 Nicholas Saladino
Nicholas Saladino Daniel J. Salamango
Daniel J. Salamango
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Virol. , 01 May 2024
Sec. Virus and Host Immunity
Volume 4 - 2024 | https://doi.org/10.3389/fviro.2024.1410258
This article is part of the Research Topic Innate and Adaptive Immune Responses to Viral Infection View all 8 articles
Viruses are obligate intracellular pathogens that hijack a myriad of host cell processes to facilitate replication and suppress host antiviral defenses. In its essence, a virus is a segment of foreign nucleic acid that engages host cell machinery to drive viral genome replication, gene transcription, and protein synthesis to generate progeny virions. Because of this, host organisms have developed sophisticated detection systems that activate antiviral defenses following recognition of aberrant nucleic acids. For example, recognition of viral nucleic acids by host DNA repair proteins results in compromised viral genome integrity, induction of antiviral inflammatory programs, cell cycle arrest, and apoptosis. Unsurprisingly, diverse viral families have evolved multiple strategies that fine-tune host DNA repair responses to suppress activation of antiviral defenses while simultaneously hijacking DNA repair proteins to facilitate virus replication. This review summarizes common molecular strategies viruses deploy to exploit host DNA repair mechanisms.
The faithful transmission of genetic information from generation-to-generation is critical for ensuring the survival of an organism. Because genetic material is constantly bombarded by endogenous and exogenous agents, organisms have evolved overlapping repair mechanisms to ensure the stability and accuracy of their genetic code. This process is called the DNA damage response (DDR), which is a highly coordinated network of sensor proteins that detect DNA lesions, kinases that propagate DNA repair signals, and numerous proteins that physically repair damaged DNA. If left unresolved, DNA lesions can progress to DNA strand breaks that can lead to chromosomal aberrations, genome instability, cell death, and even carcinogenesis.
In general, the DNA lesion itself directly dictates which repair pathway is engaged. Alterations to DNA sequence or structure (at the nucleotide level) are repaired by three main pathways: nucleotide excision repair (NER), base excision repair (BER), or mismatch repair (MMR). These pathways are activated in response to bulky DNA adducts formed due to UV radiation or chemical carcinogens, damaged bases due to spontaneous oxidation, alkylation, or deamination, or mismatched bases due to errors in DNA replication, respectively (1). Interestingly, viruses seldom hijack components of these pathways. One possible explanation is that under normal circumstances viral genomes rarely accumulate damage from these sources given the location of virus replication and the kinetics of how quickly replication occurs. In addition, individual components of these pathways don’t directly activate antiviral programs, so their functional state is likely inconsequential to the virus. The best characterized exception to this rule comes from the HIV accessory protein Vpr, as it induces the depletion of several BER and NER proteins through a proteasomal degradation mechanism (2, 3). While the functional relevance of this is debated, it likely serves to counteract the mutagenic potential of host APOBEC3 antiviral enzymes that inflict C-to-U lesions in the HIV genome. Because BER/NER proteins generate abasic sites following the excision of deaminated nucleobases, which can lead to DNA strand breaks, depleting these DNA repair proteins likely preserves HIV genome integrity.
More complex lesions require more intricate processes for repair, as is the case for DNA single-strand breaks (SSBs) and double-strand breaks (DSBs), the latter being most deleterious to a cell (4, 5). Interestingly, viruses most often hijack components of SSB and DSB repair for reasons described in more detail below (Figure 1). The repair of DNA SSBs and DSBs requires a highly coordinated signaling network that is summarized in Figure 1A. ATM (ataxia telangiectasia mutated), ATR (ATM and Rad3-related), and DNA-PK (DNA-dependent protein kinase) are essential players in this pathway and mediate the propagation of repair signals. As is the case for BER/NER/MMR, the DNA lesion dictates which pathway is utilized, with ATR-directed repair being activated upon recognition of SSBs and stalled replication forks, while ATM- and DNA-PK-directed repair pathways are activated by DSBs (6) (Figure 1A). In the case of SSBs and stalled replication forks, replication protein A (RPA) acts as a sensor protein that coats single-stranded DNA and recruits ATR interacting protein (ATRIP) and ATR to the site of damage (7) (Figure 1A). ATR activation requires complex formation with ATRIP and TOPBP1 to trigger catalytic activity and subsequent phosphorylation of downstream targets including H2AX, CHK1, and p53, resulting in cell cycle arrest, chromatin remodeling, and DNA repair (8–11) (Figure 1A).
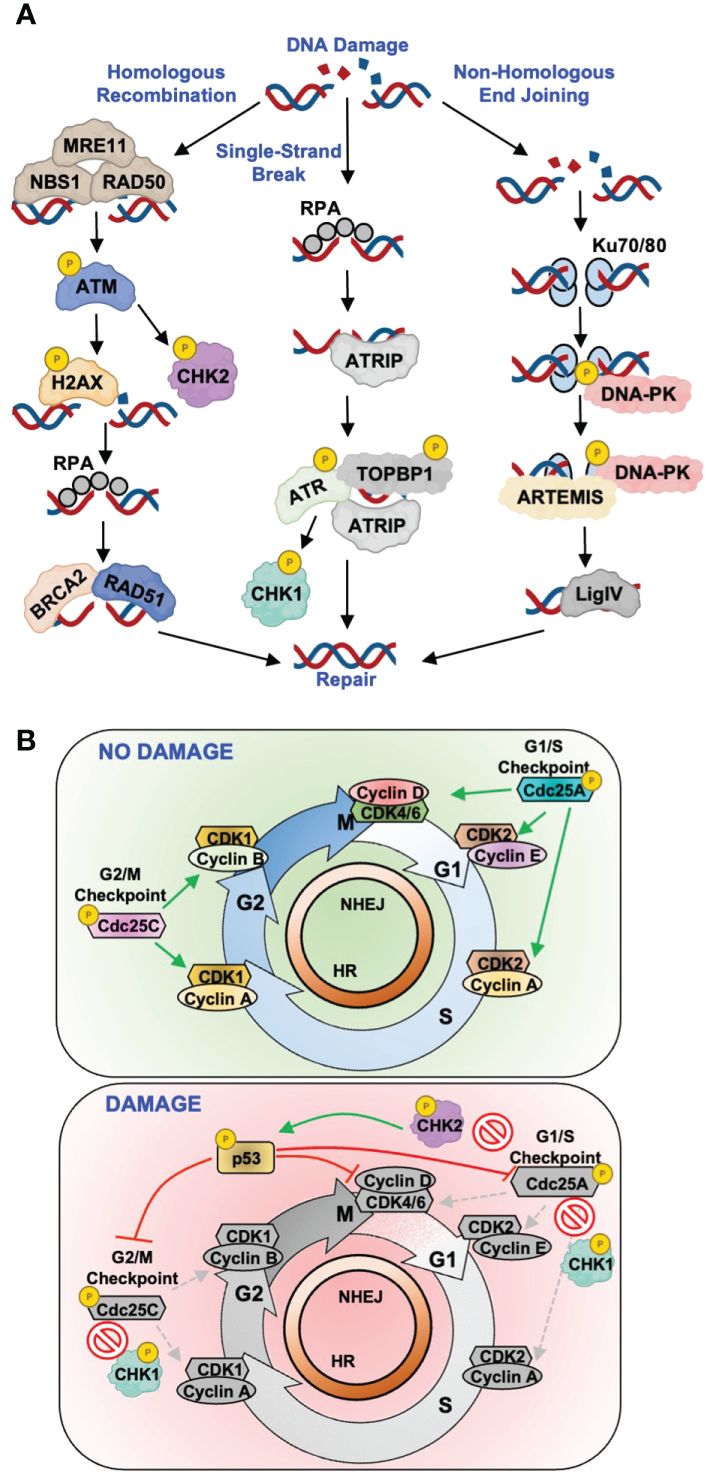
Figure 1 Host DNA break repair pathways and examples of virus-directed antagonism. (A) Simplified schematic of canonical single-strand and double-strand DNA break repair pathways. Key repair proteins known to be antagonized by viral proteins are depicted for each repair process. (B) Depiction of the cell cycle progressing from G1, to S-phase, to G2/M along with the cellular complexes that regulate these transitions. Top: in the absence of DNA damage, Cdc25A/C phosphatases regulate proper progression through the cell cycle by dephosphorylating key Cyclin/CDK complexes to activate their cell cycle regulating functions. Bottom: following the induction of DNA repair signaling, secondary DNA repair kinases CHK1 and CHK2 phosphorylate, and inactivate, Cdc25A/C phosphatases in addition to the p53 regulator.
Canonical DSB repair is mediated by two main pathways, homologous recombination (HR) and non-homologous end joining (NHEJ), with the major difference being the molecular mechanism of repair (Figure 1A). The selection of one repair mechanism over the other is largely dictated by cell cycle phase, with HR occurring more frequently during S/G2 whereas NHEJ can occur at all phases (12, 13). In the context of HR, DSBs are sensed by the MRN complex (MRE11, RAD50, and NBS1, Figure 1A). MRN mediates the recruitment and activation of ATM, which phosphorylates and activates key effector proteins H2AX, CHK2, and p53 (14–16) (Figure 1A). This leads to cell cycle arrest and recruitment of downstream repair factors BRCA2 and RAD51 (17, 18) (Figure 1A). In NHEJ, DSBs are sensed by the Ku70/80 heterodimer which recruits the catalytic subunit of DNA-PK to form an active repair complex (19) (Figure 1A). Once NHEJ has been initiated, the Artemis protein processes the broken DNA ends to allow for DNA ligase IV to join the strands and repair the DNA (20) (Figure 1A).
DDR also controls the activation of cell cycle checkpoints, which prevent cells with DNA damage from progressing into mitosis (21). Fine-tuned cell cycle regulation is critical for maintaining proper cell growth and division, and its dysregulation is a hallmark of carcinogenesis (22). There are three major cell cycle checkpoints, G1/S, S-phase, and G2/M, which are regulated by members of the cyclin and cyclin-dependent kinase (CDK) families (Figure 1B, top). Upon the induction of repair pathways, activated CHK1 and CHK2 kinases phosphorylate key cell cycle regulators such as the Cdc25A and Cdc25C phosphatases (Figure 1B, bottom). Phosphorylation of Cdc25A/C inhibits their enzymatic activity to prevent the activation of CDK-cyclin complexes that facilitate progression into the next cell cycle phase. This also prevents dephosphorylation of the tumor suppressor p53, which results in its stabilization and further inhibition of CDK-cyclin complexes (23–25) (Figure 1B, bottom).
In the context of virus infection, DDR proteins can also direct the activation of multiple proinflammatory programs. This is reviewed extensively here (26). During circumstances of abnormal or prolonged DDR signaling, ATM forms a heterodimer with the NF-κB regulator NEMO to trigger NF-κB cytoplasm-to-nucleus translocation and upregulation of proinflammatory cytokines, chemokines, and interferons (27–29). As discussed in more detail below, several viral families induce constitutive DDR signaling to exploit active repair proteins for promoting different phases of virus replication. This prolonged activation of DDR would normally trigger the pro-inflammatory ATM-NEMO-NF-κB nexus; however, many viruses deploy countermeasures to ensure this pathway is disrupted (discussed below). ATM can further mediate the activation of NF-κB signaling by complexing with ELKS, a protein necessary for the activation of the IKK-induced TAK1 kinase (28, 30). Moreover, the cGAS sensor can directly bind cytoplasmic double-stranded DNA to trigger IRF3-mediated transcription of proinflammatory genes through STING activation, in addition to serving as a central control point for integrating inflammatory signals from DNA-PK and MRE11 (31). The cGAS-STING sensor serves as a mechanism to recognize double-stranded DNA viruses that replicated in the cytoplasm, such as poxviruses. Lastly, ATM and ATR can directly bind aberrant DNA structures, such as viral replication intermediates, to trigger DDR responses and proinflammatory signaling (32–34).
Manipulation of host DDR responses is a broadly conserved activity among diverse RNA and DNA viruses, including adenoviruses (multiple serotypes), herpesviruses [herpes simplex virus-1 (HSV-1), Epstein-Barr virus (EBV), Kaposi’s sarcoma herpesvirus (KSHV)], human papilloma viruses (HPV, multiple serotypes), hepatitis viruses [hepatitis B virus (HBV), hepatitis C virus (HCV)], retroviruses [human T-lymphotropic virus-1 (HTLV-1), human immunodeficiency virus-1 (HIV-1)], and rotaviruses (select examples of DDR modulation detailed in Table 1). In general, these viruses deploy one or more proteins that inhibit the initiation or propagation of DDR signaling responses by regulating the activity of primary (ATM, ATR, DNA-PK) or secondary (CHK1 and CHK2) DDR kinases. This establishes an environment favorable for virus replication by inhibiting antiviral programs, maintaining viral genome integrity, inducing cell cycle arrest at phases that permit enhanced viral genome replication and/or gene transcription, and blocking apoptotic responses. In this section, we provide an overview of the molecular mechanisms’ viruses use to subvert DDR responses.
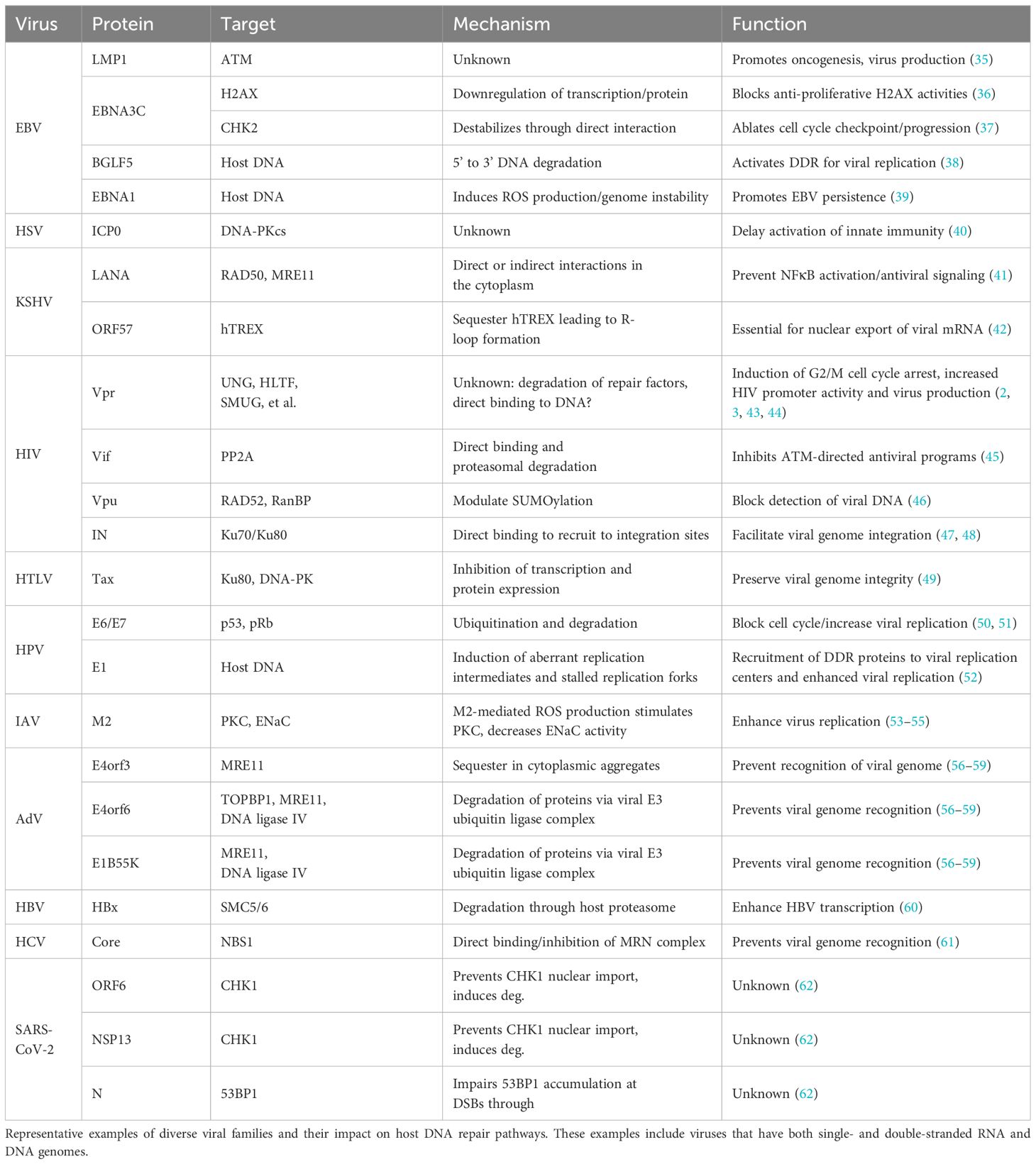
Table 1 Summary of virus-DDR antagonism mechanisms and associated functions.
Viruses antagonize DDR responses through direct and indirect mechanisms that culminate in the inactivation, depletion, or relocalization/sequestration of key DDR proteins. In many instances, repair factors that initially sense DNA damage or that directly activate DDR signaling pathways, such as the MRN complex, ATM, or DNA-PK, are downregulated transcriptionally or post-translationally. For example, herpesviruses and retroviruses deploy multiple accessory proteins that abrogate DNA-PK or ATM activity to block the initiation of DSB repair signaling, which prevents the activation of innate immune defenses and preserves genome integrity (Table 1). The ICP0 protein of HSV-1 inhibits DSB repair by inducing proteasomal degradation of the DNA-PK catalytic subunit shortly after infection (40, 63) (Table 1). While the mechanistic details are still unclear, DNA-PK restricts HSV-1 replication by affecting genome integrity and activating innate immune defenses such as cGAS-STING (64). Similarly, EBV LMP-1 inhibits DSB repair by downregulating ATM expression in vitro and in patient biopsies, which leads to DNA strand breaks, genome instability, and likely contributes to EBV-associated cancers (35, 65, 66). EBV further attenuates ATM-directed signaling by deploying the EBNA3C protein to downregulate H2AX and CHK2 transcripts and proteins, an activity that has been documented in multiple different cell types (36, 37) (Table 1). Collectively, these activities likely serve to disable cell cycle checkpoints and preserve EBV genome integrity.
In addition to antagonizing BER and NER, the HIV Vpr protein induces the depletion of roughly two-dozen cellular proteins involved in DNA repair or DNA modification (67, 68). Vpr achieves this by hijacking a host CUL4-DDB1-containing E3-ubiquitin ligase complex, which normally functions to regulate DNA repair, DNA replication, and chromatin remodeling (69–71) (Table 1). Interestingly, the HBx protein of HBV hijacks the same E3-ubiquitin ligase complex to inhibit DSB repair through degradation of the SMC5/6 repair complex, which enhances HBV transcription (60). Recent evidence indicates that HIV-1 accessory proteins Vpu and Vif can also antagonize DDR through mechanisms distinct, and independent from, Vpr. Vpu utilizes a SUMO E3-ubiquitin ligase complex to inhibit RAD52-mediated non-canonical DSB repair to preserve HIV-1 genome integrity (46) (Table 1). The ends of the double-stranded HIV genome can be recognized as broken DNA and are targeted by either NHEJ or HR for “repair”, which generates dead-end circular DNA products. Vpu prevents this while simultaneously suppressing innate immune sensing of the HIV-1 cDNA. Recent work by our group demonstrated that Vif antagonizes cellular phosphatase complexes to inhibit ATM-directed antiviral programs (45) (Table 1). We speculate that this counteracts the constitutive activation of DDR signaling induced by Vpr. For both Vif and Vpr, DDR antagonism has been linked to inducing G2/M cell cycle arrest, which has been correlated with increased HIV-1 promoter activity and virus production (43, 44, 72). Another retrovirus, HTLV-1, also targets DSB repair by deploying the Tax accessory protein to diminish DNA-PK activity through downregulation of the Ku80 DNA damage sensor, which is required for recruitment of DNA-PK to DNA strand breaks (49, 73) (Table 1). While the proviral function of this activity has yet to be elucidated, it is likely that this preserves viral genome integrity by preventing NHEJ-mediated circularization of the double-stranded DNA.
Adenoviruses also block DSB repair by inducing the degradation of multiple factors involved in HR or NHEJ. Accessory proteins E4orf3, E4orf6, and E1B55k coordinate to induce E3-ubiquitin ligase-mediated degradation of MRE11, LigIV, and TOPBP1, and can inhibit DNA-PK activity through a direct-binding mechanism (56–59) (Table 1). When functional, these pathways can recognize and fuse the free adenoviral DNA ends, creating a concatenated byproduct that is defective for replication. Interestingly, antagonizing DNA repair proteins is not constrained to viruses with DNA-based genomes. HCV, which has a positive-stranded RNA genome, utilizes its core protein to directly bind NBS1 and inhibit the formation of the MRN complex (61) (Table 1). In addition, a recent study demonstrated that SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), which also has a positive-stranded RNA genome, deploys several viral proteins to inhibit DSB repair by directing the proteasomal degradation of CHK1 and blocking 53BP1 recruitment to sites of DNA damage (62) (Table 1). In these latter two examples, it has yet to be established how viruses with RNA genomes benefit from antagonizing DNA repair pathways.
Another common strategy to modulate DDR responses is the relocalization or sequestration of DNA repair proteins. KSHV utilizes viral factors to recruit RPA and MRE11 to sites of viral DNA synthesis while deploying the LANA protein to relocalize other MRN components to the cytoplasm, thus blocking their ability to activate antiviral inflammatory programs (41, 74) (Table 1). Recruiting DDR proteins to viral replication centers is also a conserved function of HSV-1 and EBV. HSV-1 recruits the ATR/ATRIP complex as well as several MMR proteins to viral replication centers, while EBV recruits ATM and components of the MRN complex (75–78) (Table 1). Recruiting DDR proteins to viral replications centers is also prevalent among adenoviruses, rotaviruses, and HPV. Adenoviruses recruit several proteins involved in ATR-directed repair processes including ATR/ATRIP, RPA, TOPBP1, and multiple RAD proteins to viral replication centers (79, 80). In addition, adenovirus E4orf3 relocalizes MRE11 to nuclear tracts and cytoplasmic aggregates to maintain viral genome integrity during replication (81) (Table 1). In a similar fashion, several HPV serotypes recruit multiple proteins involved in ATM-directed responses, such as H2AX, 53BP1, CHK2, RAD51, and BRCA1 to replication centers (82–84) (Table 1). As was the case above, relocalization of repair factors is not exclusively used by DNA viruses, as rotaviruses, which have a double-stranded RNA genome, utilize viral proteins NSP2 and NSP5 to recruit ATM, CHK2, and the MRN complex to replication centers (85) (Table 1).
Viruses have also evolved multiple strategies to activate DDR signaling, which is counterintuitive given the interplay between DDR, antiviral responses, and viral genome integrity discussed above. In some instances, viruses directly damage host DNA to trigger the enzymatic activity of repair proteins, which are then leveraged to facilitate viral genome integration into host chromatin or to promote virus replication. In other cases, viral proteins indirectly antagonize cellular processes that regulate chromatin states or DNA integrity, which inevitably activates DDR proteins. As a bystander effect, these activities often have deleterious consequences for the host cell and can exacerbate pathogenesis. This section highlights the mechanisms deployed by diverse viruses to activate and hijack host DDR responses.
One of best-characterized examples of virus-directed DNA damage is the process of viral genome integration into host chromatin. While this process is traditionally associated with retroviruses, several other viral families including hepadnaviruses, papillomaviruses, and adeno-associated viruses undergo genome integration (86–88). However, it is worth noting that contrary to retroviruses, these latter viruses do not require genome integration for replication. Retroviral genome integration is mediated by the integrase (IN) enzyme, which directly fuses one 5’-end of a viral DNA strand to a corresponding 3’-end of a host DNA strand (89, 90). This results in a SSB for the opposing DNA strand, which is repaired through NHEJ. For many years researchers were puzzled as to why DSB repair proteins would be required for resolving a SSB. This mechanism was clarified following the recent discovery that HIV-1 IN directly binds to the Ku70/80 complex to redirect proteins involved in NHEJ to the site of integration (47, 48) (Table 1).
The natural formation of DSBs during host DNA replication and maintenance can also promote viral genome integration. HBV genomes spontaneously integrate into host chromatin during both chronic and acute infection, which can be enhanced ~10-fold by inducing DSBs in infected liver cells (91) (Table 1). Integrated HBV DNA is often observed in liver tumors and has been correlated with the onset and severity of liver disease and carcinogenesis. Similarly, integration of HPV genomes into host DNA has been correlated with invasive cervical carcinomas. The E6/E7 accessory proteins from high-risk HPV isolates induce host genome instability and spontaneous integration of HPV DNA; however, E6/E7 proteins from low-risk isolates don’t exhibit this activity, which potentially rationalizes why high-risk isolates correlate with oncogenesis (50, 51, 92) (Table 1). The HPV E1 helicase further compromises host DNA integrity by inducing aberrant replication intermediates and stalled replication forks (93). This triggers the activation of DDR proteins that are recruited into viral replication centers to facilitate genome amplification (52). Another example of this functional dichotomy is the EBV BGLF5 protein, which is essential for virus replication but also exacerbates carcinogenesis. BGLF5 is a DNase that induces DNA strand breaks to activate DNA repair proteins that are leveraged to facilitate virus replication; however, a consequence of this activity is the induction of genome instability and cellular transformation (38, 94, 95) (Table 1).
Viruses can activate DDR through indirect mechanisms as well. The KSHV ORF57 protein sequesters a host complex required for mRNA processing and stability, which drives a global loss of mRNA stability and the formation of R-loops, DSBs, and the activation of DDR signaling (42). Not only is this interaction essential for efficient export of viral mRNA from the nucleus, but it also contributes to KSHV-associated tumorigenesis (96). In the case of BK polyomaviruses, the process of virus replication itself indirectly activates ATM, ATR, and downstream signaling responses, which are required for optimal virus replication (97) (Table 1). Interestingly, the adenoviral E1A protein is required for facilitating viral genome replication, but as a consequence of its virus-associated activity also induces massive cellular DNA synthesis during S-phase, which triggers significant replication stress and the activation of DDR signaling (98). An additional consequence of these virus-directed activities is the production of ROS. EBV-encoded EBNA-1 is known to induce host genome instability, DNA damage, and DDR signaling by inducing significant amounts of ROS production (39). EBNA-1-mediated ROS production is thought to promote cellular transformation and the establishment of EBV persistence (39). This is further supported by observations that EBV-mediated B cell immortalization is promoted by oxidative stress and hindered by antioxidants (99, 100) (Table 1). Similarly, influenza A virus (IAV) infection is associated with generating high levels of ROS that contribute to severe pathogenesis (101). ROS are generated as a byproduct of the interaction between the IAV M2 ion channel and mitochondrial membranes (53, 54). Given that M2 modulates epithelial ion channel functions, which are known to influence IAV replication (55), it is likely that M2-generated ROS confers a replication advantage.
Future studies investigating the interplay between viruses and host DDR will continue to uncover novel mechanisms used to hijack these pathways and the functional outcomes for virus replication. Importantly, these studies will inevitably yield valuable insights into how host repair pathways function in general and how dysregulation of these pathways leads to human disease. In the last decade alone, research into virus-DDR interactions has led to major discoveries regarding the interconnectedness of DDR and innate immune responses, how virus-mediated subversion of DDR leads to carcinogenesis, and novel facets of DDR and cell cycle regulation. Furthermore, a more comprehensive understanding of virus-DDR interactions can inform the development of therapeutics that exacerbate defects in DNA repair to promote “synthetic lethality” and induce apoptosis of virus-infected or carcinogenic cells (102, 103). Thus, therapeutic interventions that target the virus-DDR nexus could not only suppress virus replication but alleviate the onset and severity of several associated diseases.
NS: Writing – review & editing, Writing – original draft. DS: Writing – review & editing, Funding acquisition, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIAID R00 award AI147811 (to DS) and startup funds from the University of Texas Health Science Center at San Antonio.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hakem R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. (2008) 27:589–605. doi: 10.1038/emboj.2008.15
2. Schrofelbauer B, Yu Q, Zeitlin SG, Landau NR. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol. (2005) 79:10978–87. doi: 10.1128/JVI.79.17.10978-10987.2005
3. Lahouassa H, Blondot ML, Chauveau L, Chougui G, Morel M, Leduc M, et al. HIV-1 Vpr degrades the HLTF DNA translocase in T cells and macrophages. Proc Natl Acad Sci U S A. (2016) 113:5311–6. doi: 10.1073/pnas.1600485113
4. Subba Rao K. Mechanisms of disease: DNA repair defects and neurological disease. Nat Clin Pract Neurol. (2007) 3:162–72. doi: 10.1038/ncpneuro0448
5. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol. (2010) 11:220–8. doi: 10.1038/nrm2858
6. Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. (2008) 9:619–31. doi: 10.1038/nrg2380
7. Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. (2003) 300:1542–8. doi: 10.1126/science.1083430
8. Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. (2000) 14:1448–59. doi: 10.1101/gad.14.12.1448
9. Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. (1999) 13:152–7. doi: 10.1101/gad.13.2.152
10. Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. (2001) 276:47759–62. doi: 10.1074/jbc.C100569200
11. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. (2010) 40:179–204. doi: 10.1016/j.molcel.2010.09.019
12. Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst). (2006) 5:1042–8. doi: 10.1016/j.dnarep.2006.05.026
13. Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. (2011) 30:1079–92. doi: 10.1038/emboj.2011.27
14. Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. (2001) 276:42462–7. doi: 10.1074/jbc.C100466200
15. Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U.S.A. (2000) 97:10389–94. doi: 10.1073/pnas.190030497
16. Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. (1998) 281:1677–9. doi: 10.1126/science.281.5383.1677
17. Martinez J, Georgoff I, Martinez J, Levine AJ. Cellular localization and cell cycle regulation by a temperature-sensitive p53 protein. Genes Dev. (1991) 5:151–9. doi: 10.1101/gad.5.2.151
18. Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, et al. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. (2001) 7:273–82. doi: 10.1016/S1097-2765(01)00175-7
19. Suwa A, Hirakata M, Takeda Y, Jesch SA, Mimori T, Hardin JA. DNA-dependent protein kinase (Ku protein-p350 complex) assembles on double-stranded DNA. Proc Natl Acad Sci U S A. (1994) 91:6904–8. doi: 10.1073/pnas.91.15.6904
20. Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. (2004) 16:715–24. doi: 10.1016/j.molcel.2004.10.029
21. Matellan L, Monje-Casas F. Regulation of mitotic exit by cell cycle checkpoints: lessons from Saccharomyces cerevisiae. Genes (Basel). (2020) 11:195. doi: 10.3390/genes11020195
22. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
23. Costanzo V, Robertson K, Ying CY, Kim E, Avvedimento E, Gottesman M, et al. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol Cell. (2000) 6:649–59. doi: 10.1016/S1097-2765(00)00063-0
24. Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer. (2014) 134:1013–23. doi: 10.1002/ijc.28226
25. He G, Siddik ZH, Huang Z, Wang R, Koomen J, Kobayashi R, et al. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene. (2005) 24:2929–43. doi: 10.1038/sj.onc.1208474
26. Lopez A, Nichols Doyle R, Sandoval C, Nisson K, Yang V, Fregoso OI. Viral modulation of the DNA damage response and innate immunity: two sides of the same coin. J Mol Biol. (2022) 434:167327. doi: 10.1016/j.jmb.2021.167327
27. Elkon R, Rashi-Elkeles S, Lerenthal Y, Linhart C, Tenne T, Amariglio N, et al. Dissection of a DNA-damage-induced transcriptional network using a combination of microarrays, RNA interference and computational promoter analysis. Genome Biol. (2005) 6:R43. doi: 10.1186/gb-2005-6-5-r43
28. Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science. (2006) 311:1141–6. doi: 10.1126/science.1121513
29. Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell. (2003) 115:565–76. doi: 10.1016/S0092-8674(03)00895-X
30. Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, et al. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell. (2010) 40:75–86. doi: 10.1016/j.molcel.2010.09.010
31. Zhang X, Brann TW, Zhou M, Yang J, Oguariri RM, Lidie KB, et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J Immunol. (2011) 186:4541–5. doi: 10.4049/jimmunol.1003389
32. Smith GC, Cary RB, Lakin ND, Hann BC, Teo SH, Chen DJ, et al. Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. Proc Natl Acad Sci U S A. (1999) 96:11134–9. doi: 10.1073/pnas.96.20.11134
33. Unsal-Kacmaz K, Makhov AM, Griffith JD, Sancar A. Preferential binding of ATR protein to UV-damaged DNA. Proc Natl Acad Sci U S A. (2002) 99:6673–8. doi: 10.1073/pnas.102167799
34. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. (2000) 14:927–39. doi: 10.1101/gad.14.8.927
35. Dutton A, Woodman CB, Chukwuma MB, Last JI, Wei W, Vockerodt M, et al. Bmi-1 is induced by the Epstein-Barr virus oncogene LMP1 and regulates the expression of viral target genes in Hodgkin lymphoma cells. Blood. (2007) 109:2597–603. doi: 10.1182/blood-2006-05-020545
36. Jha HC, JM A, Saha A, Banerjee S, Lu J, Robertson ES. Epstein-Barr virus essential antigen EBNA3C attenuates H2AX expression. J Virol. (2014) 88:3776–88. doi: 10.1128/JVI.03568-13
37. Choudhuri T, Verma SC, Lan K, Murakami M, Robertson ES. The ATM/ATR signaling effector Chk2 is targeted by Epstein-Barr virus nuclear antigen 3C to release the G2/M cell cycle block. J Virol. (2007) 81:6718–30. doi: 10.1128/JVI.00053-07
38. Feederle R, Bannert H, Lips H, Muller-Lantzsch N, Delecluse HJ. The Epstein-Barr virus alkaline exonuclease BGLF5 serves pleiotropic functions in virus replication. J Virol. (2009) 83:4952–62. doi: 10.1128/JVI.00170-09
39. Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci U S A. (2009) 106:2313–8. doi: 10.1073/pnas.0810619106
40. Parkinson J, Lees-Miller SP, Everett RD. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol. (1999) 73:650–7. doi: 10.1128/JVI.73.1.650-657.1999
41. Mariggio G, Koch S, Zhang G, Weidner-Glunde M, Ruckert J, Kati S, et al. Kaposi Sarcoma Herpesvirus (KSHV) Latency-Associated Nuclear Antigen (LANA) recruits components of the MRN (Mre11-Rad50-NBS1) repair complex to modulate an innate immune signaling pathway and viral latency. PloS Pathog. (2017) 13:e1006335. doi: 10.1371/journal.ppat.1006335
42. Jackson BR, Noerenberg M, Whitehouse A. A novel mechanism inducing genome instability in Kaposi’s sarcoma-associated herpesvirus infected cells. PloS Pathog. (2014) 10:e1004098. doi: 10.1371/journal.ppat.1004098
43. Izumi T, Io K, Matsui M, Shirakawa K, Shinohara M, Nagai Y, et al. HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc Natl Acad Sci U S A. (2010) 107:20798–803. doi: 10.1073/pnas.1008076107
44. Andersen JL, Le Rouzic E, Planelles V. HIV-1 Vpr: mechanisms of G2 arrest and apoptosis. Exp Mol Pathol. (2008) 85:2–10. doi: 10.1016/j.yexmp.2008.03.015
45. Wong HT, Luperchio AM, Riley S, Salamango DJ. Inhibition of ATM-directed antiviral responses by HIV-1 Vif. PloS Pathog. (2023) 19:e1011634. doi: 10.1371/journal.ppat.1011634
46. Volcic M, Sparrer KMJ, Koepke L, Hotter D, Sauter D, Sturzel CM, et al. Vpu modulates DNA repair to suppress innate sensing and hyper-integration of HIV-1. Nat Microbiol. (2020) 5:1247–61. doi: 10.1038/s41564-020-0753-6
47. Zheng Y, Ao Z, Wang B, Jayappa KD, Yao X. Host protein Ku70 binds and protects HIV-1 integrase from proteasomal degradation and is required for HIV replication. J Biol Chem. (2011) 286:17722–35. doi: 10.1074/jbc.M110.184739
48. Knyazhanskaya E, Anisenko A, Shadrina O, Kalinina A, Zatsepin T, Zalevsky A, et al. NHEJ pathway is involved in post-integrational DNA repair due to Ku70 binding to HIV-1 integrase. Retrovirology. (2019) 16:30. doi: 10.1186/s12977-019-0492-z
49. Ducu RI, Dayaram T, Marriott SJ. The HTLV-1 Tax oncoprotein represses Ku80 gene expression. Virology. (2011) 416:1–8. doi: 10.1016/j.virol.2011.04.012
50. Kessis TD, Connolly DC, Hedrick L, Cho KR. Expression of HPV16 E6 or E7 increases integration of foreign DNA. Oncogene. (1996) 13:427–31.
51. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. (1990) 63:1129–36. doi: 10.1016/0092-8674(90)90409-8
52. Swindle CS, Zou N, Van Tine BA, Shaw GM, Engler JA, Chow LT. Human papillomavirus DNA replication compartments in a transient DNA replication system. J Virol. (1999) 73:1001–9. doi: 10.1128/JVI.73.2.1001-1009.1999
53. Vlahos R, Stambas J, Bozinovski S, Broughton BR, Drummond GR, Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PloS Pathog. (2011) 7:e1001271. doi: 10.1371/journal.ppat.1001271
54. Lazrak A, Iles KE, Liu G, Noah DL, Noah JW, Matalon S. Influenza virus M2 protein inhibits epithelial sodium channels by increasing reactive oxygen species. FASEB J. (2009) 23:3829–42. doi: 10.1096/fj.09-135590
55. Hoffmann HH, Palese P, Shaw ML. Modulation of influenza virus replication by alteration of sodium ion transport and protein kinase C activity. Antiviral Res. (2008) 80:124–34. doi: 10.1016/j.antiviral.2008.05.008
56. Boyer J, Rohleder K, Ketner G. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology. (1999) 263:307–12. doi: 10.1006/viro.1999.9866
57. Baker A, Rohleder KJ, Hanakahi LA, Ketner G. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J Virol. (2007) 81:7034–40. doi: 10.1128/JVI.00029-07
58. Cheng CY, Gilson T, Dallaire F, Ketner G, Branton PE, Blanchette P. The E4orf6/E1B55K E3 ubiquitin ligase complexes of human adenoviruses exhibit heterogeneity in composition and substrate specificity. J Virol. (2011) 85:765–75. doi: 10.1128/JVI.01890-10
59. Forrester NA, Sedgwick GG, Thomas A, Blackford AN, Speiseder T, Dobner T, et al. Serotype-specific inactivation of the cellular DNA damage response during adenovirus infection. J Virol. (2011) 85:2201–11. doi: 10.1128/JVI.01748-10
60. Lee TH, Elledge SJ, Butel JS. Hepatitis B virus X protein interacts with a probable cellular DNA repair protein. J Virol. (1995) 69:1107–14. doi: 10.1128/jvi.69.2.1107-1114.1995
61. Machida K, McNamara G, Cheng KT, Huang J, Wang CH, Comai L, et al. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. J Immunol. (2010) 185:6985–98. doi: 10.4049/jimmunol.1000618
62. Gioia U, Tavella S, Martinez-Orellana P, Cicio G, Colliva A, Ceccon M, et al. SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence. Nat Cell Biol. (2023) 25:550–64. doi: 10.1038/s41556-023-01096-x
63. Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J Virol. (1996) 70:7471–7. doi: 10.1128/jvi.70.11.7471-7477.1996
64. Burleigh K, Maltbaek JH, Cambier S, Green R, Gale M Jr., James RC, et al. Human DNA-PK activates a STING-independent DNA sensing pathway. Sci Immunol. (2020) 5:eaba4219. doi: 10.1126/sciimmunol.aba4219
65. Gruhne B, Sompallae R, Masucci MG. Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene. (2009) 28:3997–4008. doi: 10.1038/onc.2009.258
66. Bose S, Yap LF, Fung M, Starzcynski J, Saleh A, Morgan S, et al. The ATM tumour suppressor gene is down-regulated in EBV-associated nasopharyngeal carcinoma. J Pathol. (2009) 217:345–52. doi: 10.1002/path.2487
67. Greenwood EJD, Williamson JC, Sienkiewicz A, Naamati A, Matheson NJ, Lehner PJ. Promiscuous targeting of cellular proteins by Vpr drives systems-level proteomic remodeling in HIV-1 infection. Cell Rep. (2019) 27:1579–96.e7. doi: 10.1016/j.celrep.2019.04.025
68. Fabryova H, Strebel K. Vpr and its cellular interaction partners: R we there yet? Cells. (2019) 8:1310. doi: 10.3390/cells8111310
69. Iovine B, Iannella ML, Bevilacqua MA. Damage-specific DNA binding protein 1 (DDB1): a protein with a wide range of functions. Int J Biochem Cell Biol. (2011) 43:1664–7. doi: 10.1016/j.biocel.2011.09.001
70. Belzile JP, Duisit G, Rougeau N, Mercier J, Finzi A, Cohen EA. HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PloS Pathog. (2007) 3:e85. doi: 10.1371/journal.ppat.0030085
71. Hrecka K, Gierszewska M, Srivastava S, Kozaczkiewicz L, Swanson SK, Florens L, et al. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc Natl Acad Sci U S A. (2007) 104:11778–83. doi: 10.1073/pnas.0702102104
72. Salamango DJ, Ikeda T, Moghadasi SA, Wang J, McCann JL, Serebrenik AA, et al. HIV-1 Vif triggers cell cycle arrest by degrading cellular PPP2R5 phospho-regulators. Cell Rep. (2019) 29:1057–65.e4. doi: 10.1016/j.celrep.2019.09.057
73. Durkin SS, Guo X, Fryrear KA, Mihaylova VT, Gupta SK, Belgnaoui SM, et al. HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J Biol Chem. (2008) 283:36311–20. doi: 10.1074/jbc.M804931200
74. Hollingworth R, Skalka GL, Stewart GS, Hislop AD, Blackbourn DJ, Grand RJ. Activation of DNA damage response pathways during lytic replication of KSHV. Viruses. (2015) 7:2908–27. doi: 10.3390/v7062752
75. Taylor TJ, Knipe DM. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J Virol. (2004) 78:5856–66. doi: 10.1128/JVI.78.11.5856-5866.2004
76. Mohni KN, Livingston CM, Cortez D, Weller SK. ATR and ATRIP are recruited to herpes simplex virus type 1 replication compartments even though ATR signaling is disabled. J Virol. (2010) 84:12152–64. doi: 10.1128/JVI.01643-10
77. Mohni KN, Mastrocola AS, Bai P, Weller SK, Heinen CD. DNA mismatch repair proteins are required for efficient herpes simplex virus 1 replication. J Virol. (2011) 85:12241–53. doi: 10.1128/JVI.05487-11
78. Hau PM, Deng W, Jia L, Yang J, Tsurumi T, Chiang AK, et al. Role of ATM in the formation of the replication compartment during lytic replication of Epstein-Barr virus in nasopharyngeal epithelial cells. J Virol. (2015) 89:652–68. doi: 10.1128/JVI.01437-14
79. Blackford AN, Bruton RK, Dirlik O, Stewart GS, Taylor AM, Dobner T, et al. A role for E1B-AP5 in ATR signaling pathways during adenovirus infection. J Virol. (2008) 82:7640–52. doi: 10.1128/JVI.00170-08
80. Reyes ED, Kulej K, Pancholi NJ, Akhtar LN, Avgousti DC, Kim ET, et al. Identifying host factors associated with DNA replicated during virus infection. Mol Cell Proteom. (2017) 16:2079–97. doi: 10.1074/mcp.M117.067116
81. Araujo FD, Stracker TH, Carson CT, Lee DV, Weitzman MD. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J Virol. (2005) 79:11382–91. doi: 10.1128/JVI.79.17.11382-11391.2005
82. Kadaja M, Isok-Paas H, Laos T, Ustav E, Ustav M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PloS Pathog. (2009) 5:e1000397. doi: 10.1371/journal.ppat.1000397
83. Gillespie KA, Mehta KP, Laimins LA, Moody CA. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol. (2012) 86:9520–6. doi: 10.1128/JVI.00247-12
84. McKinney CC, Hussmann KL, McBride AA. The role of the DNA damage response throughout the papillomavirus life cycle. Viruses. (2015) 7:2450–69. doi: 10.3390/v7052450
85. Sarkar R, Patra U, Lo M, Mukherjee A, Biswas A, Chawla-Sarkar M. Rotavirus activates a noncanonical ATM-Chk2 branch of DNA damage response during infection to positively regulate viroplasm dynamics. Cell Microbiol. (2020) 22:e13149. doi: 10.1111/cmi.13149
86. Deyle DR, Russell DW. Adeno-associated virus vector integration. Curr Opin Mol Ther. (2009) 11:442–7.
87. Yang W, Summers J. Integration of hepadnavirus DNA in infected liver: evidence for a linear precursor. J Virol. (1999) 73:9710–7. doi: 10.1128/JVI.73.12.9710-9717.1999
88. Williams VM, Filippova M, Soto U, Duerksen-Hughes PJ. HPV-DNA integration and carcinogenesis: putative roles for inflammation and oxidative stress. Future Virol. (2011) 6:45–57. doi: 10.2217/fvl.10.73
89. Miller MD, Wang B, Bushman FD. Human immunodeficiency virus type 1 preintegration complexes containing discontinuous plus strands are competent to integrate in vitro. J Virol. (1995) 69:3938–44. doi: 10.1128/jvi.69.6.3938-3944.1995
90. Daniel R, Kao G, Taganov K, Greger JG, Favorova O, Merkel G, et al. Evidence that the retroviral DNA integration process triggers an ATR-dependent DNA damage response. Proc Natl Acad Sci U S A. (2003) 100:4778–83. doi: 10.1073/pnas.0730887100
91. Bill CA, Summers J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc Natl Acad Sci U S A. (2004) 101:11135–40. doi: 10.1073/pnas.0403925101
92. Zhang B, Chen W, Roman A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc Natl Acad Sci U S A. (2006) 103:437–42. doi: 10.1073/pnas.0510012103
93. Sakakibara N, Mitra R, McBride AA. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol. (2011) 85:8981–95. doi: 10.1128/JVI.00541-11
94. Wu CC, Liu MT, Chang YT, Fang CY, Chou SP, Liao HW, et al. Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. (2010) 38:1932–49. doi: 10.1093/nar/gkp1169
95. Wu CC, Chen MS, Lee TY, Cheng YJ, Tsou HH, Huang TS, et al. Screening and identification of emodin as an EBV DNase inhibitor to prevent its biological functions. Virol J. (2023) 20:148. doi: 10.1186/s12985-023-02107-x
96. Boyne JR, Colgan KJ, Whitehouse A. Recruitment of the complete hTREX complex is required for Kaposi’s sarcoma-associated herpesvirus intronless mRNA nuclear export and virus replication. PloS Pathog. (2008) 4:e1000194. doi: 10.1371/journal.ppat.1000194
97. Verhalen B, Justice JL, Imperiale MJ, Jiang M. Viral DNA replication-dependent DNA damage response activation during BK polyomavirus infection. J Virol. (2015) 89:5032–9. doi: 10.1128/JVI.03650-14
98. Singhal G, Leo E, Setty SK, Pommier Y, Thimmapaya B. Adenovirus E1A oncogene induces rereplication of cellular DNA and alters DNA replication dynamics. J Virol. (2013) 87:8767–78. doi: 10.1128/JVI.00879-13
99. Ranjan D, Siquijor A, Johnston TD, Wu G, Nagabhuskahn M. The effect of curcumin on human B-cell immortalization by Epstein-Barr virus. Am Surg. (1998) 64:47–51; discussion -2.
100. Chen C, Jeon H, Johnston TD, Gedaly R, McHugh PP, Ranjan D. Cyclosporin A-induced lipid and protein oxidation in human B-cells and in Epstein-Barr virus-infected B-cells is prevented by antioxidants. J Invest Surg. (2008) 21:201–8. doi: 10.1080/08941930802262223
101. Oda T, Akaike T, Hamamoto T, Suzuki F, Hirano T, Maeda H. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science. (1989) 244:974–6. doi: 10.1126/science.2543070
102. Kaelin WG Jr. Synthetic lethality: a framework for the development of wiser cancer therapeutics. Genome Med. (2009) 1:99. doi: 10.1186/gm99
Keywords: antiviral signaling, DNA damage repair, Host-Pathogen Interactions, innate immunity, virus
Citation: Saladino N and Salamango DJ (2024) Wielding a double-edged sword: viruses exploit host DNA repair systems to facilitate replication while bypassing immune activation. Front. Virol. 4:1410258. doi: 10.3389/fviro.2024.1410258
Received: 31 March 2024; Accepted: 22 April 2024;
Published: 01 May 2024.
Edited by:
Stefania Varchetta, San Matteo Hospital Foundation (IRCCS), ItalyReviewed by:
Tiansheng Li, National Institutes of Health (NIH), United StatesCopyright © 2024 Saladino and Salamango. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel J. Salamango, c2FsYW1hbmdvQHV0aHNjc2EuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.