 Fatemeh Ebrahimi1,2†
Fatemeh Ebrahimi1,2† Reyhaneh Rasizadeh2,3,4†
Reyhaneh Rasizadeh2,3,4† Shabnam Sharaflou1
Shabnam Sharaflou1 Parisa Shiri Aghbash2,4
Parisa Shiri Aghbash2,4 Ali Shamekh2,4
Ali Shamekh2,4 Abolfazl Jafari-Sales3,5
Abolfazl Jafari-Sales3,5 Hossein Bannazadeh Baghi2,3,4*
Hossein Bannazadeh Baghi2,3,4*- 1Student Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran
- 2Department of Virology, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran
- 3Infectious and Tropical Diseases Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
- 4Immunology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
- 5Department of Microbiology School of Basic Sciences, Kazerun Branch, Islamic Azad University, Kazerun, Iran
The Epstein-Barr viwrus (EBV) is a common herpesvirus that affects more than 90% of people worldwide. Even while EBV infections are frequently asymptomatic, they can cause autoimmune diseases and a number of cancers, especially in those with impaired immune systems. The intricate relationships between EBV and other coinfecting pathogens, including as human immunodeficiency virus (HIV), human papilloma virus (HPV), cytomegalovirus (CMV), and Plasmodium species, are examined in this study. We investigated the fundamental processes of these coinfections, their effects on the course of the disease, and their practical ramifications. The study reviewed how co-infections with EBV might modify immune responses, promote oncogenesis, and make treatment plans more challenging. In this review, we also discussed current therapeutic strategies, such as targeted molecular interventions, EBV vaccines, and adoptive T-cell therapy. The review underscores the need for more research to provide more focused and effective therapies that address the mutually reinforcing effects of numerous infections in disorders linked with EBV.
1 Introduction
The Epstein-Barr virus (EBV) is a widespread virus with a global seroprevalence rate of almost 90% (1). This enveloped virus contains a linear, double-stranded DNA genome of approximately 170 kilo base pairs, encoding over 80 proteins and 46 functional small untranslated RNAs (2). Despite its prevalence, EBV has evolved mechanisms for long-term survival and persistence in human hosts (3). While many infected individuals remain asymptomatic throughout their lives, EBV infection in immunocompromised individuals can lead to serious health conditions, including malignancies and organ failure (3).
EBV was initially identified through the electron microscopy examination of Burkitt’s lymphoma tumor cells in 1964 (4). Subsequently, four years later, the virus was conclusively linked to infectious mononucleosis (IM); while its etiological significance in the disease was firmly established by 1973 (4). Furthermore, the initial virus directly linked to a human cancer was the widely prevalent B-lymphotropic herpesvirus, commonly referred to as EBV (5). Since its initial identification and correlation with Burkitt lymphoma (BL) fifty years ago, EBV has been associated with various lymphomas and epithelial malignancies (5).
The EBV presents a complex interplay of prevalence, clinical manifestations, and geographical considerations (6). Most of the malignancies caused by EBV are seen in immunocompromised patients; while they can also occur sporadically in individuals without apparent immunodeficiency (6). There are geographically unique zones where certain malignancies linked to EBV are mostly recorded. In regions of sub-Saharan Africa and New Guinea where holoendemic falciparum malaria is rife, endemic Burkitt lymphoma is observed (7). Native Americans and people from Southeast Asia are more likely to develop nasopharyngeal carcinoma due to environmental and dietary exposure to substances that may promote tumor growth (7). East Asia also presents a higher incidence of EBV-related T-cell/Natural killer (NK)-cell malignancies, suggesting multifactorial risks involving genetic and environmental influences (8).
Many B-cell-derived lymphoid neoplasms, such as Burkitt’s lymphoma, lymphomas developing in immunocompromised patients (including human immunodeficiency virus (HIV)-associated and post-transplant lymphomas), and Hodgkin’s lymphoma are linked to EBV (9, 10). Furthermore, EBV has been associated to nasopharyngeal cancers, a subgroup of gastric malignancies, and several T-cell lymphomas, including angioimmunoblastic T-cell lymphoma, extranodal nasal-type NK/T-cell lymphoma, and other uncommon histotypes (11). It infects squamous epithelium of the oropharynx and nasopharynx, glandular epithelium of the thyroid, stomach, and salivary glands, B and T lymphocytes, follicular dendritic cells, smooth muscle cells, and many other cells and tissues (12, 13). The majority of EBV-associated cancers originate from B cells, which constitute the main cellular reservoir for EBV persistence. EBV encodes a wide range of products that facilitate EBV infection, immortalization, and transformation by imitating or activating cytokines, signal transducers, and antiapoptotic molecules (12, 13).
In clinical settings, EBV is commonly associated with IM, characterized by systemic symptoms such as fever, sore throat, exhaustion, malaise, and lymphadenopathy (14). Over 90% of IM cases are caused by EBV (15). IM exhibits a widespread global prevalence, predominantly affecting adolescents and young adults from affluent socioeconomic backgrounds in industrialized nations, whereas primary infection typically manifests later in life, usually between the ages of 10 and 30 (16). Conversely, its occurrence is less frequent in tropical and underdeveloped regions, as well as among individuals with lower socioeconomic status. In these populations, EBV infection typically occurs asymptomatically during early childhood (4).
Moreover, EBV coinfection with other pathogens can lead to severe complications from both pathogens, which can reactivate under immunosuppression (Figure 1). This dual infection not only complicates the clinical presentation but also depresses the host immune response, increasing the risk of recurrent infections and highlighting the need for greater attention to the clinical features and management of such coinfections (17). Furthermore, EBV coinfection may exacerbate thrombocytopenia by triggering complex immune responses that lead to platelet depletion, highlighting the intricate relationship between viral infections and hematological abnormalities (18).
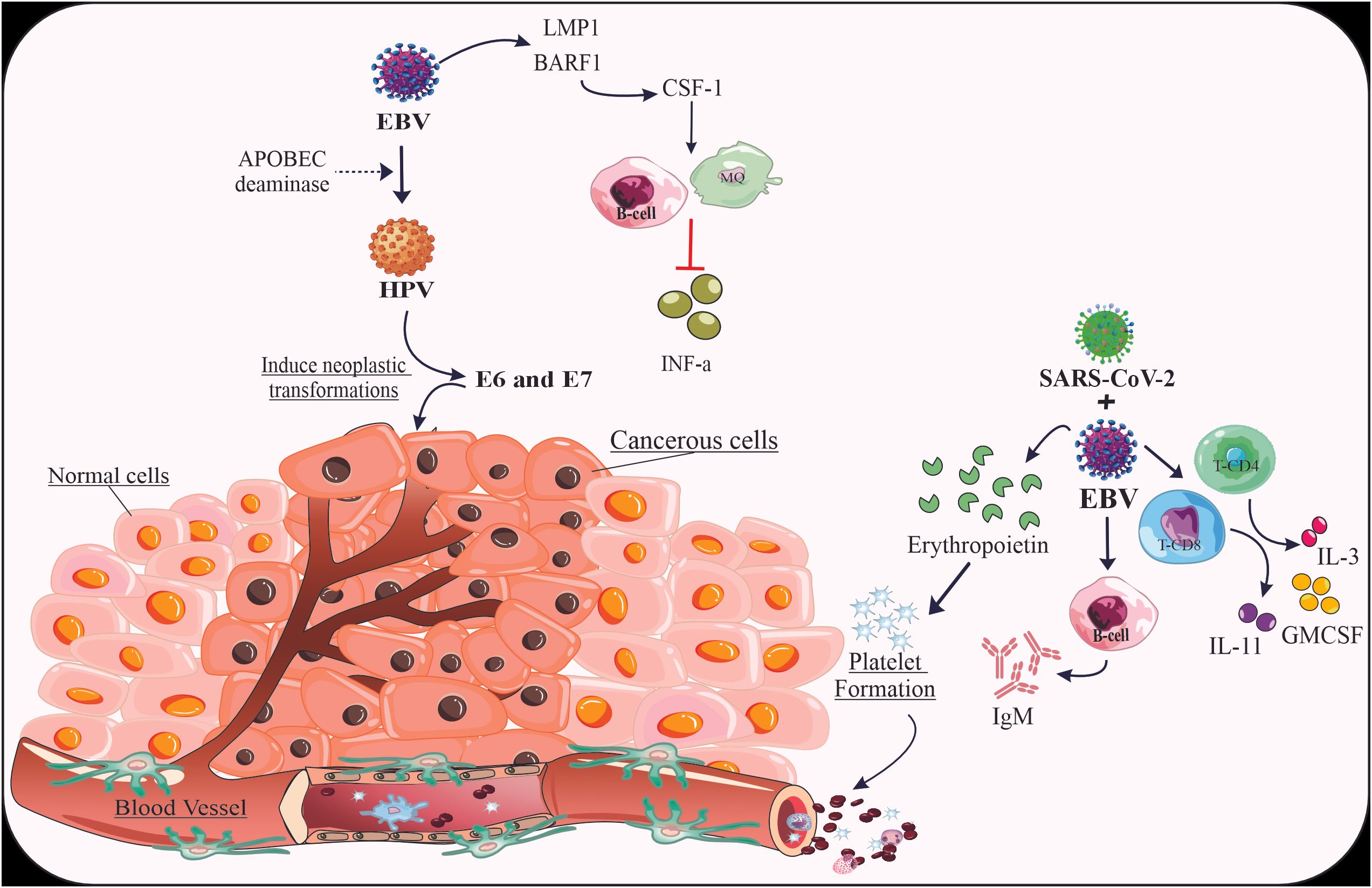
Figure 1. The mechanisms related to the co-infection of EBV virus with HPV and SARS-CoV-2. The coinfection of HPV and SARS-CoV-2 with EBV can promote the severity of malignancy by inducing the expression of genes and cytokines related to specific pathways.
The complex interactions between EBV and various coinfecting pathogens have emerged as a critical area of research in recent years. These coinfections, involving viruses such as HIV, human papilloma virus (HPV), and cytomegalovirus (CMV), as well as parasites like Plasmodium species, can significantly alter disease progression, immune responses, and clinical outcomes. The synergistic effects of EBV coinfections may contribute to the development and exacerbation of various malignancies and other associated conditions. Understanding these intricate relationships is crucial for developing more effective diagnostic, preventive, and therapeutic strategies for EBV-related diseases. The aim of this study is to provide a comprehensive review of EBV coinfections with various pathogens, focusing on their mechanisms of interaction, impact on disease progression, and clinical implications. Additionally, this study seeks to explore current and potential future treatment approaches for EBV-associated diseases in the context of coinfections, highlighting the need for targeted therapeutic strategies that address the complex interplay between multiple pathogens.
2 EBV infection
2.1 Primary infection
A primary EBV infection can result in a range of clinical symptoms, and trigger a number of cancers (19). Typically, affected individuals have peripheral blood lymphocytosis, containing abnormal lymphocytes (20). Usually symmetrical, the lymphadenopathy affects the posterior cervical chain more predominantly compared to the anterior chain (20). Moreover, along with less common signs including palate petechiae, periorbital or palpebral edema, and maculopapular or morbilliform rashes, severe weariness may be evident (21). Up to 50% of the patients develop splenomegaly; while hepatomegaly and jaundice are less common (22). Nonetheless, the disease presents in a number of various forms. Some patients with IM exhibit the so-called “glandular” form of the infection, in which the size of the lymph nodes is disproportionate to the pharyngeal symptoms; others experience a systemic form of the infection, characterized by a predominant fever and fatigue, with either mild or nonexistent lymphadenopathy and pharyngitis (23). Moreover, in several cases, hepatitis coexists with other characteristic signs of immune modulation (24). According to a study, prior viral infections, such as the influenza virus, may have an impact on the severity and commencement of host’s immune modulation, increasing the proportion of T cells that mount an immune response against the EBV infection (25). The majority of people who have a primary EBV infection recover without any adverse events and show a good level of long-lasting immunity (26). While weariness frequently lasts for weeks or months, acute symptoms usually subside within one to two weeks (26).
2.2 EBV latency
EBV establishes latency following primary infection, which is a critical aspect of its lifecycle (27). During this phase, the virus remains dormant in B lymphocytes and other cell types, allowing it to evade the host immune response (27). The establishment of latency involves several mechanisms, including the infection of naïve B cells, immune evasion strategies, and the expression of specific latency-associated genes (27).
The establishment of latency during EBV infection is a complex process that involves several key steps and mechanisms (28). Initially, EBV infects differentiated epithelial cells, particularly in the stratified squamous epithelium of the oral cavity or pharynx, which serves as the primary site for infection and viral multiplication (28). However, EBV predominantly establishes latency in B lymphocytes, which are the main reservoir for the virus (28). This preference for B cells is a distinctive feature of EBV compared to other herpesviruses, such as herpes simplex virus (HSV), which typically enter a lytic cycle upon infection of many cell types (28).
Upon infection of primary B cells, EBV triggers the expression of both latent and lytic genes during what is termed the pre-latent phase (28). This phase can last for several weeks, during which the virus expresses lytic genes without producing infectious viral particles. This initial expression of lytic genes may help the virus evade cell death and promote B cell immortalization (28). Key latent genes, including the EBV nuclear antigens (EBNAs) and latent membrane proteins (LMPs), are expressed shortly after infection (28). Notably, EBNA-2 plays a crucial role in reprogramming the resting B cell transcriptome, facilitating the transition to latency (28).
Depending on the specific viral genes expressed, there are three main types of EBV latency:
Type I Latency: In this type, only the EBNA-1 nuclear antigen and two small non-coding RNAs called EBER-1 and EBER-2 are expressed (29).
Type II Latency: In addition to EBNA-1, EBER-1, and EBER-2, the virus also expresses three membrane proteins: LMP-1, LMP-2A, and LMP-2B (29).
Type III Latency: This is the most immunogenic type, with the expression of all six nuclear antigens (EBNA-1, -2, -3A, -3B, -3C, and -LP), all three latent membrane proteins (LMP-1, -2A, -2B), and the EBER RNAs (29).
Type I latency expresses only the EBNA-1 antigen and EBERs, primarily associated with endemic Burkitt’s lymphoma and allowing immune evasion, but can reactivate under immunosuppression (30). Type II latency includes additional proteins (LMP-1 and LMP-2), linked to Hodgkin’s lymphoma and nasopharyngeal carcinoma, enhancing oncogenic potential through altered immune responses (30). Type III latency expresses all nuclear and membrane proteins, found in immunoblastic lymphomas, leading to aggressive disease due to its high immunogenicity and severe consequences in immunocompromised patients. Understanding these latency types is crucial for managing EBV-related diseases effectively (31). During latency, the viral genome is replicated only once per cell cycle, ensuring its maintenance in the host cells without causing active disease (30).
2.3 Congenital and perinatal infections
Less than 5% of pregnant women are susceptible to EBV, making intrauterine infections with the virus uncommon (32). Furthermore, there has been no evidence of congenital malformations in the offsprings of women who did develop primary EBV infection during pregnancy, according to prospective investigations of susceptible (i.e., seronegative) women (33). There have been rare reports of infants with congenital malformations (biliary atresia, congenital heart disease, hypotonia, micrognathia, cataracts, and thrombocytopenia) and their possible association with EBV infection; nevertheless, the general evidence does not support EBV as a major cause of congenital infection (34). In particular, there has been no signs of EBV infection found in cord blood samples or in extensive investigations involving children with congenital abnormalities (34).
2.4 Additional symptoms
EBV can impact almost every organ system and has been linked to a wide range of illness presentations, including pancreatitis, myocarditis, pneumonia, mesenteric adenitis, myositis, glomerulonephritis, and genital ulcers (35). Other unusual symptoms that have also been linked to primary EBV infection include: Guillain-Barré syndrome, meningoencephalitis, aseptic meningitis, transverse myelitis, peripheral neuritis, and optic neuritis (36). Hemolytic anemia, thrombocytopenia, aplastic anemia, thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome, and disseminated intravascular coagulation are examples of its associated hematologic disorders (37). Furthermore, EBV infection can lead to a variety of acute side effects as well as more chronic ones in some hosts (38). The complications of EBV (39) are summarized in Table 1. The proposed pathogenesis of EBV co-infection with other pathogens is outlined in Table 2.
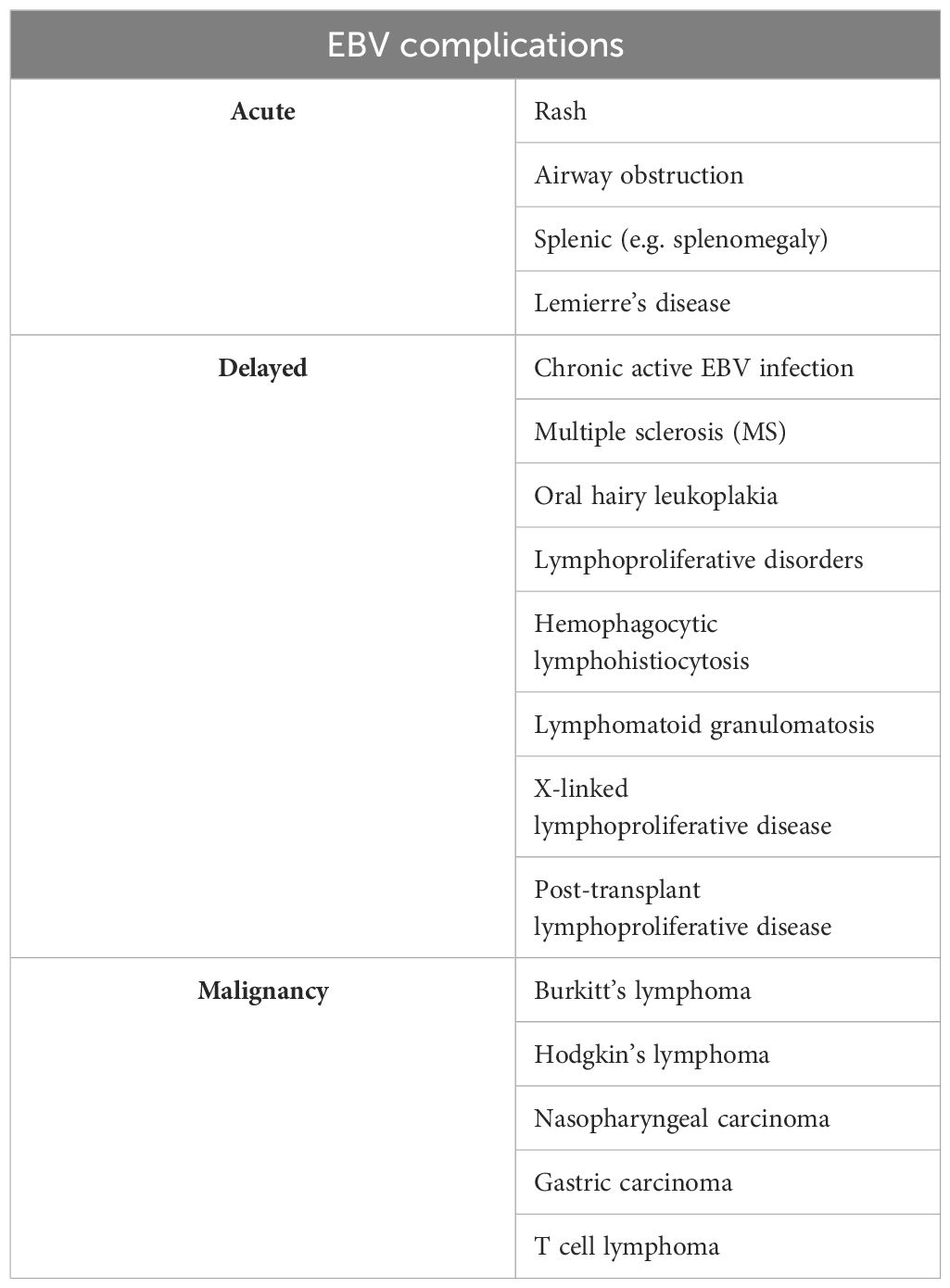
Table 1. The complications of EBV.
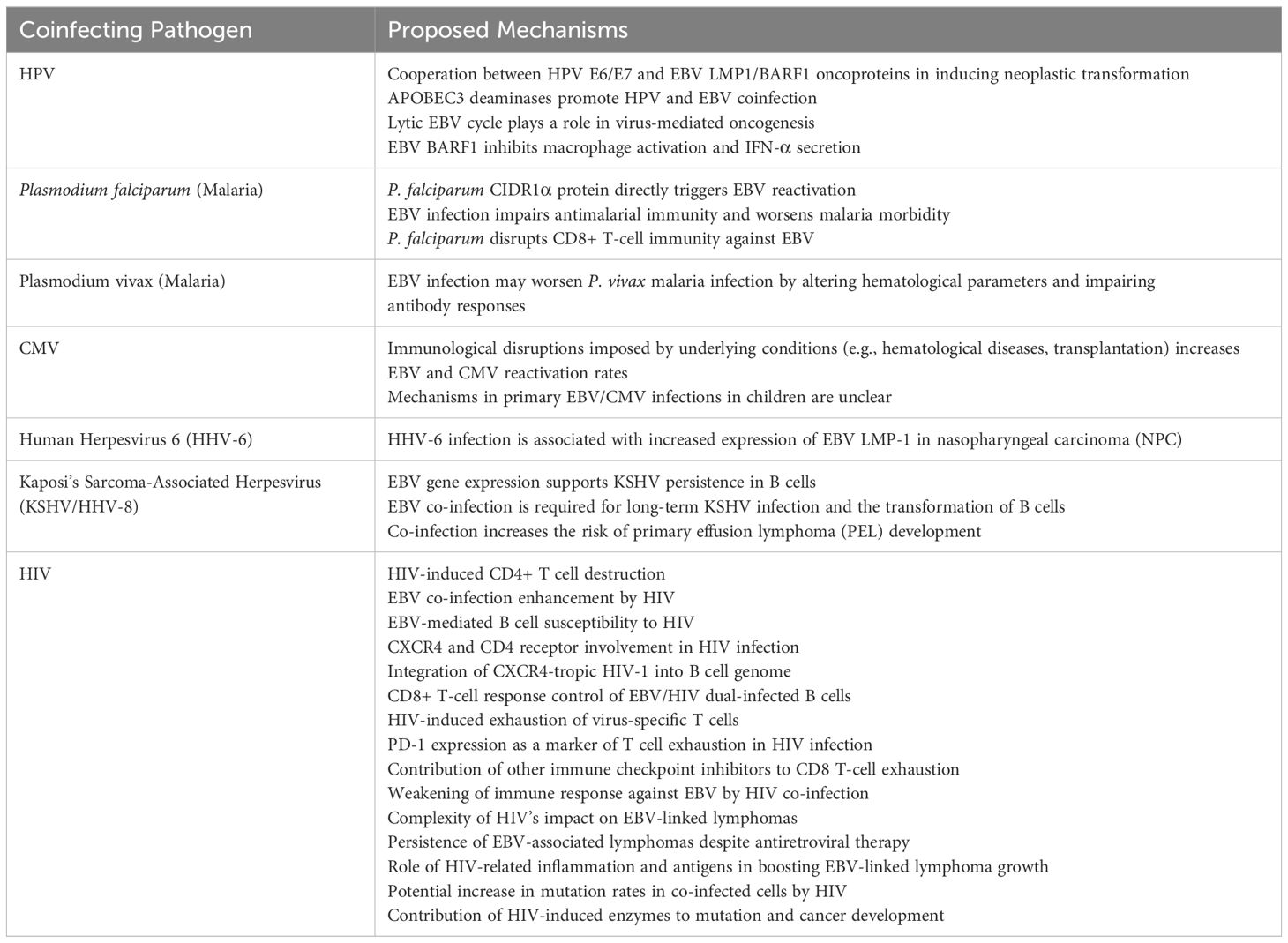
Table 2. Proposed pathogenesis of EBV coinfection with other pathogens.
3 EBV coinfection with HPV
Numerous groups of DNA and RNA viruses that are required, but are usually insufficient, for the development of cancers are classified as oncogenic viruses (40). Around 12% of malignancies globally are caused by viral infections, the great majority (>85%) of which occur in impoverished nations and regions (41, 42).
Two DNA viruses, HPV and EBV, were reported to be connected to 38% of all malignancies brought on by viruses (43). Coinfections, like HPV/EBV, can heighten the risk of cancer by impacting immune function, playing a role in cancer development, and affecting how cancer responds to treatment (44). Specifically, the presence of both HPV and EBV might influence the onset of prostate cancer by altering cellular behavior (44).
The viruses connected to the majority of cancer cases are caused by HPVs, which include cervical carcinoma as well as a number of other epithelial cancers (45, 46). There is a growing likelihood that a single patient will have infections from two or more different virus types (47, 48). It is not necessary for all the viral infections to coexist within the carcinoma cells for implementing their carcinogenic effects (48). Over the past 20 years, significant emphasis has been made on the observation that individuals with HIV infection are at a higher risk of contracting some malignancies, such cervical carcinoma as well as certain non-Hodgkin lymphomas (48). Notably, malignancies that arise in the presence of HIV-induced immunosuppression are often associated with other infections by cancer-causing viruses such as HPV and EBV (49). Nevertheless, little is known about the specific mechanism of the cooperation, and more research is required to determine whether this cooperation is essential for the growth of tumors (50–53). It is commonly known that HPV affects the prognosis and carcinogenesis of cervical cancer; while numerous researchers have discovered that HPV infection is also an etiologic factor for NPC (50–53). Furthermore, according to Whitaker et al. (54), both HPV and EBV gene sequences have been detected across normal, benign, and malignant specimens in prostate tissues. Recent experimental findings propose a collaborative role between HPV and EBV in promoting cell proliferation in cervical cells (54). However, the implications of their presence in prostate tissues remain uncertain (54). While it’s possible they are harmless, HPV type 18, known for its high oncogenic potential, may be associated with certain prostate cancers (54). Furthermore, the identification of HPV-associated koilocytes in prostate cancer specimens suggests prior HPV infection and hints at its potential oncogenic effects in the development of prostate cancer (54). Additionally, there are reports indicating the simultaneous presence of EBV and HPV in various epithelial malignancies, including some cases of breast cancer (55).
The beginning of a neoplastic transformation and carcinogenesis is significantly influenced by the co-infection of HPV and EBV (56). It’s interesting to note that there is a relationship between EBV and HPV (whole viruses) as well as a relationship between HPV and EBV oncoproteins (for example, latent membrane protein-1 (LMP1) decreases apoptosis in vitro) (57). EBV, like high-risk HPVs, can infect and immortalize human primary epithelial cells in vitro (58). Among the EBV genes consistently expressed in the infected cells are EBERs, EBNA1, LMP1, LMP2A, BARF0, and BARF1 (58). Notably, only LMP1 and BARF1 have been found to be capable of inducing malignant transformation in rodent fibroblasts, classifying them as viral oncogenes (58). BARF1 is consistently expressed at high levels in nasopharyngeal cancer, EBV-associated gastric carcinomas, and EBV-immortalized epithelial cells in vitro (58). Furthermore, BARF1’s interaction with colony-stimulating factor-1 inhibits macrophage activation and interferon-alpha secretion in EBV-infected B-cells. It’s hypothesized that EBV oncoproteins, along with HPV’s E6/E7 oncoproteins, cooperate to induce neoplastic transformations (58). Double transgenic cells and animal models are considered the best approaches to test this hypothesis effectively (58). Moreover, APOBEC3 deaminases may facilitate the co-infection of EBV and HPV (59).
According to research on the functions of chromatin, dynamic interactions between the chromatins of the virus and the host are crucial in regulating viral infections (60–62). Infections in addition to the way that proteins are encoded by viruses, interact to modify the chromatin (60–62). These interactions are particularly important for a number of epigenetic processes, including higher-order chromosomal structures that regulate the production and maintenance of viral episomes following viral infections, chromatin assembly, and histone and DNA modifications (60–62). EBV and HPV seem to cooperate in promoting cervical carcinogenesis through both direct and indirect mechanisms (63). Directly, EBV may infect epithelial cells alongside HPV, potentially synergizing with HPV oncoproteins (63). The combined effects of viral proteins like EBV’s LMP1 and HPV’s E6/E7 can lead to increased cell proliferation, resistance to apoptosis, and enhanced invasiveness (63). These proteins work together to disrupt cell cycle controls, impair DNA damage responses, and promote EMT (63). Indirectly, EBV infection of tumor-infiltrating lymphocytes may generate local immunosuppression, helping HPV-infected cells evade immune surveillance (63). Additionally, EBV proteins like BARF1 may further contribute to immune evasion and oncogenic changes (63). The coinfection appears to favor the establishment of EBV latency in cervical epithelial cells, possibly facilitated by HPV-induced DNA damage, leading to long-term viral persistence and continued oncogenic effects (63). Cancers caused by oncogenic viruses are a major worldwide concern (64). In the global battle against cancer, notable advancements are still being achieved by preventing the production and distribution of secure and efficient vaccinations against these viruses, as well as by figuring out the oncogenic pathways these viruses employ, making it possible to create novel therapeutic approaches for various human cancers linked to viruses (64). Despite the fact that HPV vaccines exist (64), there are no effective ones available for EBV (almost 50 years after EBV’s discovery).
The study conducted by Lu et al. (65) explores DZ1, a DNAzyme targeting the LMP1 gene, as a therapy for EBV-associated carcinomas. DZ1 effectively reduces LMP1 expression in B95-8 cells, inhibits cell growth, and induces apoptosis by downregulating the Bcl-2 gene (65). There is hope for DZ1 therapeutic techniques for the treatment of EBV-related malignancies as the DZ1 treatment is deemed safe and effective in several models (65). HPV and EBV coinfections increase cancer risk by altering immune responses and promoting tumor growth. HPV is linked to cervical and nasopharyngeal cancers, while both viruses may influence prostate and breast cancers. Emerging treatments, like DNAzyme, show promise in targeting EBV-related cancers, but more research is needed to understand their combined oncogenic effects.
4 EBV coinfection with SARS-CoV-2
Due to the great importance of the diseases caused by the two mentioned viruses in children, we examine this co-infection only in children and refrain from examining it in adults (66). Coronavirus disease 2019 (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (67). Most COVID-19 cases in children were mild and the treatment consisted of supportive care; only a small number needed hospitalization and mortality rate was lower than 0.1% of diagnosed children (68–71). As we are discovering more about SARS-CoV-2 infection each day, differences between adults and pediatric disease are probably due to changes within both the immune function and the angiotensin-converting enzyme (ACE) 2 receptor, used by the virus to enter type II pneumocytes in the lung (72, 73). The immune system of children is highly prepared to novel pathogens, due to high levels of innate IgM antibodies with broad reactivity, in addition to the production of the anti-inflammatory interleukin (IL)-10 by the neonatal B cells (72). Other probable explanations are alternations in T cell populations in adults due to continuous antigen stimulation and thymic involution, varied levels of ACE-2 expression in children, and the simultaneous presence of other viruses in the respiratory mucosa of children, competing with SARS-CoV-2 (74). Besides, children have fewer comorbidities and a stronger pulmonary regenerative potential than adults (75). A 5-year-old boy with prolonged fever was admitted to hospital. Initially treated for tonsillitis, his symptoms worsened. Tests revealed COVID-19 IgM antibodies and acute EBV infection (75). The child had respiratory and gastrointestinal symptoms, lymphadenitis, and thrombocytosis. Though not fully meeting criteria for multisystem inflammatory syndrome in children (MIS-C), the dual infection caused severe inflammation. With symptomatic treatment, the child’s condition improved (75).
Pediatricians find it challenging to distinguish between COVID-19 infection and other possible childhood viruses as the SARS-CoV-2 pandemic continues to spread around the world (76). Nearly all medical resources are focused on treating COVID-19 infections, while diagnosing or anticipating the illness’s consequences in children remains difficult (76). It is already well known that children have received fewer COVID-19 diagnoses than adults (76). According to the World Health Organization, most of the pediatric cases in China have been moderate; nevertheless, 2.5% of pediatric cases have been reported to have serious illness (76). For youngsters, fever, pharyngeal erythema, and cough are the most commonly reported signs and symptoms. Other less typical indications and symptoms consist of diarrhea, weariness, rhinorrhea, nausea, and congestion in the nose (71). A tiny percentage exhibit dyspnea, a high temperature that doesn’t subside, lethargy, elevated enzyme levels, and serious illness (71). A high incidence rate of MIS-C has been recorded since May 2020 in a number of highly endemic countries (77–80). Fever, increased inflammatory markers, and organ dysfunction not linked to another infectious etiology, are all included (81). 25 days is the median time between the beginning of COVID-19 symptoms and MIS (81). When compared to nasopharyngeal reverse transcription-polymerase chain reaction (RT-PCR), the greater percentage of positive serologic testing is an indication of a disease-related late consequence (66, 77, 81–83). Apart from fever, the most typical MIS manifestations are cardiovascular, mucocutaneous (rash, mucous membrane alterations, conjunctival injection), respiratory (including sore throat), headache, limb and periorbital edema, and gastrointestinal (diarrhea, vomiting, abdominal discomfort) (80–82). Laboratory results that are related to these conditions include hypertriglyceridemia, elevated D-dimer and fibrinogen, thrombocytopenia, lymphopenia, elevated troponin and N-terminal pro-B-type natriuretic peptide (NT-proBNP), and elevated inflammation markers (neutrophilia, C-reactive protein, ferritin, and erythrocyte sedimentation rate) (83). There are certain patients who fit in the category of macrophage activation syndrome (MAS) (75). The presenting child had a prolonged fever lasting more than ten days, cough, diarrhea, exhaustion, cervical lymphadenopathy, and pharyngeal erythema in addition to a negative reverse transcriptase protein chain reaction (RT-PCR) and a positive serology for COVID-19 (increased IgM levels, which are higher during weeks two and three of the illness) (75). It was first suspected that certain inflammatory markers, such as C-reactive protein (CRP) and d-Dimer, were elevated in which we in favor of MIS-C (75). Nevertheless, several confounding factors, such as lymphocytosis and thrombocytosis, were detected in laboratory data that were inconsistent with MIS-C. Children with confirmed COVID-19 do not exhibit a consistent pattern of laboratory abnormalities; however, hospitalized patients frequently have lymphopenia (caused by the destruction of infected T lymphocyte cells) and thrombocytopenia along with other abnormalities related to coagulation parameters (75). Conversely, EBV, a common pathogen that causes illness in youngsters, has similar symptoms with COVID-19. The majority of people on the planet are infected with EBV, which is common in the natural environment (75). The primary infection in young children typically manifests as asymptomatic or causes an acute illness that is frequently misdiagnosed as EBV-related (75). It can also manifest as the full-blown IM clinic, which includes fever, pharyngitis, lymphadenopathy, hepato-splenomegaly, and fatigue (75). Recovery from IM is sluggish, and the typical length is 16 days, far longer than other acute viral diseases (84). During a primary EBV infection, a strong innate and adaptive immune response takes place. The initial line of defense is the innate immune system, causing fever, exhaustion, and headache (84). Both CD4 and CD8 T cells respond well to EBV antigens; however, it is believed that CD8 T cells specific to EBV lytic antigens are primarily responsible for the extensive lymphocytosis in the blood which is a characteristic sign of IM (85). SARS-CoV-2 and EBV are two viruses that alter the immune system. When SARS-CoV-2 or EBV infections occur, thrombocytosis is not usually the case; instead, mild thrombocytopenia is seen (18, 86). Children’s thrombocytosis is typically reactive, more common during the healing stage of an infection or inflammation, and is usually transitory, going away as soon as the main stimulus stops (86). Mainly, the increased release of several cytokines in response to infections is the mechanism underlying the reactive thrombocytosis (86). Numerous cytokines, including IL-3, IL-11, granulocyte-macrophage colony-stimulating factor, and erythropoietin, can stimulate platelet formation; however, thrombopoietin and IL-6, which are first raised in response to infections, play the most important roles (86). Thrombocytosis in this instance is an amplification of the body’s reaction to the two illnesses together (86). Even with the remarkably elevated platelet count—which can occasionally surpass 1,000,000 cells/mm3—thrombotic and/or hemorrhagic consequences are very remarkable (86). In summary, distinguishing COVID-19 from EBV in children is challenging due to overlapping symptoms. Co-infection can cause immune system alterations, including reactive thrombocytosis. Careful diagnosis and further research are essential for managing these cases effectively.
5 EBV coinfection with HIV
Acquired immune deficiency syndrome (AIDS) is caused by HIV infection, which destroys CD4+ T cells, leading to immunodeficiency and susceptibility to infections (87). Moreover, the majority of individuals who contract HIV are already infected with EBV, as EBV infection typically occurs during childhood and adolescence (88). HIV co-infection boosts EBV replication and the development of EBV-related cancers (89). EBV transforms B cells, making them vulnerable to HIV-1 infection, particularly through CXCR4 and CD4 receptors. In laboratory experiments, CXCR4-tropic HIV-1 integrates into these B cells’ genome, similar to how it does in CD4+ T cells (90). A humanized mouse model has been developed to study EBV and HIV-1 interaction in vivo (90). These mice mimic the aspects of EBV and HIV infection observed in humans (90). In the mouse model, EBV/HIV dual-infected B cells were identified but were controlled by CD8+ T-cell immune response (90).
In HIV-1 infection, PD-1 expression on virus-specific T cells indicates immune exhaustion and disease progression, correlating with reduced CD8 T-cell function, higher viral load, and lower CD4 counts (91). Cytomegalovirus-specific CD8 T cells in the same individuals do not show increased PD-1 levels, suggesting HIV-specific defects due to antigen exposure (91). Long-term studies show that PD-1 levels decrease with ART, and long-term non-progressors (LTNPs) have lower PD-1 levels and better T-cell function (91). Other immune checkpoints like LAG3, TIM-3, and TIGIT also contribute to T-cell exhaustion. Co-expression of PD-1 with TIGIT is linked to disease progression, and multiple checkpoints correlate with HIV load and T-cell function (91).
The findings indicate that HIV co-infection weakens the immune response against EBV, particularly for high-immunogenic lymphomas (where all latent EBV proteins are expressed, known as latency III). However, for lymphomas linked to EBV latency I and II, like Burkitt’s lymphoma, Hodgkin’s lymphoma, and primary effusion lymphoma, the impact of HIV co-infection is more complex (92). These lymphomas also occur more frequently in individuals co-infected with HIV (92). Interestingly, their incidence has, however, not decreased, and for Burkitt’s and EBV-associated Hodgkin’s lymphoma, they have even increased after the introduction of antiretroviral therapy (93). Therefore, they now compose around half of the EBV-associated lymphomas in people living with HIV. Hodgkin’s and Burkitt’s lymphomas are believed to originate from germinal centers (93), a location where HIV can continue to proliferate even when receiving antiretroviral therapy (94). The inflammation caused by HIV, along with HIV-related antigens, might boost the growth of EBV-linked lymphoma cells, which are already prone to mutations (95, 96). This could lead to genetic changes like c-myc translocation, possibly triggered by the EBV (95, 96). In people with both viruses, HIV may increase mutation rates in certain cells, particularly those affected by specific HIV strains. Moreover, HIV infection produces enzymes that induce mutations, contributing to cancer development (95, 96). HIV boosts EBV replication and increases the risk of EBV-related cancers like Burkitt’s and Hodgkin’s lymphomas. HIV weakens the immune system, leading to higher incidence of these cancers, even with antiretroviral therapy.
6 EBV coinfection with Helicobacter
In recent years, bacterial and viral infections have been associated with the onset of gastric disorders such as gastric cancer (GC), mucosa-assisted lymphoid tissue (MALT) lymphoma, and chronic gastritis (97–101). Notably, the roles of Helicobacter pylori and EBV in stomach cancer development have been elucidated (10). The International Agency for Research on Cancer classified H. pylori as a group 1 carcinogen in 1994; while EBV keeps its genome within the host cells, eluding the immune system and preventing cell death, facilitating the process of neoplasia (102, 103). Currently, there is a discussion and developing evidence on the involvement of EBV infection in inflammatory gastrointestinal diseases (104–106). The development of gastric disorders is mostly attributed to the virulence factors of H. pylori, which include the genes cagA, vacA, and oipA. The most researched H. pylori virulence factor is called CagA, which is a polymorphic gene (107). The 3′ region of the gene has a variable number of repeat sequences. The CagA protein has Glu-Pro-IleTyr-Ala (EPIYA) motifs in all of its repeated regions. VacA ranks second most regarded as a virulence factor for H. pylori (107). The vacA gene structure is known to contain three distinct regions: the intermediate (i) region (i1 and i2), the middle (m) region (m1 and m2), and the signal (s) region (s1 and s2) (107). Ultimately, it was shown that OipA is a protein which induces a proinflammatory response and conditions the gastric epithelial cells’ ability to produce IL-8 (108). It is evident that eliminating the bacteria seems to reduce the incidence of stomach cancer in individuals who are at higher risks (109). The efficacy of eradication regimens may be impacted by resistance to antibiotics, especially Clarithromycin, a crucial medication in this process (110). Common clarithromycin resistance is explained by point mutations in the 23S rRNA gene (111).
After infecting cells in the oropharynx, EBV spreads to the lymphoid tissues and infects the present B lymphocytes (112). Research on the relationship between EBV and autoimmune diseases as well as its role as a triggering mechanism in MS have been conducted both in vitro and in vivo (113, 114). Patients with mononucleosis syndrome have also recently been reported to have secondary hemophagocytic lymphohistiocytosis (HLH) (115), which is characterized by hemophagocytosis and histiocyte proliferation as reported by Cascio et al. in cases of severe infection (115–118). Recent research demonstrating the potential of pathogenicity of co-infection with both H. pylori and EBV raises questions about the importance of the involved inflammatory responses to the tissue or the increased interaction between the EBV’s CagA protein and H. pylori, which promotes the activation of B lymphocytes passing through the stomach mucosa (119).
The study conducted in Sicily examined the prevalence of H. pylori and EBV co-infections, along with the presence of specific genetic markers and antibiotic resistance in stomach biopsies from adult patients with and without gastric illness (120). Among 24 patients with chronic gastritis (CGA) and 24 individuals without gastric disease (NGD), a significant co-infection rate was observed, with 42% of individuals harboring both pathogens, and a higher dual prevalence of 54% in CGA patients (120). The research identified key genetic markers, such as the cagA, vacA, and oipA genes, and resistance patterns in H. pylori, suggesting that childhood is a crucial period for acquiring these infections (120). This co-infection has the potential to jointly alter the gastric mucosa, thereby influencing the clinical outcomes in affected individuals (120).
Furthermore, the cagA gene is exclusively linked to s1i1m1 or s1i1m2 vacA gene mosaicism (109, 110). The study found that 80% of GCA patients and 90% of NGD patients had the “OFF” status of the oipA gene (120). High rates of H. pylori resistance to clarithromycin suggest that empirical therapy should be reconsidered in Sicily. Co-infection with H. pylori and EBV was prevalent, with 75% of NGD and 54% of GCA patients harboring both pathogens. Further research is needed to understand their role in gastric disease (120).
Moreover, using radiological and morphological investigations, the authors conclude by urging researchers to apply both new and existing knowledge within a multidisciplinary team investigation for a more clinical amplification of the microbiological features of the gastrointestinal tract (121, 122). The aim of virologist researchers is to provide an efficient vaccination-based prevention. Currently, the authors support uniform vaccination schedules as a means of promoting health (123, 124). H. pylori and EBV are linked to gastric disorders. H. pylori’s virulence factors (CagA, VacA, OipA) and EBV’s immune evasion both contribute to cancer risk. High co-infection rates and antibiotic resistance complicate treatment. More research and improved prevention strategies are needed.
7 EBV coinfection with malaria
Each year, over 200 million people worldwide contract Plasmodium infections, resulting in approximately 1 million deaths, primarily among children under the age of 5 and pregnant women (125). In sub-Saharan Africa, Plasmodium falciparum is the culprit behind over 90% of these infections. The spectrum of disease severity varies, from mild febrile illness to critical conditions like cerebral malaria and severe malarial anaemia (125).
One of the crucial aspects of P. falciparum’s biology is its capacity to make infected red blood cells (RBCs) stick to the walls of small blood vessels (126, 127). This adherence of parasitized cells leads to significant blockages in tissue blood flow (126, 127). Moreover, in severe malaria cases, there could be notable decreases in the flexibility of healthy, uninfected RBCs (126, 127).
Given their wide geographic distribution, EBV and Plasmodium coinfection is frequent in Africa (128). EBV and Plasmodium have long been associated with the emergence of endemic Burkitt’s lymphoma (eBL), the predominant pediatric cancer affecting children aged 5 to 9 in equatorial Africa (129, 130). This malignancy is a significant health concern in the region, with both pathogens playing pivotal roles in its development. While EBV is recognized as a contributing factor to eBL, particularly in its endemic form, Plasmodium’s involvement underscores the complexity of this malignancy’s etiology (129, 130). The coexistence of these two pathogens in equatorial Africa highlights the intricate interplay between infectious agents and the oncogenic processes leading to eBL, necessitating further investigation into their synergistic effects and potential therapeutic interventions (129, 130). In this region, children typically acquire EBV infection around the age of six months, coinciding with a decline in maternal antibodies that offer passive protection (131). This transition marks a period of heightened susceptibility to primary Plasmodium infection, suggesting a potential interplay between the acquisition of EBV and the vulnerability to malaria (131).
Moreover, acute P. falciparum infection has been demonstrated to affect EBV persistence. This impact includes increased EBV levels in circulation and elevated antibody titers against the Z Epstein-Barr replication activator (ZEBRA) protein and viral capsid antigen (VCA) in peripheral blood (128, 132–134). Donati et al. demonstrated that in children living in malaria-endemic regions, EBV loads can decrease to undetectable levels after the initiation of antimalarial treatment (133).
Though the precise mechanisms through which acute malaria triggers lytic EBV infection remain unclear, prolonged and severe exposure to P. falciparum malaria compromises T-cell immunity, particularly CD8+ T cells, leading to a loss of viral control (130, 135, 136). Additionally, studies have shown that the polyclonal B cell activator Cysteine-rich inter-domain region 1α (CIDR1α) of the P. falciparum membrane protein 1 directly triggers EBV reactivation during malaria infection, heightening the likelihood of BL development in children living in malaria-endemic regions (128). Recently, instances of concurrent infection involving various Plasmodium species and EBV have been reported. In the Southeast Anatolia and Cukurova regions of Turkey, where Plasmodium vivax predominates as the primary cause of malaria, a notable case occurred in 2013 involving a 5-year-old child (137). This case marked the initial documented occurrence of Plasmodium vivax malaria coupled with EBV coinfection in the region (137). In a study conducted by Wedderburn et al., co-infection with Plasmodium brasilianum and EBV in marmoset mice led to more severe glomerulonephritis and longer periods of parasitemia compared to the mice infected with P. brasilianum alone (138). Furthermore, acute EBV infection has immunosuppressive effects on the development of humoral antimalarial immunity (125). Also, EBV infection may impact the morbidity of P. vivax malaria by altering hematological parameters and impairing long-term acquired antibody responses to specific P. vivax proteins (139). These findings indicate that acute EBV infection disrupts the immune responses to malaria. Reactivated EBV, triggered by certain risk factors, may further modify malaria immunity and worsen the disease (140, 141). Extensive research has shown that unchecked EBV reactivation can exacerbate various illnesses and increase the risk of developing tumors and malignancies like nasopharyngeal carcinoma and post-transplant lymphoproliferative disorders (140, 141).
The early production of pro-inflammatory cytokines by the innate immune cells, including tumor necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-6, and others, enables quicker inhibition and clearance of the parasite, along with enhanced phagocytosis (142, 143). Dendritic Cells (DCs) infected with the gamma herpesvirus MHV68 display antigens less effectively compared to the uninfected DCs (144). Moreover, EBV can impact the functional abilities of the infected cells, hence diminishing their phagocytic ability response to other pathogens like bacteria and preventing the release of important cytokines (e.g. TNF-α) (145, 146). EBV prevents the growth of dendritic cells by inducing apoptosis in their monocytic precursors when IL-4 and granulocyte macrophage-colony-stimulating factor (GM-CSF) are present (147). Together, EBV and malaria co-infection is common in Africa and contributes to Burkitt’s lymphoma in children. Malaria can boost EBV levels and weaken immune responses, worsening both infections.
8 EBV coinfection with CMV (HHV-5)
Common among the general population, CMV infection poses a significant risk in transplantation, leading to higher chances of allograft rejection, morbidity, and mortality (148). CMV directly damages different organ systems, resulting in conditions like pneumonia, gastrointestinal tract disease, and hepatitis (149, 150). CMV infection can also induce indirect immunosuppressive effects, such as disruptions in T-cell production, alterations in major histocompatibility antigen expression, and changes in cytokine and chemokine activity (150, 151). These effects further elevate the risk of opportunistic infections (150, 151). Even with advanced treatment and prevention methods, patients undergoing allogeneic hematopoietic stem cell transplantation (HSCT) face amplified mortality risks due to double-stranded DNA (dsDNA) viruses. Reactivation of various herpes viruses, notably CMV, frequently occurs post-HSCT (152). Additionally, the reactivation of EBV is common and can lead to severe complications like post-transplant lymphoproliferative disorder (PTLD) (152).
Some studies have examined the correlation between CMV reactivation and EBV in recipients of HSCT. According to the research conducted by Fan et al., pre-infection with either CMV or EBV did not notably influence the incidence of coinfection with EBV or CMV, respectively (153). Two studies (154, 155) have explored the connection between donor/recipient serostatus and the likelihood of CMV-EBV coinfection in renal transplant recipients. In the first study, all five instances of CMV-EBV coinfection were observed in cases where both the donor and recipient tested positive for CMV. In the second study, donor CMV seropositivity emerged as the sole clinical factor significantly linked to the combined CMV-EBV reactivation. Additionally, CMV-EBV coinfection was notably associated with elevated CMV and EBV viral loads exceeding detectable levels, as well as with increased CMV and EBV DNAemia (154, 155). Another study conducted by Li et al. (152) suggests that CMV reactivation negatively impacts HSCT outcomes regardless of EBV reactivation.
Numerous studies indicate that EBV and CMV often co-infect patients alongside other pathogens, but little is known about this phenomenon in children with primary EBV/CMV infection (17). In a retrospective analysis, children with suspected IM were studied (17). Results showed a significantly higher occurrence of multiple infections in children with primary EBV, CMV, or EBV/CMV infection compared to other groups (17). Moreover, patients with multi-pathogen infection and EBV/CMV primary infection experienced more severe symptoms and longer hospital stays. These findings underscore the need for further research in this area (17).
Furthermore, the frequency of CMV and EBV infections is notably higher in patients with hematological diseases, suggesting a possible association between the two conditions (156). While Burkitt’s lymphoma stands out as the primary hematological disorder linked to EBV infection, studies also indicate a connection between EBV infection and the development of various hematological diseases, particularly AML and ALL (156). Conversely, CMV infection is often associated with the onset or worsened prognosis of ALL (156). The risk of CMV infection appears to be influenced by blood transfusions, although it remains uncertain whether such transfusions elevate the risk of EBV infection. Epidemiological surveillance of both infections in patients with hematological diseases is crucial for enhancing their clinical management (156). Moreover, the reactivation rates for both viruses tend to increase among patients with hematological diseases, potentially due to the immunological status imposed by the underlying hematological condition (156).
In summary, EBV and CMV coinfection is a significant concern, particularly in immunocompromised populations like transplant recipients and those with hematological diseases. This coinfection can lead to more severe outcomes, longer hospital stays, and increased complications. The interplay between these viruses, as well as their individual and combined effects on the immune system and disease progression, underscores the need for vigilant monitoring and tailored management strategies in at-risk populations.
9 EBV coinfection with HHV-6
The herpesvirus family includes HHV-6 viruses, of which HHV-6A and HHV-6B are the prominent subtypes that are classified based on genetic, antigenic, and possible variations in each of their pathogenicity (157, 158). Most children are infected with this virus during their early years of life, and like most other herpes viruses, it remains dormant in the body after the primary infection (159).
In EBV-related NPC, there’s a notable trend of HHV-6/EBV coinfection. In a study of 34 NPC tumor samples, 14.7% tested positive for HHV-6 DNA and 94.1% for EBV DNA (160). Conversely, none of the nasopharyngeal tissue samples from five controls showed positivity for either viruses (160). Furthermore, the presence of HHV-6 infection in NPC specimens has been associated with increased expression of EBV LMP-1 (161).
While evidence indicates that HHV-6 is present in specific cancer types, solely detecting the virus within the tumor cells does not suffice to definitively attribute a direct role to HHV-6 in tumorigenesis (162). Credible signs of a virus playing a causal role in cancer include its prevalence across a significant portion of cases, its pervasiveness within most tumor cells, and its capacity to induce cell transformation in laboratory conditions (162). Although HHV-6 may not directly induce cancer, it can act as a contributing factor, indirectly fostering tumor cell growth, potentially through collaboration with other viruses (162). Alternatively, HHV-6 might be an opportunistic virus thriving in the immunodeficient tumor microenvironment (162). Despite numerous studies, conclusively, linking HHV-6 to various human cancers remains unclear (162). Some evidence suggests that HHV-6 could collaborate with other viruses like EBV, HPV, and HHV-8 in cancer development, implicating it in conditions such as nodular sclerosis Hodgkin lymphoma, gastrointestinal cancer, glial tumors, and oral cancers. However, further research is necessary to elucidate HHV-6’s precise role in tumorigenesis (162).
Taken together, the relationship between EBV and HHV-6 is complex, particularly in the context of nasopharyngeal carcinoma (NPC) and other cancers. While HHV-6 is frequently detected alongside EBV in certain tumors, its exact role in carcinogenesis remains unclear. HHV-6 may act as a cofactor in cancer development, potentially collaborating with EBV and other viruses, but further research is needed to fully elucidate its role in tumorigenesis and its interactions with EBV.
10 EBV coinfection with KSHV (HHV-8)
Although human herpes virus 8 (HHV-8) is known to be associated with Kaposi sarcomas, it can also cause primary effusion lymphoma (PEL) and Castleman’s disease (MCD) (163). EBV and Kaposi sarcoma-associated herpesvirus (KSHV), the two human γ-herpesviruses, are classified as WHO class I carcinogens (164). Numerous neoplasms can result from HHV8 and EBV infections, either synchronously or alone (165). Most research has been done on the two human tumor viruses, KSHV and EBV, separately (166). According to recent research, KSHV persistence may require co-infection with EBV, as seen in PEL, one of their related cancers (167). The two viruses attack B cells in submucosal secondary lymphoid tissues, including the tonsils, and are assumed to be mainly spread via salivary fluids (168, 169). Since 1995, KSHV infection has been linked to PELs, and since then, detecting KSHV has been crucial in the diagnosis of PELs (170). In well-established PEL cell lines, co-infection is commonly observed, resulting in the two viral genomes being preserved, independently replicating, and being divided into the daughter cells (166, 171). In vitro studies have revealed that KSHV may infect peripheral B cells on its own, but it cannot convert them and thus cannot persist (172). Research conducted on mice have also shown that co-infection with EBV raises the likelihood of KSHV persistence (173). Co-infection with EBV stimulates B cells, promotes long-term KSHV infection, and allows for cell proliferation via transformation (172). KSHV genomes appear to be maintained by EBV co-infection; this is shown by the higher number of KSHV genomes per cell seen in the co-infected cells, although the precise mechanism is unclear (172). PEL cells require latent KSHV gene expression primarily LANA, vFLIP, and vIRF3—in addition to EBV genes in order to survive, since these genes engage with tumor suppressors and impede apoptotic mechanisms (174, 175). Through its association with KSHV terminal repeat sequences, LANA mediates the KSHV episome’s persistence (176). Additional viral genes are not necessary for this persistence, and LANA knockdown results in the loss of episomes (177). NF-κB is essentially active in PEL and can be activated by vFLIP (178). Due to its increased apoptosis and decreased proliferation upon knockdown, vIRF3 is necessary for the survival of both EBV+ and EBV− PEL cells (179). Epidemiology evidence suggests that KSHV infection is almost always associated with EBV co-infection and that one of the strongest environmental risk factors for KSHV seropositivity is EBV seropositivity (180, 181). As a result, EBV gene expression supports B cell transformation, proliferation, and survival while also aiding in KSHV persistence in the B cells. This permits EBV co-infection-induced KSHV persistence (167). Research indicates that KSHV and EBV both have a role in the development of primary effusion lymphomas, and that co-infection can raise the risk of developing tumors by influencing the tumor microenvironment and promoting its development and longevity (182). The interplay between EBV and KSHV is significant in several malignancies, particularly PEL. EBV appears to play a crucial role in supporting KSHV persistence and transformation in B cells. Their co-infection can enhance viral genome maintenance, cell survival, and tumor development. This synergistic relationship highlights the importance of considering viral co-infections in the pathogenesis and management of certain lymphoproliferative disorders.
11 Treatment approaches
Recent advancements in understanding the pathobiology of EBV oncogenesis have paved the way for novel therapeutic strategies (11). These approaches capitalize on insights into the virus’ immunogenic and transforming properties, as well as its role in immunological dysregulation (11). Among these techniques, adoptive T-cell therapies, EBV vaccines, inhibition of EBV-induced carcinogenic signaling pathways, and the induction of lytic viral infection alongside antiviral drugs are the most investigated ones (11). Although there have been significant advancements in the treatment of EBV-associated cancers, there are still a number of obstacles to overcome (183).
Research on drug discovery of viral target inhibitors and EBV-activated cellular targets, disruption of latent infection, and development of immunotherapeutic agents, such as vaccines and adoptive cellular therapies, are all included in the scope of current and future studies (184).
Target-based therapy development offers a promising approach for treating cancers associated with EBV. Specifically, T cell-based immunotherapy targets viral proteins produced by tumor cells, which serve as immunogenic tumor-specific targets (185, 186). Since oncogenic viral proteins manipulate cell signaling pathways involved in cell survival and proliferation, potential targets for therapy can be readily identified. Initial attempts to modify immune responses to the virus for treating EBV-associated malignancies were focused on PTLD (185, 186). These efforts involved infusing unmodified donor mononuclear cells into post-transplant patients, yielding high response rates, however, unselected cell populations carry a risk of graft-versus-host disease (GVHD) (185, 186). Subsequent research found that using cytotoxic T lymphocytes (CTLs) specific for EBV-encoded antigens may provide more potent anti-tumor effects without increasing the risk of GVHD (187, 188).
Currently, preemptive CTL therapy is preferred because it seems to work better than direct PTLD therapy, however, treating advanced PTLD with CTLs can cause significant side effects due to strong inflammatory responses. Despite this, this method can still lead to complete remission (187).
In the last decade, our understanding of how EBV triggers cell transformation and evades host immune defenses has greatly improved (187). The use of polyclonal EBV-specific CTLs has shown success in preventing and treating PTLD patients (187). This experience highlights the need for further research to validate the effectiveness of CTL-based therapies in treating diseases associated with EBV, such as NPC (189). While initial results are promising, enhancing T-cell responses to less immunogenic targets like LMP-1 and LMP-2 is crucial to improve treatment outcomes (189). CTL treatment shows promise in the prevention of PTLD, although specific intervention criteria remain unclear. Monitoring EBV DNA load can be helpful, but its predictive accuracy is limited, necessitating additional parameters to evaluate EBV-specific T cell responses (189). Various other virus-targeted treatment approaches are also under investigation, including triggering the EBV tumor cells’ lytic cycle; although challenges exist due to the highly controlled promoters of key lytic genes (189). Clinical trials focusing on understanding the viral and cellular mechanisms regulating the transcription of these genes can help confirm the practical applicability of this method. Additionally, treatment strategies aiming to eliminate EBV episomes from tumor cells, such as hydroxyurea administration, hold clinical significance (189).
As more clinics adopt immunotherapy to treat EBV-related cancers, new advanced strategies are poised to move swiftly from research to bedside (190). One such approach is RNA interference (RNAi), a recently discovered process that targets specific genes by degrading mRNAs within cells (190). This natural defense mechanism is found in animals and plants, offering potential effectiveness against viral infections (190). RNAi involves cleaving double-stranded RNA, typical of viruses, into small segments (siRNA), about 21–23 nucleotides long. These siRNAs are processed by Dicer and incorporated into the RNA Induced Silencing Complex (RISC), which degrades single-stranded RNA molecules in the cytoplasm, like mRNAs. RNAi holds promise for treating genetic diseases and gene-related disorders, including cancer (191–193). Specifically, recent studies have shown the effectiveness of RNA interference (RNAi) in inhibiting cancer growth in virus-associated cancers (191–193). In these cases, the silencing of a crucial viral protein necessary for cell transformation has been found to hinder the growth of cancerous cells, while leaving healthy cellular networks intact (191–193). Moreover, recent studies focusing on EBV-related cancers have revealed that silencing LMP-1 in NPC cells notably diminishes tumor cell metastasis, offering promising therapeutic strategies for treating not only nasopharyngeal carcinoma but also other EBV-related cancers (193). The full use of RNAi’s therapeutic potential will be possible if issues with siRNA delivery efficiency are resolved (194).
Treatment strategies for EBV-associated cancers have diversified, including T-cell therapies, vaccines, and targeting of viral pathways. While cytotoxic T lymphocyte therapy shows promise, especially for PTLD, emerging techniques like RNA interference offer new possibilities. Despite progress, challenges in efficacy and delivery persist, driving ongoing research to refine these approaches.
12 Conclusion
In conclusion, the prevalent EBV is known to coexist and coinfect with various other pathogens, leading to intricate interactions that can contribute to the development and progression of several cancers and other diseases. The coinfection of EBV with HPV is proposed to involve cooperation between viral oncoproteins, chromatin interactions, and the potential involvement of the EBV lytic cycle in oncogenesis. EBV’s coinfection with Plasmodium species, particularly P. falciparum and P. vivax, has been implicated in the pathogenesis of endemic Burkitt’s lymphoma and the exacerbation of malaria morbidity through mechanisms such as the disruption of antimalarial immunity and CD8+ T-cell responses. Coinfections with CMV and other herpesviruses, including HHV-6 and KSHV/HHV-8, have been associated with increased viral loads, altered disease progression, and the facilitation of viral persistence and transformation of infected cells. These intricate coinfection scenarios highlight the need for further research to elucidate the underlying mechanisms and develop targeted therapeutic strategies that address the synergistic effects of multiple pathogens. Understanding the complex interplay between EBV and other microbial agents will be crucial in combating the oncogenic potential of these coinfections and thus improving the clinical outcomes for affected patients.
Author contributions
FE: Investigation, Writing – original draft, Writing – review & editing. RR: Investigation, Writing – original draft, Writing – review & editing. SS: Investigation, Writing – original draft, Writing – review & editing. PSA: Writing – review & editing. AS: Writing – review & editing. AJS: Writing – review & editing. HBB: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
This project was supported by the Infectious and Tropical Diseases Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kangro H, Osman H, Lau Y, Heath R, Yeung C, Ng M. Seroprevalence of antibodies to human herpesviruses in England and Hong Kong. J Med Virol. (1994) 43:91–6. doi: 10.1002/jmv.1890430117
2. Dasari V, Sinha D, Neller MA, Smith C, Khanna R. Prophylactic and therapeutic strategies for epstein–barr virus-associated diseases: emerging strategies for clinical development. Expert Rev Vaccines. (2019) 18:457–74. doi: 10.1080/14760584.2019.1605906
3. Dowd JB, Palermo T, Brite J, McDade TW, Aiello A. Seroprevalence of epstein-barr virus infection in us children ages 6-19, 2003-2010. PloS One. (2013) 8:e64921. doi: 10.1371/journal.pone.0064921
4. Grotto I, Mimouni D, Huerta M, Mimouni M, Cohen D, Robin G, et al. Clinical and laboratory presentation of ebv positive infectious mononucleosis in young adults. Epidemiol Infect. (2003) 131:683–9. doi: 10.1017/S0950268803008550
6. de-Thé G, Day NE, Geser A, Lavoué MF, Ho JH, Simons MJ, et al. Sero-epidemiology of the epstein-barr virus: preliminary analysis of an international study-a review. IARC Sci publications. (1975) 11 Pt 2:3–16.
7. Niederman JC. Infectious mononucleosis: observations on transmission. Yale J Biol Med. (1982) 55:259.
8. Hudnall SD. Epstein–barr virus: epidemiology and clinical features of related cancer. Viruses Hum Cancer. (2014), 25–50. doi: 10.1007/978-1-4939-0870-7
9. Saha A, Robertson ES. Epstein-barr virus–associated B-cell lymphomas: pathogenesis and clinical outcomes. Clin Cancer Res. (2011) 17:3056–63. doi: 10.1158/1078-0432.CCR-10-2578
10. Ebadi A, Rasizadeh R, Aghbash PS, Hemmat N, Baghi HB. Investigating the role of micrornas, inflammation, and helicobacter pylori in epstein-barr virus associated gastric cancer. J Exp Clin Med. (2023) 40:646–58. doi: 10.52142/omujecm.40.3.37
11. Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. The pathogenesis of epstein–barr virus persistent infection. Curr Opin Virol. (2013) 3:227–32. doi: 10.1016/j.coviro.2013.04.005
12. Ersing I, Bernhardt K, Gewurz BE. Nf-Kb and irf7 pathway activation by epstein-barr virus latent membrane protein 1. Viruses. (2013) 5:1587–606. doi: 10.3390/v5061587
13. Aghbash PS, Hemmat N, Nahand JS, Shamekh A, Memar MY, Babaei A, et al. The role of th17 cells in viral infections. Int Immunopharmacol. (2021) 91:107331. doi: 10.1016/j.intimp.2020.107331
14. Jenson HB, Nelson WE, Behrman RE, Kliegman R. Nelson Textbook of Pediatrics. Philadelphia, PA: Saunders (2004).
15. De-Thé G, Geser A, Day N, Tukei P, Williams E, Beri D, et al. Epidemiological evidence for causal relationship between epstein-barr virus and Burkitt’s lymphoma from Ugandan prospective study. Nature. (1978) 274:756–61. doi: 10.1038/274756a0
16. Godshall SE, Kirchner JT. Infectious mononucleosis. Complexities of a common syndrome. Postgraduate Med. (2000) 107:175–9, 83. doi: 10.3810/pgm.2000.06.1130
17. Wang X, Yang K, Wei C, Huang Y, Zhao D. Coinfection with ebv/cmv and other respiratory agents in children with suspected infectious mononucleosis. Virol J. (2010) 7:1–10. doi: 10.1186/1743-422X-7-247
18. Rasizadeh R, Ebrahimi F, Zamani A, Sorkhabi AD, Sarkesh A, Nahand JS, et al. Viruses and thrombocytopenia. Heliyon. (2024) 10(6). doi: 10.1016/j.heliyon.2024.e27844
19. Odumade OA, Hogquist KA, Balfour HH Jr. Progress and problems in understanding and managing primary epstein-barr virus infections. Clin Microbiol Rev. (2011) 24:193–209. doi: 10.1128/CMR.00044-10
20. Dunmire S, Balfour HK. Infectious mononucleosis. Curr Top Microbiol Immunol. (2015) 390:211–40. doi: 10.1007/978-3-319-22822-8_9
21. Aslan N, Watkin LB, Gil A, Mishra R, Clark FG, Welsh RM, et al. Severity of acute infectious mononucleosis correlates with cross-reactive influenza cd8 T-cell receptor repertoires. MBio. (2017) 8:10.1128/mbio.01841-17. doi: 10.1128/mbio.01841-17
22. Horwitz CA, Henle W, Henle G, Goldfarb M, Kubic P, Gehrz RC, et al. Clinical and laboratory evaluation of infants and children with epstein-barr virus-induced infectious mononucleosis: report of 32 patients (Aged 10–48 months). Blood. (1981) 57:933–8. doi: 10.1182/blood.V57.5.933.933
23. Goldberg GN, Fulginiti VA, Ray CG, Ferry P, Jones JF, Cross H, et al. In utero epstein-barr virus (Infectious mononucleosis) infection. Jama. (1981) 246:1579–81. doi: 10.1001/jama.1981.03320140067034
24. Chang RS, Seto DY. Perinatal infection by epstein-barr virus. Lancet. (1979) 314:201. doi: 10.1016/S0140-6736(79)91469-7
25. Hudson LB, Perlman SE. Necrotizing genital ulcerations in a premenarcheal female with mononucleosis. Obstet Gynecol. (1998) 92:642–4.
26. Tselis A, Duman R, Storch GA, Lisak RP. Epstein-barr virus encephalomyelitis diagnosed by polymerase chain reaction: detection of the genome in the csf. Neurology. (1997) 48:1351–5. doi: 10.1212/WNL.48.5.1351
27. Kempkes B, Robertson ES. Epstein-barr virus latency: current and future perspectives. Curr Opin Virol. (2015) 14:138–44. doi: 10.1016/j.coviro.2015.09.007
28. Murata T, Sugimoto A, Inagaki T, Yanagi Y, Watanabe T, Sato Y, et al. Molecular basis of epstein–barr virus latency establishment and lytic reactivation. Viruses. (2021) 13:2344. doi: 10.3390/v13122344
29. Kliszczewska E, Jarzyński A, Boguszewska A, Pasternak J, Polz-Dacewicz M. Epstein-barr virus-pathogenesis, latency and cancers. J Pre-Clinic Clin Res. (2017) 11:142–6. doi: 10.26444/jpccr/81214
30. Crawford DH. Biology and disease associations of epstein–barr virus. Philos Trans R Soc London Ser B: Biol Sci. (2001) 356:461–73. doi: 10.1098/rstb.2000.0783
31. Tempera I, Klichinsky M, Lieberman PM. Ebv latency types adopt alternative chromatin conformations. PloS Pathog. (2011) 7:e1002180. doi: 10.1371/journal.ppat.1002180
32. Straus SE. The chronic mononucleosis syndrome. J Infect Dis. (1988) 157:405–12. doi: 10.1093/infdis/157.3.405
33. Cohen JI, Jaffe ES, Dale JK, Pittaluga S, Heslop HE, Rooney CM, et al. Characterization and treatment of chronic active epstein-barr virus disease: A 28-year experience in the United States. Blood J Am Soc Hematol. (2011) 117:5835–49. doi: 10.1182/blood-2010-11-316745
34. Kimura H, Cohen JI. Chronic active epstein–barr virus disease. Front Immunol. (2017) 8:1867. doi: 10.3389/fimmu.2017.01867
35. Ohshima K, Kimura H, Yoshino T, Kim CW, Ko YH, Lee SS, et al. Proposed categorization of pathological states of ebv-associated T/natural killer-cell lymphoproliferative disorder (Lpd) in children and young adults: overlap with chronic active ebv infection and infantile fulminant ebv T-lpd. Pathol Int. (2008) 58:209–17. doi: 10.1111/j.1440-1827.2008.02213.x
36. Okuno Y, Murata T, Sato Y, Muramatsu H, Ito Y, Watanabe T, et al. Defective epstein–barr virus in chronic active infection and haematological Malignancy. Nat Microbiol. (2019) 4:404–13. doi: 10.1038/s41564-018-0334-0
37. Meedt E, Weber D, Bonifacius A, Eiz-Vesper B, Maecker-Kolhoff B, Delecluse S, et al. Chronic active epstein-barr virus (Ebv) infection controlled by allogeneic stem cell transplantation and ebv-specific T cells. Clin Infect Dis. (2023) 76:2200–2. doi: 10.1093/cid/ciad131
38. Bollard CM, Cohen JI. How I treat T-cell chronic active epstein-barr virus disease. Blood J Am Soc Hematol. (2018) 131:2899–905. doi: 10.1182/blood-2018-03-785931
39. Sullivan JL. Clinical manifestations and treatment of epstein-barr virus infection. Доступно. (2019). http://www.uptodate.com/contents/clinical-manifestations-and-treatment-of-epstein-barr-virus-infection.
40. Schiller JT, Lowy DR. Virus infection and human cancer: an overview. Viruses Hum Cancer: Basic Sci to Clin Prev. 2014):1–10. doi: 10.1007/978-3-642-38965-8_1
41. Zur Hausen H. The search for infectious causes of human cancers: where and why. Virology. (2009) 392:1–10. doi: 10.1016/j.virol.2009.06.001
42. De Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. (2012) 13:607–15. doi: 10.1016/S1470-2045(12)70137-7
43. Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe. (2014) 15:266–82. doi: 10.1016/j.chom.2014.02.011
44. Nahand JS, Khanaliha K, Mirzaei H, Moghoofei M, Baghi HB, Esghaei M, et al. Possible role of hpv/ebv coinfection in anoikis resistance and development in prostate cancer. BMC Cancer. (2021) 21:1–19. doi: 10.1186/s12885-021-08658-y
45. McLaughlin-Drubin ME, Meyers J, Munger K. Cancer associated human papillomaviruses. Curr Opin Virol. (2012) 2:459–66. doi: 10.1016/j.coviro.2012.05.004
46. Baghi HB, Aghbash PS, Rasizadeh R, Poortahmasebi V, Alinezhad F. Cancers associated with human papillomavirus: an overview of prevalence in Iran and the Middle East. Exploratory Res Hypothesis Med. (2024) 9:115–27. doi: 10.14218/ERHM.2023.00053
47. Trivedi P, Takazawa K, Zompetta C, Cuomo L, Anastasiadou E, Carbone A, et al. Infection of hhv-8+ Primary effusion lymphoma cells with a recombinant epstein-barr virus leads to restricted ebv latency, altered phenotype, and increased tumorigenicity without affecting tcl1 expression. Blood. (2004) 103:313–6. doi: 10.1182/blood-2003-05-1710
48. de Oliveira DE. DNA viruses in human cancer: an integrated overview on fundamental mechanisms of viral carcinogenesis. Cancer Lett. (2007) 247:182–96. doi: 10.1016/j.canlet.2006.05.010
49. Rickinson A ed. Co-Infections, Inflammation and Oncogenesis: Future Directions for Ebv Research. In: Seminars in cancer biology. Elsevier.
50. Giannoudis A, Ergazaki M, Segas J, Giotakis J, Adamopoulos G, Gorgoulis V, et al. Detection of epstein-barr virus and human papillomavirus in nasopharyngeal carcinoma by the polymerase chain reaction technique. Cancer Lett. (1995) 89:177–81. doi: 10.1016/0304-3835(94)03667-8
51. Punwaney R, Brandwein MS, Zhang DY, Urken ML, Cheng R, Park CS, et al. Human papillomavirus may be common within nasopharyngeal carcinoma of Caucasian Americans: investigation of epstein-barr virus and human papillomavirus in eastern and western nasopharyngeal carcinoma using ligation-dependent polymerase chain reaction. Head Neck: J Sci Special Head Neck. (1999) 21:21–9. doi: 10.1002/(SICI)1097-0347(199901)21:1<21::AID-HED3>3.0.CO;2-Z
52. Singhi AD, Califano J, Westra WH. High-risk human papillomavirus in nasopharyngeal carcinoma. Head Neck. (2012) 34:213–8. doi: 10.1002/hed.21714
53. Rassekh CH, Rady PL, Arany I, Tyring SK, Knudsen S, Calhoun KH, et al. Combined epstein-barr virus and human papillomavirus infection in nasopharyngeal carcinoma. Laryngosc. (1998) 108:362–7. doi: 10.1097/00005537-199803000-00010
54. Whitaker NJ, Glenn WK, Sahrudin A, Orde MM, Delprado W, Lawson JS. Human papillomavirus and epstein barr virus in prostate cancer: koilocytes indicate potential oncogenic influences of human papillomavirus in prostate cancer. Prostate. (2013) 73:236–41. doi: 10.1002/pros.22562
55. Glenn WK, Heng B, Delprado W, Iacopetta B, Whitaker NJ, Lawson JS. Epstein-barr virus, human papillomavirus and mouse mammary tumour virus as multiple viruses in breast cancer. PloS One. (2012) 7:e48788. doi: 10.1371/journal.pone.0048788
56. Al Moustafa A-E, Chen D, Ghabreau L, Akil N. Association between human papillomavirus and epstein-barr virus infections in human oral carcinogenesis. Med Hypotheses. (2009) 73:184–6. doi: 10.1016/j.mehy.2009.02.025
57. Ammatuna P, Giovannelli L, Giambelluca D, Mancuso S, Rubino E, Colletti P, et al. Presence of human papillomavirus and epstein-barr virus in the cervix of women infected with the human immunodeficiency virus. J Med Virol. (2000) 62:410–5. doi: 10.1002/1096-9071(200012)62:4<410::AID-JMV3>3.0.CO;2-J
58. Wei K-R, Zheng R-S, Zhang S-W, Liang Z-H, Li Z-M, Chen W-Q. Nasopharyngeal carcinoma incidence and mortality in China, 2013. Chin J Cancer. (2017) 36:1–8. doi: 10.1186/s40880-017-0257-9
59. Nikitin PA, Yan CM, Forte E, Bocedi A, Tourigny JP, White RE, et al. An atm/chk2-mediated DNA damage-responsive signaling pathway suppresses epstein-barr virus transformation of primary human B cells. Cell Host Microbe. (2010) 8:510–22. doi: 10.1016/j.chom.2010.11.004
60. Woellmer A, Hammerschmidt W. Epstein–barr virus and host cell methylation: regulation of latency, replication and virus reactivation. Curr Opin Virol. (2013) 3:260–5. doi: 10.1016/j.coviro.2013.03.005
61. Lieberman PM. Keeping it quiet: chromatin control of gammaherpesvirus latency. Nat Rev Microbiol. (2013) 11:863–75. doi: 10.1038/nrmicro3135
62. Knipe DM, Lieberman PM, Jung JU, McBride AA, Morris KV, Ott M, et al. Snapshots: chromatin control of viral infection. Virology. (2013) 435:141–56. doi: 10.1016/j.virol.2012.09.023
63. Blanco R, Carrillo-Beltrán D, Osorio JC, Calaf GM, Aguayo F. Role of epstein-barr virus and human papillomavirus coinfection in cervical cancer: epidemiology, mechanisms and perspectives. Pathogens. (2020) 9:685. doi: 10.3390/pathogens9090685
64. Schiller JT, Davies P. Delivering on the promise: hpv vaccines and cervical cancer. Nat Rev Microbiol. (2004) 2:343–7. doi: 10.1038/nrmicro867
65. Lu Z-X, Ye M, Yan G-R, Li Q, Tang M, Lee LM, et al. Effect of ebv lmp1 targeted dnazymes on cell proliferation and apoptosis. Cancer Gene Ther. (2005) 12:647–54. doi: 10.1038/sj.cgt.7700833
66. Organization WH. Coronavirus Disease 2019 (Covid-19): Situation Report, 73. Geneva: World Health Organization (2020) 2020-04-02. Report No.
67. Rasizadeh R, Baghi HB. Increase in rabies cases during covid-19 pandemic: is there a connection? J Infect Develop Countries. (2023) 17:335–6. doi: 10.3855/jidc.17537
68. Dong Y, Mo X, Hu Y, Qi X, Jiang F, Jiang Z, et al. Epidemiology of covid-19 among children in China. Pediatrics. (2020) 145:e20200702. doi: 10.1542/peds.2020-0702
69. Team CC-R, Team CC-R, Team CC-R, Bialek S, Gierke R, Hughes M, et al. Coronavirus disease 2019 in children—United states, February 12–April 2, 2020. Morbid Mortal Week Rep. (2020) 69:422–6. doi: 10.15585/mmwr.mm6914e4
70. Livingston E, Bucher K. Coronavirus disease 2019 (Covid-19) in Italy. Jama. (2020) 323:1335–. doi: 10.1001/jama.2020.4344
71. Lu X, Zhang L, Du H, Zhang J, Li YY, Qu J, et al. Sars-cov-2 infection in children. New Engl J Med. (2020) 382:1663–5. doi: 10.1056/NEJMc2005073
72. Carsetti R, Quintarelli C, Quinti I, Mortari EP, Zumla A, Ippolito G, et al. The immune system of children: the key to understanding sars-cov-2 susceptibility? Lancet Child Adolesc Health. (2020) 4:414–6. doi: 10.1016/s2352-4642(20)30135-8
73. Rasizadeh R, Milani ES, Aghbash PS, Arefi V, Fathi H, Nahand JS, et al. Beyond the virus: exploring coinfections in the covid-19 pandemic. Open Microbiol J. (2023) 17. doi: 10.2174/0118742858274177231110050202
74. Yuki K, Fujiogi M, Koutsogiannaki S. Covid-19 pathophysiology: A review. Clin Immunol. (2020) 215:108427. doi: 10.1016/j.clim.2020.108427
75. Skenderi E, Sulovari A, Kuli-Lito G, Shahini N, Toci G, Pema A. Covid-19 and ebv co-infection in a child. J Biosci Medicines. (2021) 9:20–7. doi: 10.4236/jbm.2021.95003
76. Toubiana J, Poirault C, Corsia A, Bajolle F, Fourgeaud J, Angoulvant F, et al. Kawasaki-like multisystem inflammatory syndrome in children during the covid-19 pandemic in Paris, France: prospective observational study. bmj. (2020) 369:m2094. doi: 10.1136/bmj.m2094
77. Verdoni L, Mazza A, Gervasoni A, Martelli L, Ruggeri M, Ciuffreda M, et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the sars-cov-2 epidemic: an observational cohort study. Lancet. (2020) 395:1771–8. doi: 10.1016/S0140-6736(20)31103-X
78. Rauf A, Vijayan A, John ST, Krishnan RA, Latheef A. Multisystem inflammatory syndrome with features of atypical Kawasaki disease during covid-19 pandemic: report of a case from India. (2020) 87(9):745-. doi: 10.21203/rs.3.rs-29369/v1
79. Riphagen S, Gomez X, Gonzalez-Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during covid-19 pandemic. Lancet. (2020) 395:1607–8. doi: 10.1016/S0140-6736(20)31094-1
80. Godfred-Cato S, Bryant B, Leung J, Oster ME, Conklin L, Abrams J, et al. Covid-19–associated multisystem inflammatory syndrome in children—United states, March–July 2020. Morbid mortal week Rep. (2020) 69:1074. doi: 10.15585/mmwr.mm6932e2
81. Feldstein LR, Rose EB, Horwitz SM, Collins JP, Newhams MM, Son MBF, et al. Multisystem inflammatory syndrome in us children and adolescents. New Engl J Med. (2020) 383:334–46. doi: 10.1056/NEJMoa2021680
82. Whittaker E, Bamford A, Kenny J, Kaforou M, Jones CE, Shah P, et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with sars-cov-2. Jama. (2020) 324:259–69. doi: 10.1001/jama.2020.10369
83. Nakra NA, Blumberg DA, Herrera-Guerra A, Lakshminrusimha S. Multi-system inflammatory syndrome in children (Mis-C) following sars-cov-2 infection: review of clinical presentation, hypothetical pathogenesis, and proposed management. Children. (2020) 7:69. doi: 10.3390/children7070069
84. Schiller JH, Storer BE, Witt PL, Nelson BE, Brown RR, Horisberger MA, et al. Biological and clinical effects of the combination of B-and Γ-interferons administered as a 5-day continuous infusion. Cancer Res. (1990) 50:4588–94.
85. Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from epstein-barr virus. Annu Rev Immunol. (2007) 25:587–617. doi: 10.1146/annurev.immunol.25.022106.141553
86. Zheng S-Y, Xiao Q-Y, Xie X-H, Deng Y, Ren L, Tian D-Y, et al. Association between secondary thrombocytosis and viral respiratory tract infections in children. Sci Rep. (2016) 6:22964. doi: 10.1038/srep22964
87. Khan MM, Khan MM. Acquired immune deficiency syndrome. Immunopharmacology. (2016), 293–330. doi: 10.1007/978-3-319-30273-7
88. Hjalgrim H, Friborg J, Melbye M. The Epidemiology of Ebv and Its Association with Malignant Disease. Cambridge: Cambridge University Press (2007) 2007.
89. Whitehurst CB, Rizk M, Teklezghi A, Spagnuolo RA, Pagano JS, Wahl A. Hiv co-infection augments ebv-induced tumorigenesis in vivo. Front Virol. (2022) 2:861628. doi: 10.3389/fviro.2022.861628
90. McHugh D, Myburgh R, Caduff N, Spohn M, Kok YL, Keller CW, et al. Ebv renders B cells susceptible to hiv-1 in humanized mice. Life Sci alliance. (2020) 3(8):e202000640. doi: 10.26508/lsa.202000640
91. Fenwick C, Joo V, Jacquier P, Noto A, Banga R, Perreau M, et al. T-cell exhaustion in hiv infection. Immunol Rev. (2019) 292:149–63. doi: 10.1111/imr.v292.1
92. Carbone A, Vaccher E, Gloghini A. Hematologic cancers in individuals infected by hiv. Blood J Am Soc Hematol. (2022) 139:995–1012. doi: 10.1182/blood.2020005469
93. Shannon-Lowe C, Rickinson A. The global landscape of ebv-associated tumors. Front Oncol. (2019) 9:713. doi: 10.3389/fonc.2019.00713
94. Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, et al. Pd-1+ and follicular helper T cells are responsible for persistent hiv-1 transcription in treated aviremic individuals. Nat Med. (2016) 22:754–61. doi: 10.1038/nm.4113
95. Cervantes-Gracia K, Gramalla-Schmitz A, Weischedel J, Chahwan R. Apobecs orchestrate genomic and epigenomic editing across health and disease. Trends Genet. (2021) 37:1028–43. doi: 10.1016/j.tig.2021.07.003
96. Mertz TM, Collins CD, Dennis M, Coxon M, Roberts SA. Apobec-induced mutagenesis in cancer. Annu Rev Genet. (2022) 56:229–52. doi: 10.1146/annurev-genet-072920-035840
97. de Souza CRT, de Oliveira KS, Ferraz JJS, Leal MF, Calcagno DQ, Seabra AD, et al. Occurrence of helicobacter pyloriand epstein-barr virus infection in endoscopic and gastric cancer patients from Northern Brazil. BMC Gastroenterol. (2014) 14:1–9. doi: 10.1186/1471-230X-14-179
98. Nagini S. Carcinoma of the stomach: A review of epidemiology, pathogenesis, molecular genetics and chemoprevention. World J gastrointest Oncol. (2012) 4:156. doi: 10.4251/wjgo.v4.i7.156
99. Luzzi I, Covacci A, Censini S, Pezzella C, Crotti D, Facchini M, et al. Detection of a vacuolating cytotoxin in stools from children with diarrhea. Clin Infect Dis. (1996) 23:101–6. doi: 10.1093/clinids/23.1.101
100. Forte GI, Cala C, Scola L, Crivello A, Gullo A, Marasa L, et al. Role of environmental and genetic factor interaction in age-related disease development: the gastric cancer paradigm. Rejuv Res. (2008) 11:509–12. doi: 10.1089/rej.2008.0678
101. Iarc L. Schistosomes, liver flukes and helicobacter pylori. IARC Monogr Evalutaion Carcinogen Risks to Humans. (1994) 61:1–241.
102. Na SJ, Park HL, Lee SY, Song KY, Kim SH. Correlation between infection status of epstein-barr virus and 18f-fluorodeoxyglucose uptake in patients with advanced gastric cancer. In Vivo. (2017) 31:749–53. doi: 10.21873/invivo.11126
103. Sohn BH, Hwang J-E, Jang H-J, Lee H-S, Oh SC, Shim J-J, et al. Clinical significance of four molecular subtypes of gastric cancer identified by the cancer genome atlas project. Clin Cancer Res. (2017) 23:4441–9. doi: 10.1158/1078-0432.CCR-16-2211
104. Carrasco-Avino G, Riquelme I, Padilla O, Villaseca M, Aguayo FR, Corvalan AH. The conundrum of the epstein-barr virus-associated gastric carcinoma in the americas. Oncotarget. (2017) 8:75687. doi: 10.18632/oncotarget.18497
105. Iaria C, Arena L, Di Maio G, Fracassi MG, Leonardi MS, Famulari C, et al. Acute acalculous cholecystitis during the course of primary epstein–barr virus infection: A new case and a review of the literature. Int J Infect Dis. (2008) 12:391–5. doi: 10.1016/j.ijid.2007.10.005
106. Cascio A, Iaria C, Ruggeri P, Fries W. Cytomegalovirus pneumonia in patients with inflammatory bowel disease: A systematic review. Int J Infect Dis. (2012) 16:e474–e9. doi: 10.1016/j.ijid.2012.03.008
107. Archampong TN, Asmah RH, Aidoo EK, Wiredu EK, Gyasi RK, Adjei DN, et al. Helicobacter pylori caga and vaca genes in dyspeptic Ghanaian patients. BMC Res Notes. (2017) 10:1–5. doi: 10.1186/s13104-017-2542-8
108. Dabiri H, Jafari F, Baghaei K, Shokrzadeh L, Abdi S, Pourhoseingholi MA, et al. Prevalence of helicobacter pylori vaca, caga, cage, oipa, icea, baba2 and babb genotypes in Iranian dyspeptic patients. Microb pathogen. (2017) 105:226–30. doi: 10.1016/j.micpath.2017.02.018
109. Chiarini A, Calà C, Bonura C, Gullo A, Giuliana G, Peralta S, et al. Prevalence of virulence-associated genotypes of helicobacter pylori and correlation with severity of gastric pathology in patients from Western Sicily, Italy. Eur J Clin Microbiol Infect Dis. (2009) 28:437–46. doi: 10.1007/s10096-008-0644-x
110. Fasciana T, Calà C, Bonura C, Di Carlo E, Matranga D, Scarpulla G, et al. Resistance to clarithromycin and genotypes in helicobacter pylori strains isolated in Sicily. J Med Microbiol. (2015) 64:1408–14. doi: 10.1099/jmm.0.000163
111. Tamayo E, Montes M, Fernández-Reyes M, Lizasoain J, Ibarra B, Mendarte U, et al. Clarithromycin resistance in helicobacter pylori and its molecular determinants in northern Spain, 2013–2015. J Global antimicrob resist. (2017) 9:43–6. doi: 10.1016/j.jgar.2016.12.019
112. Kim H-J, Ko YH, Kim JE, Lee S-S, Lee H, Park G, et al. Epstein-barr virus–associated lymphoproliferative disorders: review and update on 2016 who classification. J Pathol Trans Med. (2017) 51:352. doi: 10.4132/jptm.2017.03.15
113. Iovene MR, Bombace F, Maresca R, Sapone A, Iardino P, Picardi A, et al. Intestinal dysbiosis and yeast isolation in stool of subjects with autism spectrum disorders. Mycopathologia. (2017) 182:349–63. doi: 10.1007/s11046-016-0068-6
114. Giardina A, Rizzo A, Ferrante A, Capra G, Triolo G, Ciccia F. Giant cell arteritis associated with chronic active epstein-barr virus infection. Reumatismo. (2013) 65:36–9. doi: 10.4081/reumatismo.2013.36
115. Thompson G, Pepperell D, Lawrence I, McGettigan BD. Crohn’s disease complicated by epstein-barr virus-driven haemophagocytic lymphohistiocytosis successfully treated with rituximab. Case Rep. (2017) 2017):bcr2016218578. doi: 10.1136/bcr-2016-218578
116. Cascio A, Pernice LM, Barberi G, Delfino D, Biondo C, Beninati C, et al. Secondary hemophagocytic lymphohistiocytosis in zoonoses. A systematic review. Eur Rev Med Pharmacol Sci. (2012) 16.
117. Colomba C, Di Carlo P, Scarlata F, Iaria C, Barberi G, Famà F, et al. Visceral leishmaniasis, hypertriglyceridemia and secondary hemophagocytic lymphohistiocytosis. Infection. (2016) 44:391–2. doi: 10.1007/s15010-016-0881-3
118. Cascio A, Giordano S, Dones P, Venezia S, Iaria C, Ziino O. Haemophagocytic syndrome and rickettsial diseases. J Med Microbiol. (2011) 60(1):537–42. doi: 10.1099/jmm.0.025833-0
119. Singh S, Jha HC. Status of epstein-barr virus coinfection with helicobacter pylori in gastric cancer. J Oncol. (2017) 2017(1):3456264. doi: 10.1155/2017/3456264
120. Fasciana T, Capra G, Calà C, Zambuto S, Mascarella C, Colomba C, et al. Helicobacter pylori and epstein–barr co-infection in gastric. Helicobacter. (2017) 1:73–82.
121. Bonventre S, Inviati A, Di Paola V, Morreale P, Di Giovanni S, Di Carlo P, et al. Evaluating the efficacy of current treatments for reducing postoperative ileus: A randomized clinical trial in a single center. Minerva Chir. (2014) 69:47–55.
122. Bellevicine C, Migliatico I, Vigliar E, Serra N, Troncone G. Intra-institutional second opinion diagnosis can reduce unnecessary surgery for indeterminate thyroid fna: A preliminary report on 34 cases. Cytopathology. (2017) 28:254–8. doi: 10.1111/cyt.2017.28.issue-4
123. Gabutti G, Bergamini M, Bonanni P, Guido M, Fenoglio D, Giammanco A, et al. Assessment of humoral and cell-mediated immunity against bordetella pertussis in adolescent, adult, and senior subjects in Italy. Epidemiol Infect. (2008) 136:1576–84. doi: 10.1017/S0950268807000192
124. Giammanco A, Taormina S, Chiarini A, Dardanoni G, Stefanelli P, Salmaso S, et al. Analogous igg subclass response to pertussis toxin in vaccinated children, healthy or affected by whooping cough. Vaccine. (2003) 21:1924–31. doi: 10.1016/S0264-410X(02)00823-X
125. Matar CG, Jacobs NT, Speck SH, Lamb TJ, Moormann AM. Does ebv alter the pathogenesis of malaria? Parasite Immunol. (2015) 37(9):433–45. doi: 10.1111/pim.12212
126. English M, Sauerwein R, Waruiru C, Mosobo M, Obiero J, Lowe B, et al. Acidosis in severe childhood malaria. QJM: month J Assoc Phys. (1997) 90:263–70. doi: 10.1093/qjmed/90.4.263
127. Dondorp AM, Kager PA, Vreeken J, White NJ. Abnormal blood flow and red blood cell deformability in severe malaria. Parasitol Today. (2000) 16:228–32. doi: 10.1016/S0169-4758(00)01666-5
128. Chêne A, Donati D, Guerreiro-Cacais AO, Levitsky V, Chen Q, Falk KI, et al. A molecular link between malaria and epstein–barr virus reactivation. PloS Pathog. (2007) 3(6):e80. doi: 10.1371/journal.ppat.0030080
129. Mawson AR, Majumdar S. Malaria, epstein–barr virus infection and the pathogenesis of Burkitt’s lymphoma. Int J Cancer. (2017) 141:1849–55. doi: 10.1002/ijc.v141.9
130. Moormann AM, Snider CJ, Chelimo K. The company malaria keeps: how co-infection with epstein-barr virus leads to endemic burkitt lymphoma. Curr Opin Infect Dis. (2011) 24:435. doi: 10.1097/QCO.0b013e328349ac4f
131. Piriou E, Asito AS, Sumba PO, Fiore N, Middeldorp JM, Moormann AM, et al. Early age at time of primary epstein–barr virus infection results in poorly controlled viral infection in infants from western Kenya: clues to the etiology of endemic Burkitt lymphoma. J Infect Dis. (2012) 205:906–13. doi: 10.1093/infdis/jir872
132. Piriou E, Kimmel R, Chelimo K, Middeldorp JM, Odada PS, Ploutz-Snyder R, et al. Serological evidence for long-term epstein–barr virus reactivation in children living in a holoendemic malaria region of Kenya. J Med Virol. (2009) 81:1088–93. doi: 10.1002/jmv.21485
133. Donati D, Espmark E, Kironde F, Mbidde EK, Kamya M, Lundkvist Å, et al. Clearance of circulating epstein-barr virus DNA in children with acute malaria after antimalaria treatment. J Infect Dis. (2006) 193:971–7. doi: 10.1086/jid.2006.193.issue-7
134. Rasti N, Falk K, Donati D, Gyan B, Goka B, Troye-Blomberg M, et al. Circulating epstein–barr virus in children living in malaria-endemic areas. Scandinavian J Immunol. (2005) 61:461–5. doi: 10.1111/j.1365-3083.2005.01589.x
135. Chattopadhyay PK, Chelimo K, Embury PB, Mulama DH, Sumba PO, Gostick E, et al. Holoendemic malaria exposure is associated with altered epstein-barr virus-specific cd8+ T-cell differentiation. J Virol. (2013) 87:1779–88. doi: 10.1128/JVI.02158-12
136. Njie R, Bell AI, Jia H, Croom-Carter D, Chaganti S, Hislop AD, et al. The effects of acute malaria on epstein-barr virus (Ebv) load and ebv-specific T cell immunity in Gambian children. J Infect Dis. (2009) 199:31–8. doi: 10.1086/594373
137. Akin F, Kocaoglu C, Solak ES, Ozdemir H, Pektas B, Arslan S. Coinfection of plasmodium vivax and epstein-barr virus: case report. Asian Pacific J Trop Dis. (2013) 3:74–5. doi: 10.1016/S2222-1808(13)60016-X
138. Wedderburn N, Davies DR, Mitchell GH, Desgranges C, de Thé G. Glomerulonephritis in common marmosets infected with plasmodium brasilianum and epstein-barr virus. J Infect Dis. (1988) 158:789–94. doi: 10.1093/infdis/158.4.789
139. Dias MHF. Influência da infecção pelo epstein-barr vírus na malária humana causada por plasmodium vivax. (2018).
140. Chen Y, Xu Y, Zhao W, Xiao X, Zhou X, Lin L, et al. Lack of association between cigarette smoking and epstein barr virus reactivation in the nasopharynx in people with elevated ebv iga antibody titres. BMC Cancer. (2018) 18:1–7. doi: 10.1186/s12885-018-4110-6
141. Das B, Morrow R, Huang R, Fixler D. Persistent epstein-barr viral load in epstein-barr viral naïve pediatric heart transplant recipients: risk of late-onset post-transplant lymphoproliferative disease. World J Transplant. (2016) 6:729. doi: 10.5500/wjt.v6.i4.729
142. Ayimba E, Hegewald J, Segbena A, Gantin R, Lechner C, Agosssou A, et al. Proinflammatory and regulatory cytokines and chemokines in infants with uncomplicated and severe plasmodium falciparum malaria. Clin Exp Immunol. (2011) 166:218–26. doi: 10.1111/j.1365-2249.2011.04474.x
143. Oyegue-Liabagui SL, Bouopda-Tuedom AG, Kouna LC, Maghendji-Nzondo S, Nzoughe H, Tchitoula-Makaya N, et al. Pro-and anti-inflammatory cytokines in children with malaria in Franceville, Gabon. Am J Clin Exp Immunol. (2017) 6(2):9–20.
144. Smith CM, Gill MB, May JS, Stevenson PG. Murine gammaherpesvirus-68 inhibits antigen presentation by dendritic cells. PloS One. (2007) 2:e1048. doi: 10.1371/journal.pone.0001048
145. Savard M, Gosselin J. Epstein-barr virus immunossuppression of innate immunity mediated by phagocytes. Virus Res. (2006) 119:134–45. doi: 10.1016/j.virusres.2006.02.008
146. Lin YL, Li M. Human cytomegalovirus and epstein–barr virus inhibit oral bacteria-induced macrophage activation and phagocytosis. Oral Microbiol Immunol. (2009) 24:243–8. doi: 10.1111/j.1399-302X.2009.00504.x
147. Abrial C. Caractérisation de la Polarisation Des Macrophages Pulmonaires Humains Et Voies De Régulation: Versailles-St Quentin en Yvelines. (2014) Français.
148. Sia IG, Patel R. New strategies for prevention and therapy of cytomegalovirus infection and disease in solid-organ transplant recipients. Clin Microbiol Rev. (2000) 13:83–121. doi: 10.1128/CMR.13.1.83
149. Azevedo LS, Pierrotti LC, Abdala E, Costa SF, Strabelli TMV, Campos SV, et al. Cytomegalovirus infection in transplant recipients. Clinics. (2015) 70:515–23. doi: 10.6061/clinics/2015(07)09
150. Kotton CN, Fishman JA. Viral infection in the renal transplant recipient. J Am Soc Nephrol. (2005) 16:1758–74. doi: 10.1681/ASN.2004121113
151. Snydman DR, Singh N. Interactions between viruses in transplant recipients. Clin Infect Dis. (2005) 40:430–6. doi: 10.1086/427214
152. Li S-s, Zhang N, Jia M, Su M. Association between cytomegalovirus and epstein-barr virus co-reactivation and hematopoietic stem cell transplantation. Front Cell Infect Microbiol. (2022) 12:818167. doi: 10.3389/fcimb.2022.818167
153. Fan J, Jing M, Yang M, Xu L, Liang H, Huang Y, et al. Herpesvirus infections in hematopoietic stem cell transplant recipients seropositive for human cytomegalovirus before transplantation. Int J Infect Dis. (2016) 46:89–93. doi: 10.1016/j.ijid.2016.03.025
154. Blazquez-Navarro A, Dang-Heine C, Wittenbrink N, Bauer C, Wolk K, Sabat R, et al. Bkv, cmv, and ebv interactions and their effect on graft function one year post-renal transplantation: results from a large multi-centre study. EBioMedicine. (2018) 34:113–21. doi: 10.1016/j.ebiom.2018.07.017
155. Shivanesan P, Minz M, Minz RW, Kumar Y, Sharma A, Kanwar DB, et al. Utility of quantitative real time pcr in detection and monitoring of viral infections in post renal transplant recipients. Indian J Transplant. (2016) 10:9–14. doi: 10.1016/j.ijt.2016.03.007
156. de Melo Silva J, Pinheiro-Silva R, Dhyani A, Pontes GS. Cytomegalovirus and epstein-barr infections: prevalence and impact on patients with hematological diseases. BioMed Res Int. (2020) 2020(1):1627824. doi: 10.1155/2020/1627824
157. Rasa S, Nora-Krukle Z, Henning N, Eliassen E, Shikova E, Harrer T, et al. Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (Me/cfs). J Trans Med. (2018) 16:268. doi: 10.1186/s12967-018-1644-y
158. Ablashi D, Agut H, Alvarez-Lafuente R, Clark DA, Dewhurst S, DiLuca D, et al. Classification of hhv-6a and hhv-6b as distinct viruses. Arch Virol. (2014) 159:863–70. doi: 10.1007/s00705-013-1902-5
159. Traore L, Nikiema O, Ouattara AK, Compaore TR, Soubeiga ST, Diarra B, et al. Ebv and hhv-6 circulating subtypes in people living with hiv in Burkina Faso, impact on cd4 T cell count and hiv viral load. Mediterr J Hematol Infect Dis. (2017) 9:e2017049. doi: 10.4084/mjhid.2017.049
160. Kositanont U, Kondo K, Chongkolwatana C, Metheetrairut C, Puthavathana P, Chantarakul N, et al. Detection of epstein-barr virus DNA and hhv-6 DNA in tissue biopsies from patients with nasopharyngeal carcinoma by polymerase chain reaction. Southeast Asian J Trop Med Public Health. (1993) 24:455–60.
161. Chen X, Jian S, Wang H. The study on clinical significance of human herpesvius 6 infection of npc tissues. Zhonghua shi yan he lin Chuang Bing du xue za zhi= Zhonghua Shiyan he Linchuang Bingduxue Zazhi= Chin J Exp Clin Virol. (1999) 13(3):262–5.
162. Eliassen E, Lum E, Pritchett J, Ongradi J, Krueger G, Crawford JR, et al. Human herpesvirus 6 and Malignancy: A review. Front Oncol. (2018) 8:512. doi: 10.3389/fonc.2018.00512
163. Oksenhendler E, Boutboul D, Galicier L. Kaposi sarcoma–associated herpesvirus/human herpesvirus 8–associated lymphoproliferative disorders. Blood. (2019) 133:1186–90. doi: 10.1182/blood-2018-11-852442
164. de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob Health. (2020) 8:e180–e90. doi: 10.1016/s2214-109x(19)30488-7
165. Crane GM, Ambinder RF, Shirley CM, Fishman EK, Kasamon YL, Taube JM, et al. Hhv-8-positive and ebv-positive intravascular lymphoma: an unusual presentation of extracavitary primary effusion lymphoma. Am J Surg Pathol. (2014) 38:426–32. doi: 10.1097/pas.0000000000000128
166. Sugden AU, Hayes M, Sugden B. How epstein-barr virus and Kaposi’s sarcoma-associated herpesvirus are maintained together to transform the same B-cell. Viruses. (2021) 13(8):1478. doi: 10.3390/v13081478
167. Böni M, Rieble L, Münz C. Co-infection of the epstein-barr virus and the kaposi sarcoma-associated herpesvirus. Viruses. (2022) 14:2709. doi: 10.3390/v14122709
168. Casper C, Corey L, Cohen JI, Damania B, Gershon AA, Kaslow DC, et al. Kshv (Hhv8) vaccine: promises and potential pitfalls for a new anti-cancer vaccine. NPJ Vaccines. (2022) 7:108. doi: 10.1038/s41541-022-00535-4
169. Münz C. Latency and lytic replication in the oncogenesis of the epstein barr virus. Nat Rev Micobiol. (2019) 17:691–700. doi: 10.1038/s41579-019-0249-7
170. Baidoun F, Moustafa MA, Tun HW, Hill BT. Clinical characteristics and survival outcomes of primary effusion lymphoma: A national cancer database study. Clin Lymph Myeloma Leukemia. (2022) 22:e485–e94. doi: 10.1016/j.clml.2022.01.006
171. Bigi R, Landis JT, An H, Caro-Vegas C, Raab-Traub N, Dittmer DP. Epstein–barr virus enhances genome maintenance of kaposi sarcoma-associated herpesvirus. Proc Natl Acad Sci. (2018) 115:E11379–E87. doi: 10.1073/pnas.1810128115
172. Faure A, Hayes M, Sugden B. How Kaposi’s sarcoma-associated herpesvirus stably transforms peripheral B cells towards lymphomagenesis. Proc Natl Acad Sci. (2019) 116:16519–28. doi: 10.1073/pnas.1905025116
173. Caduff N, McHugh D, Rieble L, Forconi CS, Ong’echa JM, Oluoch PO, et al. Kshv infection drives poorly cytotoxic cd56-negative natural killer cell differentiation in vivo upon kshv/ebv dual infection. Cell Rep. (2021) 35. doi: 10.1016/j.celrep.2021.109056
174. Dittmer DP, Damania B. Kaposi sarcoma–associated herpesvirus: immunobiology, oncogenesis, and therapy. J Clin Invest. (2016) 126:3165–75. doi: 10.1172/JCI84418
175. Godfrey A, Anderson J, Papanastasiou A, Takeuchi Y, Boshoff C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin rna. Blood. (2005) 105:2510–8. doi: 10.1182/blood-2004-08-3052
176. Ballestas ME, Kaye KM. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (Tr) sequence and specifically binds tr DNA. J Virol. (2001) 75:3250–8. doi: 10.1128/JVI.75.7.3250-3258.2001
177. Zhang Y-J, Wang K-Y, Stein DA, Patel D, Watkins R, Moulton HM, et al. Inhibition of replication and transcription activator and latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus by morpholino oligomers. Antiviral Res. (2007) 73:12–23. doi: 10.1016/j.antiviral.2006.05.017
178. Maecker HT, McCoy JP Jr. A model for harmonizing flow cytometry in clinical trials. Nat Immunol. (2010) 11:975–8. doi: 10.1038/ni1110-975
179. Wies E, Mori Y, Hahn A, Kremmer E, Stürzl M, Fleckenstein B, et al. The viral interferon-regulatory factor-3 is required for the survival of kshv-infected primary effusion lymphoma cells. Blood J Am Soc Hematol. (2008) 111:320–7. doi: 10.1182/blood-2007-05-092288
180. Labo N, Marshall V, Miley W, Davis E, McCann B, Stolka KB, et al. Mutual detection of Kaposi’s sarcoma-associated herpesvirus and epstein–barr virus in blood and saliva of Cameroonians with and without Kaposi’s sarcoma. Int J Cancer. (2019) 145:2468–77. doi: 10.1002/ijc.v145.9
181. Sallah N, Miley W, Labo N, Carstensen T, Fatumo S, Gurdasani D, et al. Distinct genetic architectures and environmental factors associate with host response to the Γ 2-herpesvirus infections. Nat Commun. (2020) 11:3849. doi: 10.1038/s41467-020-17696-2
182. Bentz GL, Bheda-Malge A, Wang L, Shackelford J, Damania B, Pagano JS. Kshv lana and ebv lmp1 induce the expression of uch-L1 following viral transformation. Virology. (2014) 448:293–302. doi: 10.1016/j.virol.2013.10.018
183. Meij P, van Esser JW, Niesters HG, van Baarle D, Miedema F, Blake N, et al. Impaired Recovery of Epstein-Barr Virus (Ebv)—Specific Cd8+ T Lymphocytes after Partially T-Depleted Allogeneic Stem Cell Transplantation May Identify Patients at Very High Risk for Progressive Ebv Reactivation and Lymphoproliferative Disease. Blood. (2003) 101:4290–7. doi: 10.1182/blood-2002-10-3001
184. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. (2014) 12:358–71.
185. Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, et al. Infusions of donor leukocytes to treat epstein-barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. New Engl J Med. (1994) 330:1185–91. doi: 10.1056/NEJM199404283301703
186. Gottschalk S, Heslop H, Rooney C. Adoptive immunotherapy for ebv-associated Malignancies. Leukemia lymphoma. (2005) 46(1):1–10. doi: 10.1080/10428190400002202
187. Rooney CM, Smith CA, Ng CY, Loftin SK, Sixbey JW, Gan Y, et al. Infusion of cytotoxic T cells for the prevention and treatment of epstein-barr virus–induced lymphoma in allogeneic transplant recipients. Blood J Am Soc Hematol. (1998) 92(5):1549–55. doi: 10.1182/blood.V92.5.1549
188. Cm R. Use of gene-modified virus-specific T lymphocytes to control epstein-barr-virus-related lymphoproliferation. Lancet. (1995) 345:9–13. doi: 10.1016/S0140-6736(95)91150-2
189. Duraiswamy J, Sherritt M, Thomson S, Tellam J, Cooper L, Connolly G, et al. Therapeutic lmp1 polyepitope vaccine for ebv-associated hodgkin disease and nasopharyngeal carcinoma. Blood J Am Soc Hematol. (2003) 101:3150–6. doi: 10.1182/blood-2002-10-3092
190. Ding S-W, Li H, Lu R, Li F, Li W-X. Rna silencing: A conserved antiviral immunity of plants and animals. Virus Res. (2004) 102:109–15. doi: 10.1016/j.virusres.2004.01.021
191. Butz K, Ristriani T, Hengstermann A, Denk C, Scheffner M, Hoppe-Seyler F. Sirna targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene. (2003) 22:5938–45. doi: 10.1038/sj.onc.1206894
192. Hall AH, Alexander KA. Rna interference of human papillomavirus type 18 E6 and E7 induces senescence in hela cells. J Virol. (2003) 77:6066–9. doi: 10.1128/JVI.77.10.6066-6069.2003
193. Li X-P, Li G, Peng Y, Kung H-f, Lin MC. Suppression of epstein–barr virus-encoded latent membrane protein-1 by rna interference inhibits the metastatic potential of nasopharyngeal carcinoma cells. Biochem Biophys Res Commun. (2004) 315:212–8. doi: 10.1016/j.bbrc.2004.01.045
Keywords: EBV, coinfection, HPV, SARS-CoV-2, HHV-6, Helicobacter
Citation: Ebrahimi F, Rasizadeh R, Sharaflou S, Aghbash PS, Shamekh A, Jafari-Sales A and Bannazadeh Baghi H (2024) Coinfection of EBV with other pathogens: a narrative review. Front. Virol. 4:1482329. doi: 10.3389/fviro.2024.1482329
Received: 18 August 2024; Accepted: 16 October 2024;
Published: 08 November 2024.
Edited by:
Patricia A. Barril, National Scientific and Technical Research Council (CONICET), ArgentinaReviewed by:
Takahiro Watanabe, Nagoya University, JapanLijun Ling, University of Alabama at Birmingham, United States
Copyright © 2024 Ebrahimi, Rasizadeh, Sharaflou, Aghbash, Shamekh, Jafari-Sales and Bannazadeh Baghi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hossein Bannazadeh Baghi, aGJhbm5hemFkZWhAdGJ6bWVkLmFjLmly; aGIuemFkZWhAZ21haWwuY29t
†These authors have contributed equally to this work