Department of Biology, Institute for Systems Research, and Program in Neuroscience and Cognitive Science, University of Maryland, College Park, MD, USA
The developing cerebral cortex contains a distinct class of cells, subplate neurons, which form one of the first functional cortical circuits. Subplate neurons reside in the cortical white matter, receive thalamic inputs and project into the developing cortical plate, mostly to layer 4. Subplate neurons are present at key time points during development. Removal of subplate neurons profoundly affects cortical development. Subplate removal in visual cortex prevents the maturation of thalamocortical synapse, the maturation of inhibition in layer 4, the development of orientation selective responses in individual cortical neurons, and the formation of ocular dominance columns. In addition, monocular deprivation during development reveals that ocular dominance plasticity is paradoxical in the absence of subplate neurons. Because subplate neurons projecting to layer 4 are glutamatergic, these diverse deficits following subplate removal were hypothesized to be due to lack of feed-forward thalamic driven cortical excitation. A computational model of the developing thalamocortical pathway incorporating feed-forward excitatory subplate projections replicates both normal development and plasticity of ocular dominance as well as the effects of subplate removal. Therefore, we postulate that feed-forward excitatory projections from subplate neurons into the developing cortical plate enhance correlated activity between thalamus and layer 4 and, in concert with Hebbian learning rules in layer 4, allow maturational and plastic processes in layer 4 to commence. Thus subplate neurons are a crucial regulator of cortical development and plasticity, and damage to these neurons might play a role in the pathology of many neurodevelopmental disorders.
Subplate neurons are among the earliest generated neurons in the cerebral cortex of mammals and are located in the developing white matter of all cortical regions (Luskin and Shatz, 1985
; Valverde and Facal-Valverde, 1987
, 1988
; Mrzljak et al., 1988
; Kostovic and Rakic, 1990
; Allendoerfer and Shatz, 1994
; Reep, 2000
; Kostovic et al., 2002
; Kostovic and Judas, 2006
; Perkins et al., 2008
). In humans subplate neurons comprise up to 50% of the cortical neurons in the second trimester and are present in the first few years of life (depending on cortical area) (Luskin and Shatz, 1985
; Valverde and Facal-Valverde, 1987
, 1988
; Mrzljak et al., 1988
; Kostovic and Rakic, 1990
; Allendoerfer and Shatz, 1994
; Reep, 2000
; Kostovic et al., 2002
; Kostovic and Judas, 2006
; Perkins et al., 2008
). In rodents some subplate neurons can remain into adulthood forming layer 6b (Woo et al., 1991
; Wood et al., 1992
; Price et al., 1997
; Arias et al., 2002
; Torres-Reveron and Friedlander, 2007
). Subplate neurons thus comprise additional cortical circuits that are only present during cortical development, and these circuits appear to play a major role in development and early cortical function, but are only beginning to be characterized.
The cell bodies of subplate neurons are located in the cerebral white matter (Mrzljak et al., 1988
; Kostovic and Rakic, 1990
). A diagram of the early cortical circuitry that the subplate participates in has been established from physiological and anatomical studies (Figure 1
). Subplate neurons are a diverse neuropil encompassing glutamatergic and GABAergic neurons and receiving glutamatergic, GABAergic, cholinergic and glycinergic inputs (Wahle et al., 1987
, 1994
; Chun and Shatz, 1989a
,b
; Cobas et al., 1991
; Meinecke and Rakic, 1992
; Matute et al., 1993
; Zecevic and Milosevic, 1997
; Hanganu et al., 2002
; Hanganu and Luhmann, 2004
; Hirsch and Luhmann, 2008
; Kilb et al., 2008
).
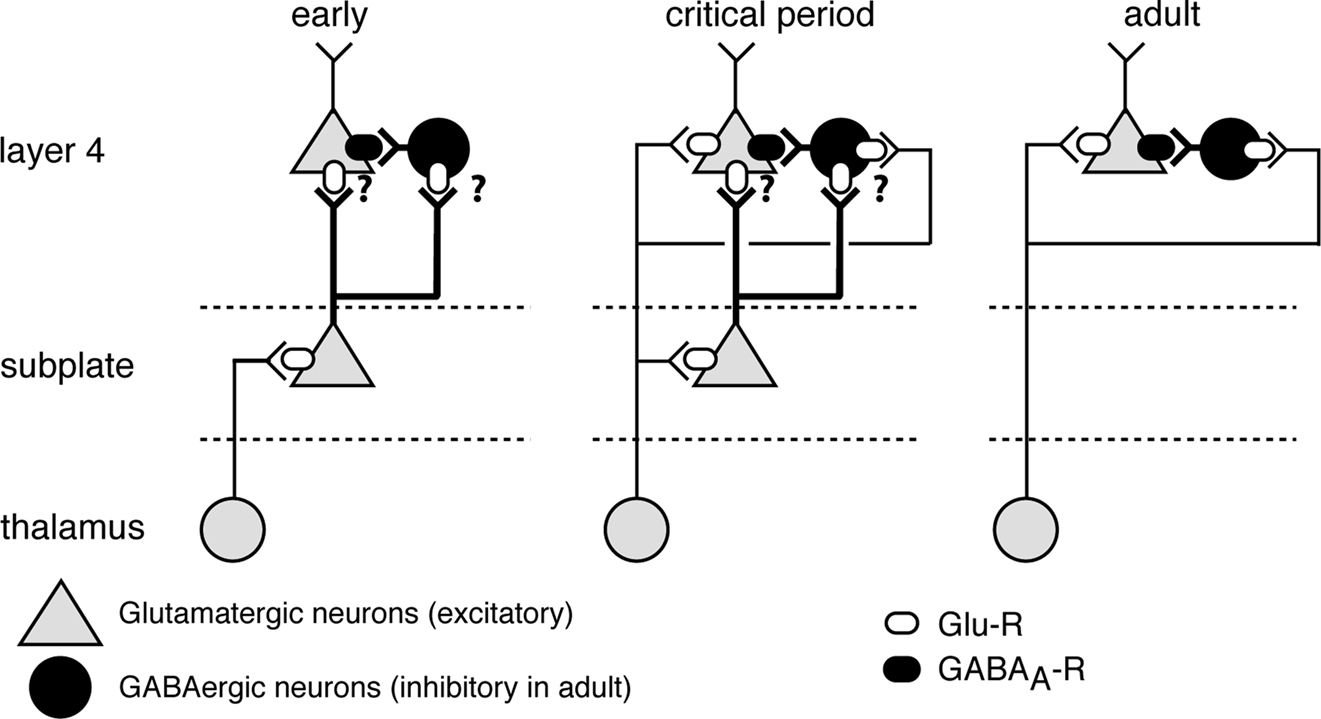
Figure 1. Subplate neurons and their connectivity during development. Early: subplate neurons (green) receive inputs from thalamus and subplate neuron axons project to layer 4, but precise targets are unknown (‘?’). Onset of critical period (coincides with time of subplate ablation in lesion studies): Both subplate neurons and thalamus project to layer 4. Adult: SPNs have been eliminated by programmed cell death and layer 4 neurons receive inputs from thalamus. Subplate neurons can depolarize layer 4 neurons via two pathways: directly via excitatory inputs and indirectly via exciting GABAergic neurons (red) and driving GABAergic depolarization.
Subplate neurons receive glutamatergic input from the thalamus before these thalamic axons grow to their targets in layer 4 (Friauf et al., 1990
; Allendoerfer and Shatz, 1994
; Hanganu et al., 2002
; Higashi et al., 2002
; Molnar et al., 2003
; Torres-Reveron and Friedlander, 2007
). Subplate axons mainly project to cortical layer 4 (Friauf and Shatz, 1991
; Allendoerfer and Shatz, 1994
; Pinon et al., 2009
), thus there is a time period when subplate neurons are in a key position to relay thalamic input to layer 4 (Figure 1
left) (Valverde and Facal-Valverde, 1987
, 1988
; Robinson and Dreher, 1990
; Catalano et al., 1991
; Friauf and Shatz, 1991
; Allendoerfer and Shatz, 1994
; Molnar and Blakemore, 1995
; Clancy et al., 2001
; Pinon et al., 2009
). After thalamic axons grow into layer 4, thalamocortical synapses and GABAergic circuits in layer 4 undergo refinement and maturation and over this time are particularly influenced by sensory experience (defining the “critical period”) (Friauf and Shatz, 1991
; Allendoerfer and Shatz, 1994
; Clancy et al., 2001
; Chen et al., 2001a
; Kanold et al., 2003
; Kanold and Shatz, 2006
). During this time subplate neurons are still present, receive direct thalamic input, and project to layer 4 (Figure 1
middle). The majority of subplate neurons are gradually eliminated postnatally by programmed cell death (Figure 1
right) and remaining neurons are retained as interstitial neurons (Allendoerfer and Shatz, 1994
; Arias et al., 2002
; Kanold et al., 2003
; Torres-Reveron and Friedlander, 2007
).
Functional evidence for these changing circuits was provided by current source density analysis in developing cat visual cortex (V1) (Friauf and Shatz, 1991
). White matter stimulations in V1 at early ages (P0) show short latency sinks in the subplate and long latency sinks in layer 4. The difference in latencies suggests that subplate neurons make excitatory connections to layer 4 neurons and drive their activity (Friauf and Shatz, 1991
). The presence of a disynaptic sink in layer 4 also implies that subplate to layer 4 connections are relatively strong. At later ages short latency sinks emerge in layer 4 after white matter stimulation. Thus now thalamic activity could directly activate layer 4 neurons indicating that thalamocortical circuits had matured. Similar results were obtained from recordings in rodent somatosensory system (Higashi et al., 2002
; Molnar et al., 2003
). These recordings show that thalamic stimulation activates subplate neurons by E18-19 while cortical plate activation is seen at E21. The difference in timing (prenatal in rodent vs. postnatal in cat) might reflect the early maturation of the somatosensory system relative to the visual systems or a difference between rodent and cat. Retrograde labeling studies show that most of the subplate neurons projecting to layer 4 are glutamatergic (Finney et al., 1998
). Thus, subplate neurons are thought to provide excitatory input to layer 4. Since subplate neurons receive GABAergic, cholinergic and glycinergic inputs (Wahle et al., 1987
, 1994
; Chun and Shatz, 1989a
,b
; Cobas et al., 1991
; Meinecke and Rakic, 1992
; Matute et al., 1993
; Zecevic and Milosevic, 1997
; Hanganu et al., 2002
; Hanganu and Luhmann, 2004
; Hirsch and Luhmann, 2008
; Kilb et al., 2008
) this feed-forward excitation to layer 4 can be modified by processing within the subplate.
Subplate neurons can be selectively ablated in early development by excitotoxic kainic acid injections (Ghosh et al., 1990
; Ghosh and Shatz, 1992
, 1993
, 1994
). Ablation of subplate neurons before thalamic axons invade layer 4 (Figure 1
left) causes these axons to bypass the ablated area and grow into layer 4 at areas that contain subplate neurons (Ghosh et al., 1990
). Thus subplate neurons seem to provide a guidance role in targeting thalamic axons to layer 4. Since subplate neurons project radially to layer 4 they might provide a scaffold that enables thalamic axons, which travel tangentially below their eventual target layer, to find their targets.
After thalamic axons grow into layer 4 they make synaptic connections with layer 4 neurons and build up these connections over time to adult strength. The strengthening of the thalamocortical synapses from an initially weak state occurs while there is already strong input from subplate neurons (Friauf and Shatz, 1991
) (Figure 1
) and possibly intracortical connections.
Recent experiments indicate that subplate neurons play a major role in the developmental strengthening of thalamocortical projections. Subplate ablation after thalamic afferents have grown into layer 4 but before these afferents have made a strong synapse with layer 4 neurons prevents the strengthening of thalamocortical connections (Kanold et al., 2003
). In addition, the frequency – but not amplitude – of spontaneous excitatory synaptic events in layer 4 cells is increased (Kanold et al., 2003
), which is consistent with a lack of functional refinement of cortical connections. Together these data indicated that without subplate neurons, there is a failure of appropriate synapses to strengthen and others to weaken, and the visual cortex becomes functionally decoupled from its thalamic inputs.
The maturation of intracortical inhibition is central to normal cortical function. In addition GABAergic activity is thought to be involved in the maturation of glutamatergic circuits (Ben-Ari, 2002
; Ben-Ari et al., 2004
). Despite this importance of inhibition, the cells and circuits that control inhibitory development are unknown.
Key processes of inhibitory maturation occur postsynaptically by changes in the subunit composition of the GABAA receptor (Figure 2
A) and the intracellular Cl−-concentration (which affects the ion flow through the GABAA receptor). The Cl−-reversal potential (ECl) controls if GABAA-ergic activity is depolarizing or hyperpolarizing. ECl is mediated by Cl− transporters such as KCC2 and NKCC1 that control Cl− levels in the cytosol (Shimizu-Okabe et al., 2002
, 2007
; Yamada et al., 2004
; Blaesse et al., 2009
). KCC2 levels are low (ECl high) in early development, thus GABA can be depolarizing (Rivera et al., 1999
; Ganguly et al., 2001
; Owens and Kriegstein, 2002
; Kanold and Shatz, 2006
; Blaesse et al., 2009
). Depending on the amount of depolarization, depolarizing GABA can be excitatory or have a shunting inhibitory influence (Blaesse et al., 2009
). Over development, KCC2 levels increase (decreasing ECl), rendering GABA inhibitory (Rivera et al., 1999
; Ganguly et al., 2001
; Owens and Kriegstein, 2002
; Shimizu-Okabe et al., 2002
; Yamada et al., 2004
; Kanold and Shatz, 2006
; Blaesse et al., 2009
). The strengthening of both excitatory and inhibitory circuits while maintaining “appropriate” activity levels might be achieved by wiring up GABAergic circuits first (Ben-Ari et al., 2004
) and then utilizing depolarizing GABA to aid in maturing glutamatergic connections (Ben-Ari, 2002
; Ben-Ari et al., 2004
).
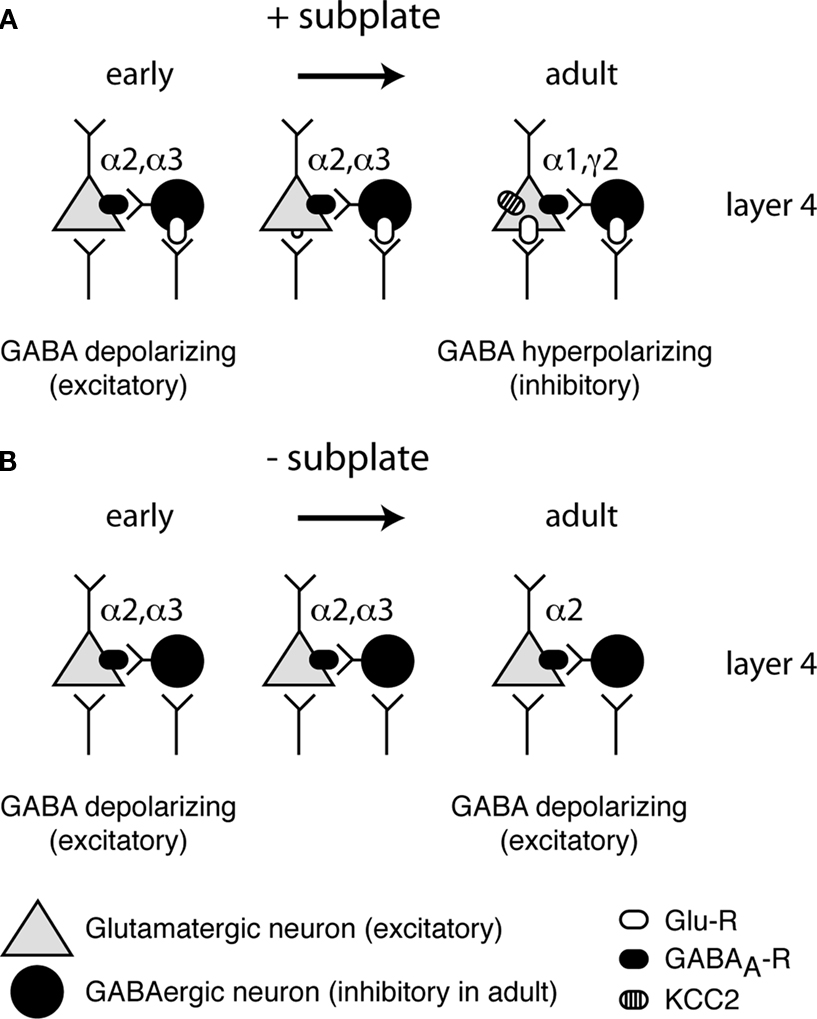
Figure 2. Maturation of GABA(A)ergic circuitry. (A) Normal development with subplate present. Early in development (left) GABAergic inputs to neurons are depolarizing due to the lack of KCC2. As development progresses (middle) glutamatergic synapses appear and further depolarize neurons. Glutamatergic synapses strengthen and at a certain point in development a critical threshold of depolarization is reached (right) and KCC2 levels are increased, rendering GABA hyperpolarizing (inhibitory). In addition, mature GABAergic receptor subunits are expressed. (B) Abnormal development with subplate absent. KCC2 levels remain low and immature GABA receptor subunits remain expressed, while levels of mature subunits remain low.
The maturation of inhibition depends on normal sensory experience. Sensory deprivations (i.e. dark rearing, deafness, whisker trimming) prevent inhibitory maturation and high expression levels of BNDF, which is involved in inhibitory maturation (Fuchs and Salazar, 1998
; Huang et al., 1999
; Lein and Shatz, 2000
; Chen et al., 2001b
; Morales et al., 2002
; Gianfranceschi et al., 2003
; Vale et al., 2003
; Jiang et al., 2005
; Kotak et al., 2005
; Jiao et al., 2006
; Huang, 2009
). Because subplate neurons form a crucial relay of sensory information, and because subplate neurons provide excitation to developing circuits, subplate neurons are in a key position to regulate the maturation of cortical GABAergic inhibition. In particular since subplate neurons are driven by thalamic afferents, strong synaptic inputs between subplate neurons and cortical neurons might amplify the action of sensory inputs.
Removal of subplate neurons at early ages, when inhibition is immature, prevents both the developmental increase in KCC2 expression and the expression of a mature complement of GABAA receptor subunits (Kanold and Shatz, 2006
) (Figure 2
B). Consistent with these molecular abnormalities, electrophysiological recordings showed that GABAergic circuits remain depolarizing (Kanold and Shatz, 2006
). How then is KCC2 regulated, and how are subplate neurons involved? Recent experiments have led to the hypothesis that KCC2 expression can be regulated by GABAergic depolarization (Ganguly et al., 2001
; Leitch et al., 2005
), while others report no influence of GABAergic signaling on KCC2 expression (Ludwig et al., 2003
; Titz et al., 2003
; Sipila et al., 2009
). However, there are other sources for depolarization. Blocking glutamatergic signaling during early ages in vivo is sufficient to prevent the developmental increase in KCC2 (Kanold and Shatz, 2006
). Thus early glutamatergic activity might be required for GABAergic maturation in layer 4 (Kanold and Shatz, 2006
). There are three sources of glutamatergic inputs to cortical layer 4: intracortical, thalamus and subplate neurons. Subplate removal by itself prevents inhibitory development despite the presence of intracortical and thalamic inputs (Kanold and Shatz, 2006
). Thus, together these data suggest that glutamatergic excitation from subplate neurons is needed for inhibitory maturation (Figure 1
). Such a role of subplate neurons in inhibitory maturation would require that subplate neurons depolarize layer 4 neurons, which can be achieved either by exciting GABAergic neurons and increasing early depolarizing GABAergic activity or by exciting the targets of GABAergic neurons directly. Thus by providing feed-forward excitation to the developing cortical circuits subplate neurons can regulate both the maturation of glutamatergic thalamocortical and GABAergic intracortical synapses.
By controlling cortical inhibition, subplate neurons might also play a role in regulating cortical activity levels after GABAergic circuits have matured. Since subplate ablation prevents inhibitory maturation, maybe by directly activating GABAergic circuits, subplate activation is likely able to dampen cortical activity levels. Thus even temporary depression of subplate activity could lead to cortical hyperexcitability, which might underlie pathophysiological conditions (see below).
Lesioning subplate at a time when thalamocortical axons are present in layer 4 (Figure 1
middle), but before these projections have refined into a mature pattern, revealed a role for subplate neurons in thalamocortical patterning. The organizational pattern observed in the visual cortex is that of the ocular dominance columns (ODCs). These columns are formed by the segregation of thalamic afferents innervated by either eye into alternating bands of left eye or right eye dominance. In cat ODCs form during the postnatal period from an initially non-segregated state (see Figure 3
A left). Analysis of the ODCs following ablation of subplate neurons in V1 with either kainic acid injections (Ghosh et al., 1990
; Ghosh and Shatz, 1992
, 1993
, 1994
) or immunoablation (Kanold et al., 2003
) shows that subplate ablation prevents the formation of ODCs (Ghosh and Shatz, 1992
, 1993
, 1994
; Kanold et al., 2003
). This deficit in thalamocortical patterning is present even though both thalamic axons and their target neurons are present in layer 4 (Ghosh and Shatz, 1992
, 1993
, 1994
; Kanold et al., 2003
). Thus subplate neurons are necessary for the patterned organization of the cerebral cortex.
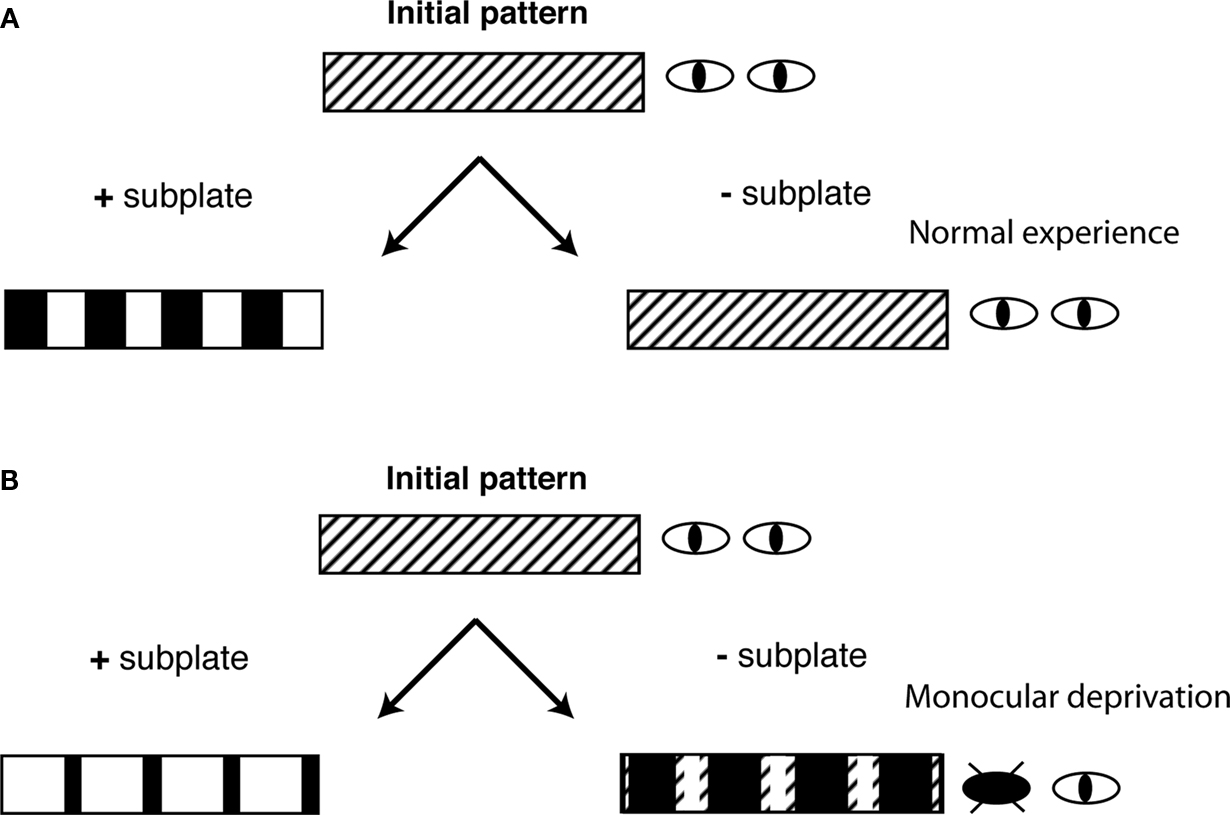
Figure 3. Subplate neurons control the sign of OD plasticity. Shown are schematic diagrams of the ocular dominance columns in cat visual cortex under 4 conditions. ODCs form from an initial non-segregated pattern at early stages of development. (A) With normal experience and with subplate present (+subplate), each eye is represented in equally large regions of visual cortex. If subplate is removed (−subplate), ODCs do not form and thalamic afferents representing both eyes are intermingled. (B) If one eye is closed (monocular deprivation) and if subplate is present then the open eye representation expands. If subplate is absent there is a paradoxical removal of open eye projections.
Activity dependent mechanisms have been known to be required for normal ODC formation (Shatz and Stryker, 1978
; Chapman et al., 1986
; Reiter et al., 1986
; Stryker and Harris, 1986
; Reiter and Stryker, 1988
; Cang et al., 2005
; Huberman et al., 2006
). Indeed failure of thalamocortical strengthening after subplate removal is paralleled by decreased visual responsiveness and the lack of functional refinement of visual responses (Kanold et al., 2003
). Thus, the fidelity of visual evoked responses in visual cortex is severely impaired and it is likely that these functional deficits in cortical processing subsequent to subplate ablation underlie the lack of ODCs consequent to subplate ablation (Ghosh and Shatz, 1992
, 1993
, 1994
; Kanold et al., 2003
).
In addition to thalamocortical and intracortical inhibitory circuits, long-range excitatory intracortical circuits also refine with sensory experience (Innocenti and Frost, 1980
; Frost et al., 1990
; Callaway and Katz, 1991
; Sur et al., 1999
). Thus, while the status of intracortical excitatory connections following subplate lesions has not been investigated, it is likely that subplate lesions, which abolish normal responses to patterned stimuli, will also affect the development of long-range intracortical circuits. Cognitive defects present after subplate damage, as discussed below, might be due from both deficits in thalamocortical and intracortical connections. A lack of refinement in intracortical excitatory circuits might functionally be evident as a larger number of smaller synaptic contacts which would be consistent with the observation that rates of spontaneous EPSC’s but not their amplitude were increased in ablated areas (Kanold et al., 2003
). However, these recordings were performed in layer 4, thus the source of the spontaneous EPSCs could also be from non-refined thalamocortical projections.
The function of inhibitory circuits is crucial for critical period plasticity in the visual cortex (Hensch, 2004
; Kanold and Shatz, 2006
). Sensory manipulations that alter inhibition also result in impaired synaptic plasticity mechanisms that underlie critical period plasticity. Thus, there is a co-regulation of inhibition and critical period plasticity (Kirkwood et al., 1995
, 1996
; Kotak et al., 2007
; Kanold et al., 2009
). Because subplate neurons play a crucial role in maturation of inhibition, it is likely that they also mediate cortical plasticity during the critical period.
In normal animals, if visual experience is altered, for example by monocular eye closure (monocular deprivation, MD) during the critical period then the pattern of ODCs is perturbed (Wiesel and Hubel, 1963
; Hubel and Wiesel, 1970
; Shatz and Stryker, 1978
). MD causes a rearrangement of thalamocortical projections such that projections representing the open eye occupy a larger territory in layer 4, while projections representing the deprived eye occupy a smaller area (Figure 3
B left). There is also a matching shift in physiological ocular dominance in cortical neurons towards the open eye.
As described above, under normal visual experience, removal of subplate neurons prevents the formation of ODCs from initially non-segregated inputs (Ghosh and Shatz, 1992
, 1993
, 1994
; Kanold et al., 2003
) (Figure 3
A right). These results could be interpreted as thalamocortical projections remaining in their early non-segregated projection pattern. However, after subplate ablation, following MD, there is a “paradoxical” form of OD plasticity (Kanold and Shatz, 2006
), meaning that the sign of the OD change is reversed. In these experiments subplate neurons were ablated before OD columns formed, meaning that projections from both eyes were present in all areas of the visual cortex. Eye closure is performed at the time of ablation, before the normal opening of the eyes. Transneuronal labeling at later ages shows that, in subplate ablated cortex, the deprived eye projections occupy a larger area than in cortical areas where the subplate was intact while projections representing the open eye occupy a smaller area in ablated compared to non-ablated areas (Kanold and Shatz, 2006
) (Figure 3
B right). Thus, paradoxically projections representing the more active eye have been removed while those representing the less active closed eye have been spared removal. A similar paradoxical removal of the more active inputs has been observed when cortical activity was pharmacologically silenced (Hata and Stryker, 1994
; Hata et al., 1999
).
Genetic manipulations that decrease inhibitory efficacy prevent OD plasticity entirely and it is thought that the maturation of inhibition is a key process enabling critical period plasticity (Hensch, 2004
; Kanold and Shatz, 2006
). Computational modeling studies (Kanold and Shatz, 2006
) suggest that the outcome of OD plasticity following MD might exist on a continuum between “normal” and “paradoxical” and that the levels of inhibition might control where on this continuum the circuit operates. By regulating both the maturation of excitation and the maturation of inhibition, subplate neurons seem to control processes in layer 4 that are required for normal plasticity.
The disappearance of subplate neurons might ensure that certain processes, such as critical period plasticity, occur only once. While critical period plasticity might be distinct from adult plasticity by its extent and transience, the underlying mechanisms might be similar to processes underlying adult learning via attention based mechanisms (Hensch, 2004
; Keuroghlian and Knudsen, 2007
). This attentional modulation of cortical circuits develops postnatally, thus subplate neurons could provide a circuit that enables large-scale cortical plasticity mechanisms before attention is functioning.
The three developmental deficits observed after subplate ablation might have a common explanation when considering that each of these processes can be driven by depolarization. Synaptic strengthening in the brain is governed by synaptic plasticity rules such as long-term potentiation (LTP) and long-term depression (LTD) (Bear and Malenka, 1994
; Malenka and Bear, 2004
). While LTP at a particular synapse can be evoked by the activity of that synapse (homosynaptic LTP), this strengthening requires that this synapse is already of sufficient strength to modulate activity levels in the postsynaptic neuron. An alternative way of inducing LTP is associative LTP. Associative LTP of a weak synapse can be induced by simultaneous activation of another synapse, which is sufficiently strong to induce activity increases in the postsynaptic neuron.
Subplate activity can influence layer 4 synapses and enable thalamocortical strengthening to occur. In particular, subplate input to layer 4 can strongly depolarize layer 4 cells. Since subplate neurons are driven by thalamic activity, this subplate mediated depolarization of layer 4 cells occurs at the same time as direct thalamocortical input to layer 4 and may lead to a strengthening of thalamocortical synapses by associative LTP. The strength of the subplate to layer 4 connection is evidenced by the evoked disynaptic sinks in layer 4 during white matter stimulation (Friauf and Shatz, 1991
). Therefore, subplate neurons can act somewhat like a “teacher” entraining layer 4 neurons to respond to appropriate thalamic inputs.
Formal implementation of this intuitive model using a computational simulation can replicate a large body of experimental data in normal development and after subplate removal (Kanold and Shatz, 2006
). The topology of the computational model is based on the circuits present in development (Figure 1
). A layer 4 neuron receives input from two LGN cells representing inputs from each eye. In addition the layer 4 neuron receives input from one subplate neuron. The subplate neuron receives input from the two LGN cells. All synapses on the layer 4 neuron can be modified according to spike-time dependent plasticity (STDP) rules (Abbott and Nelson, 2000
; Bi and Poo, 2001
) that are found at many synapses in the brain. In STDP synapses between neurons that are active within a certain time window prior to postsynaptic firing are strengthened while synapses that are active at other times are weakened.
At the start of the simulations LGN inputs to layer 4 are too weak to drive spiking in layer 4 and are not biased towards either eye. In contrast LGN inputs to subplate and subplate inputs to layer 4 are strong. The simulations show that subplate input to layer 4 induces correlations between thalamic firing and layer 4 activity. Since subplate neurons fire action potentials after thalamocortical synapses are active there is a positive time delay between the thalamocortical EPSCs and layer 4 firing. This time delay falls within the LTP window of the STDP rule and over time weak thalamocortical synapses are strengthened to adult strength (Kanold and Shatz, 2006
).
Simulations of the effects of subplate removal show that correlations between the thalamic and layer 4 activity are not present. Thus, thalamic input to layer 4 does not strengthen, and sensory driven activity is absent in layer 4 (Kanold and Shatz, 2006
), which is also observed in physiological experiments (Kanold et al., 2003
). This is because the STDP window for LTD is longer than that for LTP (Abbott and Nelson, 2000
; Feldman, 2000
; Bi and Poo, 2001
) and therefore if pre- and postsynaptic activity are uncorrelated, synaptic weakening occurs. Since in these simulations the activity levels in both eyes are equal, no refinement of ocular dominance is observed. In fact, these simulation results suggest that thalamic inputs will be weakened from their initial values over long periods of time.
Activity manipulations, such as MD reveal the explanatory power of this simple model. In MD the activity between the two eyes is unequal: the pathway driven by the open eye is more active while the pathway from the closed eye is less active. Simulations show that thalamic fibers driven by the open eye have a larger amount of uncorrelated activity with layer 4 neurons than fibers driven by the closed eye (Kanold and Shatz, 2006
). Thus, in models with an asymmetric STDP rule, the more active inputs to layer 4 are weakened at a much faster rate than the less active inputs. Thus over time the projections representing the open eye disappear while projections representing the closed eye will be retained (Kanold and Shatz, 2006
). This parallels experimental observations (Kanold and Shatz, 2006
). Additionally, homeostatic mechanisms known to be present in the cortex (Turrigiano, 1999
), can influence inputs from both eyes and thereby amplify this difference.
These simulation results support the view that subplate neurons can promote strengthening of thalamocortical connections by enabling correlations between thalamus and cortex that lead to synaptic strengthening. By this feed-forward mechanism, subplate neurons could also impart their stimulus selectivity to layer 4 neurons. Hence, cortical maps might be partially set up in the subplate and then transferred to and refined in layer 4 as has been proposed previously (Grossberg and Seitz, 2003
).
Given the central role of subplate neurons in the maturation of cortical circuits, damage to subplate neurons at any point during development could lead to neurological diseases. Subplate neurons are particularly prone to injury (especially hypoxic-ischemic injuries) during development and are especially vulnerable at time points when injuries are associated with many neurodevelopmental disorders (second trimester) (Volpe, 1996, 2000
; du Plessis and Volpe, 2002
; McQuillen et al., 2003
; McQuillen and Ferriero, 2005
). The enhanced vulnerability of subplate neurons may be due to their early maturation and therefore higher metabolic requirements. This vulnerability of subplate neurons might be more pronounced in infants born prematurely that are at a higher risk for neurodevelopmental disorders disorders as a large period of development occur ex utero. In animals, neonatal hypoxia damages subplate neurons and prevents normal critical period plasticity (McQuillen et al., 2003
; McQuillen and Ferriero, 2004
; Failor et al., 2006
), supporting the idea that such injuries damage circuits needed for the development of normal tuning and plasticity. In humans, such hypoxic-ischemic injuries, especially in the second trimester are associated with various neurodevelopmental disorders such as cerebral palsy and epilepsy (Volpe, 1996
, 2000
; Cioni et al., 1997
; Schatz et al., 1997
; Lanzi et al., 1998
; Krageloh-Mann et al., 1999
; Jacobson and Dutton, 2000
; Deukmedjian et al., 2004
; McQuillen and Ferriero, 2004
; Meberg and Broch, 2004
; Ozduman et al., 2004
; Robinson, 2005
; Robinson et al., 2006
).
Subplate ablation in animals is followed by a period of seizures (Lein et al., 1999
) indicating hyperactivity. These seizures develop a couple days after the time of ablation and thus are likely reflecting adjustments of the cortical network (Lein et al., 1999
). The origin of these seizures is unclear. Seizures could be generating by depolarizing GABAergic activity (Kanold and Shatz, 2006
) or alternatively be generated by glutamatergic activity that is not balanced appropriately by inhibitory GABAergic circuits. The different possible origins of seizures after subplate lesion are of clinical relevance. GABAA agonists can be used to treat seizures if they decrease firing probability. However, a GABAergic origin of seizures would also indicate that GABAA agonists would increase seizures instead of preventing them.
Many neurodevelopmental disorders are characterized by abnormal neuronal activity, hyperexcitability, and learning impairments due to impaired inhibition (Mathern et al., 2000
; Lewis and Levitt, 2002
; Cepeda et al., 2003
; Christ et al., 2003
; Lewis et al., 2004
; Robinson et al., 2006
), suggesting that altered inhibitory development underlies these disorders. Thus, a common outcome for early injuries and deprivations is that both alter inhibitory development, which in turn might alter critical period plasticity and normal development.
In addition to subplate lesions, the activity of subplate neurons can be altered by neuromodulators such as GABA, acetylcholine and glycine (see above). The subplate is also innervated by serotonergic fibers (Nakazawa et al., 1992
) and subplate neurons selectively express progestin receptor (Lopez and Wagner, 2009
). Thus, maternal or neonatal exposure to drugs (ranging from nicotine to sedatives and antidepressants) or hormones might alter subplate activity and thereby potentially disrupt cortical development. Therefore monitoring the status of subplate neurons in human infants is of high clinical relevance. Animal studies have shown that sensory activity can trigger cortical spindle bursts (Hanganu et al., 2006
). While, the involvement of subplate neurons in the generation of spindle bursts is unclear, subplate neurons are thought to play a role in driving oscillatory activity in cortex (Dupont et al., 2006
; Hanganu et al., 2009
). In vitro MRI has been used to identify the subplate in humans (Rados et al., 2006
), but functional MRI studies of subplate neurons have not been performed. However, such oscillatory activity can be detected using EEG, which has been used to monitor spontaneous and sensory evoked activity in human preterm and full-term infants (Vanhatalo et al., 2005
; Vanhatalo and Kaila, 2006
; Colonnese et al., 2008
). These EEG studies identify the emergence of spontaneous activity transients (SATs) when thalamocortical fibers start to innervate the cortical plate (∼30 GW) (Vanhatalo and Kaila, 2006
) and also show bipolar responses to early sensory stimuli (Colonnese et al., 2008
). Thus by quantifying the emergence and characteristics of EEG features and studying the alteration of such features in subplate lesioned cortex one might be able to clinically assess the function of the early thalamocortical network of which the subplate is a key component.
Subplate neurons are present in the cerebral cortex of all mammals (Luskin and Shatz, 1985
; Valverde and Facal-Valverde, 1987
, 1988
; Allendoerfer and Shatz, 1994
; Reep, 2000
). Subplate neurons are more prominent in species with increased radial and tangential cortical connectivity such as cat, monkey and human (Mrzljak et al., 1988
; Kostovic and Rakic, 1990
; Kostovic et al., 2002
; Kostovic and Judas, 2006
), suggesting that subplate neurons might be needed for the establishment of more complex processing capabilities. The disappearance of subplate neurons over development suggests that their role is purely developmental. As discussed above, subplate neurons enable the functional maturation of cortical circuits. However, other areas in the brain (such as subcortical areas) seem mature without neurons equivalent to subplate neurons. Thus one can speculate that the role of subplate neurons might have to do with unique properties of the cerebral cortex. One hallmark of the cerebral cortex is complex interconnectivity and its ability to adjust its connectivity during early development in response to altered patterns of spontaneous and sensory inputs. This capability for rewiring of the cerebral cortex is greatly diminished after the critical period. Thus removing these enabling (or “teacher”) circuits is one way to ensure that plasticity occurs only once and only during early development and might allow the development of higher cognitive processes at later stages of cortical processing at later ages.
Subplate neurons are an integral part of the developing cerebral cortex but their role in cortical development has been enigmatic. Recent progress has shown that they are required for the functional maturation of cortical circuits and for cortical plasticity. Because of their vulnerabilities, subplate neurons provide a key link between early brain injury and altered cortical function in the adult.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
POK is supported by NIDCD R01DC009607, NIDCD R21DC009454 and by the International Cerebral Palsy Research Association. The author thanks members of the Kanold lab and Dr. Dan Butts for helpful comments.
Cepeda, C., Hurst, R. S., Flores-Hernandez, J., Hernandez-Echeagaray, E., Klapstein, G. J., Boylan, M. K., Calvert, C. R., Jocoy, E. L., Nguyen, O. K., Andre, V. M., Vinters, H. V., Ariano, M. A., Levine, M. S., and Mathern, G. W. (2003). Morphological and electrophysiological characterization of abnormal cell types in pediatric cortical dysplasia. J. Neurosci. Res. 72, 472–486.