both groups (Figure
2
B). The average amplitude of postsynaptic responses 10–15 min after 100 Hz stimulation was significantly enhanced in α1
−/− mice (140 ± 4.8% of baseline;
n = 4, eight slices) compared to the floxed controls (123 ± 2.8% of baseline;
n = 4, eight slices) (t
(14) = 3.02,
p < 0.01), and 25–30 min after 100 Hz stimulation the numeric difference between groups approached statistical significance (141 ± 6.2% of baseline in slices from α1
−/− mice and 126 ± 4.0% of baseline in floxed control slices) (t
(14) = 2.01,
p = 0.064). Thus, deletion of the α1 protein produces an enhancement in activity-dependent synaptic plasticity in the lateral amygdala. In a second set of experiments, population responses were recorded in the BLA after activation of presynaptic fibers by a stimulating electrode placed in the LA (Figure
2
C). The 100 Hz stimulation protocol produced a lasting increase in the amplitude of the BLA field potential in both groups (Figure
2
D). α1
−/− (
n = 5, eight slices) and floxed (
n = 5, nine slices) mice showed no differences in fPSP amplitude after 100 Hz stimulation [(10–15 min post stimulation: α1
−/− = 154 ± 9.7% of baseline; floxed = 157 ± 15.5% of baseline (t
(15) = −0.21,
p = 0.84)] [(25–30 min post stimulation: α1
−/− = 153 ± 9.8% of baseline; floxed = 152 ± 16.8% of baseline) (t
(15) = −0.51,
p = 0.62)]. Thus, deletion of the α1 protein did not modulate activity-dependent plasticity in the BLA.
Figure 2. (A) Schematic describing the electrode placement for experiments involving the thalamo-LA pathway. (B) The amount of lateral amygdala activity-dependent plasticity induced by 100 Hz stimulation of the thalamo-LA pathway in slices from floxed+/+ controls (black circles) and α1−/− (white circles) mice. Inset shows sample extracellular traces elicited during baseline (smaller response) and 25–30 min after 100 Hz stimulation in slices from floxed+/+ controls (left side of panel) and α1−/− mice (right side of panel). Calibration bars: 1 ms, 0.1 mV. (C) Schematic describing the electrode placement for experiments involving the LA-BLA pathway. (D) The amount of basolateral amygdala activity-dependent plasticity induced by 100 Hz stimulation of the LA-BLA pathway in slices from floxed+/+ (black circles) and α1−/− (white circles) mice. Inset shows sample extracellular traces elicited during baseline (smaller response) and 25–30 min after 100 Hz stimulation in slices from floxed+/+ (left side of panel) and α1−/− mice (right side of panel). Calibration bars: 1 ms, 0.1 mV.
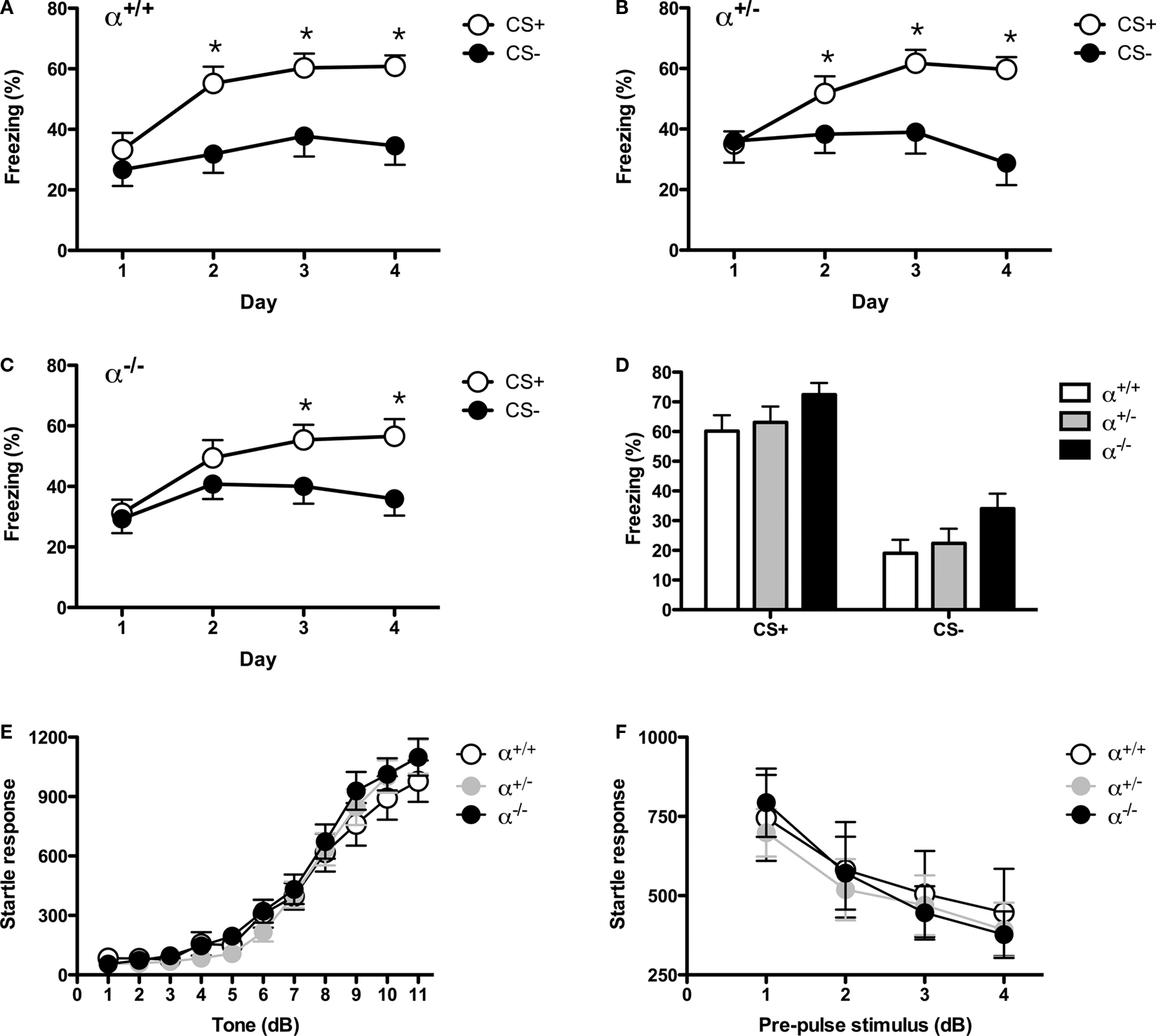
Deletion of α1 Enhances Auditory Fear Learning
We examined the effects of α1 deletion on fear learning by training α1
+/+, α1
+/− and α1
−/− animals with three shocks, each signaled by a 20 s tone. The day after fear conditioning, the animals were tested in a novel environment where they received a 5-min tone presentation. All animals exhibited an increase in freezing during the tone relative to baseline [Main effect of stimulus,
F(1, 66) = 77.71,
p < 0.05].
Post hoc tests (Fisher’s PLSD) revealed that α1
−/− mice showed significantly enhanced fear to the auditory stimulus relative to controls (
p < 0.05). Heterozygote knockouts showed a similar trend although it did not reach significance (
p > 0.05) (Figure
3
A). The next day all animals were placed back in the original training environment and received a 5-min context test. For this test, we also included separate groups of animals that received unsignaled training (i.e. shocks but no tone). In contrast to auditory learning, we found that context conditioning was normal in homozygous and heterozygous knockouts after signaled and unsignaled training (No main effect of genotype,
F < 1; No genotype × training condition interaction,
F < 1) (Figure
3
B). These data are consistent with our immunohistochemical and electrophysiological results demonstrating selective loss of α1 in the lateral amygdala of knockout mice and increased plasticity in this region.
Figure 3. (A) Mice were placed in the training context and received three tone-shock pairings. The next day the animals were placed in a novel environment and received a tone test. All mice showed low levels of baseline freezing and a substantial increase in freezing after the tone was presented. α1−/− mice froze significantly more during the tone than α1+/+ control mice. (B) Mice were placed back into the training environment for a 5-min context test. In addition to the mice that received tone-shock parings (Signaled) we also examined context fear in mice that received three unsignaled shocks during training (Unsignaled). There was no genotype difference in the amount of context freezing. (C) One group of mice were placed in the training context and received five tone-shock pairings (Signaled). Another group of animals received five unsignaled shocks (Unsignaled). The next day the mice received a tone test in a novel environment. All animals showed low levels of baseline freezing and a significant increase in freezing during the tone presentations. The increase in freezing was significantly larger in mice that received signaled training. α1 knockout mice that received signaled training showed substantially more tone fear than α1+/+ control mice. In addition, this fear was specific to the tone paired with shock as α1 knockouts that received unsignaled training froze substantially less. (D) Mice were placed back into the training environment for a 5-min context test. There was no genotype difference in the amount of context freezing.
To determine if enhanced auditory fear in homozygous knockouts was associative in nature we ran a similar experiment and this time also tested the tone in animals that were trained with unsignaled shocks. The goal was to determine if the enhanced tone freezing observed in homozygous knockout mice was contingent on the auditory stimulus being paired with shock. The day after training all animals received a tone test in a novel environment. Across genotypes, groups that received signaled conditioning showed significantly more auditory fear than those receiving unsignaled training [Main effect of training
F(1, 65) = 21.42,
p < 0.05; No training × genotype interaction,
F < 1] demonstrating that auditory learning was associative in nature (Figure
3
C). Similar to the first experiment, tone conditioning was enhanced in α1
−/− mice that received paired training relative to control mice (Fisher’s PLSD,
p < 0.05). In contrast, α1
−/− mice that received unsignaled conditioning showed the same amount of fear as control animals (Fisher’s PLSD,
p > 0.05). This suggests that associative learning is selectively enhanced in mice lacking the α1 receptor in the lateral amygdala. Once again, context conditioning was similar in all groups [No main effect of procedure
F(1, 65) = 1.882,
p > 0.05; No main effect of genotype
F < 1] (Figure
3
D).
Auditory Discrimination and Startle Responding are Normal in α1−/− Mice
In the next experiment, we determined if the specificity of auditory fear was altered in homozygous knockout mice. It is possible that these animals show enhanced associative learning that is less specific because an increased number of auditory inputs are associated with shock. This would result in increased generalization and reduced discrimination. To test this idea we used an auditory discrimination procedure. Mice received an auditory stimulus (CS+) that was reliably paired with shock during four training sessions. In the same sessions, a different auditory stimulus (CS−) was presented but never paired with shock. Across the course of training, all four groups learned to discriminate between the stimuli [Stimulus × Training Day Interaction
F(3, 126) = 13.487,
p < 0.05; No stimulus × Training × Genotype Interaction,
F < 1] (Figures
4
A–C). This suggests that auditory discrimination learning is normal in knockout mice. At the end of training, an extinction test was conducted where the CS+ and CS− were presented in the absence of shock. Once again, all groups were able to discriminate between these stimuli [Main effect of stimulus
F(1, 42) = 156.71,
p < 0.05; No stimulus × genotype interaction,
F < 1] (Figure
4
D). In addition, there was an overall effect of genotype [Main effect of genotype,
F(2, 42) = 3.13,
p = 0.05] driven by the fact that homozygous knockouts showed more auditory fear than control animals (Fisher’s PLSD,
p < 0.05). These data suggest that homozygous knockouts exhibit an enhancement in auditory conditioning but remain capable of discriminating between distinct auditory stimuli.
Figure 4. (A) Mice were trained with a white noise and clicker auditory stimulus. For each animal, one of these stimuli was paired with shock (CS+) and the other was not (CS−). Mice received discrimination training across 4 days. Each day, the CS+ and CS− were presented four times in a pseudorandom order (3 min ITI). α1+/+ control mice learned to discriminate across training days and eventually froze more to the CS+ than the CS−. (B) Heterozygous knockout mice learned to discriminate across training days and eventually froze more to the CS+ than the CS−. (C) Homozygous knockout mice learned to discriminate across training days and eventually froze more to the CS+ than the CS−. (D) The day after discrimination training ended mice were given an extinction test. During this test, the CS+ and CS− were each presented four times in the absence of shock. As observed during training, all mice froze significantly more to the CS+ than the CS−. In addition, there was an overall effect of genotype as homozygous knockout mice froze significantly more than heterozygous knockouts and control mice. This increase in freezing did not interact with stimulus type (CS+ or CS−). (E) Acoustic startle was measured over a range of stimuli (0–120 dB) using the MED-ASR-310 testing system. There was a significant effect of volume as startle amplitude increased systematically with increasing dB level. The responses of control mice, heterozygous knockouts and homozygous knockouts were the same across all test stimuli. (F) Mice were next tested on pre-pulse inhibition (PPI). Three pre-pulse intensities were tested: 70, 75, 80 dB. The onset of the pre-pulse preceded the onset of the startle stimulus by 100 ms. The amplitude of the startle response decreased systematically with the intensity of the pre-pulse stimulus. This decrease was the same for all genotypes.
To determine if the detection of auditory stimuli is altered in homozygous knockout mice we conducted an acoustic startle test. We found a systematic increase in the startle response as the intensity of the auditory stimulus was increased [Main effect of stimulus,
F(10, 480) = 167.56,
p < 0.05] that did not differ between any of the genotypes (No stimulus × genotype interaction,
F < 1) (Figure
4
E). This suggests that basic auditory function is normal in α1 homozgous knockout mice, a fact that is consistent with previously published data on these animals (
Maison et al., 2006
).
Startle is mediated solely by a brainstem reflex and therefore only assesses auditory processing at this low level. Therefore, to assess auditory processing at higher levels of the central nervous system we used a prepulse inhibition task (PPI). This version of the startle task measures the ability of the forebrain to gate acoustic reflexes (
Fendt et al., 2001
). If enhanced auditory conditioning in knockout mice results from altered sensory processing of auditory signals then we should see a significant change in PPI in these animals. Following an acclimation period of 5 min, mice were presented with 20 noise bursts that were preceded by three different pre-pulse auditory stimuli of different intensities (70, 75 and 80 dB). As previously reported, the pre-pulse stimulus reduced the magnitude of the startle response to the noise bursts and this effect was similar across genotypes (
Fendt et al., 2001
;
Frankland et al., 2004
) [Main effect of Pre-Pulse
F(3, 147) = 63.76,
p < 0.05; No Pre-Pulse × Genotype Interaction
F(6, 147) = 1,
p > 0.05] (Figure
4
F). This suggests that forebrain sensory processing of acoustic stimuli is normal in α1 knockout mice.
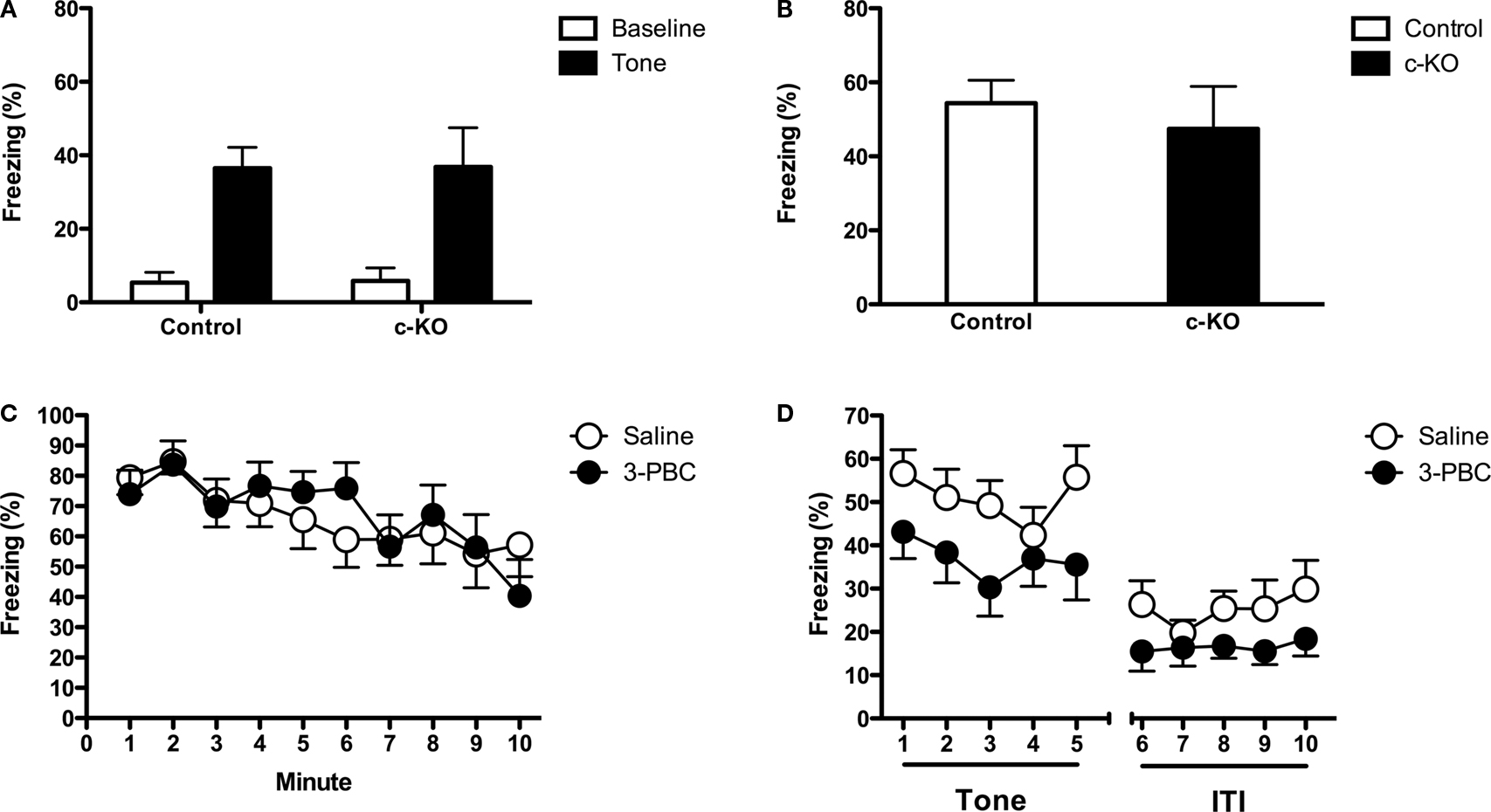
Forebrain Deletion of α1 in CaMKII-Containing Neurons does not Alter Fear Conditioning
α1 containing GABARs are present in both excitatory and inhibitory neurons. Global knockout mice lack these receptors in both types of neurons making it difficult to determine which cells are responsible for their phenotype (Figure
1
B). Therefore, in this experiment we examined auditory fear learning in conditional α1 knockout mice (cKO) generated using the Cre-loxP system (
Sonner et al., 2005
). In these animals, Cre recombinase is under the control of the αCaMKII promoter, which in the lateral and basolateral amygdala is expressed exclusively in excitatory neurons (
McDonald et al., 2002
). Previous studies with these mice have shown that α1 is deleted in the amygdala and that benzodiazepine and TBPS binding sites are reduced in cortex to approximately 75% and 30% of normal, respectively (
Sonner et al., 2005
).
Using the same conditioning parameters as experiment 1, we found no change in auditory fear learning in conditional α1 knockout mice (No effect of genotype,
F < 1) (Figure
5
A). Context conditioning was also normal in these animals (Main effect of genotype,
F < 1) (Figure
5
B). These results demonstrate that deletion of α1 in αCaMKII-containing neurons does not increase learned fear. Given that αCaMKII is selectively expressed in excitatory cells in the lateral and basolateral amygdala (
McDonald et al., 2002
) these results suggest that the loss of α1 in inhibitory neurons is likely responsible for the increased auditory fear conditioning observed in global knockout mice. This finding can help explain the reduced inhibitory tone that has previously been observed in global α1 knockout mice and the increased plasticity that we observed in the lateral amygdala (
Kralic et al., 2002a
). Genetic deletion of the α1 subunit in GABAergic neurons should decrease the decay rate of IPSCs in these cells and prevent them from inhibiting excitatory neurons.
Figure 5. (A) Controls and conditional KO mice were placed in the training context and received three tone-shock pairings. The next day the animals were placed in a novel environment and received a tone test. Both genotypes mice showed low levels of baseline freezing and a substantial increase in freezing after the tone was presented. There was no genotype difference in the amount of tone fear. (B) Mice were placed back into the training environment for a 5-min context test. There was no genotype difference in the amount of context freezing. (C) Mice received intra-amygdala infusions of saline or 3-PBC and were trained 20 min later. During training the mice received 10 tone-shock. The next day mice were placed back into the training environment for a 10-min context test. There was no difference in the amount of context freezing in mice that were trained with saline and those trained with 3-PBC. (D) The following day the animals were placed in a novel environment and received a tone test. During this test, mice that were trained with 3-PBC froze significantly less than animals trained with saline.
Intra-Amygdala Infusion of 3-PBC Selectively Reduces Auditory Fear Conditioning
Because the forebrain selective α1 knockouts also have reduced α1 expression in hippocampus and cortex (
Sonner et al., 2005
), it was possible that the loss of α1 outside of the amygdala was responsible for the observed phenotype in our knockout animals. We addressed this issue by infusing a highly selective α1 antagonist (3-PBC) into the lateral amygdala during fear learning. 3-PBC has a 10- to 20-fold increased selectivity for GABA(A) receptors containing the α1 subunit (
Huang et al., 2000
;
Harvey et al., 2002
;
Gourley et al., 2005
). If α1-containing GABARs in the lateral amygdala have a specific role in auditory conditioning then this drug treatment should modulate tone but not context fear. 3-PBC was infused bilaterally into the amygdala 20 min before signaled fear conditioning. The next day all animals received a context test. There was no difference in the amount of freezing between groups suggesting that α1-containing receptors in the lateral amygdala are not essential for context fear conditioning (No effect of genotype,
F < 1) (Figure
5
C). This result also suggests that the infusion of 3-PBC into the amygdala does not alter fear learning or expression. In contrast, there was a significant reduction in conditioned fear during the auditory test in mice trained with 3-PBC [Main effect of drug,
F(1, 22) = 5.325,
p < 0.05] (Figure
5
D). This result demonstrates that α1 receptors in the lateral amygdala are selectively involved in the acquisition of auditory fear learning. One explanation for this effect is that infusions of 3-PBC prevent the inhibition of GABAergic cells in the lateral amygdala and allows them to reduce the activity of excitatory neurons. This result would be consistent with the fact that the α1 subunit is highly expressed on interneurons in the lateral amygdala (
McDonald and Mascagni, 2004
;
Meguro et al., 2004
).
The current data demonstrate that the α1 subunit of the GABA(A) receptor regulates plasticity and auditory fear learning in the lateral amygdala. Our immunohistochemical results revealed that this subunit is highly expressed in the lateral amygdala, moderately expressed in the basolateral amygdala and completely absent from the central nucleus of the amygdala. Consistent with this expression pattern, deletion of α1 significantly enhanced activity-dependent plasticity in the lateral but not basolateral amygdala. This enhancement was correlated with selective increases in auditory fear learning, which is known to depend on plasticity in the lateral amygdala (
Fanselow and Kim, 1994
;
Maren et al., 1996
;
Rodrigues et al., 2001
;
Rumpel et al., 2005
;
Han et al., 2007
). In addition, intra-amygdala infusions of 3-PBC led to a selective impairment in auditory conditioning. This drug has previously been shown to act as an antagonist with preferential action at GABA(A) receptors with α1 subunits (
Gourley et al., 2005
).
Recent work has demonstrated that gene expression profiles can be used to determine the unique functions of neural circuits both within and across brain structures (
Lein et al., 2007
;
Thompson et al., 2008
;
Dong et al., 2009
;
Ng et al., 2009
). Our α1 observations in the amygdala support this idea. Previous lesion work has indicated that the lateral, basolateral and central amygdala play distinct roles in fear learning. Auditory and other sensory inputs terminate in the lateral amygdala while spatial and contextual information are sent from the hippocampus to the basolateral complex (
Maren and Fanselow, 1995
;
LeDoux, 2000
). Consistent with this fact, damage to the lateral amygdala impairs auditory fear learning while basolateral damage affects contextual conditioning (
Nader et al., 2001
;
Calandreau et al., 2005
). However, it should be noted, that there is more overlap in these circuits than previously believed (
Goosens and Maren, 2001
;
Malkani and Rosen, 2001
). Our data extends this work by showing that the α1 subunit of the GABA(A) receptor is highly expressed in the lateral amygdala and contributes specifically to auditory but not contextual fear learning.
The functional significance of selective α1 expression in the lateral amygdala is currently unknown. However, this subunit does impart unique properties on GABA(A) receptors that may be particularly important for learning about discrete cues that predict danger. For example, GABA(A) receptors with α1 exhibit very rapid decay of IPSCs while receptors without this subunit show prolonged IPSC decay rates (
Vicini et al., 2001
;
Goldstein et al., 2002
). The rapid activation and deactivation of these receptors provides a plausible mechanism for animals to detect and learn about acute sensory cues that predict danger. Unlike spatial environments, discrete cues tend to be phasic and more proximal to threat (
Konorski, 1967
). Consistent with this fact, behavioral data has demonstrated that animals learn about discrete cues much better if they are short in duration (seconds as opposed to many minutes) (
Kamin, 1965
;
Lubow, 1973
;
Mackintosh, 1983
). The fact that α1 containing GABA(A) receptors show extremely fast IPSC decay might afford them the capacity to associate these cues with shock. An incoming sensory signal could briefly inhibit GABAergic cells with α1 and produce activation of excitatory pyramidal cells. Given the properties of α1 containing GABA(A) receptors, this activation would be short-lived and provide a brief window for cues to be associated with shock.
In contrast, contextual cues have different learning requirements. They tend to be more prolonged and diffuse in nature. Consistent with this fact, behavioral studies have shown that short presentations of contextual cues produce little or no learning, while prolonged presentations produce optimal learning (
Fanselow, 1986
,
1990
;
Wiltgen et al., 2006
). GABA(A) receptors without α1, like those found in the basolateral and central amygdala, provide a possible mechanism for this type of learning as they afford a longer period during which contextual cues could coincide with shock presentation.
The current results demonstrate the importance of inhibitory transmission in the amygdala for both LTP and learning. These findings complement current work that is beginning to define the critical function of GABAergic transmission in learning, memory and plasticity (
Costa et al., 2002
;
Kleschevnikov et al., 2004
;
Fernandez et al., 2007
;
Cui et al., 2008
;
Ehninger et al., 2008
;
Ehrlich et al., 2009
). In the amygdala, recent experiments have also uncovered changes in GABA(A) receptor subunit expression following fear learning and extinction. These results suggest that inhibitory transmission may be altered in response to learning and play an important role in the plasticity changes that underlie memory (
Heldt and Ressler, 2007
). Together with our results, these studies all point to the critical role of GABA(A) receptor in plasticity and learning in multiple brain regions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Mareike Kuypers for maintaining the mouse colony and Carolyn Ferguson for genotyping. This work was supported by NIH grants AA10422 to GEH, NS051311 to CRH, A016933 to HLJ and P01NS35985 to MSF.
Anagnostaras, S. G., Josselyn, S. A., Frankland, P. W., and Silva, A. J. (2000). Computer-assisted behavioral assessment of Pavlovian fear conditioning in mice.
Learn. Mem. 7, 58–72.
Blaeser, F., Sanders, M. J., Truong, N., Ko, S., Wu, L. J., Wozniak, D. F., Fanselow, M. S., Zhuo, M., and Chatila, T. A. (2006). Long-term memory deficits in Pavlovian fear conditioning in Ca
2+/calmodulin kinase kinase alpha-deficient mice.
Mol. Cell. Biol. 26, 9105–9115.
Blanchard, R. J., and Blanchard, D. C. (1989). Attack and defense in rodents as ethoexperimental models for the study of emotion.
Prog. Neuropsychopharmacol. Biol. Psychiatry 13(Suppl.), S3–S14.
Calandreau, L., Desmedt, A., Decorte, L., and Jaffard, R. (2005). A different recruitment of the lateral and basolateral amygdala promotes contextual or elemental conditioned association in Pavlovian fear conditioning.
Learn. Mem. 12, 383–388.
Costa, R. M., Federov, N. B., Kogan, J. H., Murphy, G. G., Stern, J., Ohno, M., Kucherlapati, R., Jacks, T., and Silva, A. J. (2002). Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1.
Nature 415, 526–530.
Cui, Y., Costa, R. M., Murphy, G. G., Elgersma, Y., Zhu, Y., Gutmann, D. H., Parada, L. F., Mody, I., and Silva, A. J. (2008). Neurofibromin regulation of ERK signaling modulates GABA release and learning.
Cell 135, 549–560.
Davis, M. (1992). The role of the amygdala in fear and anxiety.
Annu. Rev. Neurosci. 15, 353–375.
Davis, M. (1997). Neurobiology of fear responses: the role of the amygdala.
J. Neuropsychiatry Clin. Neurosci. 9, 382–402.
Davis, M., Rainnie, D., and Cassell, M. (1994). Neurotransmission in the rat amygdala related to fear and anxiety.
Trends Neurosci. 17, 208–214.
Dong, H. W. (2009). The Allen Reference Atlas. Wiley.
Dong, H. W., Swanson, L. W., Chen, L., Fanselow, M. S., and Toga, A. W. (2009). Genomic-anatomic evidence for distinct functional domains in hippocampal field CA1.
Proc. Natl. Acad. Sci. U.S.A. 106, 11794–11799.
Ehninger, D., Li, W., Fox, K., Stryker, M. P., and Silva, A. J. (2008). Reversing neurodevelopmental disorders in adults.
Neuron 60, 950–960.
Ehrlich, I., Humeau, Y., Grenier, F., Ciocchi, S., Herry, C., and Luthi, A. (2009). Amygdala inhibitory circuits and the control of fear memory.
Neuron 62, 757–771.
Fanselow, M. S. (1986). Associative vs topographical accounts of the immediate shock-freezing deficit in rats: implications for the response selection rules governing species-specific defensive reactions.
Learn. Motiv. 17, 16–39.
Fanselow, M. S. (1990). Factors governing one-trial contextual conditioning.
Anim. Learn. Behav. 18, 264–270.
Fanselow, M. S., and Kim, J. J. (1994). Acquisition of contextual Pavlovian fear conditioning is blocked by application of an NMDA receptor antagonist d,l-2-amino-5-phosphonovaleric acid to the basolateral amygdala.
Behav. Neurosci. 108, 210–212.
Fendt, M., and Fanselow, M. S. (1999). The neuroanatomical and neurochemical basis of conditioned fear.
Neurosci. Biobehav. Rev. 23, 743–760.
Fendt, M., Li, L., and Yeomans, J. S. (2001). Brain stem circuits mediating prepulse inhibition of the startle reflex.
Psychopharmacology (Berl.) 156, 216–224.
Fernandez, F., Morishita, W., Zuniga, E., Nguyen, J., Blank, M., Malenka, R. C., and Garner, C. C. (2007). Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome.
Nat. Neurosci. 10, 411–413.
Frankland, P. W., Wang, Y., Rosner, B., Shimizu, T., Balleine, B. W., Dykens, E. M., Ornitz, E. M., and Silva, A. J. (2004). Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice.
Mol. Psychiatry 9, 417–425.
Fritschy, J. M., and Mohler, H. (1995). GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits.
J. Comp. Neurol. 359, 154–194.
Goldstein, P. A., Elsen, F. P., Ying, S. W., Ferguson, C., Homanics, G. E., and Harrison, N. L. (2002). Prolongation of hippocampal miniature inhibitory postsynaptic currents in mice lacking the GABA(A) receptor alpha1 subunit.
J. Neurophysiol. 88, 3208–3217.
Goosens, K. A., and Maren, S. (2001). Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats.
Learn. Mem. 8, 148–155.
Gourley, S. L., Debold, J. F., Yin, W., Cook, J., and Miczek, K. A. (2005). Benzodiazepines and heightened aggressive behavior in rats: reduction by GABA(A)/alpha(1) receptor antagonists.
Psychopharmacology (Berl.) 178, 232–240.
Han, J. H., Kushner, S. A., Yiu, A. P., Cole, C. J., Matynia, A., Brown, R. A., Neve, R. L., Guzowski, J. F., Silva, A. J., and Josselyn, S. A. (2007). Neuronal competition and selection during memory formation.
Science 316, 457–460.
Han, J. H., Kushner, S. A., Yiu, A. P., Hsiang, H. L., Buch, T., Waisman, A., Bontempi, B., Neve, R. L., Frankland, P. W., and Josselyn, S. A. (2009). Selective erasure of a fear memory.
Science 323, 1492–1496.
Harvey, S. C., Foster, K. L., McKay, P. F., Carroll, M. R., Seyoum, R., Woods, J. E., 2nd, Grey, C., Jones, C. M., McCane, S., Cummings, R., Mason, D., Ma, C., Cook, J. M., and June, H. L. (2002). The GABA(A) receptor alpha1 subtype in the ventral pallidum regulates alcohol-seeking behaviors.
J. Neurosci. 22, 3765–3775.
Heldt, S. A., and Ressler, K. J. (2007). Training-induced changes in the expression of GABAA-associated genes in the amygdala after the acquisition and extinction of Pavlovian fear.
Eur. J. Neurosci. 26, 3631–3644.
Huang, Q., He, X., Ma, C., Liu, R., Yu, S., Dayer, C. A., Wenger, G. R., McKernan, R., and Cook, J. M. (2000). Pharmacophore/receptor models for GABA(A)/BzR subtypes (alpha1beta3gamma2, alpha5beta3gamma2, and alpha6beta3gamma2) via a comprehensive ligand-mapping approach.
J. Med. Chem. 43, 71–95.
Jechlinger, M., Pelz, R., Tretter, V., Klausberger, T., and Sieghart, W. (1998). Subunit composition and quantitative importance of hetero-oligomeric receptors: GABAA receptors containing alpha6 subunits.
J. Neurosci. 18, 2449–2457.
Kamin, L. J. (1965). Temporal and intensity characteristics of the conditioned stimulus. In Classical Conditioning, W. F. Prokasy, ed. (Norwalk, CT, Appleton and Lange), pp. 118–147.
Kaufmann, W. A., Humpel, C., Alheid, G. F., and Marksteiner, J. (2003). Compartmentation of alpha 1 and alpha 2 GABA(A) receptor subunits within rat extended amygdala: implications for benzodiazepine action.
Brain Res. 964, 91–99.
Keir, W. J., Kozak, C. A., Chakraborti, A., Deitrich, R. A., and Sikela, J. M. (1991). The cDNA sequence and chromosomal location of the murine GABAA alpha 1 receptor gene.
Genomics 9, 390–395.
Kleschevnikov, A. M., Belichenko, P. V., Villar, A. J., Epstein, C. J., Malenka, R. C., and Mobley, W. C. (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome.
J. Neurosci. 24, 8153–8160.
Konorski, J. (1967). Integrative Activity of the Brain. University of Chicago Press. Chicago.
Kralic, J. E., Korpi, E. R., O’Buckley, T. K., Homanics, G. E., and Morrow, A. L. (2002a) Molecular and pharmacological characterization of GABA(A) receptor alpha1 subunit knockout mice.
J. Pharmacol. Exp. Ther. 302, 1037–1045.
Kralic, J. E., O’Buckley, T. K., Khisti, R. T., Hodge, C. W., Homanics, G. E., and Morrow, A. L. (2002b) GABA(A) receptor alpha-1 subunit deletion alters receptor subtype assembly, pharmacological and behavioral responses to benzodiazepines and zolpidem.
Neuropharmacology 43, 685–694.
Kralic, J. E., Sidler, C., Parpan, F., Homanics, G. E., Morrow, A. L., and Fritschy, J. M. (2006). Compensatory alteration of inhibitory synaptic circuits in cerebellum and thalamus of gamma-aminobutyric acid type A receptor alpha1 subunit knockout mice.
J. Comp. Neurol. 495, 408–421.
LeDoux, J. E. (2000). Emotion circuits in the brain.
Annu. Rev. Neurosci. 23, 155–184.
LeDoux, J. E., Iwata, J., Cicchetti, P., and Reis, D. J. (1988). Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear.
J. Neurosci. 8, 2517–2529.
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., Boe, A. F., Boguski, M. S., Brockway, K. S., Byrnes, E. J., Chen, L., Chen, L., Chen, T. M., Chin, M. C., Chong, J., Crook, B. E., Czaplinska, A., Dang, C. N., Datta, S., Dee, N. R., Desaki, A. L., Desta, T., Diep, E., Dolbeare, T. A., Donelan, M. J., Dong, H. W., Dougherty, J. G., Duncan, B. J., Ebbert, A. J., Eichele, G., Estin, L. K., Faber, C., Facer, B. A., Fields, R., Fischer, S. R., Fliss, T. P., Frensley, C., Gates, S. N., Glattfelder, K. J., Halverson, K. R., Hart, M. R., Hohmann, J. G., Howell, M. P., Jeung, D. P., Johnson, R. A., Karr, P. T., Kawal, R., Kidney, J. M., Knapik, R. H., Kuan, C. L., Lake, J. H., Laramee, A. R., Larsen, K. D., Lau, C., Lemon, T. A., Liang, A. J., Liu, Y., Luong, L. T., Michaels, J., Morgan, J. J., Morgan, R. J., Mortrud, M. T., Mosqueda, N. F., Ng, L. L., Ng, R., Orta, G. J., Overly, C. C., Pak, T. H., Parry, S. E., Pathak, S. D., Pearson, O. C., Puchalski, R. B., Riley, Z. L., Rockett, H. R., Rowland, S. A., Royall, J. J., Ruiz, M. J., Sarno, N. R., Schaffnit, K., Shapovalova, N. V., Sivisay, T., Slaughterbeck, C. R., Smith, S. C., Smith, K. A., Smith, B. I., Sodt, A. J., Stewart, N. N., Stumpf, K. R., Sunkin, S. M., Sutram, M., Tam, A., Teemer, C. D., Thaller, C., Thompson, C. L., Varnam, L. R., Visel, A., Whitlock, R. M., Wohnoutka, P. E., Wolkey, C. K., Wong, V. Y., Wood, M., Yaylaoglu, M. B., Young, R. C., Youngstrom, B. L., Yuan, X. F., Zhang, B., Zwingman, T. A., and Jones, A. R. (2007). Genome-wide atlas of gene expression in the adult mouse brain.
Nature 445, 168–176.
Lubow, R. E. (1973). Latent inhibition.
Psychol. Bull. 79, 398–407.
Mackintosh, N. J. (1983). Conditioning and Associative Learning. Oxford, Claredon.
Maison, S. F., Rosahl, T. W., Homanics, G. E., and Liberman, M. C. (2006). Functional role of GABAergic innervation of the cochlea: phenotypic analysis of mice lacking GABA(A) receptor subunits alpha 1, alpha 2, alpha 5, alpha 6, beta 2, beta 3, or delta.
J. Neurosci. 26, 10315–10326.
Malkani, S., and Rosen, J. B. (2001).
N-Methyl-
D-aspartate receptor antagonism blocks contextual fear conditioning and differentially regulates early growth response-1 messenger RNA expression in the amygdala: implications for a functional amygdaloid circuit of fear.
Neuroscience 102, 853–861.
Maren, S. (2005). Synaptic mechanisms of associative memory in the amygdala.
Neuron 47, 783–786.
Maren, S., Aharonov, G., Stote, D. L., and Fanselow, M. S. (1996).
N-methyl-
D-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats.
Behav. Neurosci. 110, 1365–1374.
Maren, S., and Fanselow, M. S. (1995). Synaptic plasticity in the basolateral amygdala induced by hippocampal formation stimulation in vivo.
J. Neurosci. 15, 7548–7564.
Marowsky, A., Fritschy, J. M., and Vogt, K. E. (2004). Functional mapping of GABA A receptor subtypes in the amygdala.
Eur. J. Neurosci. 20, 1281–1289.
McDonald, A. J., and Mascagni, F. (2004). Parvalbumin-containing interneurons in the basolateral amygdala express high levels of the alpha1 subunit of the GABAA receptor.
J. Comp. Neurol. 473, 137–146.
McDonald, A. J., Muller, J. F., and Mascagni, F. (2002). GABAergic innervation of alpha type II calcium/calmodulin-dependent protein kinase immunoreactive pyramidal neurons in the rat basolateral amygdala.
J. Comp. Neurol. 446, 199–218.
Meguro, R., Lu, J., Gavrilovici, C., and Poulter, M. O. (2004). Static, transient and permanent organization of GABA receptor expression in calbindin-positive interneurons in response to amygdala kindled seizures.
J. Neurochem. 91, 144–154.
Mohler, H. (2007). Molecular regulation of cognitive functions and developmental plasticity: impact of GABAA receptors.
J. Neurochem. 102, 1–12.
Nader, K., Majidishad, P., Amorapanth, P., and LeDoux, J. E. (2001). Damage to the lateral and central, but not other, amygdaloid nuclei prevents the acquisition of auditory fear conditioning.
Learn. Mem. 8, 156–163.
Ng, L., Bernard, A., Lau, C., Overly, C. C., Dong, H. W., Kuan, C., Pathak, S., Sunkin, S. M., Dang, C., Bohland, J. W., Bokil, H., Mitra, P. P., Puelles, L., Hohmann, J., Anderson, D. J., Lein, E. S., Jones, A. R., and Hawrylycz, M. (2009). An anatomic gene expression atlas of the adult mouse brain.
Nat. Neurosci. 12, 356–362.
Pare, D., Quirk, G. J., and Ledoux, J. E. (2004). New vistas on amygdala networks in conditioned fear.
J. Neurophysiol. 92, 1–9.
Peng, Z., Hauer, B., Mihalek, R. M., Homanics, G. E., Sieghart, W., Olsen, R. W., and Houser, C. R. (2002). GABA(A) receptor changes in delta subunit-deficient mice: altered expression of alpha4 and gamma2 subunits in the forebrain.
J. Comp. Neurol. 446, 179–197.
Peng, Z., Huang, C. S., Stell, B. M., Mody, I., and Houser, C. R. (2004). Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy.
J. Neurosci. 24, 8629–8639.
Pirker, S., Schwarzer, C., Wieselthaler, A., Sieghart, W., and Sperk, G. (2000). GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain.
Neuroscience 101, 815–850.
Rodrigues, S. M., Schafe, G. E., and LeDoux, J. E. (2001). Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning.
J. Neurosci. 21, 6889–6896.
Rodrigues, S. M., Schafe, G. E., and LeDoux, J. E. (2004). Molecular mechanisms underlying emotional learning and memory in the lateral amygdala.
Neuron 44, 75–91.
Rumpel, S., LeDoux, J., Zador, A., and Malinow, R. (2005). Postsynaptic receptor trafficking underlying a form of associative learning.
Science 308, 83–88.
Schafe, G. E., and LeDoux, J. E. (2000). Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala.
J. Neurosci. 20, RC96.
Schneider Gasser, E. M., Duveau, V., Prenosil, G. A., and Fritschy, J. M. (2007). Reorganization of GABAergic circuits maintains GABAA receptor-mediated transmission onto CA1 interneurons in alpha1-subunit-null mice.
Eur. J. Neurosci. 25, 3287–3304.
Sonner, J. M., Cascio, M., Xing, Y., Fanselow, M. S., Kralic, J. E., Morrow, A. L., Korpi, E. R., Hardy, S., Sloat, B., Eger, E. I., 2nd, and Homanics, G. E. (2005). Alpha 1 subunit-containing GABA type A receptors in forebrain contribute to the effect of inhaled anesthetics on conditioned fear.
Mol. Pharmacol. 68, 61–68.
Sperk, G., Schwarzer, C., Tsunashima, K., Fuchs, K., and Sieghart, W. (1997). GABA(A) receptor subunits in the rat hippocampus I: immunocytochemical distribution of 13 subunits.
Neuroscience 80, 987–1000.
Sur, C., Wafford, K. A., Reynolds, D. S., Hadingham, K. L., Bromidge, F., Macaulay, A., Collinson, N., O’Meara, G., Howell, O., Newman, R., Myers, J., Atack, J. R., Dawson, G. R., McKernan, R. M., Whiting, P. J., and Rosahl, T. W. (2001). Loss of the major GABA(A) receptor subtype in the brain is not lethal in mice.
J. Neurosci. 21, 3409–3418.
Thompson, C. L., Pathak, S. D., Jeromin, A., Ng, L. L., MacPherson, C. R., Mortrud, M. T., Cusick, A., Riley, Z. L., Sunkin, S. M., Bernard, A., Puchalski, R. B., Gage, F. H., Jones, A. R., Bajic, V. B., Hawrylycz, M. J., and Lein, E. S. (2008). Genomic anatomy of the hippocampus.
Neuron 60, 1010–1021.
Tsien, J. Z., Chen, D. F., Gerber, D., Tom, C., Mercer, E. H., Anderson, D. J., Mayford, M., Kandel, E. R., and Tonegawa, S. (1996). Subregion- and cell type-restricted gene knockout in mouse brain.
Cell 87, 1317–1326.
Vicini, S., Ferguson, C., Prybylowski, K., Kralic, J., Morrow, A. L., and Homanics, G. E. (2001). GABA(A) receptor alpha1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons.
J. Neurosci. 21, 3009–3016.
Wiltgen, B. J., Sanders, M. J., Ferguson, C., Homanics, G. E., and Fanselow, M. S. (2005). Trace fear conditioning is enhanced in mice lacking the delta subunit of the GABAA receptor.
Learn. Mem. 12, 327–333.
Wiltgen, B. J., Sanders, M. J., Anagnostaras, S. G., Sage, J. R., and Fanselow, M. S. (2006). Context fear learning in the absence of the hippocampus.
J. Neurosci. 26, 5484–5491.