
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol. , 24 January 2025
Sec. Antivirals and Vaccines
Volume 5 - 2025 | https://doi.org/10.3389/fviro.2025.1520109
This article is part of the Research Topic Development and Optimization of in vitro Antiviral Drug Screening Assays for Emerging and Re-Emerging Viral Pathogens View all articles
 Donya Ghorbani1,2†
Donya Ghorbani1,2† Masoumeh Beig1,3†
Masoumeh Beig1,3† Narjes Noori Goodarzi4
Narjes Noori Goodarzi4 Mohammad Sholeh1,3
Mohammad Sholeh1,3 Behzad Shahbazi5,6Yaser Moghaddam1
Behzad Shahbazi5,6Yaser Moghaddam1 Farzad Badmasti1*
Farzad Badmasti1*Background: Multidrug-resistant Burkholderia cenocepacia and Burkholderia multivorans have emerged as significant pathogens, particularly in patients with cystic fibrosis (CF) and chronic granulomatous disease (CGD).
Objective: Given the absence of approved vaccines, this study aimed to identify potential vaccine candidates against these pathogens.
Methods: The complete genomes of B. cenocepacia and B. multivorans were retrieved from the GenBank. Surface-exposed proteins that were antigenic, non-allergenic, and non-homologous to human proteins were selected for further analysis. The conserved domains of the selected proteins were analyzed, and their presence was examined across 68 genomes. Subsequently, linear and conformational B-cell epitopes and human MHC II binding sites were identified. Highly conserved and immunogenic B-cell epitopes from outer membrane proteins (OMPs) were incorporated into a multi-epitope vaccine (MEV). Molecular docking analysis was performed to assess the interaction of the selected proteins. Finally, molecular dynamics (MD) simulations were conducted using GROMACS 2019 to evaluate the feasibility and dynamics of the interactions between the chimeric MEV and Toll-like receptor complexes, TLR2 and TLR4.
Results: Of 16,723 proteins identified in B. multivorans and B. cenocepacia strains, nine proteins (six OMPs and three extracellular) were selected as ideal candidates based on established criteria. These proteins had a molecular weight of 110 kDa and were present in ≥ 75% of the dataset of B. multivorans and B. cenocepacia genomes. In addition, molecular docking and MD indicated stable and feasible interactions between MEV and TLRs. The MEV-TLR4 system demonstrates the greatest stability and tightly bound interaction, with minimal fluctuations and high structural integrity. In contrast, the MEV-only system exhibits significant flexibility and dynamic behavior as a free ligand, while the MEV-TLR2 system balances stability and flexibility, showing a dynamic but stable interaction.
Conclusion: Nine potential immunogenic proteins were identified as viable targets for vaccine development. An optimized MEV was explicitly designed for B. multivorans and B. cenocepacia. The novel MEV platform exhibited high binding affinity to immune receptors and favorable molecular docking characteristics. Although these findings are encouraging, additional in vitro and in vivo testing is necessary to validate the vaccine’s effects.
Chronic respiratory infections in cystic fibrosis (CF) patients lead to a gradual decline in lung function, mainly due to bacterial infections and CF’s unique pathophysiology (1, 2). Among these pathogens, Burkholderia cepacia complex (Bcc) is a significant contributor, particularly Burkholderia cenocepacia and Burkholderia multivorans (3). Bcc consists of at least 24 related species that cause severe respiratory infections in patients with CF, impairing lung function (3). B. cenocepacia and B. multivorans are responsible for 85-97% of Bcc infections in patients with CF, contributing to declining lung function and increased hospitalization rates (4, 5). Their expansion and high antibiotic resistance make treatment challenging. Antibiotic resistance limits treatment options, leading to poor outcomes, such as increased morbidity and mortality (2). Traditional therapies often fail, highlighting the need for alternative approaches, such as vaccination and novel interventions. Vaccination is a promising preventive strategy that can reduce antibiotic reliance and resistance development (5, 6).
Several previous studies have identified potential vaccine candidates that can elicit robust immune responses against Bcc. These efforts have explored multiple vaccine strategies, including live-attenuated, subunit, and extracellular proteins.
A notable approach is the development of live-attenuated vaccines. For instance, a mutant strain of B. cenocepacia lacking tonB demonstrated promising results in murine models, leading to a significant survival rate following acute infection (7). This suggests that live-attenuated vaccines could be a viable strategy for inducing protective immunity.
Another research focus has been on subunit vaccines that target specific proteins, such as outer membrane proteins (OMPs) and polysaccharides (8). Identifying surface-exposed membrane proteins using genomic and bioinformatics approaches is essential. For example, in experimental models, OMPs paired with mucosal adjuvants elicit balanced Th1/Th2 immune responses, thereby protecting against B. multivorans and B. cenocepacia (9). Specific proteins, such as the OmpA-like protein BCAL2958, have demonstrated strong reactivity with sera from patients with CF, leading to IgG production and enhanced neutrophil activation (10). Additionally, proteins like OmpW and BCAL2645 are involved in immune responses, adhesion, and host cell invasion and have also shown potential as vaccine targets (9).
Research has further highlighted the potential of extracellular proteins as vaccine candidates, with several of these proteins demonstrating immunogenicity in the sera of patients with CF (10). Mutant strains deficient in key virulence genes, such as peptidoglycan-associated lipoprotein, have exhibited decreased virulence and reduced immune responses, suggesting their potential as therapeutic targets (7). Moreover, proteins from the trimeric autotransporter adhesin family, including BCAM2418 and BCAM0224, are crucial for adhesion, biofilm formation, and immune evasion, making them attractive targets for vaccine development (9).
Building on these findings, the present study applied a reverse vaccinology approach to identify novel B. cenocepacia and B. multivorans vaccine candidates. For the first time, we aim to discover and evaluate potential antigens, including OMPs, that could be developed into a multi-epitope vaccine (MEV). By integrating comprehensive genome analysis with diverse immunoinformatics tools, this research will significantly advance vaccine development for these specific pathogens, with broader implications for combating other Gram-negative bacteria.
In total, 132 complete genome sequences of Bcc strains were initially retrieved from the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/). These genome sequences were converted into proteomes using CLC Genomics Workbench Software (Qiagen, Hilden, Germany) (11). CLC Genomics Workbench is a user-friendly software suite that provides various bioinformatics tools, including sequence alignment, variant analysis, and functional annotation, making it suitable for proteome analysis. The resulting proteomes were then subjected to core/pan-genome analysis using the BPGA (Bacterial Pan Genome Analysis) software (12). BPGA is designed to analyze genome sequences and classify genes into core and accessory genomes, helping identify conserved and variable genes across bacterial strains. Following this evaluation, B. multivorans and B. cenocepacia were selected as reference strains owing to their clinical relevance. These strains are particularly notable for their high prevalence in CF infections, increased virulence, multi-drug resistance, biofilm-forming abilities, and prioritization for vaccine development (13). A comprehensive list of the strains and their corresponding accession numbers is provided in the Supplementary Table S1.
The proteins were submitted to the PSORTb v3.0.2 database (https://www.psort.org/psortb/) and CELLO (http://cello.life.nctu.edu.tw/) to predict subcellular localization, with particular attention given to identifying extracellular, secreted, and surface-exposed proteins (14, 15). PSORTb is a web-based tool that uses sequence-based information to predict the subcellular localization of bacterial proteins, while CELLO provides an accurate prediction of protein localization based on a multi-class classifier system. To validate these predictions, TMHMM Server v.2.0 (https://services.healthtech.dtuthe.dk/service.php?TMHMM-2.0) was used to confirm the surface-exposed regions (16). The TMHMM is a tool for predicting transmembrane helices, enabling the identification of surface-exposed regions of proteins.
To identify potential immunogenic proteins, antigenicity was assessed using the VaxiJen v2.0 tool (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) with a threshold score of ≥ 0.5 (17). VaxiJen uses an alignment-free method based on amino acid composition to predict antigenicity, which is essential for identifying proteins that may trigger immune responses. Additionally, allergenicity was predicted for vaccine safety using AlgPred v2.0 (https://www.ddg-harmfac.net/AllerTOP/) with a cutoff value of ≥ 0.3 (17). The AlgPred uses a prediction model based on physicochemical properties to evaluate the likelihood of an allergenic protein.
The selected proteins were screened for sequence similarity to the human proteome (Homo sapiens, Taxid: 9606) using the PSI-BLAST tool in the BLASTp database (https://blast.ncbi.nlm.nih.gov/Blast.cgi?SIDE=protein) (18). PSI-BLAST is an advanced version of the BLAST algorithm that identifies sequence homology by iteratively searching protein databases. This ensures that proteins similar to human proteins are excluded to avoid potential cross-reactivity. Any proteins identical to the host proteome were excluded from further analysis to prevent potential cross-reactivity or adverse effects.
The VICMpred tool (https://webs.iiitd.edu.in/raghava/vicmpred/submission.html) was used to classify the function of the selected proteins (19). The VICMpred provides functional annotations based on protein sequences and known biological functions. Subsequently, these proteins’ amino acid composition and molecular weight were determined using the Expasy ProtParam server (https://web.expasy.org/protparam/) (20). The ProtParam is a tool for calculating various physical-chemical properties of proteins, such as molecular weight and amino acid composition.
Protein domains were analyzed using the Conserved Domain Database (CDD) (https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) and the EggNOG (http://eggnog5.embl.de/#/app/home). CDD, integrated into NCBI’s Entrez search system, provides comprehensive annotations of protein sequences, emphasizing conserved domain regions by identifying evolutionary conserved functional motifs and structural features in protein fields (21, 22). The EggNOG categorizes proteins into orthologous groups and performs functional predictions based on evolutionary relationships, leveraging its extensive database of orthologous groups to assign functional annotations and infer domain structures.
The occurrence of each identified protein was evaluated across 68 B. multivorans and B. cenocepacia strains. This evaluation involved analyzing the presence of protein sequences across genome data to determine their conservation levels. Proteins in ≥ 75% of the strains were categorized as potential vaccine candidates, as high conservation across strains indicates their potential as broad-spectrum vaccine targets (23).
BepiPred v2.0 (https://services.healthtech.dtu.dk/service.php?BepiPred-2.0) was used to identify linear B-cell epitopes with a threshold of ≥ 0.6 (24). The BepiPred employs a machine learning algorithm trained on known epitopes to predict linear B-cell epitopes based on amino acid sequences, highlighting regions likely to elicit immune responses. The B-cell epitope ratio was calculated for each protein by dividing the total number of amino acids in all epitopes by the total number of amino acids in the protein. Proteins with ratios above average were selected.
To evaluate the vaccine candidate’s immune recognition and global applicability, we performed population coverage and MHC binding site analyses using TepiTool (http://tools.iedb.org/tepitool/help/) from IEDB. Population coverage analysis utilized a predefined set of representative alleles from MHC class I supertypes to assess the vaccine’s applicability across diverse ethnic and geographical populations. We focused on the top 5% of peptides ranked by binding affinity for MHC-II binding site prediction, identifying epitopes with a high likelihood of immune recognition. The ratio of MHC-II binding sites was calculated by dividing the predicted binding sites by the total amino acids in each protein. These analyses provide critical insights into the vaccine’s population coverage and ability to elicit immune responses, ensuring the robustness and relevance of the candidate for broad application.
The selected proteins were evaluated using a quartile scoring approach, considering antigenicity, linear B-cell epitope abundance, and MHC-II binding site ratios. The final score was the sum of individual metric scores.
The predicted immunogenic protein tertiary (3D) structure was modeled using the Robetta tool (https://robetta.bakerlab.org/) (25). The Robetta is an automated tool that predicts protein structures using comparative modeling and de novo modeling methods, depending on the availability of homologous templates. The ProSA web server (https://prosa.services.came.sbg.ac.at/prosa.php) was used to assess the quality of the 3D model (26). ProSA evaluates the structural quality of protein models by comparing them to a statistical potential derived from experimentally solved protein structures, identifying regions with potential errors. The ElliPro tool (http://tools.iedb.org/ellipro/) was used to detect conformational B-cell epitopes with a threshold of ≥0.8 (27). The ElliPro identifies discontinuous epitopes based on protein shape and surface protrusion, providing an epitope score reflecting antigenicity’s likelihood. The predicted conformational B-cell epitopes were visualized in various colors on the surface of each protein using the Jmol software (28). This open-source molecular visualization tool enables interactive 3D representation of protein structures.
Linear B-cell epitopes were predicted on the extracellular loops of selected OMPs using BepiPred with a threshold of ≥ 0.6. The BepiPred identifies linear B-cell epitopes based on a combination of propensity scale methods and machine learning techniques, which analyze amino acid sequences to predict regions likely to interact with antibodies. Additionally, the conservation of these epitopes was evaluated using the ConSurf web tool (https://consurf.tau.ac.il/consurf_index.php) (29). The ConSurf assesses the evolutionary conservation of amino acid residues in proteins by comparing sequences across homologous proteins, helping to identify conserved and functionally important regions.
MEVs were generated using proteins isolated from B. multivorans and B. cenocepacia. The 3D structure was modeled using the Robetta web tool, which predicts protein tertiary structures based on comparative modeling or ab initio methods when no homologous structure is available. This tool generates structural models by threading the sequence onto known templates and refining regions without template matches. The 3D structure was verified using the ProSA web server (https://prosa.services.came.sbg.ac.at/prosa.php). This tool evaluates the quality of a protein structure by analyzing its energy distribution and detecting any potential errors within the model. Further assessment was conducted using the Ramachandran plot (https://zlab.umassmed.edu/bu/rama/), which visualizes dihedral angles of amino acid residues in the protein to confirm the stereochemical quality and identify favored, allowed, and disallowed regions of the structure.
The safety of the MEV was assessed using the ToxinPred web server (http://crdd.osdd.net/raghava/toxinpred/) (30), which predicts peptide toxicity based on amino acid composition and motifs. The ToxinPred employs a machine learning-based approach that analyzes peptide sequences to identify toxic regions by evaluating physicochemical properties and motifs associated with toxicity. The MEV construct was confirmed to be non-toxic, with all identified epitopes scoring below the toxicity threshold, ensuring the vaccine’s suitability for further development.
Three multi-epitope-based vaccines were generated using the peptides of nine selected proteins obtained from B. multivorans and B. cenocepacia: porin (WP_176035635.1, WP_105763988.1, AYZ01212.1, ABK11981.1, and CDN62452.1), TonB-dependent receptor (WP_181146976.1), coagulation factor 5/8 type domain protein (ABK11820.1), glutamate synthase [NADPH] large chain (ARF89570.1), and alkaline phosphatase family protein (WP_006487965.1).
Molecular dockings and the binding affinities of MEV to human TLR2 (PDB: 2Z7X) and TLR4 (PDB: 3FXI) were assessed with the pyDockWEB (https://life.bsc.es/pid/pydockweb/default/index) (31). The PyDockWEB is a web-based molecular docking tool that utilizes a rigid-body docking approach combined with energy-scoring functions, including electrostatics, desolvation, and van der Waals interactions, to predict the most favorable protein-protein binding conformations.
In addition, the C-ImmSim (https://kraken.iac.rm.cnr.it/C-IMMSIM/index.php) was used to predict the simulation of the immunoreactivity of MEV (32). Finally, The docked complexes were validated using the PDBsum server (https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/) (33), which provides a detailed graphical representation of the interactions, including hydrogen bonds, hydrophobic contacts, and residue-level interface analysis.
MD simulations were performed to assess the feasibility of interaction between the chimeric MEV and TLR2 and TLR4 complexes using GROMACS 2019 software (34). (Groningen Machine for Chemical Simulations) is a widely used MD simulation software that calculates the movements of atoms and molecules over time by solving Newton’s equations of motion. It allows for simulating biomolecular interactions under physiological conditions, making it an essential tool for studying protein-ligand and protein-protein interactions.
The simulations employed the Optimized Potential for Liquid Simulations force field to evaluate the stability and conformational dynamics of MEV both in its unbound form and in complex with its receptors. Each complex was placed in a 10 Å solvent box filled with simple point-charge water molecules, and system neutrality was achieved by adding appropriate amounts of Na+ and Cl- ions. Energy minimization was followed by two-phase equilibration: the systems were equilibrated for 100 ps under a constant number of particles, volume, and temperature and a continuous number of particles, pressure, and temperature. The Parrinello–Rahman barostat was used to maintain a stable temperature of 300 K and pressure of 1.0 bar. Long-range electrostatic interactions were handled by the particle mesh Ewald method with a 10 Å cutoff and grid spacing of 0.16 nm. In contrast, van der Waals interactions were calculated using a 1 nm cutoff. The Linear Constraint Solver (LINCS) algorithm was applied to constrain covalent bond lengths.
MD simulations are instrumental in elucidating the stability and dynamics of vaccine complexes. This study investigates the stability of these complexes over a 100-ns simulation period, focusing on key metrics, including root mean square fluctuation (RMSF), root mean square deviation (RMSD), radius of gyration (Rg), and the number of hydrogen bonds within each protein-protein complex, to assess the stability of interactions within the vaccine complex (35).
Core-pan genome analysis was performed on B. multivorans and B. cenocepacia strains to investigate the genetic basis of potential vaccine candidates. The study revealed a pan-proteome comprising 16,723 proteins, of which 1,058 proteins were conserved across all strains, forming the core proteome (Figure 1A). KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway mapping categorized these proteins into six functional groups, with the majority associated with essential metabolic processes, such as carbohydrate and amino acid metabolism. Proteins involved in environmental adaptation and genetic information processing also formed a significant fraction, reflecting the bacteria’s capability to survive in diverse niches and its adaptability within-host environments (Figure 1B). These results underscore the core proteome’s potential as a reservoir of conserved targets for vaccine development, given its critical role in bacterial physiology and survival.
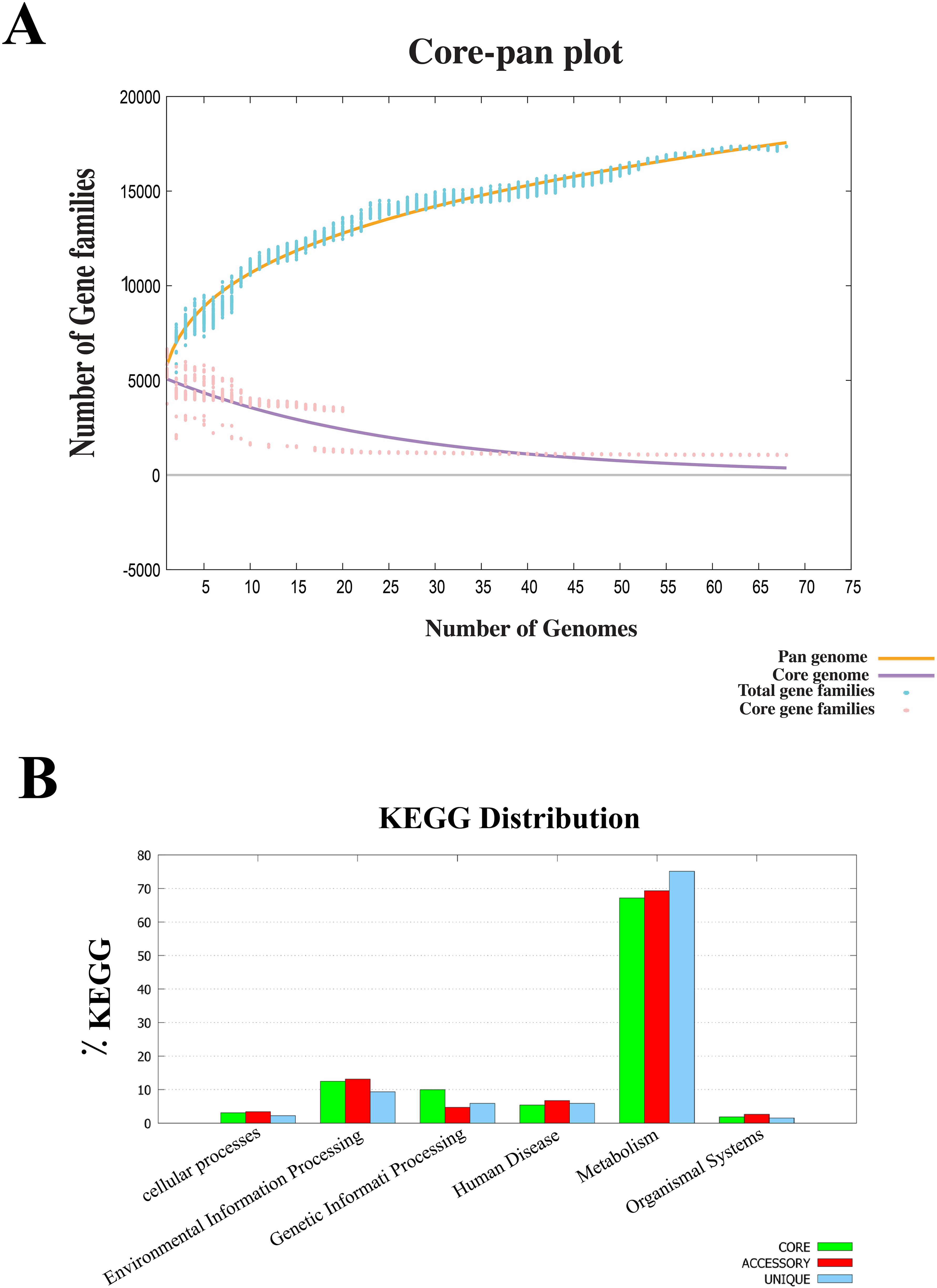
Figure 1. Analysis of core proteomes using the BPGA database. (A) Core/pan-genome analysis of 68 B. multivorans and B. cenocepacia strains identified 1,058 core proteins. The core genome (purple line) represents conserved genes that stabilize as more genomes are included in the analysis. In contrast, the pan-genome (orange line). The plot also depicts the total gene families (blue line) and core gene families (red line), highlighting the dynamic interplay between shared and strain-specific genes. (B) KEGG analysis of core, accessory, and unique genes from B. multivorans and B. cenocepacia strains. Most genes in each category were mainly involved in the metabolic pathways of the pathogen.
The next step aimed to narrow potential vaccine targets from the core proteome. Out of the 1,058 core proteins, subcellular localization predictions identified 43 as surface-exposed, comprising 25 OMPs and 18 extracellular proteins. These surface proteins are particularly promising as vaccine targets due to their accessibility to the host immune system.
The antigenicity and allergenicity of these proteins were evaluated to ensure they could elicit an immune response without causing adverse effects. This analysis revealed 25 proteins with high antigenicity and non-allergenic properties, making them suitable candidates for further investigation. Homology analysis excluded four proteins that shared significant similarities with human proteome sequences to minimize the risk of autoimmunity. Further refinement based on molecular weight (≤ 110 kDa) and prevalence (≥ 75% across strains) resulted in a final list of 21 proteins distributed across various functional categories: six involved in virulence, 12 in cellular processes, one in metabolism, and two related to genetic information processing (Supplementary Table S2). These findings demonstrate the methodical approach taken to identify conserved and immunogenic proteins.
The 21 selected proteins were subjected to quartile-based scoring, which ranked their potential as vaccine candidates based on properties such as antigenicity, solubility, and stability. This process prioritized nine proteins for detailed structural and functional analysis. These included six OMPs, such as porins (WP_176035635.1, WP_105763988.1, AYZ01212.1, ABK11981.1, and CDN62452.1) and a TonB-dependent receptor (WP_181146976.1), along with three secreted proteins, including a coagulation factor 5/8 type domain protein (ABK11820.1), glutamate synthase (ARF89570.1), and an alkaline phosphatase family protein (WP_006487965.1).
Structural prediction of these proteins provided critical insights into their immunogenic potential. For example, porins exhibited large extracellular loops known to interact with host immune factors. Similarly, the TonB-dependent receptor showed a characteristic barrel-shaped structure, enabling nutrient transport and contributing to bacterial survival in nutrient-depleted environments. These structural features highlight their role in pathogen-host interactions and reinforce their vaccine potential. Table 1 presents the physiological properties of nine putative immunogenic proteins from B. multivorans and B. cenocepacia strains. Conserved domains identified through CD-search and EggNOG confirmed their evolutionary conservation and functional importance (Table 2).
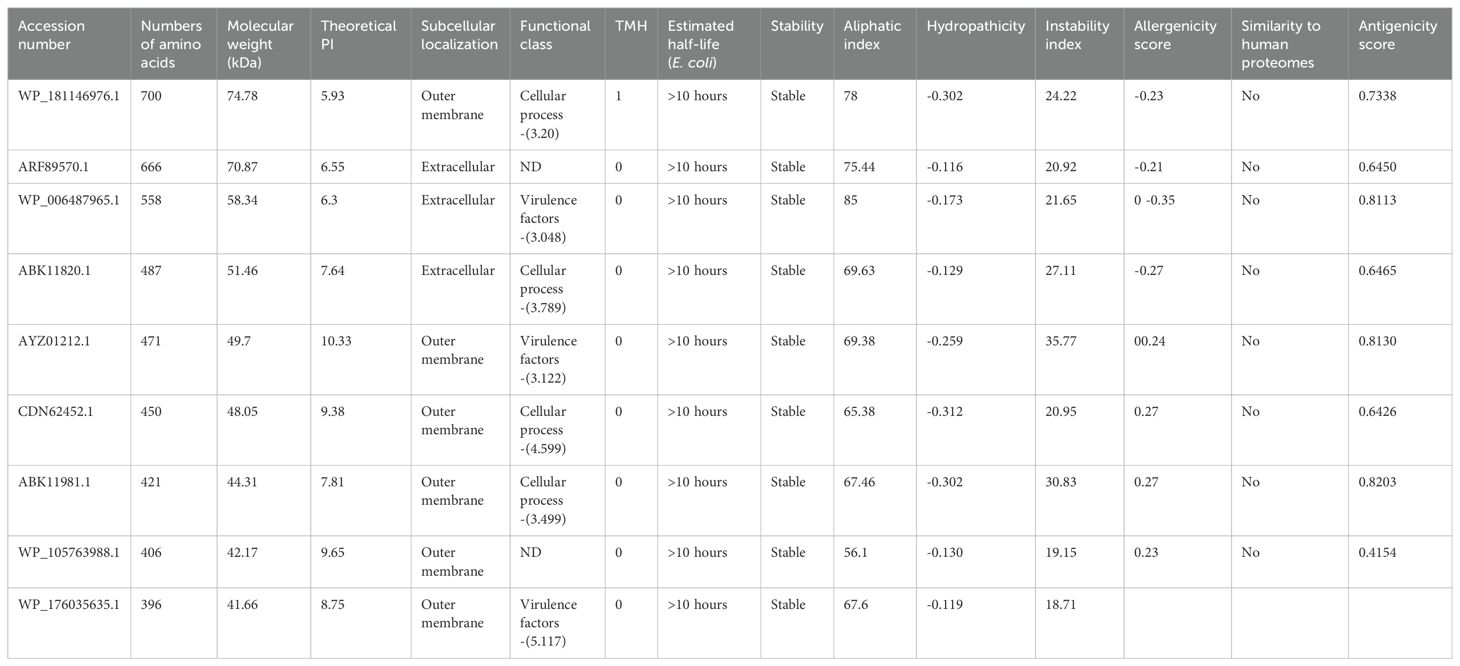
Table 1. Physicochemical analysis of nine prospective immunogenic proteins isolated from B. multivorans and B. cenocepacia strains.
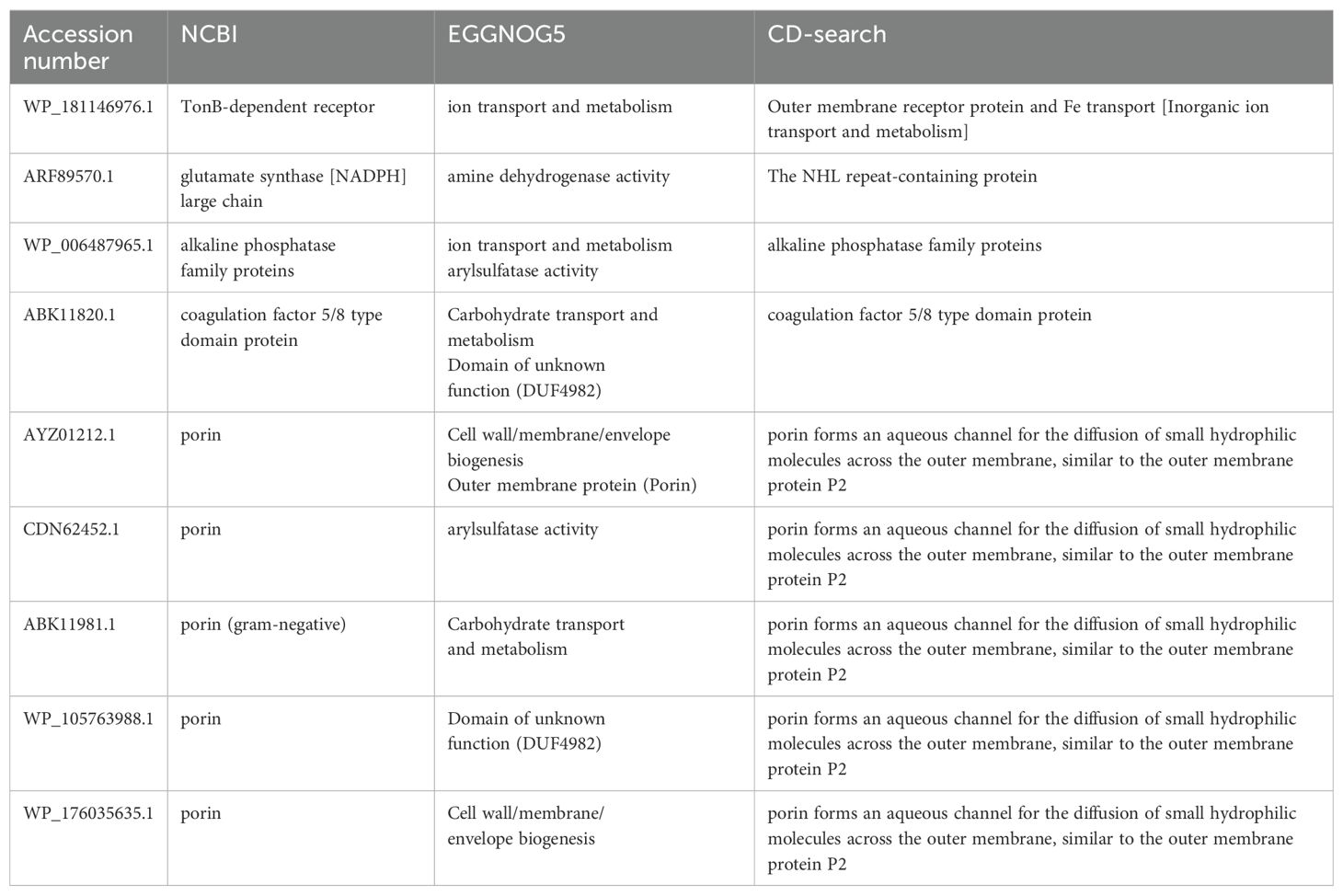
Table 2. Identification of conserved immunogenic targets in B. multivorans and B. cenocepacia for vaccine development.
The workflow illustrates the identification of novel immunogenic targets and the subsequent development of a MEV against B. multivorans and B. cenocepacia (Figure 2). The process encompasses core genome analysis, subcellular localization, antigenicity and allergenicity screening, homology exclusion, and molecular weight filtration. Prioritized proteins underwent epitope mapping, structural modeling, and characterization to construct and validate the MEV.
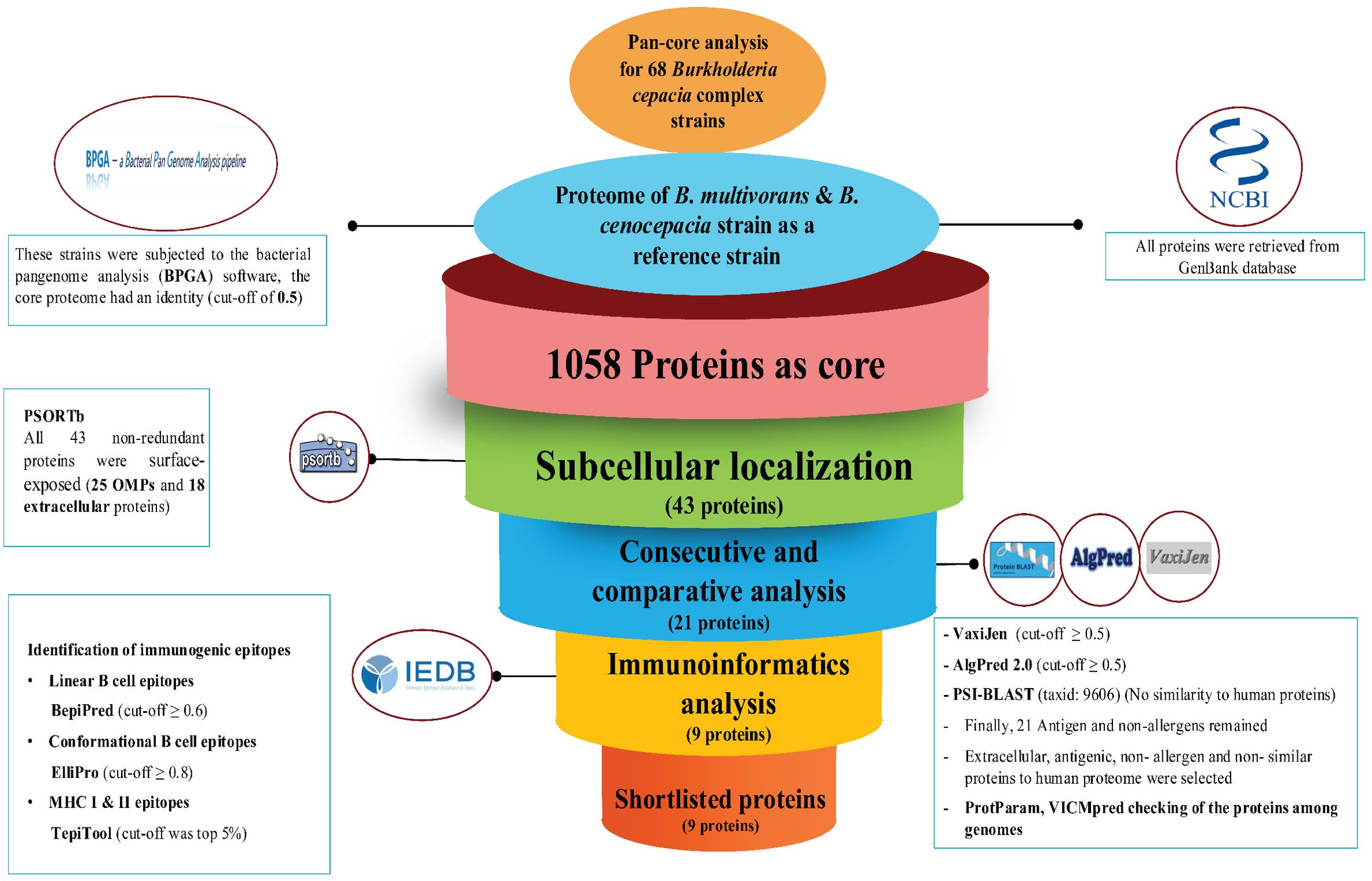
Figure 2. A schematic depiction of the process of selecting and validating potential immunogenic targets and developing a Multi-Epitope Vaccine (MEV) for B. multivorans and B. cenocepacia strains using reverse vaccinology techniques and advanced bioinformatics tools.
To better understand the structural and functional attributes of the nine shortlisted proteins, tertiary structure predictions were conducted using homology modeling tools. The resulting 3D models revealed detailed structural features critical to their immunogenic potential (Figure 3). These structures were further refined and validated using the ProSA-web, confirming their overall quality with Z-scores consistent with experimentally solved structures.
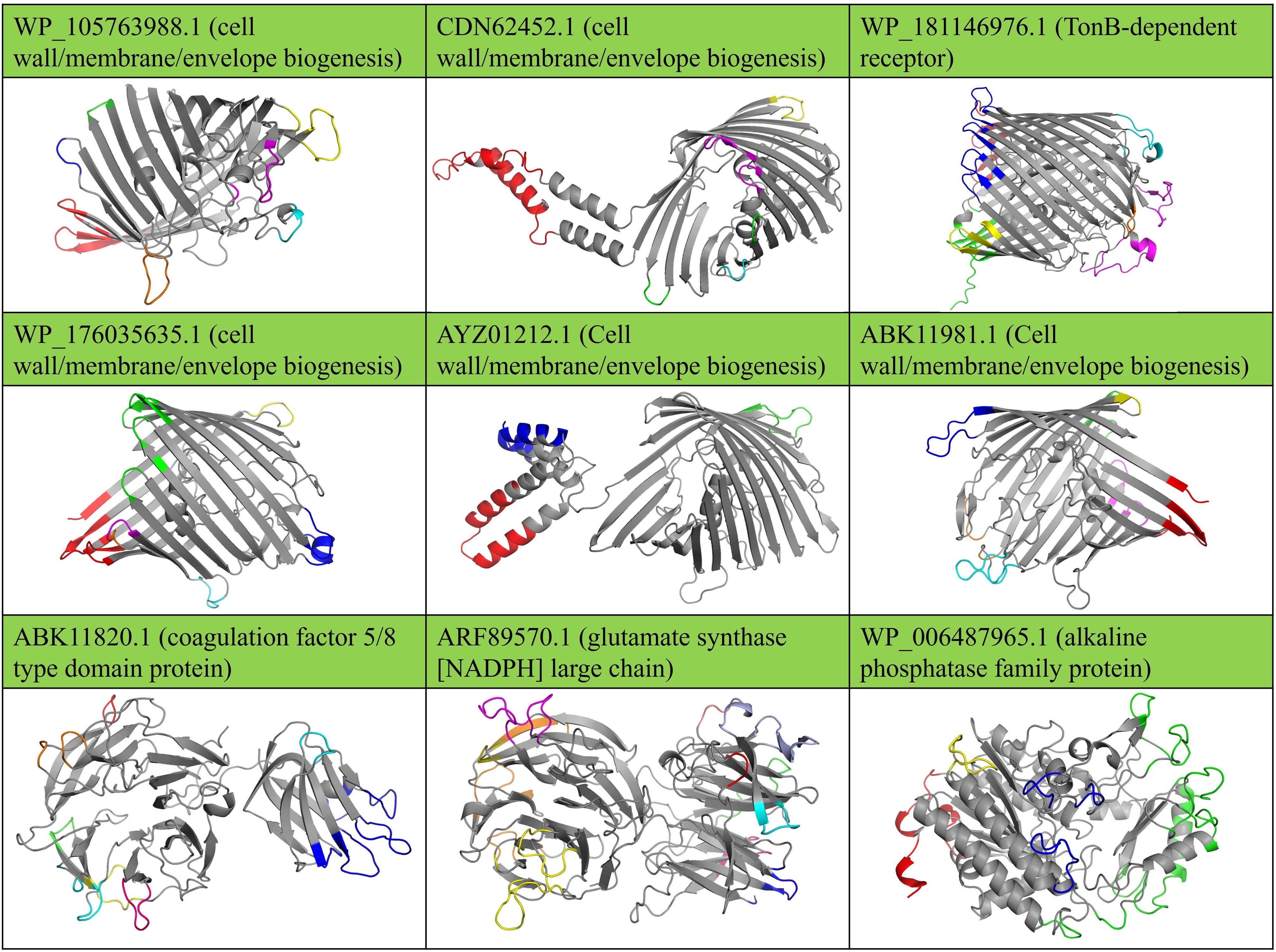
Figure 3. Conformational B cell epitopes on the surface of immunogenic targets against B. multivorans and B. cenocepacia are shown in distinct colors. The protein tertiary structures were predicted using Robetta, and the surface-exposed epitopes were identified and visualized using 3D models illustrated using Jmol software.
Additionally, Figure 3 shows the 3D structure predictions of the nine shortlisted proteins and their surface-exposed conformational B-cell epitopes. The Supplementary Table S3 provides detailed information regarding the epitopes of the nine selected OMPs from B. multivorans and B. cenocepacia. At the same time, the Supplementary Table S4 contains detailed information on the conformational B-cell epitopes. These analyses provide deeper insights into the structural and immunogenic properties of the selected proteins.
The OMPs analyzed in this study include porins (WP_176035635.1, WP_105763988.1, AYZ01212.1, ABK11981.1, CDN62452.1), which exhibited classical β-barrel structures with extracellular loops extending beyond the membrane surface. These loops are critical for their functional interactions with host immune factors. For example, WP_176035635.1 demonstrated a prominent extracellular loop containing conserved residues that could be potential antibody binding sites. The β-barrel architecture provides structural stability, ensuring these proteins remain accessible and functional under varying environmental conditions, including those encountered during host-pathogen interactions. Another significant OMP, the TonB-dependent receptor (WP_181146976.1), displayed a sizeable β-barrel domain with a plug-like structure inside the barrel, a configuration crucial for substrate transport. The receptor’s outer loops contained several antigenic regions, including epitopes predicted to interact with host Toll-like receptors (TLRs), highlighting its potential as an immunogenic target.
Among the secreted proteins, the coagulation factor 5/8 type domain protein (ABK11820.1) exhibited a conserved 5/8 domain characterized by repeated beta-sheet motifs forming a stable scaffold. These surface-exposed motifs make this protein accessible to immune recognition. Similarly, the glutamate synthase (ARF89570.1) displayed a large, multi-domain arrangement with a cleft housing the active site. Conformational B-cell epitope mapping identified residues within this cleft as highly immunogenic. Lastly, the alkaline phosphatase family protein (WP_006487965.1) exhibited a compact α/β fold with a surface-exposed catalytic region. The catalytic residues formed a conserved and antigenic epitope, further solidifying this protein as a strong candidate for vaccine design.
Structural modeling tools mapped Conformational B-cell epitopes on the predicted 3D structures. The analysis revealed surface-exposed regions with high antigenicity scores, likely to elicit a strong immune response.
Epitope mapping was performed on the nine prioritized proteins to identify B-cell epitopes capable of eliciting an immune response. This analysis identified nine highly conserved and antigenic epitopes linked using GPGPG linkers to construct an MEV.
The epitopes were linked using a GPGPG linker. The conserved and highly antigenic B-cell epitopes identified included YSGYESNYGSVAEDDVRL (TonB-dependent receptor, WP_181146976.1), LDTTTGIKGQV (glutamate synthase [NADPH] large chain, ARF89570.1), and QDGNAQGGDNGRA (alkaline phosphatase family protein, WP_006487965.1). Additionally, epitopes such as SGGADQIYAKTADPASTPS, DTRAGQT, EQRTPDGGTQAAQASIGSYGYGG, SDVDGIDN, SQTTLGSTAGGH, and DATGSSLDQAYIPGAADLSST were identified in the porin proteins (ABK11820.1, AYZ01212.1, CDN62452.1, ABK11981.1, WP_105763988.1, and WP_176035635.1, respectively) (Table 3). The 3D structure of the MEV is shown in Figure 4A.
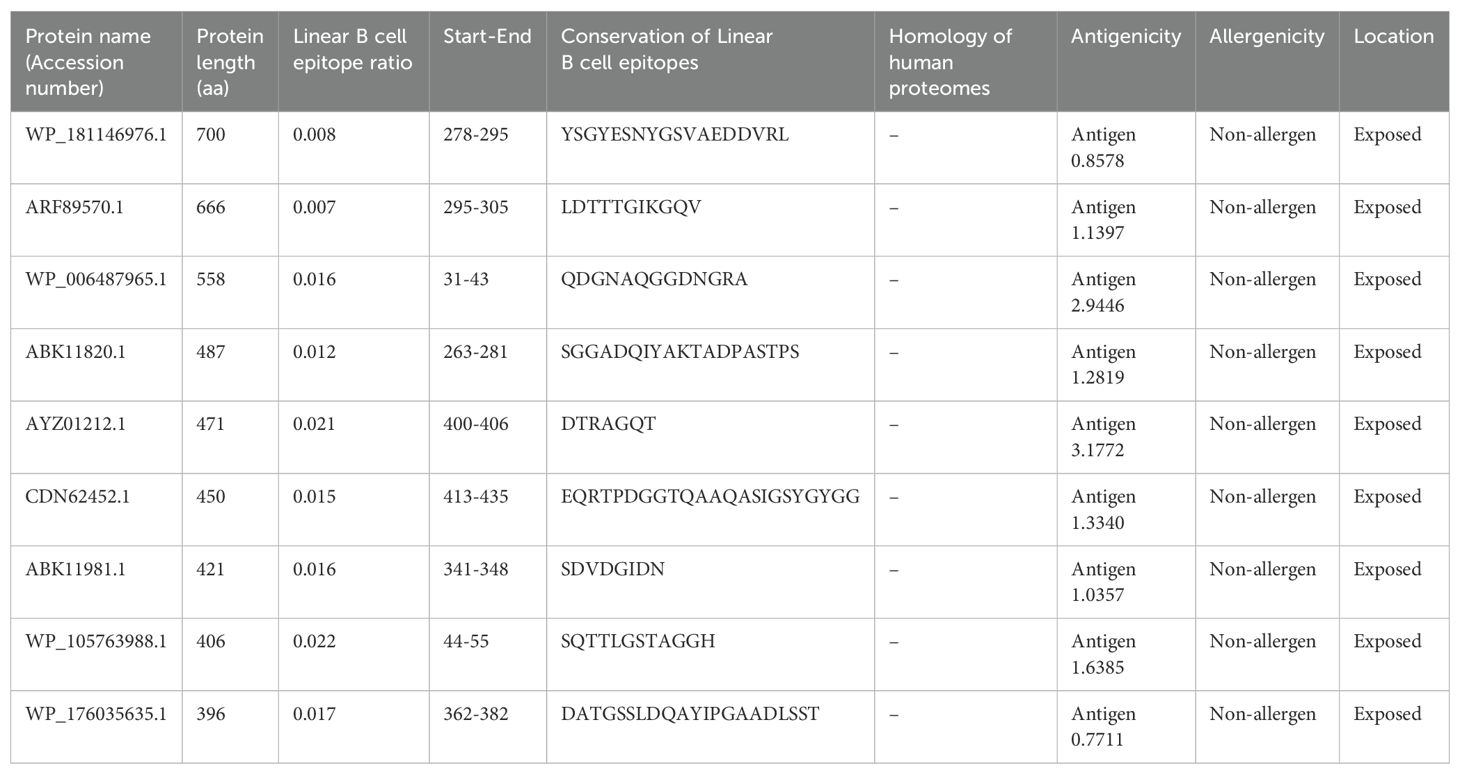
Table 3. Analysis of linear B cell epitopes of nine proteins of B. multivorans and B. cenocepacia strains.
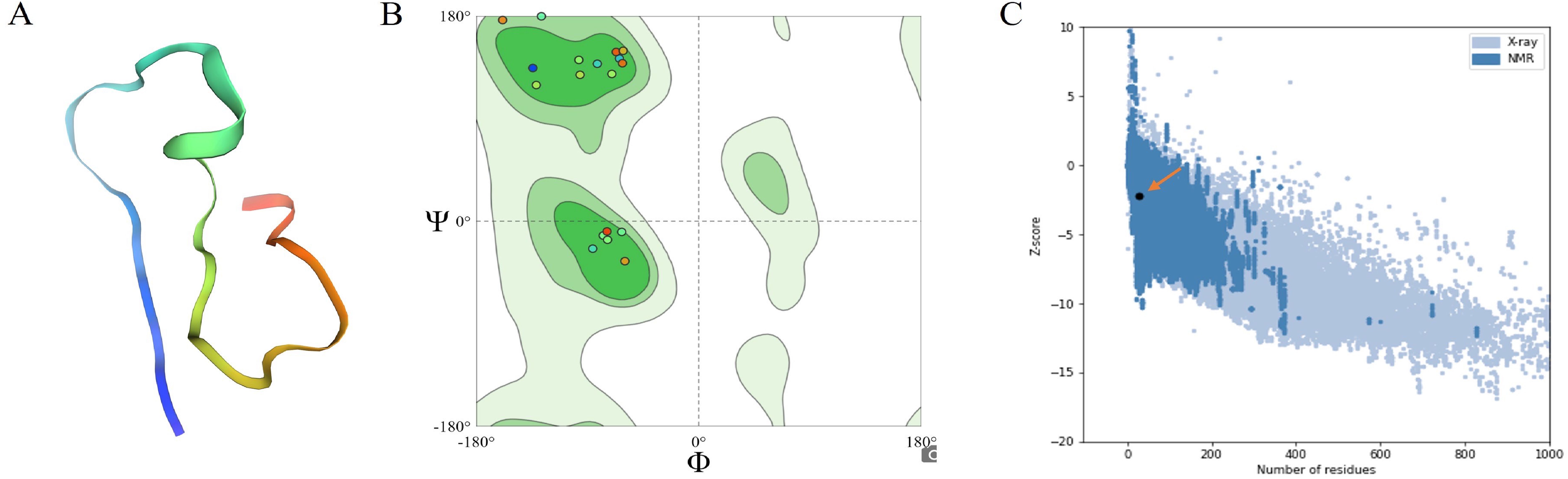
Figure 4. Predicted 3D Structures of Multi-Epitope Vaccines Using Robetta and Validation by ProSA Web Server (A) Schematic of the designed MEV. (B) Structural validation of the predicted MEV was performed using a Ramachandran plot, which evaluates the stereochemical quality of the protein model by displaying the phi (Φ) and psi (Ψ) dihedral angles of the amino acid residues. The green regions represent the most favored conformations in the plot, whereas lighter green regions indicate permissible regions. Colored dots correspond to MEV protein residues, with 91.3% located in favored regions and 8.7% falling within allowed regions. This distribution confirmed the vaccine design’s proper folding, structural stability, and reliability. (C) PROSA web analysis was used to validate the overall quality and accuracy of the MEV constructs. The Z-scores of the protein structures, determined by X-ray crystallography (light blue) and NMR spectroscopy (dark blue), are plotted against the number of residues. Z-scores, which measure model quality, indicate that values closer to zero represent better-quality structures. The orange arrow indicates the Z-score of the specific protein of interest. The clustering of data points reflects the typical Z-score distributions of various structural methods across a range of protein sizes, supporting the reliability of MEV constructs.
Various configurations of the selected epitopes were evaluated, and the most common antigenic combinations were chosen to develop the MEV. The selection process was guided by several essential criteria, including toxin prediction, antigenicity, non-allergenicity to avoid toxigenic, allergic reactions, solubility, and a lack of similarity to human proteins to minimize the risk of autoimmunity. The toxicity of the MEV construct was evaluated using the ToxinPred server. Each peptide sequence in the MEV was analyzed for potential toxic effects using SVM-based (Support Vector Machine) prediction. The results confirmed that all epitopes and the final vaccine construct are non-toxic, with SVM scores below the toxicity threshold. For example, the peptide YSGYESNYGSVAEDDVRL scored -0.59, and LDTTTGIKGQV scored -0.84, indicating non-toxicity. These findings ensure that the vaccine is safe for further experimental validation without adverse effects due to toxicity (Supplementary Table S5).
Structural validation revealed that 91.3% of the vaccine residues were located in the favored regions of the Ramachandran plot. In contrast, the remaining 8.7% fell within permissible areas, confirming the structural reliability of the design (Figure 4B). Additionally, ProSA-web analysis further validated the overall quality and accuracy of the MEV constructs (Figure 4C). The VaxiJen server predicted an antigenicity score of 1.4514 for MEV, affirming its non-allergenic properties, further confirmed by AlgPred 2.0 and AllerTOP v2.0. The selected vaccine candidate also exhibited the highest solubility score (0.844). With a molecular weight of 13.69 kDa, MEV exhibits high thermotolerance, as indicated by its aliphatic index 42.99. The isoelectric point (PI) was calculated to be 4.53, and a negative GRAVY score of -1.036 classified it as a hydrophilic molecule. The instability index of the vaccine was 27.28, indicating that the polypeptide was stable. The estimated half-lives of MEVs in vitro are 1.9 h in mammalian reticulocytes and >10 h in vivo in E. coli.
The interaction of the MEV with host immune receptors was evaluated through molecular docking.
The pyDockWEB results demonstrated that MEV had the most robust interactions with TLR2 (-18.708 kcal/mol) and TLR4 (-33.215 kcal/mol) (Supplementary Table S6). Furthermore, the interactions between MEV, TLR2, and TLR4 are illustrated in Figures 5A, B. These interactions indicate that the MEV can effectively activate innate immune responses, crucial for initiating downstream adaptive immunity.
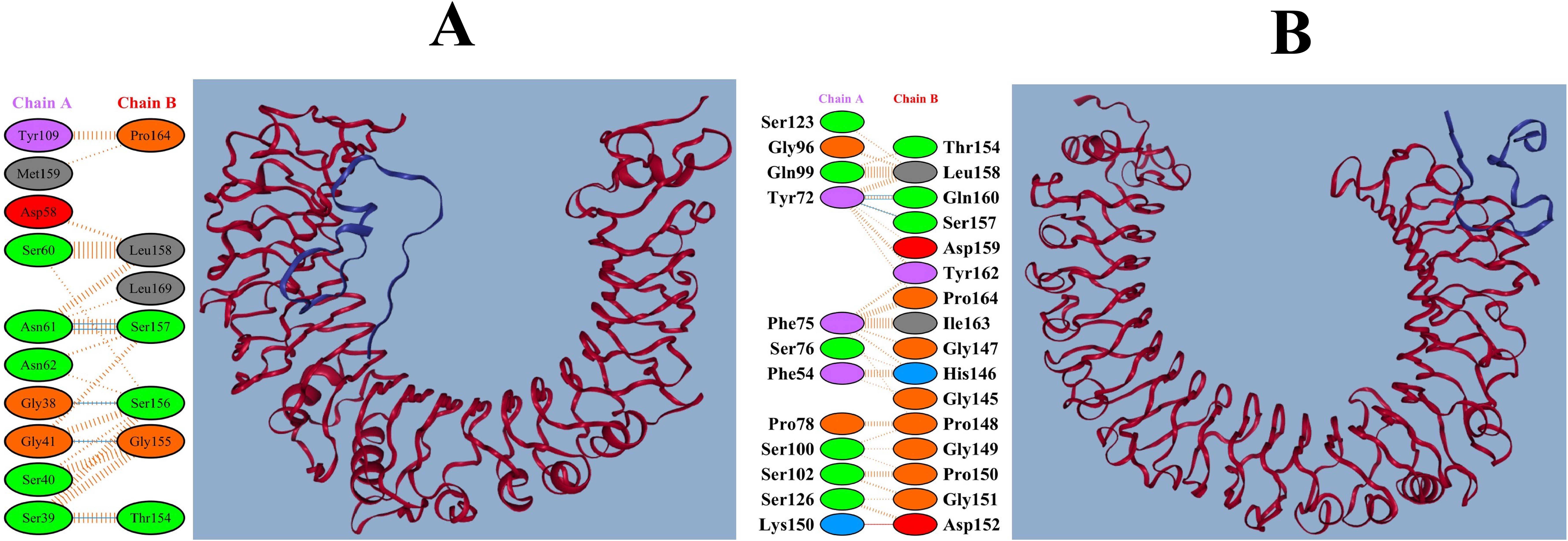
Figure 5. Detailed interactions of MEV with TLR2 and TLR4. (A) Interaction of the MEV construct with human TLR2, depicting a vaccine candidate in complex with TLR2. (B) Interaction of the MEV construct with human TLR4, indicating that the vaccine candidate binds to TLR4.
The immune simulation analysis conducted using the C-ImmSim tool provided valuable insights into the immunogenic potential of the MEV. The data revealed that the MEV elicited a strong immune response characterized by elevated levels of immunoglobulins, T-helper cell populations, and cytokine production.
Specifically, the simulation showed that the MEV induced higher levels of IgM and IgG1 antibodies than baseline, with IgM peaking early (~day 5) and rapidly declining. At the same time, IgG1 appeared later at moderate levels (Figure 6A).
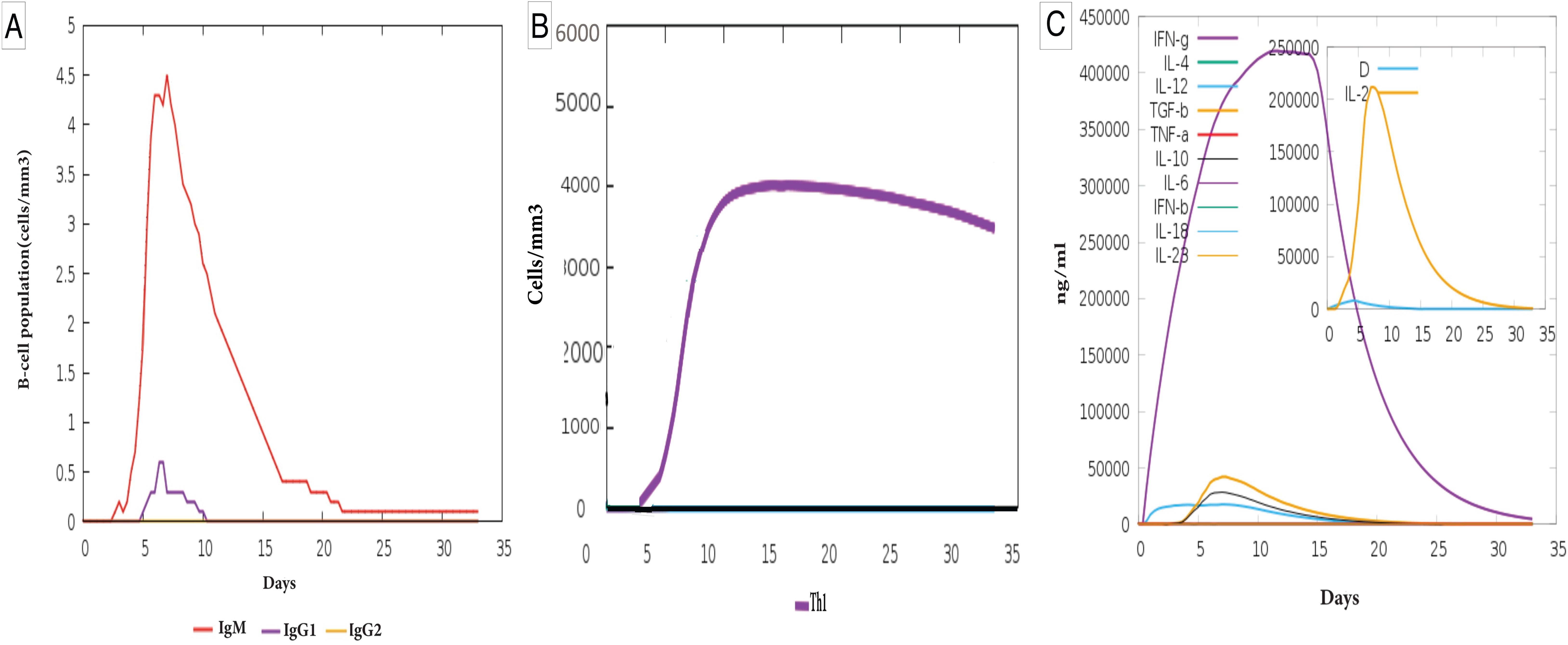
Figure 6. Immune response simulation of the designed vaccine. (A) B-cell population and antibody production: The graph illustrates the dynamics of B-cell populations and immunoglobulin (Ig) production in response to the vaccine. The MEV induced higher levels of IgM (red line), which peaked early (~day 5) and declined rapidly. IgG1 (purple line) appeared later at lower levels, while IgG2 (yellow line) remained negligible throughout the response. (B) T-helper 1 (Th1) cell population dynamics: Th1 cell levels increased steadily from ~day 3, peaking around day 15, and gradually declined thereafter. The MEV stimulated a higher production of Th1 cells. (C) Cytokine production dynamics: The graph shows cytokine concentrations over time, demonstrating their roles in immune regulation. IFN-γ (purple line) peaked sharply (~day 5) and declined steadily, while IL-2 (yellow line) exhibited an early peak around day 5, followed by a rapid decline. Other cytokines, including IL-6, IL-10, and TNF-α, showed smaller early peaks, contributing to immune modulation. The inset plot highlights the early peaks of the danger signal and the leukocyte growth factor IL-2, demonstrating elevated levels of IL-2 and IFN-γ induced by the MEV.
In addition, the vaccine promoted a significant increase in Th1 cell populations, which steadily rose from day 3, peaked around day 15, and then gradually declined, indicative of a robust adaptive immune response (Figure 6B).
Furthermore, the MEV stimulated increased production of key cytokines, such as IFN-γ and IL-2, critical for immune system activation and regulation. IFN-γ exhibited a sharp peak around day 5, while IL-2 showed an early surge, highlighting the vaccine’s ability to enhance immune signaling and modulate immune cell proliferation. Other cytokines, such as IL-6, IL-10, and TNF-α, displayed smaller peaks, contributing to the overall immune response (Figure 6C). In silico immunization studies further validated the MEV’s efficacy. Simulations showed that the vaccine could elicit a robust immune response characterized by high levels of immunoglobulins and memory cell formation. These findings establish the MEV as a promising candidate for experimental testing.
Analyzing the RMSD, RMSF, and Rg profiles for the MEV-TLR4, MEV-only, and MEV-TLR2 systems provides a comprehensive understanding of their structural dynamics and stability during MD simulations. The MEV-TLR4 system demonstrated the highest structural stability, with RMSD values consistently fluctuating between 0.25 nm and 0.35 nm, indicating a robust and well-maintained interaction between the MEV ligand and the TLR4 receptor. The early stabilization of RMSD values further underscores the strong binding interface, suggesting that the interaction forms quickly and remains stable throughout the simulation. In contrast, the MEV-only system exhibited the greatest flexibility, with RMSD values ranging from 0.2 nm to 0.4 nm, reflecting the expected behavior of a small molecule freely exploring its conformational space without receptor constraints. Meanwhile, the MEV-TLR2 system displayed intermediate behavior, with an initial gradual increase in RMSD values as the ligand adjusted to the receptor, followed by stabilization between 0.3 nm and 0.4 nm, signifying a less rigid but still stable receptor-ligand interaction compared to MEV-TLR4 (Figure 7A).
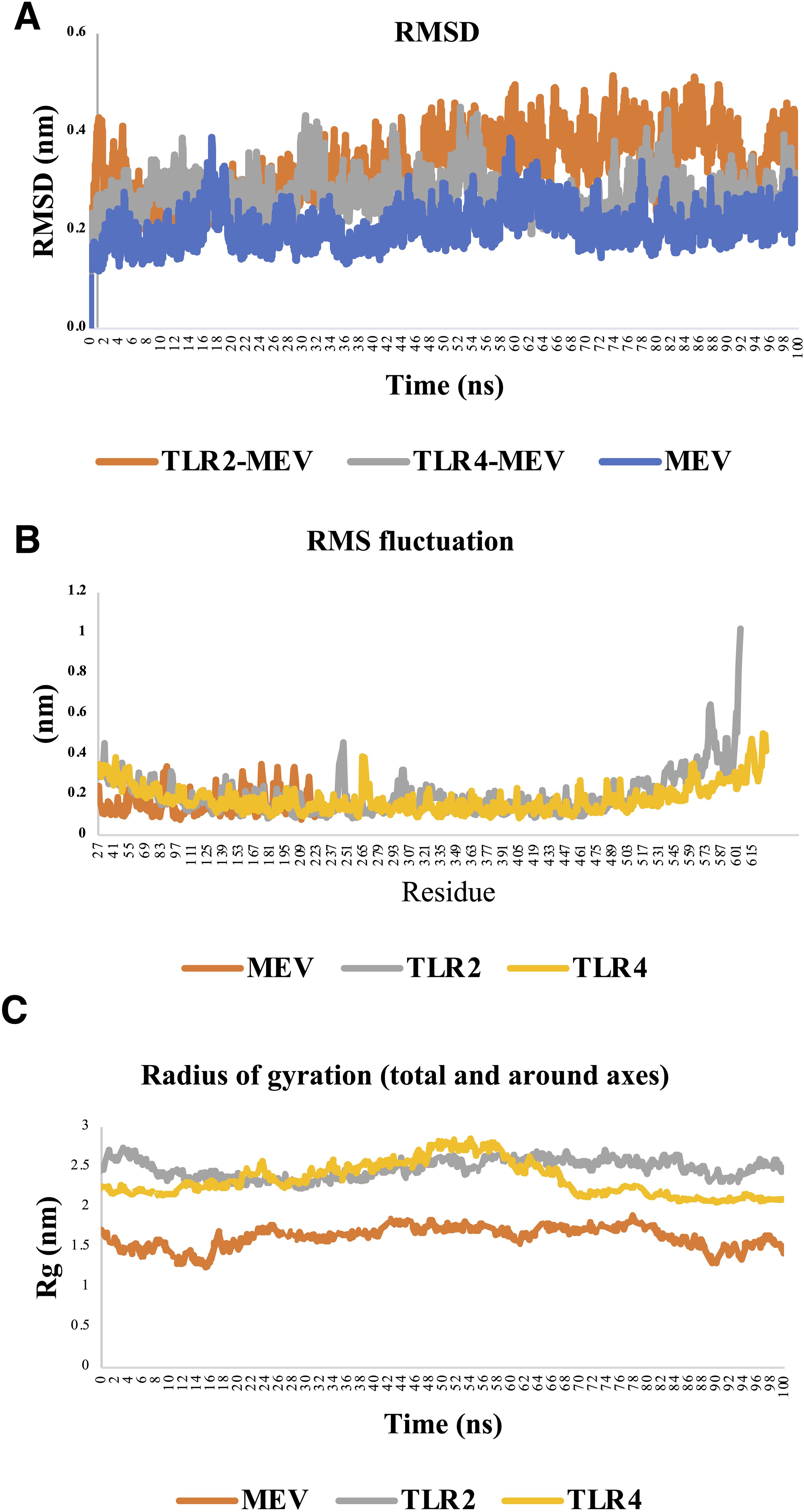
Figure 7. Structural stability and flexibility analysis of TLR4-MEV, TLR2-MEV, and MEV systems during molecular dynamics simulation. (A) Root mean square deviation (RMSD): The RMSD analysis highlights the structural stability of the systems. The MEV-TLR4 complex shows the most stable behavior, with minimal fluctuations, while the MEV-only system exhibits the highest variability due to its flexibility. The MEV-TLR2 complex displays intermediate stability, indicating a dynamic but stable interaction. (B) Root mean square fluctuation (RMSF): The RMSF results reveal residue-level flexibility. The MEV-TLR4 complex has the lowest fluctuations, suggesting strong stabilization by the receptor. In contrast, the MEV-only system shows significant flexibility, and the MEV-TLR2 complex exhibits moderate fluctuations, particularly in dynamic regions like loops. (C) Radius of gyration (Rg): The Rg analysis reflects structural compactness. The MEV-TLR4 complex maintains the most significant and stable Rg, indicating a compact and stable structure. The MEV-only system has the smallest and most variable Rg, reflecting its flexibility, while the MEV-TLR2 complex shows intermediate compactness with some dynamic behavior.
The RMSF analysis further highlights the differences among the three systems. The MEV-TLR4 complex exhibited the lowest overall fluctuations, with most residues showing RMSF values below 0.3 nm, indicating high stability, particularly in the backbone regions. The minimal flexibility observed corresponds to strong and stable interactions between MEV and TLR4, with only minor peaks in loop regions or terminal segments reflecting natural conformational movements. The MEV-only system displayed the highest RMSF values, reaching up to 0.35 nm, indicative of significant conformational variability and freedom to explore different structural states. The MEV-TLR2 complex showed intermediate flexibility, with most residues maintaining low RMSF values but with noticeable peaks near loop regions and the binding site, reflecting the dynamic adjustments necessary for accommodating the ligand (Figure 7B).
The Rg analysis complements these findings by revealing differences in structural compactness. The MEV-TLR4 complex maintained Rg values fluctuating narrowly between 3.2 and 3.5 nm, indicating a stable and compact structure throughout the simulation. This compactness reflects the stabilizing effect of the strong MEV-TLR4 binding interaction. Conversely, the MEV-only system showed Rg values ranging from 1.85 nm to 2.1 nm, with significant variability reflecting the unconstrained and flexible nature of the free ligand. The MEV-TLR2 complex displayed Rg values between 2.9 nm and 3.2 nm, representing a moderately stable and compact structure with slight dynamic adjustments, indicative of a more flexible receptor-ligand interaction than MEV-TLR4 (Figure 7C).
This study presents a novel approach for identifying putative Bcc vaccine candidates using reverse vaccination. The Bcc poses significant challenges in clinical settings because it is resistant to multiple antibiotics and can potentially cause life-threatening infections, particularly in immunocompromised individuals such as patients with CF and chronic granulomatous disease (CGD) (3). Traditional antibiotic treatments are often ineffective against Bcc, necessitating the development of new therapeutic strategies, including vaccines (36).
Reverse vaccinology leverages computational tools to analyze genomic data and identify proteins likely to elicit strong immune responses, focusing on surface-exposed proteins that play critical roles in immune recognition and bacterial survival (37). In this study, we aimed to explore novel vaccine candidates against B. multivorans and B. cenocepacia, two of the most clinically significant species in the Bcc. Our analysis involved comprehensive screening of core proteins shared across these strains to identify those that were both immunogenic and specific to bacterial cells, thereby avoiding unwanted cross-reactivity with human proteins. The reverse vaccinology approach targeted proteins conserved across strains of B. multivorans and B. cenocepacia, which are highly adaptable and resistant to multiple antibiotics, contributing to severe and often fatal infections in patients with CF (13). B. multivorans is commonly associated with chronic infections, leading to a gradual decline in lung function and often resulting in a severe condition known as cepacia syndrome (38). In contrast, B. cenocepacia is the most virulent species in the Bcc, and it is associated with rapid deterioration of lung function, bacteremia, and septicemia (39). The aggressive behavior of B. cenocepacia highlights the critical need for preventive measures, such as vaccines. Many infected patients are deemed ineligible for lung transplantation, making vaccination an essential preventive strategy (40).
Our reverse vaccinology process involved identifying surface-exposed proteins accessible to the host immune system capable of eliciting strong immune responses. Specifically, OMPs were prioritized because of their roles in pathogen-host interactions and immune recognition (41). Our core proteome analysis identified 1,058 shared proteins across 68 B. multivorans and B. cenocepacia strains, among which 43 proteins were predicted as surface-exposed candidates. Identifying these surface-exposed proteins is critical because they often play significant roles in immune recognition and host-pathogen interactions, making them ideal candidates for vaccine development. This finding aligns with previous studies highlighting the importance of OMPs in eliciting immune responses, as they are accessible to the immune system and often involved in critical pathogenic functions (42, 43). A significant consideration in our candidate selection process was to ensure that the selected proteins were pathogen-specific and did not share homology with human proteins, as similarity with human proteins could increase the risk of autoimmune responses. Using the PSI-BLAST, we systematically excluded proteins homologous to the human proteome, ultimately retaining 21 proteins. This filtering step aligns with best practices in vaccine development. The aim of therapy is to address the potential for adverse immune responses, as demonstrated in other studies focusing on human-specific pathogens (44, 45). The antigenicity and allergenicity of these 21 proteins further narrowed our selection to nine potential vaccine candidates, including OMPs, TonB-dependent receptors, coagulation factor 5/8 type domain proteins, alkaline phosphatase family proteins, and glutamate synthase NADPH large chains. These proteins play crucial roles in bacterial survival, pathogenicity, and immune evasion, making them ideal candidates for vaccine development.
OMPs in Burkholderia species perform various essential functions, including transport of nutrients, adhesion to host tissues, and immune evasion. Their surface exposure makes them prime candidates for vaccine development, as demonstrated by successful applications in other Gram-negative pathogens like Neisseria meningitidis, in which OMPs have been effectively integrated into vaccine formulations (46). OMPs are the first molecules encountered by the immune system, highlighting their potential as effective vaccine targets (7, 45, 47).
TonB-dependent receptors are critical for iron acquisition, bacterial growth, and virulence. Research on pathogens such as Vibrio cholerae and Pseudomonas aeruginosa has shown that targeting these receptors can significantly impair bacterial survival by disrupting iron uptake (48). In Burkholderia species, the uptake of ferric siderophore complexes relies on various outer membrane receptors that interact with the inner membrane TonB complex, and disruption of the tonB gene reduces virulence in Burkholderia mallei and B. cenocepacia (49).
Coagulation factor 5/8 type domain protein facilitates evasion by modulating host immune responses. Similar proteins in another pathogen, such as Staphylococcus aureus, have beennowing to play significant roles in immune modulation, making them attractive targets for vaccine development (50). By targeting this protein, it may be possible to enhance the host’s ability to detect and eliminate Burkholderia species, thereby improving vaccine efficacy. The alkaline phosphatase family of proteins is involved in phosphate metabolism, which is crucial for bacterial growth and virulence. Studies have indicated that targeting metabolic enzymes such as alkaline phosphatase reduces bacterial fitness and enhances immune recognition (51). This suggests that vaccines incorporating alkaline phosphatase could stimulate a robust immune response while impairing the pathogen’s metabolic capabilities, thus reducing its virulence. Glutamate synthase is required for nitrogen metabolism and bacterial survival under nutrient-limited conditions. Targeting this enzyme has been shown to reduce the pathogenicity of other intracellular pathogens, such as Mycobacterium tuberculosis (52).
Collectively, these proteins represent a strategic focus for Bcc vaccine development. The aim is to effectively enhance the host’s ability to detect and eliminate opportunistic pathogens.
Several studies have employed reverse vaccinology to identify candidate vaccine candidates against Bcc. Muruato (2017) explored the potential of reverse vaccinology to identify immunogenic proteins from Bcc (53). Their study emphasized the importance of bioinformatics tools in predicting protective antigens, which can serve as candidates for vaccine development. By analyzing the genomic data of Burkholderia species, the researchers identified several surface-exposed proteins that could elicit an immune response, thus laying the groundwork for further vaccine design efforts. In a recent study, Alsowayeh et al. (2022) used reverse vaccinology and immunoinformatic to design an MEV targeting nosocomial B. cepacia (43). They identified 19 virulence proteins in the virulence factor database. They linked immunodominant epitopes using GPGPG linkers, creating an MEV related to the cholera toxin B subunit as an adjuvant. This approach enhances the immunogenicity of vaccines and addresses the challenge of poor immunogenicity, which is often associated with peptide vaccines. Shahab et al. (2022) investigated the immunogenic potential of various epitopes from Bcc (54). Their research focused on identifying T-cell and B-cell epitopes using immunoinformatics, which is crucial for developing effective vaccines. This study highlighted the significance of computational tools for predicting epitopes that stimulate robust immune responses, thereby facilitating the design of MEVs.
Another study investigated the immunogenic potential of various Bcc epitopes (55). Their research focused on identifying T-cell and B-cell epitopes using immunoinformatics, which is crucial for developing effective vaccines. This study highlighted the significance of computational tools for predicting epitopes that stimulate robust immune responses, thereby facilitating the design of MEVs. Irudal et al. (2023) further advanced the understanding of Bcc vaccine development by employing a comprehensive reverse vaccinology approach (56). Their study identified several novel antigens that may serve as potential vaccine candidates. By integrating genomic and proteomic data, researchers can pinpoint conserved regions within the Burkholderia genome, which are used to elicit strong and lasting immune responses.
Identifying B-cell epitopes and MHC-II-binding sites is pivotal for understanding proteins’ immunogenic potential, particularly in vaccine development.
Linear and conformational B cell epitopes are crucial for eliciting humoral immune responses. At the same time, MHC-II binding sites are essential for stimulating T cell-mediated immunity, which is critical for long-term protection against pathogens. Jankowski et al.’s study findings (57) underscore the necessity of integrating both B-cell and T-cell epitopes in vaccine design to enhance efficacy (57). Predicting the binding affinities of these epitopes to MHC class II molecules is also vital because it helps assess the capacity of vaccine candidates to stimulate T-cell responses, thereby contributing to sustained immunity (57).
We used a quartile scoring system to evaluate the immunogenic potential of the shortlisted proteins based on criteria such as epitope distribution, MHC binding, and allergenic properties. This systematic approach ensures the selection of proteins capable of eliciting immediate and long-lasting immune responses. We also characterized the 3D structures of the selected proteins and assessed their interactions with TLRs, which are crucial for initiating adaptive immune responses. Our molecular docking results indicated strong interactions between the proteins and TLRs, suggesting that our vaccine constructs effectively engaged in innate immunity and supported robust adaptive immunity.
The MD simulation results provide valuable insights into the interaction dynamics and stability of the chimeric MEV with Toll-like receptors TLR2 and TLR4. The MEV-TLR4 complex exhibited the highest structural stability, as evidenced by minimal RMSD and RMSF fluctuations and compact Rg values, indicating a robust and tightly bound interaction. This suggests that MEV forms a well-defined and stable interface with TLR4, likely contributing to strong receptor-ligand engagement. In contrast, the MEV-TLR2 complex displayed moderate stability with higher RMSD and RMSF values, reflecting a more dynamic interaction. These results highlight that while TLR2 forms a stable complex with MEV, it allows for greater flexibility than TLR4. The free MEV system, characterized by significant conformational variability, reinforces the role of receptor binding in stabilizing the ligand’s structure. These findings underscore the distinct binding dynamics of MEV with TLR2 and TLR4, with TLR4 providing a more rigid and stable interaction, which could have implications for receptor-specific therapeutic strategies. The designed vaccine construct exhibited favorable properties, including high solubility, non-allergenicity, and strong antigenicity, critical characteristics of an effective vaccine. Immunoinformatics analysis, including ProSA-web and Ramachandran plot validation, confirmed that the vaccine construct’s 3D structure was stable and suitable for immunogenic purposes. The MEV construct incorporated epitopes from all nine selected proteins and was designed to stimulate both humoral and cellular immune responses. This vaccine can offer immediate and long-lasting protection against Bcc by effectively targeting both B and T cells.
This study highlights the potential of reverse vaccinology and computational methods for identifying and refining vaccine candidates, offering a promising direction for combating severe Bcc infections.
In conclusion, this study identified nine essential immunogenic proteins, including OMPs and TonB-dependent receptors, as promising vaccine candidates against B. multivorans and B. cenocepacia. MD simulations revealed that the chimeric MEV interacts more stably with TLR4 than TLR2, as evidenced by lower RMSD, RMSF, and compact Rg values, indicating a robust TLR4-MEV interface. At the same time, TLR2-MEV exhibited moderate stability and flexibility, highlighting receptor-specific binding dynamics with therapeutic implications. As a result of this study, MEV incorporating conserved B-cell epitopes demonstrated strong antigenicity, stability, and robust interactions with immune receptors such as TLR2 and TLR4 in molecular docking studies. The MEV construct has the potential to stimulate both humoral and cellular immune responses, offering broad protection against multidrug-resistant pathogens. Further in vitro and in vivo validation is required to confirm the vaccine’s efficacy and safety for clinical use, particularly in vulnerable populations such as patients with CF.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
DG: Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Methodology. MB: Data curation, Writing – original draft, Writing – review & editing, Software, Validation. NN: Writing – original draft, Investigation, Methodology. MS: Writing – review & editing, Software, Formal analysis, Methodology. BS: Methodology, Software, Writing – review & editing. YM: Formal analysis, Methodology, Writing – review & editing. FB: Writing – original draft, Conceptualization, Project administration, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors thank the personnel at the Pasteur Institute of Iran for their spiritual support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2025.1520109/full#supplementary-material
1. Yu C, Kotsimbos T. Respiratory infection and inflammation in cystic fibrosis: A dynamic interplay among the host, microbes, and environment for the ages. Int J Mol Sci. (2023) 24:4052. doi: 10.3390/ijms24044052
2. Scoffone VC, Chiarelli LR, Trespidi G, Mentasti M, Riccardi G, Buroni S. Burkholderia cenocepacia infections in cystic fibrosis patients: drug resistance and therapeutic approaches. Front Microbiol. (2017) 8. doi: 10.3389/fmicb.2017.01592
3. Tavares M, Kozak M, Balola A, Sá-Correia I. Burkholderia cepacia complex bacteria: a feared contamination risk in water-based pharmaceutical products. Clin Microbiol Rev. (2020) 33:e00139-19. doi: 10.1128/CMR.00139-19
4. Drevinek P, Mahenthiralingam E. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin Microbiol Infection. (2010) 16:821–30. doi: 10.1111/j.1469-0691.2010.03237.x
5. Mil-Homens D, Pinto SN, Matos RG, Arraiano CM, Fialho AM. Burkholderia cenocepacia K56-2 trimeric autotransporter adhesin bcaA binds TNFR1 and contributes to induce airway inflammation. Cell Microbiol. (2016) 19. doi: 10.1111/cmi.12677
6. Aubert D, Xu H, Yang JL, Shi X, Gao W, Li L, et al. A burkholderia type VI effector deamidates rho GTPases to activate the pyrin inflammasome and trigger inflammation. Cell Host Microbe. (2016) 19:664–74. doi: 10.1016/j.chom.2016.04.004
7. Pradenas GA, Myers JN, Torres AG. Characterization of the burkholderia cenocepacia tonB mutant as a potential live attenuated vaccine. Vaccines. (2017) 5:33. doi: 10.3390/vaccines5040033
8. Blanchard AC, Tang L, Tadros M, Muller MP, Spilker T, Waters V, et al. Burkholderia cenocepacia ET12 transmission in adults with cystic fibrosis. Thorax. (2019) 75:88–90. doi: 10.1136/thoraxjnl-2019-214098
9. Wang G, Zarodkiewicz P, Valvano MA. Current advances in burkholderia vaccines development. Cells. (2020) 9:2671. doi: 10.3390/cells9122671
10. Shinoy M, Dennehy R, Coleman LV, Carberry S, Schaffer K, Callaghan M, et al. Immunoproteomic analysis of proteins expressed by two related pathogens, burkholderia multivorans and burkholderia cenocepacia, during human infection. PloS One. (2013) 8:e80796. doi: 10.1371/journal.pone.0080796
11. Hashempour A, Khodadad N, Akbarinia S, Ghasabi F, Ghasemi Y, Nazar MMKA, et al. Reverse vaccinology approaches to design a potent multiepitope vaccine against the HIV whole genome: immunoinformatic, bioinformatics, and molecular dynamics approaches. BMC Infect Diseases. (2024) 24:873. doi: 10.1186/s12879-024-09775-2
12. Jalal K, Khan K, Ahmad D, Hayat A, Basharat Z, Abbas MN, et al. Pan-Genome Reverse Vaccinology Approach for the Design of Multi-Epitope Vaccine Construct against Escherichia albertii. Int J Mol Sci. (2021) 22:12814. doi: 10.3390/ijms222312814
13. Hassan AA, dos Santos SC, Cooper VS, Sá-Correia I. Comparative Evolutionary Patterns of Burkholderia cenocepacia and B. multivorans During Chronic Co-infection of a Cystic Fibrosis Patient Lung. Front Microbiol. (2020) 11:574626. doi: 10.3389/fmicb.2020.574626
14. Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. (2010) 26:1608–15. doi: 10.1093/bioinformatics/btq249
15. Yu CS, Lin CJ, Hwang JK. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. (2004) 13:1402–6. doi: 10.1110/ps.03479604
16. Hallgren J, Tsirigos KD, Pedersen MD, Almagro Armenteros JJ, Marcatili P, Nielsen H, et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. BioRxiv. (2022) 2022.04.08.487609. doi: 10.1101/2022.04.08.487609
17. Sharma N, Patiyal S, Dhall A, Pande A, Arora C, Raghava GP. AlgPred 2.0: an improved method for predicting allergenic proteins and mapping of IgE epitopes. Briefings Bioinf. (2021) 22:bbaa294. doi: 10.1093/bib/bbaa294
18. Mahram A, Herbordt MC. NCBI BLASTP on high-performance reconfigurable computing systems. ACM Trans Reconfigurable Technol Syst (TRETS). (2015) 7:1–20. doi: 10.1145/2629691
19. Saha S, Raghava G. VICMpred: an SVM-based method for the prediction of functional proteins of Gram-negative bacteria using amino acid patterns and composition. Genomics Proteomics Bioinf. (2006) 4:42–7. doi: 10.1016/S1672-0229(06)60015-6
20. Roy S, Maheshwari N, Chauhan R, Sen NK, Sharma A. Structure prediction and functional characterization of secondary metabolite proteins of Ocimum. Bioinformation. (2011) 6:315–9. doi: 10.6026/97320630006315
21. Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, et al. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. (2005) 33::D192–6. doi: 10.1093/nar/gki069
22. Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. (2016) 44:D286–93. doi: 10.1093/nar/gkv1248
23. Chaudhari NM, Gupta VK, Dutta C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci Rep. (2016) 6:24373. doi: 10.1038/srep24373
24. Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. (2017) 45:W24–W9. doi: 10.1093/nar/gkx346
25. Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. (2003) 31:3381–5. doi: 10.1093/nar/gkg520
26. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. (2007) 35:W407–W10. doi: 10.1093/nar/gkm290
27. Bemani P, Jalili S, Hassanpour K, Faraji F, Gholijani N, Barazesh M, et al. Designing and characterization of tregitope-based multi-epitope vaccine against multiple sclerosis: an immunoinformatic approach. Curr Drug Safety. (2023) 18:79–92. doi: 10.2174/1574886317666220429105439
28. Herraez A. Biomolecules in the computer: Jmol to the rescue. Biochem Mol Biol Education. (2006) 34:255–61. doi: 10.1002/bmb.2006.494034042644
29. Glaser F, Pupko T, Paz I, Bechor D, Martz E, Ben-Tal N. ConSurf: A server for the identification of functional regions in proteins by surface-mapping of phylogenetic information. doi: 10.1093/bioinformatics/19.1.163
30. Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Open Source Drug Discovery C, et al. In silico approach for predicting toxicity of peptides and proteins. PloS One. (2013) 8:e73957. doi: 10.1371/journal.pone.0073957
31. Jiménez-García B, Pons C, Fernández-Recio J. pyDockWEB: a web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics. (2013) 29:1698–9. doi: 10.1093/bioinformatics/btt262
32. Rapin N, Lund O, Bernaschi M, Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PloS One. (2010) 5:e9862. doi: 10.1371/journal.pone.0009862
33. Laskowski RA. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res. (2001) 29:221–2. doi: 10.1093/nar/29.1.221
34. da Silva TU, de Carvalho Pougy K, Albuquerque MG, da Silva Lima CH, de Paula MaChado S. Development of parameters compatible with the CHARMM36 force field for [Fe4S4] 2+ clusters and molecular dynamics simulations of adenosine-5’-phosphosulfate reductase in GROMACS 2019. J Biomolecular Structure Dynamics. (2022) 40:3481–91. doi: 10.1080/07391102.2020.1847687
35. Albekairi TH, Alshammari A, Alharbi M, Alshammary AF, Tahir Ul Qamar M, Anwar T, et al. Design of a multi-epitope vaccine against tropheryma whipplei using immunoinformatics and molecular dynamics simulation techniques. Vaccines. (2022) 10:691. doi: 10.3390/vaccines10050691
36. Brazzoli M, Piccioli D, Marchetti F. Challenges in development of vaccines directed toward antimicrobial resistant bacterial species. Hum Vaccin Immunother. (2023) 19:2228669. doi: 10.1080/21645515.2023.2228669
37. Confer AW, Ayalew S. The OmpA family of proteins: roles in bacterial pathogenesis and immunity. Veterinary Microbiol. (2013) 163:207–22. doi: 10.1016/j.vetmic.2012.08.019
38. Coutinho CP, Dos Santos SC, Madeira A, Mira NP, Moreira AS, Sá-Correia I. Long-term colonization of the cystic fibrosis lung by Burkholderia cepacia complex bacteria: epidemiology, clonal variation, and genome-wide expression alterations. Front Cell infection Microbiol. (2011) 1:12. doi: 10.3389/fcimb.2011.00012
39. Loeven NA, Perault AI, Cotter PA, Hodges CA, Schwartzman JD, Hampton TH, et al. The burkholderia cenocepacia type VI secretion system effector tecA is a virulence factor in mouse models of lung infection. mBio. (2021) 12:e0209821. doi: 10.1128/mBio.02098-21
40. De Soyza A, Meachery G, Hester KL, Nicholson A, Parry G, Tocewicz K, et al. Lung transplantation for patients with cystic fibrosis and Burkholderia cepacia complex infection: a single-center experience. J Heart Lung Transplant. (2010) 29:1395–404. doi: 10.1016/j.healun.2010.06.007
41. Lin J, Huang S, Zhang Q. Outer membrane proteins: key players for bacterial adaptation in host niches. Microbes infection. (2002) 4:325–31. doi: 10.1016/S1286-4579(02)01545-9
42. Khalid K, Poh CL. The promising potential of reverse vaccinology-based next-generation vaccine development over conventional vaccines against antibiotic-resistant bacteria. Vaccines. (2023) 11:1264. doi: 10.3390/vaccines11071264
43. Aslowayeh N, Albutti A, Al-Shouli ST. Reverse vaccinology and immunoinformatic assisted designing of a multi-epitopes based vaccine against nosocomial burkholderia cepacia. Front Microbiol. (2022) 13. doi: 10.3389/fmicb.2022.929400
44. Chandler JC, Sutherland MD, Harton M, Molins CR, Anderson R, Heaslip DG, et al. Francisella tularensis LVS surface and membrane proteins as targets of effective post-exposure immunization for tularemia. J Proteome Res. (2014) 14:664–75. doi: 10.1021/pr500628k
45. Leow CY, Kazi A, Ismail CMKH, Chuah C, Lim BH, Singh KKB. Reverse vaccinology approach for the identification and characterization of outer membrane proteins of shigella flexneri as potential cellular- and antibody-dependent vaccine candidates. Clin Exp Vaccine Res. (2020) 9:15. doi: 10.7774/cevr.2020.9.1.15
46. Soltan MA, Elbassiouny N, Gamal H, Elkaeed EB, Eid RA, Eldeen MA, et al. In silico prediction of a multitope vaccine against moraxella catarrhalis: reverse vaccinology and immunoinformatics. Vaccines. (2021) 9:669. doi: 10.3390/vaccines9060669
47. Irfan M, Khan S, Hameed AR, Al-Harbi AI, Abideen SA, Ismail S, et al. Computational based designing of a multi-epitopes vaccine against burkholderia mallei. Vaccines. (2022) 10:1580. doi: 10.3390/vaccines10101580
48. Panta P, Kumar S, Stafford CF, Billiot CE, Douglass MV, Herrera CM, et al. A dedA family membrane protein is required for burkholderia Thailandensis colistin resistance. Front Microbiol. (2019) 10. doi: 10.3389/fmicb.2019.02532
49. Khakhum N, Bharaj P, Myers JN, Tapia D, Kilgore PB, Ross BN, et al. Burkholderia Pseudomallei Δ tonB Δ hcp1 Live Attenuated Vaccine Strain Elicits Full Protective Immunity Against Aerosolized Melioidosis Infection. Msphere. (2019) 4: e00570-18. doi: 10.1128/mSphere.00570-18
50. Paul S, Mukherjee T, Das K. Coagulation protease-driven cancer immune evasion: potential targets for cancer immunotherapy. Cancers (Basel). (2024) 16: 1568. doi: 10.3390/cancers16081568
51. Lee DH, Choi SL, Rha E, Kim SJ, Yeom SJ, Moon JH, et al. A novel psychrophilic alkaline phosphatase from the metagenome of tidal flat sediments. BMC Biotechnol. (2015) 15:1. doi: 10.1186/s12896-015-0115-2
52. Parveen S, Shen J, Lun S, Zhao L, Koleske B, Leone RD, et al. Glutamine metabolism inhibition has dual immunomodulatory and antibacterial activities against Mycobacterium tuberculosis. bioRxiv. (2023) 14:7427. doi: 10.1101/2023.02.23.529704
53. Muruato LA, Tapia D, Hatcher CL, Kalita M, Brett PJ, Gregory AE, et al. Use of reverse vaccinology in the design and construction of nanoglycoconjugate vaccines against Burkholderia pseudomallei. Clin Vaccine Immunol. (2017) 24:e00206-17. doi: 10.1128/CVI.00206-17
54. Shahab M, Hayat C, Sikandar R, Zheng G, Akter S. In silico designing of a multi-epitope vaccine against Burkholderia pseudomallei: reverse vaccinology and immunoinformatics. J Genet Eng Biotechnol. (2022) 20:100. doi: 10.1186/s43141-022-00379-4
55. Grund ME, Kramarska E, Choi SJ, McNitt DH, Klimko CP, Rill NO, et al. Predictive and experimental immunogenicity of burkholderia collagen-like protein 8-derived antigens. Vaccines (Basel). (2021) 9:1219. doi: 10.3390/vaccines9111219
56. Irudal S, Scoffone VC, Trespidi G, Barbieri G, D’Amato M, Viglio S, et al. Identification by reverse vaccinology of three virulence factors in burkholderia cenocepacia that may represent ideal vaccine antigens. Vaccines (Basel). (2023) 11: 1039. doi: 10.3390/vaccines11061039
Keywords: Bcc, reverse vaccinology, in silico, multi-epitope vaccine, toll-like receptors
Citation: Ghorbani D, Beig M, Noori Goodarzi N, Sholeh M, Shahbazi B, Moghaddam Y and Badmasti F (2025) In silico development of a multi-epitope-based vaccine against Burkholderia cepacia complex using reverse vaccinology. Front. Virol. 5:1520109. doi: 10.3389/fviro.2025.1520109
Received: 01 November 2024; Accepted: 06 January 2025;
Published: 24 January 2025.
Edited by:
Rinki Kumar, The Pennsylvania State University, United StatesReviewed by:
Chandru Subramani, University of Texas Medical Branch at Galveston, United StatesCopyright © 2025 Ghorbani, Beig, Noori Goodarzi, Sholeh, Shahbazi, Moghaddam and Badmasti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Farzad Badmasti, ZmJhZG1hc3RpMjAwOEBnbWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.