Department of Psychiatry and Behavioral Sciences, Stanford University School of Medicine, Palo Alto, CA, USA
How does the brain regulate the sleep–wake cycle? What are the temporal codes of sleep- and wake-promoting neural circuits? How do these circuits interact with each other across the light/ dark cycle? Over the past few decades, many studies from a variety of disciplines have made substantial progress in answering these fundamental questions. For example, neurobiologists and basal forebrain. Sleep-promoting circuits have been found in the preoptic area and hypothalamus. One of the greatest challenges in recent years has been to selectively record and manipulate these sleep–wake centers in vivo with high spatial and temporal resolution. Recent developments in microbial opsin-based neuromodulation tools, collectively referred to as “optogenetics,” have provided a novel method to demonstrate causal links between neural activity and specifi c behaviors. Here, we propose to use optogenetics as a fundamental tool to probe the necessity, suffi ciency, and connectivity of defi ned neural circuits in the regulation of sleep and wakefulness.
Sleep is commonly defined as “a rapidly reversible state of (behavioral) immobility and greatly reduced sensory responsiveness to environmental stimuli” (Siegel, 2008
). Although the parameters of the sleep–wake cycle vary within and between species, all animals show rest-activity patterns or sleep-like states. In addition to mammalian, reptilian, and avian species, recent studies report the existence of “sleep-like” states in lower organisms including the fish, fly, and worm (Shaw et al., 2000
; Yokogawa et al., 2007
; Allada and Siegel, 2008
; Raizen et al., 2008
; Zimmerman et al., 2008
).
In mammals, there are three states of vigilance: wakefulness, slow-wave sleep (also called non-rapid eye movement “NREM” sleep), and rapid eye movement (REM) sleep, defined by electroencephalographic (EEG) and electromyographic criteria. Wakefulness consists of fast EEG oscillations (5–12 Hz) of low amplitude with frequent and sustained motor activity. NREM sleep is characterized by slow oscillations (1–4 Hz) of high amplitude and can be divided into four stages in human (two in rats, one in mice), corresponding to increasing depth of sleep. REM sleep is characterized by relatively fast oscillations (6–12 Hz) of low amplitude and persistent muscle atonia. REM sleep has also been coined “paradoxical sleep” due to the simultaneous occurrence of cortical activity similar to wakefulness and a lack of a muscle tone (Jouvet, 1965
).
Sleep naturally follows a period of wakefulness. Prolonging the wake period increases the propensity to sleep, revealing a homeostatically regulated process (Borbely, 1982
, 2001
). In addition to homeostatic drive, sleep is regulated by circadian rhythms, which determine the timing of sleep, as well as other physiological variables including core body temperature and production of hormones (such as cortisol or melatonin). Thus, sleep homeostasis and circadian rhythms both influence the ultradian processes that governs the architecture of the sleep–wake cycle (Borbely, 2001
; Achermann and Borbely, 2003
; Saper et al., 2005
). These homeostatic and circadian factors govern a complex, yet partially defined, balance between sub-cortical excitatory and inhibitory neural populations in the brain (Pace-Schott and Hobson, 2002
; Fort et al., 2009
). Although these wake- and sleep-active neurons are distributed throughout the brain, most of the wake-promoting neurons are found in restricted brain areas including the brainstem, the hypothalamus and the basal forebrain. Interestingly, relatively few sleep-active nuclei have been discovered, mostly restricted to the lateral hypothalamus and preoptic area.
Brain arousal and sleep centers contain overlapping neuronal and non-neuronal cell types of different chemical composition and electrical properties (Gerashchenko and Shiromani, 2004
), that may influence the sleep–wake cycle and sleep-dependent processes (Halassa et al., 2009
). This heterogeneity has severely hampered our ability to record from and manipulate the neural dynamics of sleep and wake circuitry. For example, using a microelectrode to record or stimulate neural activity in brain regions known to promote sleep or wakefulness is incredibly difficult or even impossible given the 3D architecture of some neural populations spread out over millimeters, even in small rodents. Furthermore, the presence of multiple cell types that overlap within the same volume of tissue prevents an electrode from selectively stimulating specific cells of interest. New technology is clearly needed to establish causal, functional relationships between identified sleep- and wake-promoting neural circuits and how these circuits interact to govern the sleep/wake cycle.
We have recently used a new technology called optogenetics to selectively stimulate or inhibit specific neural circuits in the brain. Optogenetic tools combine the temporal precision of a microelectrode with the spatial precision of a genetically encodable probe, allowing the stimulation or inhibition of complete populations of specific neuronal subtypes. This technology has allowed us to gain insight into a population of wake-promoting neurons, hypocretin neurons, and has the potential to answer multiple questions about many other neural populations, as well as the sleep/wake cycle as a whole.
Here, we review the neural substrates of sleep and wakefulness in small and large mammals. These neural circuits have been identified and studied throughout the past few decades using a combination of histological, pharmacological, genetic, and in vitro and in vivo electrophysiology techniques. We focus on the use of these tools to study the hypocretin system, demonstrating the strengths and limitations of traditional electrophysiological and transgenic technologies. Then, we describe the use of optogenetic technology to probe the function of the hypocretin system in sleep regulation and how it has allowed us to make progress in our understanding of the role of these neurons in vivo. Finally, we propose to use optogenetics as a fundamental tool to investigate the necessity, sufficiency, and connectivity of defined neural circuits in the regulation of sleep and wakefulness.
The neural substrates of sleep-to-wake transitions are governed by distinct neural populations in the brain. Activity in these nuclei is correlated with wakefulness: not only does their activity increase when an animal is awake compared to asleep, but this activity also increases during states of enhanced arousal, such as moments of high alertness or stress. These arousal systems include:
• The noradrenergic locus coeruleus (LC), located in the pontine brainstem (Aston-Jones and Bloom, 1981
)
• The neuropeptide S cells, located in the pontine brainstem (Xu et al., 2004
)
• The serotinergic raphe neurons, located in the medial brainstem (McGinty and Harper, 1976
; Guzman-Marin et al., 2000
; Sakai and Crochet, 2000
)
• The dopaminergic neurons in the ventral mesencephalic midbrain (Dahan et al., 2007
) and the brainstem (Lu et al., 2006
)
• The histaminergic tuberomammillary nucleus, located in the posterior hypothalamus (Parmentier et al., 2002
; Takahashi et al., 2006
)
• The hypocretin neurons, located in the lateral hypothalamus (Peyron et al., 2000
; Lee et al., 2005
; Mileykovskiy et al., 2005
)
• The cholinergic neurons, located in the basal forebrain (Hassani et al., 2009a
) and pontine brainstem.
These arousal centers each send widespread ascending projections to the cerebral cortex, stimulating cortical desynchronization characterized by high frequency gamma and low frequency theta rhythmic activity (Steriade et al., 1993
).
In addition, these systems have descending projections that enhance or modulate muscle tone, sensory-motor responsiveness, and physiological activity. These multiple arousal systems show obvious redundancy, since triple lesions of the main arousal centers in the brain (basal forebrain, tuberomammillary nucleus, LC) have no major effect of the architecture of the sleep–wake cycle (Blanco-Centurion et al., 2007
), yet they can be differentiated based on their unique activity patterns throughout the sleep–wake cycle, as well as their contribution to other complex behaviors (such as food and drug seeking behaviors, attention, decision making, and the response to stress). During sleep, they are under an inhibitory tone from sleep-active neurons.
In contrast to the multiple, redundant arousal systems, there are relatively few identified sleep-promoting neural populations. These nuclei were identified on the basis of immunoreactivity to the immediate early gene c-Fos – a biomarker of neuronal activity –, single unit recordings, neurotoxic lesions, neurochemical, and thermal stimulation studies. Sleep-promoting nuclei include: the ventrolateral preoptic area (VLPO), located in the preoptic area and the median preoptic nucleus (MnPN), located in the preoptic area (Alam et al., 1995
; Gallopin et al., 2000
; Lu et al., 2002
; McGinty et al., 2004
).
Both the VLPO and MnPN neuronal populations express GABA, and send descending projections to arousal-promoting cell groups. Thus, they may inhibit noradrenergic, serotonergic, cholinergic, histaminergic and hypocretinergic neurons, as suggested by the “reciprocal inhibitory” model of the sleep–wake switch (Pace-Schott and Hobson, 2002
; McGinty and Szymusiak, 2003
) as recently suggested by Suntsova et al. (2007)
. The role of these neurons in promoting or maintaining sleep is not well understood and will require future investigation.
Brain structures responsible for REM sleep onset and maintenance were first identified in the brainstem in the pericoeruleus region in cats using a physical lesion approach (Jouvet and Michel, 1959
; Fort et al., 2009
) and many neurons in this brain area are selectively active during REM sleep (Sakai et al., 2001
). According to the “reciprocal-interaction model”, noradrenergic and serotonergic, histaminergic and hypocretin (Hcrt) neurons cease firing during REM sleep (Aston-Jones and Bloom, 1981
; Lee et al., 2005
; Mileykovskiy et al., 2005
; Takahashi et al., 2006
). Finally, pericoeruleus cholinergic and GABAergic neurons located in the dorsal paragigantocellular reticular nucleus (DPGi) and the ventrolateral periaqueductal gray have recently been identified as additional neuronal populations regulating the onset and maintenance of REM sleep events (Jones, 2004
); Pace-Schott, 2002 #17; Fort, 2009 #3122.
Recent studies identified additional sleep modulating neuronal populations in the brain (Verret et al., 2003
; Gerashchenko et al., 2008
; Hassani et al., 2009b
). One of those includes the hypothalamic neurons expressing the melanin-concentrating hormone (MCH) that are active mainly during REM sleep (Verret et al., 2003
; Hassani et al., 2009b
). However, contrasting results from experimental approaches using different techniques (immunodetection of c-Fos, pharmacology, knockout mouse models) are found in the literature (Verret et al., 2003
; Modirrousta et al., 2005
; Adamantidis et al., 2008
; Willie et al., 2008
).
In addition to classical neurotransmitters and neuropeptides described above, numerous endogenous factors modulate sleep, including prostaglandin D2 (Ueno et al., 1982
; Scammell et al., 1998
), adenosine (Virus et al., 1983
; Porkka-Heiskanen et al., 1997
), growth hormone-releasing hormone and interleukins (Krueger et al., 1999
; Kushikata et al., 1999
), all of which also have other physiological functions.
Now that we have examined the key components of the neural circuitry modulating sleep and wake states, we focus on the hypocretin system as an example of how sleep nuclei are studied and manipulated during experiments.
In 1998, two groups independently reported a novel pair of neuropeptides called the Hcrts (also known as “orexins”) exclusively expressed in a population of glutamatergic neurons in the lateral hypothalamus. These neuropeptides are cleaved from the same genetic precursor (preprohypocretin) (de Lecea et al., 1998
; Sakurai et al., 1998
). There are two hypocretin receptors (Hcrtrs) distributed throughout multiple brain areas that match the afferent fiber projections from Hcrt neurons. The projection pattern of afferent projections and receptor expression suggests that the Hcrt system is involved in multiple brain function (Sakurai, 2007
).
Hypocretin neurons are activated by neurotransmitters that promote arousal including glutamate (Li et al., 2002
; Yamanaka et al., 2003
), CRF (Winsky-Sommerer et al., 2004
), ATP (Wollmann et al., 2005
), noradrenaline and charbachol (acethycholine agonist) (Bayer et al., 2005
), A subpopulation of Hcrt neurons are activated by Ach (Sakurai et al., 2005
). Importantly, sleep-promoting neurotransmitters inhibit Hcrt neurons, including GABA (through GABAa,b; Li et al., 2002
; Yamanaka et al., 2003
; Xie et al., 2006
), and adenosine (A1) (Liu and Gao, 2007
).
In the 10 years since their discovery, much has been learned about the Hcrt system by correlating neural activity with behavioral output, as well as gain-of-function and loss-of-function studies. Experiments that manipulate Hcrt function typically involve pharmacology, in which an agonist or antagonist (or Hcrt peptides themselves) are injected into the ventricular system or discrete brain regions. Alternatively, many studies employ genetic manipulation of the Hcrt system using transgenic or knockout technologies in the mouse. We briefly summarize these studies below.
Recently, technically challenging in vivo single unit recordings of identified Hcrt neurons confirmed their high discharge activity during arousal, including behavior accompanied with a strong locomotor activity (Lee et al., 2005
; Mileykovskiy et al., 2005
; Takahashi et al., 2008
). In contrast to their oscillatory activity in brain slices (Eggermann et al., 2003
), Hcrt neurons are completely silent during quiet wakefulness, NREM and REM sleep and are reactivated during REM sleep-to-wake transitions (Lee et al., 2005
; Mileykovskiy et al., 2005
).
Many gain-of-function studies have been applied to the lateral hypothalamus as a whole. In addition to promoting food intake (Anand and Brobeck, 1951
; Bernardis and Bellinger, 1996
), electrical stimulation of lateral hypothalamic region decreased REM sleep duration in rats and cats (Suntsova et al., 2000
), possibly through a Hcrt-mediated inhibition of neurons in the oral nucleus of the pons (Dergacheva et al., 2005
; Nunez et al., 2006
), which is an important structure in the generation and maintenance of REM sleep. Disinhibition of LH cells by GABAA antagonists (bicuculline or gabazine ) injections into the LH area induced a continuous quiet waking state associated with a robust muscle tone in head-restrained rats (Goutagny et al., 2005
), partly via activation of Hcrt neurons (Lu et al., 2007
). However, none of these approaches have selectively targeted the Hcrt system.
Other studies used icv infusion of hcrt peptides or hcrt agonists (Akanmu and Honda, 2005
), as well as local injection of the peptide in the LC (Bourgin et al., 2000
), LH (Methippara et al., 2000
), laterodorsal tegmental nucleus (Xi et al., 2001
), basal forebrain structures (Espana et al., 2001
), in rodents and cats enhanced wakefulness and locomotor activity which was accompanied by a marked reduction in REM and non-REM sleep. Local Hcrt1 injections in cholinergic nuclei of the pons (nucleus pontis oralis) have promoted wakefulness, suppressed SWS and “defacilitated” REM sleep, whereas it directly inhibited REM sleep when injected in the ventral part of the NPO vs RPO (Xi et al., 2002
; Moreno-Balandran et al., 2008
; Watson et al., 2008
). Interestingly, Hcrt administration can reverse behavioral attacks in narcoleptic dogs (see below) (John et al., 2000
; Fujiki et al., 2003
). More recently, genetic disinhibition of Hcrt neurons using a selective GABAB receptor gene deletion only in Hcrt neurons in mice were found to induce severe fragmentation of sleep/wake states during both the light and dark periods, without showing an abnormality in total sleep time or signs of cataplexy (Matsuki et al., 2009
).
Although electrical or chemical anatomical lesions of the LH (the “lateral hypothalamic syndrome”) are not specific to the arousal-promoting Hcrt neurons, such lesions were reported to induced aphagia and adipsia for several days during which they showed a disorganized EEG characterized by rapid low voltage activity and high voltage low frequency waves (Danguir and Nicolaidis, 1980
). Sleep increased gradually and normal amounts of both NREM and REM sleep was observed during “stage 4” of recovery, when rats eat and drink as normal.
Other lesional studies found that extensive LH lesions caused either insomnia (Trojniar et al., 1990
; Jurkowlaniec et al., 1996
) or transient hypersomnia (Denoyer et al., 1991
) with disturbed hippocampal theta activity both during waking and paradoxical sleep (Jurkowlaniec et al., 1989
) in rodents and cats. In human, patients with cataplexy (Schwartz et al., 1984
), disrupted temporal patterning of the sleep–wake cycle (Cohen and Albers, 1991
) and increased total sleep time (Eisensehr et al., 2003
) have been reported secondary to a surgical lesion that involved the perichiasmal, the rostral hypothalamus, or bilateral posterior hypothalamus, respectively.
To selectively target Hcrt-responsive neurons, saporin-coupled Hcrt molecules were used to suppress Hcrt neurons in vivo. Saporin is a ribosome inactivating protein that kills target cells once internalized. Thus, the use of saporin toxin conjugated to the Hcrtr binding ligand, Hcrt2, lesions Hcrtr-expressing neurons. This has the effect of inducing sleep when injected into the LH (Gerashchenko et al., 2001
, 2003
) independently of adenosine levels in the basal forebrain (Murillo-Rodriguez et al., 2008
) and insomnia when injected in the VTA and substantia nigra (Gerashchenko et al., 2006
). Unfortunately, saporin alone has toxic properties on Hcrt neurons themselves, thus constituting weak control conditions.
Hypocretin receptor antagonists have been used to target the Hcrt system with receptor selectivity. Blockade of Hcrt-2R is sufficient to initiate and prolong sleep and co-administration of Hcrt-1R with Hcrt-2R antagonists greatly attenuated the sleep-promoting effects of the Hcrt2R antagonist (Dugovic et al., 2009
). Hcrt-R1 antagonist delays emergence from anesthesia, without changing anesthetic induction (Kelz et al., 2008
). Dual Hcrt receptor antagonists increased both non-REM and REM sleep in rats (Whitman et al., 2009
), somnolence and increased surrogate markers of REM sleep in dogs, and electrophysiological signs of sleep in human (Brisbare-Roch et al., 2007
). However, these antagonists differ in their affinities for HcrtR-1 and HcrtR-2. Additionally, they may, at very low binding concentrations, modulate other receptors when administered in vivo.
The most significant loss-of-function studies demonstrate that deficiency of the Hcrt system is linked to narcolepsy in humans (Peyron et al., 2000
), dogs (Lin et al., 1999
) and mice (Chemelli et al., 1999
; Blumberg et al., 2007
). Narcoleptic patients with cataplexy have non- or barely-detectable levels of Hcrt in the cerebro-spinal fluid, in addition to the absence of preproHcrt gene transcripts in the hypothalamus (Peyron et al., 2000
). Doberman narcoleptic dogs bear a mutation in Hcrt-R2, and all genetically engineered rodents with either a deletion of the Hcrt (Chemelli et al., 1999
; Hunsley et al., 2006
; Blumberg et al., 2007
), HcrtR-2 gene (Willie et al., 2003
) or Hcrt cells (Beuckmann et al., 2004
; Zhang et al., 2007b
; Fujiki et al., 2009
) present behavioral arrests that resemble cataplexy, the hallmark of narcolepsy. However, HcrtR-2 KO mice are less affected with cataplexy-like attacks of REM sleep compared to Hcrt KO mice that are more severely affected (Willie et al., 2003
), suggesting that the altered REM sleep control in narcolepsy-cataplexy syndrome emerges from loss of signaling through both HcrtR-2-dependent and Hcrt-R2-independent pathways (Willie et al., 2003
). Finally, alternative approaches to genetic technologies include the use of short interfering RNAs (siRNA) targeting prepro-orexin mRNA. Once injected into the rat LH, animals exhibited a transient increase in REM sleep (few days) compared to scrambled siRNA-treated animals (Chen et al., 2006
).
Collectively, these studies support a role for the Hcrt system in “lowering the arousal treshold” (Sutcliffe and de Lecea, 2002
), resulting in a facilitation of wakefulness when animal are asleep. Furthermore, the ability of Hcrts to modulate the reward system of the brain (Boutrel and de Lecea, 2008
) also suggest that it could promote hyperarousal (defined as a transient, hyper-alertness state triggered by salient stress or reward) when the animal is awake (DiLeone et al., 2003
; Winsky-Sommerer et al., 2004
; Boutrel et al., 2005
).
Selectively stimulating or inhibiting Hcrt function represents the next step in understanding Hcrt cell function. Although the studies mentioned above provide substantial evidence for the role of Hcrt neurons in promoting wakefulness and arousal, the 3D architecture of the Hcrt field, as well as the heterogeniety of cell types found in the lateral hypothalamus, have imposed limitations using traditional microstimulation or pharmacological techniques to perform loss-of-function or gain-of function studies. Indeed, in addition to non-neuronal cells, lateral hypothalamic cells include neurons expressing MCH, CRF, TRH, substance P or Neurotensin (Gerashchenko and Shiromani, 2004
). The most common way of inducing electrical activity in neurons for the past century has been injecting current through a microelectrode (Figure 1
A). Although temporally precise, a microelectrode cannot distinguish between cell types in the stimulated area. Furthermore, it may inadvertently stimulate other, unintended regions that are adjacent to the electrode. Pharmacological techniques lack both spatial and temporal precision, as psychoactive substances can spread throughout the brain and require minutes to hours to clear the system (Bittencourt and Sawchenko, 2000
) (Figure 1
A). Modern biochemical techniques, such as the delivery of photosensitive “caged glutamate” to neurons, improve upon the spatial and temporal properties of pharmacological methods. However, restriction of the caged glutamate to specific neurons of interest cannot be guaranteed, and the time-course of photostimulation is on the order of seconds to minutes. It is possible to deliver genetically encodable probes to neurons that can be activated to depolarize discrete subsets of neurons. For example, specific targeting of a ligand-gated ion channel that is not normally expressed in the brain may help to stimulate neural activity by just adding its selective ligand. Unfortunately, this strategy has a poor temporal resolution, as endogenous ligand must be added and then eventually cleared from the local area of interest. Finally, the crudest method of silencing Hcrt neural activity in the brain has been to ablate these neurons, either physically with a sharp instrument, pharmacologically with a toxin, or genetically with a genetically encodable toxin. Of course, these non-reversible lesions cause a total loss of neural function, with no chance for recovery.
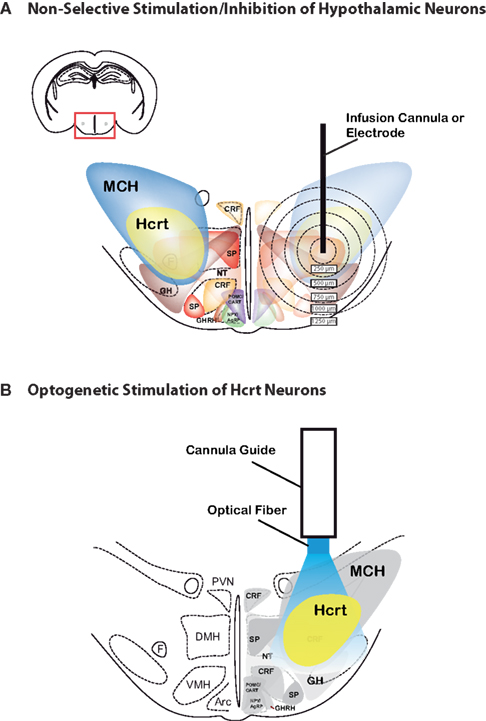
Figure 1. Comparison between electrical/pharmacological activation or inhibition and optogenetic activation of Hcrt neurons in the lateral hypothalamus. 2D architecture of the Hcrt field showing the heterogeniety of cell types found in the lateral hypothalamus, including MCH, Hcrt, corticotropin-releasing factor (CRF), NPY/AgRP, POMC/CART, growth hormone-releasing hormone (GHRH), growth hormone (GH), neurotensin (NT), and substance P. Note that, in addition to these defined cell types, the hypothalamus contain glutamatergic and GABAergic neurons. For clarity, Nesfatin-1, thyrotropin-releasing hormone (TRH), vasopressin and oxytocin are not represented. (A) Limitations of traditional microstimulation or pharmacological techniques to perform loss-of-function or gain-of function studies include confounding effect on neuronal and non-neuronal cells surrounding the tarted cells. For instance, infusion of Hcrt peptide or non-peptide agonists or antagonists can spread up to 1000 μm away from the infusion site, therefore activating or inhibiting several neuronal populations in addition to the Hcrt neurons. Alternatively, temporally precise microelectrode stimulation cannot distinguish between cell types in the stimulated area, and thus may inadvertently stimulate other, unintended regions that are adjacent to the electrode. Concentric circles represent distance (in μm) from possible injection, stimulation or lesion site. (B) Optogenetic technology allow selective stimulation (using ChR2) or inhbition (using NpHR) of genetically targeted Hcrt neurons, with no confounding modulation of surrounding cells that may regulate the same brain function.
Recently, our lab has employed optogenetic technology (Deisseroth et al., 2006
) to selectively manipulate the Hcrt system in vivo (Figure 1
B). Optogenetics can be thought of as a perfect combination of an electrode, which has high temporal precision, with a genetically encodable probe, which has high spatial resolution. Genetic delivery of a light-sensitive ion channel called channelrhodopsin-2 (ChR2) (Nagel et al., 2003
) into specific populations of neurons can be used to stimulate neural activity upon illumination in several animal species including rodents and non-human primates (Boyden et al., 2005
; Li et al., 2005
; Lima and Miesenbock, 2005
; Nagel et al., 2005
; Ishizuka et al., 2006
; Schroll et al., 2006
; Adamantidis et al., 2007
; Arenkiel et al., 2007
; Petreanu et al., 2007
; Wang et al., 2007
; Huber et al., 2008
; Gradinaru et al., 2009
; Han et al., 2009a
; Tsai et al., 2009
). This channel is naturally expressed in green algae and normally absent in animals. Because ChR2 is genetically encoded, specific promoter and enhancer elements can be used to express the channel in discrete populations of neurons in the brain. Then, ChR2-expressing neurons can be activated by delivering blue light using thin fiber-optic cables, implanted into the brain for long-term experiments over days or weeks (Adamantidis et al., 2007
; Aravanis et al., 2007
).
Alternatively, a separate light-sensitive protein, Halorhodopsin (NpHR), naturally expressed by archaebacteria and normally absent in animals, has been recently used to inhibit neural activity. NpHR is a chloride pump that hyperpolarizes neurons (and thus inhibits neural activity) upon stimulation with yellow light (Han and Boyden, 2007
; Zhang et al., 2007a
; Gradinaru et al., 2008
; Han et al., 2009b
). Importantly, ChR2 and NpHR both have a temporal resolution of milliseconds, allowing stimulation or silencing of single action potentials. Thus, optogenetic technology allows for temporally precise manipulation of specific populations of neurons in an awake, behaving animal.
In an attempt to better understand the role of Hcrt neurons on arousal, we used optogenetics to study the effect of Hcrt neuronal activation on SWS and REM sleep transitions to wakefulness. First, we genetically targeted the light-activated channel ChR2 to Hcrt neurons using a well-characterized Hcrt specific promoter in a lentiviral delivery system (Adamantidis et al., 2007
). Although the lentivirus infected multiple hypothalamic cells types, only the cells with the endogenous cellular machinery to express Hcrt were found to express ChR2. Therefore, deep brain optical stimulation activated only the Hcrt neurons and not the surrounding cells. Second, the fast ON–OFF kinetics (i.e., millisecond timescale) of ChR2 were found to induce single action potentials in brain slices in ChR2-expressing Hcrt neurons. Deep brain delivery of high frequency light pulse trains were found to activate Hcrt neurons (as measured by c-Fos immunohistochemistry) in vivo. Importantly, the millisecond timescale temporal resolution mimicked the physiological range of neuronal spiking rate, overcoming the limitations of previous techniques (e.g., uncontrolled persistence of Hcrt peptide in the brain after local infusion).
We found that direct, deep brain optical stimulation of hypocretin neurons in the hypothalamus increased the probability of transitions to wakefulness from either NREM or REM sleep (Adamantidis et al., 2007
). Interestingly, photostimulation using 5–30 Hz light pulse trains reduced latency to wakefulness, whereas 1 Hz trains did not. We also asked whether Hcrt-mediated sleep-to-wake transitions are affected by light/dark period and sleep pressure. We found that stimulation of Hcrt neurons increased the probability of an awakening event throughout the entire light/dark period but that this effect was diminished with sleep pressure induced by 2 or 4 h of sleep deprivation (Carter et al., 2009
). These results suggest that the Hcrt system promotes wakefulness throughout the light/dark period by activating multiple downstream targets, which themselves are inhibited with increased sleep pressure. Finally, stimulation of Hcrt neurons was still sufficient to increase the probability of an awakening event in histidine decarboxylase-deficient knockout animals, suggesting that histamine neurons of the TMN are not the main downstream target of Hcrt-mediated increase of arousal, as suggested by other studies (Carter et al., 2009
). These studies demonstrate that we now have the right tools to establish causal links between frequency-dependent activation of restricted neuronal population and specific behavioral state transitions.
A series of recent findings has begun to identify the important neuroanatomical substrates of sleep and wakefulness. However, important questions remain unanswered about how the brain integrates homeostatic sleep pressure and circadian rhythms at the neuronal circuit level. For example,
• Are known neural populations (e.g., catecholaminergic, noradrenergic neurons) sufficient or permissive to promote either sleep, wakefulness or state transitions?
• What are the kinetics of neurotransmission between sleep- and wake-promoting circuits in the brain in an unrestrained, behaving animal?
• What are the consequences of neurotransmitter vs neuropeptide release from synapses of sleep- or wake-promoting circuits on the sleep–wake cycle and associated brain functions (e.g., metabolism, cognition, brain plasticity)?
Addressing these questions or probing other neural circuits will require genetic targeting of optogenetic tools to specific neuronal populations. Current targeting technologies include the identification of minimal promoters compatible with viral-based approaches (Adamantidis et al., 2007
) and Cre-assisted transgenic tools (Atasoy et al., 2008
; Tsai et al., 2009
). Over the next several years, it will be important to use these different targeting strategies to apply optogenetic studies to neural populations implicated in sleep/wake circuitry. It will also be interesting to apply optogenetic manipulation to multiple sleep–wake populations to investigate circuit interactions and stimulate, for example, Hcrt neurons while simultaneously inhibiting other cell populations in order to determine their necessity in Hcrt-mediated sleep-to-wake transitions.
Current light delivery methods to activate optogenetic tools include optical fibers coupled to pulsed lasers or light emitted diodes. These methods of light delivery should be customized for the specific experiment (e.g. cortical or deep brainstimulation, unilateral or bilateral stimulation, etc.). It is currently difficult to target diffuse structures, such as the hippocampus or cerebral cortex. However, future tools may allow light delivery to large-scale neuronal structures.
Novel, synergistic combinations of recently-developed approaches (Sjulson and Miesenbock, 2008
), including in vivo optogenetics, genetic tagging of neural circuits (e.g., brainbow technology) (Livet et al., 2007
), optogenetic assisted neural circuit mapping (Petreanu et al., 2007
, 2009
; Atasoy et al., 2008
) and in vivo state-of-the-art imaging techniques now can address the questions raised above. This will expand the experimental possibilities available and allow in vivo deconstruction of previously inaccessible genetically defined sleep–wake neuronal circuits located in the hypothalamus, basal forebrain and brainstem with temporal and spatial resolutions relevant to physiological dynamics. These approaches will help to understand the function of sleep and identify new therapeutical targets to cure sleep disorders and sleep-associated neuropsychiatric disorders, including metabolic imbalance, mood-related pathology and cognitive impairment.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
A. Adamantidis is supported by fellowships from the Fonds National de la Recherche Scientifique (“Charge de Recherche”), NIH (K99) and NARSAD. M.E.C. is supported by fellowships from the National Science Foundation and National Institutes of Health (award number F31MH83439). L.d.L. is supported by grants from the National Institute on Drug Abuse, Defense Advanced Research Projects Agency, and National Alliance for Research on Schizophrenia and Depression.
Brisbare-Roch, C., Dingemanse, J., Koberstein, R., Hoever, P., Aissaoui, H., Flores, S., Mueller, C., Nayler, O., van Gerven, J., de Haas, S. L., Hess, P., Qiu, C., Buchmann, S., Scherz, M., Weller, T., Fischli, W., Clozel, M., and Jenck, F. (2007). Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat. Med. 13, 150–155.
Chemelli, R. M., Willie, J. T., Sinton, C. M., Elmquist, J. K., Scammell, T., Lee, C., Richardson, J. A., Williams, S. C., Xiong, Y., Kisanuki, Y., Fitch, T. E., Nakazato, M., Hammer, R. E., Saper, C. B., and Yanagisawa, M. (1999). Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98, 437–451.
de Lecea, L., Kilduff, T. S., Peyron, C., Gao, X., Foye, P. E., Danielson, P. E., Fukuhara, C., Battenberg, E. L., Gautvik, V. T., Bartlett, F. S., 2nd, Frankel, W. N., van den Pol, A. N., Bloom, F. E., Gautvik, K. M., and Sutcliffe, J. G. (1998). The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. U.S.A. 95, 322–327.
Dugovic, C., Shelton, J. E., Aluisio, L. E., Fraser, I. C., Jiang, X., Sutton, S. W., Bonaventure, P., Yun, S., Li, X., Lord, B., Dvorak, C. A., Carruthers, N. I., and Lovenberg, T. W. (2009). Blockade of orexin-1 receptors attenuates orexin-2 receptor antagonism-induced sleep promotion in the rat. J. Pharmacol. Exp. Ther. 330, 142–151.
Li, X., Gutierrez, D. V., Hanson, M. G., Han, J., Mark, M. D., Chiel, H., Hegemann, P., Landmesser, L. T., and Herlitze, S. (2005). Fast noninvasive activation and inhibition of neural and network activity by vertebrate rhodopsin and green algae channelrhodopsin. Proc. Natl. Acad. Sci. U.S.A. 102, 17816–17821.
Matsuki, T., Nomiyama, M., Takahira, H., Hirashima, N., Kunita, S., Takahashi, S., Yagami, K., Kilduff, T. S., Bettler, B., Yanagisawa, M., and Sakurai, T. (2009). Selective loss of GABA(B) receptors in orexin-producing neurons results in disrupted sleep/wakefulness architecture. Proc. Natl. Acad. Sci. U.S.A. 106, 4459–4464.
Nunez, A., Moreno-Balandran, M. E., Rodrigo-Angulo, M. L., Garzon, M., and De Andres, I. (2006). Relationship between the perifornical hypothalamic area and oral pontine reticular nucleus in the rat. Possible implication of the hypocretinergic projection in the control of rapid eye movement sleep. Eur. J. Neurosci. 24, 2834–2842.
Parmentier, R., Ohtsu, H., Djebbara-Hannas, Z., Valatx, J. L., Watanabe, T., and Lin, J. S. (2002). Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. J. Neurosci. 22, 7695–7711.
Peyron, C., Faraco, J., Rogers, W., Ripley, B., Overeem, S., Charnay, Y., Nevsimalova, S., Aldrich, M., Reynolds, D., Albin, R., Li, R., Hungs, M., Pedrazzoli, M., Padigaru, M., Kucherlapati, M., Fan, J., Maki, R., Lammers, G. J., Bouras, C., Kucherlapati, R., Nishino, S., and Mignot, E. (2000). A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med. 6, 991–997.
Sakurai, T., Amemiya, A., Ishii, M., Matsuzaki, I., Chemelli, R. M., Tanaka, H., Williams, S. C., Richardson, J. A., Kozlowski, G. P., Wilson, S., Arch, J. R., Buckingham, R. E., Haynes, A. C., Carr, S. A., Annan, R. S., McNulty, D. E., Liu, W. S., Terrett, J. A., Elshourbagy, N. A., Bergsma, D. J., and Yanagisawa, M. (1998). Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92, 573–585.
Wang, H., Peca, J., Matsuzaki, M., Matsuzaki, K., Noguchi, J., Qiu, L., Wang, D., Zhang, F., Boyden, E., Deisseroth, K., Kasai, H., Hall, W. C., Feng, G., and Augustine, G. J. (2007). High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 104, 8143–8148.
Whitman, D. B., Cox, C. D., Breslin, M. J., Brashear, K. M., Schreier, J. D., Bogusky, M. J., Bednar, R. A., Lemaire, W., Bruno, J. G., Hartman, G. D., Reiss, D. R., Harrell, C. M., Kraus, R. L., Li, Y., Garson, S. L., Doran, S. M., Prueksaritanont, T., Li, C., Winrow, C. J., Koblan, K. S., Renger, J. J., and Coleman, P. J. (2009). Discovery of a potent, CNS-penetrant orexin receptor antagonist based on an n,n-disubstituted-1,4-diazepane scaffold that promotes sleep in rats. ChemMedChem 4, 1069–1074.
Willie, J. T., Chemelli, R. M., Sinton, C. M., Tokita, S., Williams, S. C., Kisanuki, Y. Y., Marcus, J. N., Lee, C., Elmquist, J. K., Kohlmeier, K. A., Leonard, C. S., Richardson, J. A., Hammer, R. E., and Yanagisawa, M. (2003). Distinct narcolepsy syndromes in orexin receptor-2 and orexin null mice: molecular genetic dissection of non-REM and REM sleep regulatory processes. Neuron 38, 715–730.
Winsky-Sommerer, R., Yamanaka, A., Diano, S., Borok, E., Roberts, A. J., Sakurai, T., Kilduff, T. S., Horvath, T. L., and de Lecea, L. (2004). Interaction between the corticotropin-releasing factor system and hypocretins (orexins): a novel circuit mediating stress response. J. Neurosci. 24, 11439–11448.