 Shreoshri Bhattacharjee
Shreoshri Bhattacharjee Amit Gaba
Amit Gaba Linda Chelico
Linda Chelico
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol. , 24 January 2024
Sec. Virus and Host Immunity
Volume 4 - 2024 | https://doi.org/10.3389/fviro.2024.1343037
The human family of APOBEC3 enzymes are primarily studied as single-stranded DNA deoxycytidine deaminases that act as host restriction factors for a number of viruses and retroelements. The deamination of deoxycytidine to deoxyuridine causes inactivating mutations in target DNA and the nucleic acid binding ability may also cause deamination independent restriction. There are seven APOBEC3 enzymes in humans, named A-H, excluding E, each of which has restriction activity against a subset of viruses or retroelements. There are primarily four, APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H that have been found to restrict replication of HIV-1, however their restriction activity varies and they have primarily been studied individually despite co-expression in the cells that HIV-1 infects. It is known that APOBEC3F hetero-oligomerizes with APOBEC3G and APOBEC3H and that this influences host restriction outcomes during HIV-1 infection in tissue culture. Here, we examined if APOBEC3F interacts with APOBEC3D and the functional outcomes. We found that APOBEC3D mRNA expression was similar to or higher than APOBEC3F mRNA in multiple donors, suggesting that the proteins would be co-expressed, allowing for interactions to occur. We determined that APOBEC3F and APOBEC3D interacted primarily through an RNA intermediate; however, this interaction resulted in APOBEC3D competitively excluding APOBEC3F from virions. Although HIV-1 restriction still occurred when APOBEC3F and APOBEC3D were co-expressed, it was due to primarily APOBEC3D-mediated deamination-independent restriction. The APOBEC3D-mediated exclusion of APOBEC3F from HIV-1 encapsidation could be recapitulated in vitro through RNA capture experiments in which APOBEC3D decreased or abrogated the ability of APOBEC3F to bind to HIV-1 protease or 5’UTR RNA, respectively. Overall, the data suggest that there are mechanisms at the protein level that segregate APOBEC3s into different virus particles.
The APOBEC3 enzymes are a family of polynucleotide cytosine deaminases that can form uracil in single-stranded (ss) DNA or RNA (1, 2). The formation of uracil is used by APOBEC3 enzymes as an anti-viral and anti-retroelement defense. The uracils cause mutations that lead to functional inactivation or cause DNA repair-mediated degradation of the viral or retroelement genome (1, 3). The APOBEC3 genes are present in all placental mammals with differing numbers of paralogs (4). Most primates have seven APOBEC3 paralogs (4). For humans, these genes are all located on chromosome 22 and named APOBEC3A-H (A3A-H), excluding E (5, 6). This locus was formed by gene duplication events in primate ancestors and coincided with a requirement to suppress endogenous retroelements (7).
Although A3 enzymes have been found to deaminate the genomes of RNA and DNA viruses, the most well studied function of these enzymes is the restriction of retroviruses and retroelements (1, 2, 8–10). Retroviral restriction occurs when an A3 enzyme becomes encapsidated into the budding virion (1). Upon infection of another cell by these virions, the A3 enzymes can access the (–) DNA during proviral DNA synthesis (1). A3 enzymes can deaminate deoxycytidine to deoxyuracil, which causes errors during (+) DNA synthesis since the viral reverse transcriptase is forced to use the deoxyuracil as a template (1). In addition, several A3 enzymes have been found to physically inhibit viral reverse transcriptase by binding to the RNA or DNA template or directly to reverse transcriptase (1–3). The uracils either cause functional inactivation of the provirus through mutations or enable host DNA repair-mediated degradation of the proviral DNA (3). Retroviruses have mechanisms to suppress A3-induced mutagenesis or restriction. For example, HIV-1 (referred to as HIV) produces a protein, Vif, that hijacks a Cullin 5 E3 ligase to act as the substrate receptor and recruit A3 enzymes for ubiquitination and proteasomal degradation (2).
The seven APOBEC3 enzymes in humans have several distinguishing characteristics. The family is made up of single zinc-coordinating deaminase domain (ZDD) enzymes (A3A, A3C, A3H) and double ZDD enzymes (A3B, A3D, A3F, A3G) (11). Originally A3 enzymes were identified by bioinformatics analysis and that led to the annotation of A3D as two genes, A3D and A3E (5). After functional analysis, it was determined that A3D was a double ZDD enzyme, not two individual enzymes (12). A3B is the only family member with a nuclear localization signal (13, 14). Other A3 enzymes are primarily localized to the cytoplasm or if a single ZDD protein, can diffuse into the nucleus (15). The A3 family was functionally discovered after A3G was identified as a host restriction factor for HIV-1 (16). Thereafter, several labs characterized the family and determined that for HIV restriction, A3G, A3F and A3H stable haplotypes (Haplotypes II, V, and VII) were most active (17–20). A3A and A3B do not restrict HIV and A3C weakly restricts HIV, although a more active A3C S188I polymorphism exists in 10% of people of African descent (17, 21, 22). A3D has been the most challenging A3 enzyme to study and several incongruous reports have led to an unclear understanding of A3D function in HIV restriction (12, 18, 23).
In comparison to non-human primate A3D, human A3D has lower deamination activity, which was determined to be caused by a Y320C change (23). Although A3D in humans was initially identified to restrict HIV through a deamination-dependent mechanism, the detection of mutations may have been due to excessive overexpression of A3D in these studies since later studies did not detect strong or sometimes detected no deamination induced mutations in proviral genomes (12, 18, 23). A3D has also been found to aggregate strongly in cells and requires urea added to Laemmeli buffer to ensure denaturation and resolution on SDS-PAGE (23). Prior to this discovery, publications had reported detecting strong mRNA expression of A3D, sometimes higher than A3F and A3G, but no visible protein on immunoblots (12, 17). However, there may have been insufficient denaturing of the protein for resolution by PAGE. Similar to A3F, A3G, and A3H, A3D binds RNA in the cytoplasm and exists in large molecular mass ribonucleoprotein complexes (24).
A3D was identified by Bouzidi et al. to interact with both A3G and A3F (25). This is interesting since our group identified that A3G and A3F interact through a direct protein-protein interaction and this increases the ability of each A3 to restrict HIV and increases the resistance of A3F to Vif-mediated degradation (26, 27). However, in Bouzidi et al., the authors do not report if the co-immunoprecipitation used to characterize the A3 interactions used RNAse A or not (25). If not, the interaction may be mediated by RNA and not be a specific protein-protein interaction. In addition, for the pararetrovirus Hepatitis B, A3D was found to antagonize primarily A3F and to a lesser extent A3G restriction by inhibiting encapsidation into Hepatitis B particles, which is necessary for restriction, similar to the mechanism of inhibition of HIV (25). Overall, the authors found that A3D decreased Hepatitis B restriction by A3F and A3G (25). Since A3D mRNA expression was previously found to exceed A3F (12), we sought to determine if A3D had an effect on A3F-mediated restriction of HIV and if they interacted in the absence or presence of cellular RNA. We found that A3D and A3F interacted through an RNA intermediate and that this enabled A3D to antagonize A3F encapsidation into HIV. In contrast to the Hepatitis B scenario, restriction of HIV still occurred from the A3D that was encapsidated in lieu of A3F.
To express two A3 transcripts on a single-cell basis, we used an expression plasmid, pVIVO2 (Invivogen), with two transcription units in a single vector. Constructs contained one HA-tagged A3 or one V5-tagged A3 or both. All V5-tagged A3s (A3F and A3G) were cloned using an XbaI site in MCS1. The cDNA for A3F and A3G were amplified from pcDNA constructs and a previously established cloning strategy was used to obtain V5-tagged versions of these cDNA in pVIVO2 (22, 26, 28, 29). The HA-tagged A3D was subcloned from a pcDNA construct into the MCS2 of pVIVO2 using NheI (30). The following reagents were obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: pcDNA3.1-APOBEC3G-HA Expressing Human APOBEC3G with C-Terminal Triple HA Tag, ARP-9952, contributed by Dr. Warner C. Greene and pcDNA3.1-APOBEC3DE-V5-6xHIS, ARP-11433, contributed by Dr. Yong-Hui Zheng.
Whole blood was collected from HIV- donors and Primary Blood Mononuclear Cells (PBMCs) were isolated. Ethics approval was obtained by the University of Saskatchewan Biomedical Research Ethics Board (Ethics Number 14-62). PBMCs were isolated using SepMate tubes (Stemcell Technologies), following the Ficoll-Paque method. Primary cells were cultured in RPMI supplemented with 10% Fetal Bovine Serum, Penicillin/Streptomycin, 5 mM HEPES, non-essential amino acids, sodium pyruvate, 50µM β-mercaptoethanol and stimulated to proliferate with 50 U/ml recombinant interleukin-2 (Sigma) and 10 µg/ml phytohemagglutinin (Fisher Scientific) for 72 h. The RNA preparation, cDNA synthesis and qPCR were carried out according to Refsland et al., except that a SYBR Green based qPCR method rather than the probe and primer method was used. The primers and qPCR cycle used for A3D, A3F, A3G, A3H and TBP were as reported in Refsland et al. (31).
To produce virus particles, 1 × 105 293T cells per well of a 12-well plate were co-transfected with 500 ng of HIV-1 LAI ΔEnv ΔVif (HIV -Vif) or HIV-1 LAI ΔEnv (HIV +Vif), 180 ng of VSV-G, and pVIVO2 plasmid (empty or A3 expressing) at 0 ng, 25 ng and 50 ng (A3F-V5, A3D-HA), or 50 ng and 100 ng (A3F-V5/A3D-HA). GeneJuice (Novagen/EMD Millipore) transfection reagent was used according to the manufacturer’s instructions. Forty-eight hours post-transfection virus containing supernatants were harvested and filtered through a 0.45 µm polyvinylidene difluoride syringe filter. Filtered virus was used for both infection of TZM-bl (HeLa CD4 + CCR5 + LRT lacZ cells) reporter cells and concentration and lysis for use in immunoblots. Virus producing cells were collected for use in immunoblots. To measure the infectivity, 1 × 104 TZM-bl cells per well of a 96-well plate were infected with a dilution series of filtered virus in presence of 8 µg/mL of polybrene. Then, 48 h post-infection, the infectivity was measured through colorimetric detection using β-galactosidase assay reagent (Pierce). The infectivity of each virus was compared to the No A3 condition that was set to 100%.
Immunoblotting was conducted to measure the A3 encapsidation in virions and steady state protein levels in cell lysates. Rabbit anti-HA (1:4000, Sigma) and Mouse anti-V5 (1:1000, Sigma) were used for detection of HA-tagged A3D and V5-tagged A3F and A3G respectively. Rabbit anti-α-tubulin (1:5000, Sigma) and Mouse anti-p24 (1:1000, Cat#ARP-3537, NIH AIDS Reagent Program) were used as the loading controls for cell lysates and viral lysates, respectively. Secondary detection was performed using Licor IRDye antibodies produced in goat (1;10000, IRDye 680 labeled anti-Rabbit, and IRDye 800 labeled anti-Mouse). Blots were scanned via LICOR CLx. For quantification, Image Studio was used to detect the pixel intensity of the experimental and loading control bands. Each sample was normalized to its own loading control before comparison to other lanes. Cropped blots are shown in the manuscript for clarity. Original blot images are in Supplementary File 1.
Relative levels of integrated proviral DNA was determined as previously described (32–34). In brief, 1 × 105 293T cells per well of a 12-well plate were infected by spinoculation (1 h at 800×g) in the presence of polybrene (8 µg/mL) with HIV produced from the single-cycle replication assays. DNA was extracted after 24 h using DNazol according to the manufacturer’s instructions (Invitrogen). The DNA was then treated with DpnI and integrated pro-viral DNA was amplified using HIV-specific primers with Alu-specific primers to ensure only integrated and not episomal HIV proviral DNA was amplified (32–34). This PCR product was diluted and used for qPCR to determine the amount of integrated proviral DNA relative to RNaseP (32–34).
For proviral sequencing, 1 × 105 293T cells per well of a 12-well plate were infected by spinoculation (1 h at 800×g) in the presence of polybrene (8 µg/mL) with HIV produced from the single-cycle replication assays. DNA was extracted after 48 h using DNazol according to manufacturer’s instructions (Invitrogen). The PCR amplification of a 582 bp region of the polymerase (pol) region of HIV and treatment of DNA with DpnI was carried out as previously described (32, 35). The PCR product was cloned using CloneJET PCR cloning kit (Thermo Scientific) and DNA sent for sequencing. Sequences were analyzed with Clustal Omega (36) and Hypermut (37). The number of clones analyzed for each condition was: No A3: 5; A3F: 6; A3D: 5; A3F/A3D: 7.
The 293T cells (2.5 × 106 per T75 flask) were transfected with 2 µg of DNA. The transfections consisted of 1µg of pVIVO2 A3D-HA expression plasmid and 1µg of A3F-V5, or A3G-V5 expression plasmid. GeneJuice (Novagen/EMD Millipore) transfection reagent was used according to the manufacturer’s instructions. After 40 h of transfection, cells were washed with cold PBS, lysed with cold immunoprecipitation buffer (50 mM Tris-Cl [pH 7.4], 1% Nonident-P40, 0.1% sodium deoxycholate, 150 mM NaCl, 10% glycerol, complete, EDTA-free Protease Inhibitor Cocktail (Roche)) and clarified by centrifugation at 4°C. Clarified supernatants were split in two and treated or not treated with RNase A (0.1 mg/mL, Roche) and polyclonal anti-HA rabbit antibody (4 µL, Sigma, St. Louis, MO, USA) at 4°C during incubation with the lysate, and protein A/G agarose (Santa Cruz Biotechnology) for 90 min. The mock condition had no anti-HA antibody. The agarose resin was then washed with immunoprecipitation buffer three times before resuspending in 2× Laemmli buffer and then resolving on SDS-PAGE and transferring to nitrocellulose for immunoblotting. The blot then was probed with anti-V5 mouse antibody (1:1000, Sigma) for detecting HA-immunoprecipitated lysates. For detecting the expression of A3s in cell lysate, the nitrocellulose membrane was probed with anti-HA mouse (1:4000, Sigma), anti-V5 mouse (1:1000, Sigma) and anti-α-tubulin rabbit (1:5000, Sigma). Secondary detection was performed using Licor IRDye antibodies produced in goat (1:10000, IRDye 680 labeled anti-Rabbit, and IRDye 800 labeled anti-Mouse).
The DNA sequences for the HIV 5’UTR (1-497 nt in HIV genome) and a portion of the protease gene (2282-2402 in HIV genome) from the HIV-1 clone 93th253.3 (GenBank accession number U51189) were cloned in the pSP72 plasmid downstream of a T7 promotor using EcoRI and BglII restriction sites. Purified pSP72 plasmid was used as the template to produce 5’UTR and protease RNA in vitro according to the instructions for the mMessage mMachine T7 Ultra Kit (Life Technologies), but with the following modifications. The 5’UTR reaction was incubated overnight. The protease reaction was incubated for 4 h. To enhance the downstream reactions, the optional step of 3′ poly-A-tail was conducted, but used half the recommended E-PAP enzyme and a 30 min incubation. The RNA was then ethanol-precipitated and resuspended in nuclease-free water. For biotinylation, 5 µM RNA, 1 mM ATP, 1% (v/v) DMSO, 5 µM pCp-Biotin (cytidine 5′phosphate 3’-(6-aminohexyl), MJS BioLynx), 100 units of T4 RNA ligase 1 (NEB), and 1x NEB reaction buffer, were incubated at 37°C for 60 min. To remove free pCp-Biotin, the reaction was applied to a Micro Bio-Spin 6 size exclusion column (Bio-Rad) and the flow through collected.
The 293T cells (2.5 × 106 per T75 flask) were transfected with 1 µg of pVIVO2 A3D-HA, A3G-V5, A3F-V5/A3D-HA, or A3G-V5/A3D-HA or 0.5 µg of A3F-V5 expression plasmid. GeneJuice (Novagen/EMD Millipore) transfection reagent was used according to the manufacturer’s instructions. After 40 h of transfection, cells were washed with cold PBS and lysed with cold immunoprecipitation buffer. S-protein agarose (EMD Millipore) was incubated with 400 µg of cell lysate and 500 µg biotinylated RNA for 90 min at 4°C. The S-protein agarose was washed three times with PBS before elution in 1× Laemmli buffer, resolution by SDS-PAGE, and transferring to nitrocellulose for immunoblotting. The blot then was probed with anti-V5 mouse antibody (1:1000, Sigma) and anti-HA rabbit antibody (1:5000, Sigma). For detecting the expression of A3s in cell lysate, the nitrocellulose membrane was probed with anti-HA rabbit, anti-V5 mouse and anti-α-tubulin rabbit (1:5000, Sigma). Secondary detection was performed using Licor IRDye antibodies produced in goat (1:10000, IRDye 680 labeled anti-Rabbit, and IRDye 800 labeled anti-Mouse).
To determine if A3D could contribute to restriction of HIV, we first determined the levels of A3D mRNA relative to A3F, A3G, and A3H in PBMCs of HIV- donors (Figure 1). Using TBP mRNA to normalize the qPCR data, we observed that the A3 transcript levels between donors varied by an order of magnitude. Additionally, from each donor, the A3 transcript levels varied widely. Donor 10 showed similar levels of A3D and A3G mRNA, but had more than 100-fold less of A3F and A3H mRNA (Figure 1A). This was similar to Donor 17, except that A3D mRNA was approximately 2-fold more than A3G mRNA (Figure 1B). Donor 18 showed the lowest level of A3D mRNA (Figure 1C). Overall, A3F mRNA was always lower than A3G mRNA, similar to what was reported by Refsland et al. (31). However, in the donors tested we found that A3D mRNA levels can be as high as A3G mRNA, which has not been reported previously. Refsland et al. determined that there was good correlation between mRNA and protein levels for A3G and A3F, but for other A3s suitable native antibodies precluded this type of analysis (31). Although A3D catalytic activity has previously been reported to be low (38, 39), it may restrict HIV by deamination independent mechanisms when these high mRNA expression level occur, assuming this correlates with protein expression as has been found for A3G and A3F (31). In addition, hetero-oligomerization of A3D with highly related family member A3F may increase HIV restriction activity, similar to what has been reported for A3F and A3G (27, 35). That A3D mRNA levels were higher or similar to A3F mRNA suggest that the proteins when co-expressed could potentially influence each other’s restriction activity.
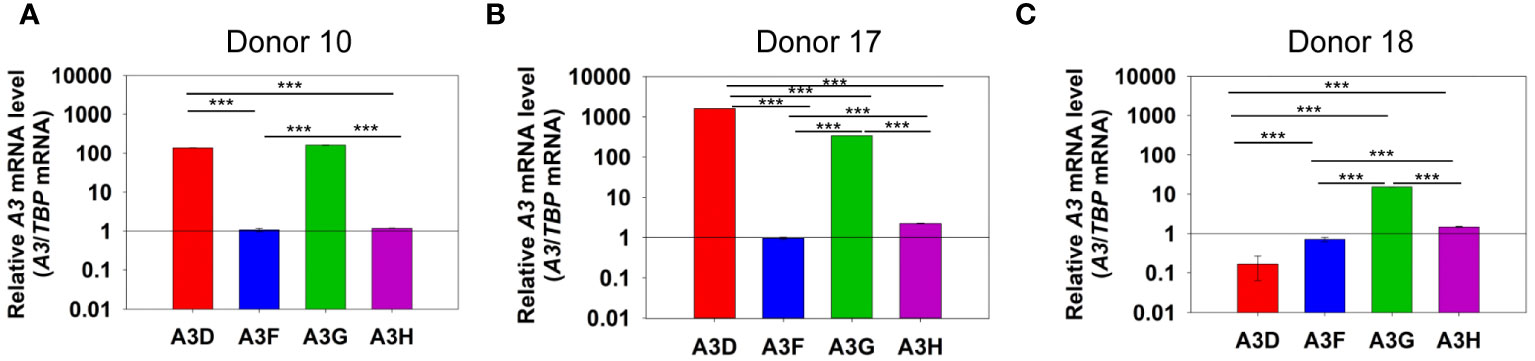
Figure 1 mRNA levels of A3D, A3F, A3G and A3H in PBMCs. PBMCs from three HIV- donors were collected, purified from other blood components, and stimulated with IL-2 and PHA for 72 h. RNA was then extracted and mRNA levels determined by qPCR using the ΔΔCt method with TBP as the expression control. We found that (A) Donor 10, (B) Donor 17, and (C) Donor 18 each had unique expression levels of A3 mRNA. Error bars represent the standard deviation of the mean from three independent experiments. Designations for significant differences between values were determined using a t-test and are shown as: p ≤ 0.001 (***), p ≤ 0.01 (**), or p ≤ 0.05 (*).
We examined the ability of A3D to become encapsidated into and restrict HIV -Vif and +Vif virions (Figure 2). We compared the A3D restriction ability to A3F, which is similar to A3D in domain structure (40). We used two plasmid transfection amounts to observe a dose dependent effect and ensure we were not saturating the experimental system with overexpression of A3 enzymes (Figures 2C, D).We also used a vector with two MCS (pVIVO2) to express A3D-HA and A3F-V5 within the same cell (see Materials and methods). To be able to detect A3F and A3D that resolve by SDS-PAGE to the same apparent molecular weight, we used distinct tags (V5 or HA). These tags enable comparisons to be made between conditions for A3F only and A3D only, but not to directly compare A3F to A3D because the immunogenicity of the V5 and HA tags may differ.
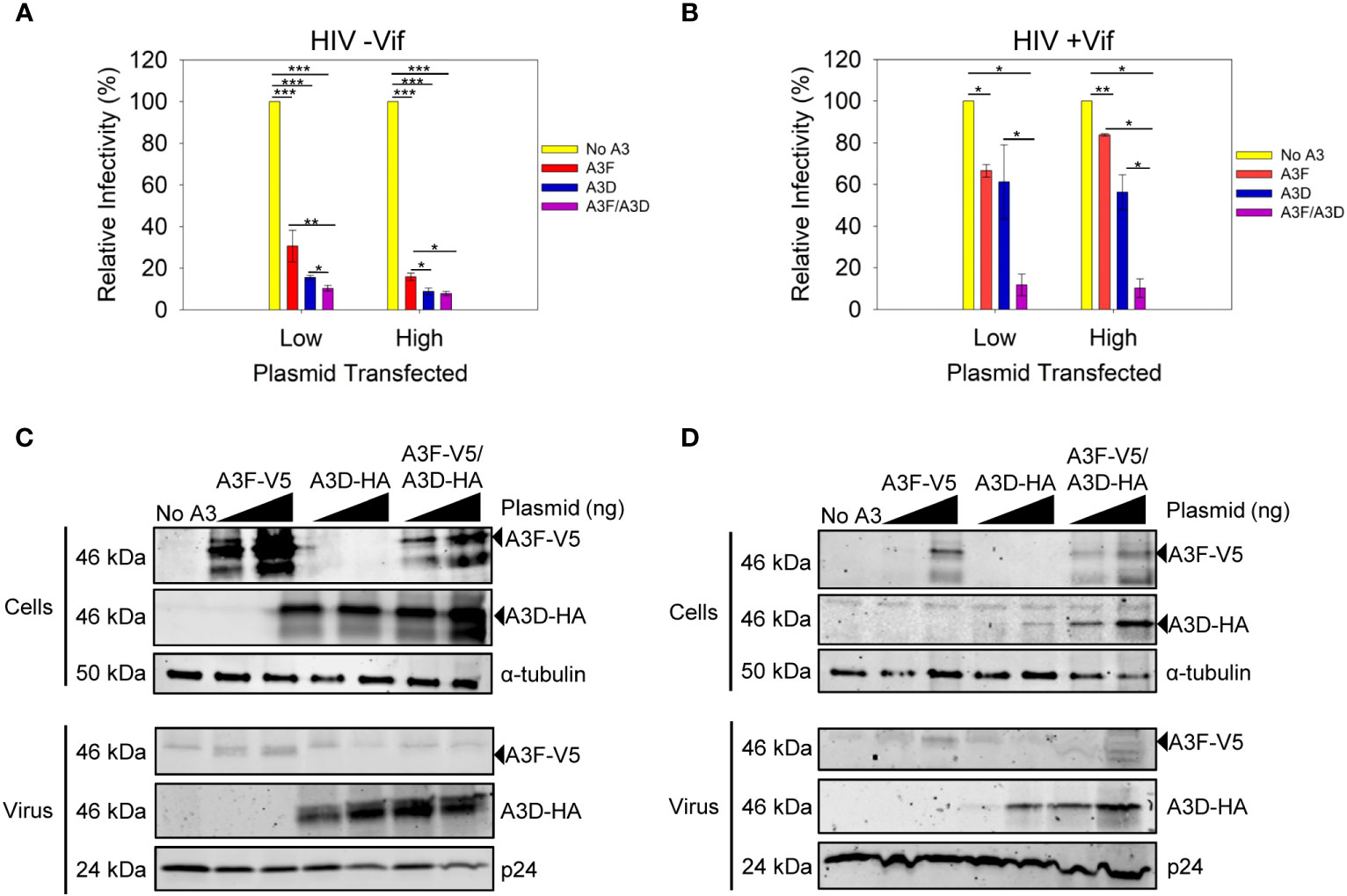
Figure 2 Restriction of HIV by A3F and A3D alone and when co-expressed. (A, B) Infectivity was measured by β-galactosidase produced from TZM-bl reporter cells infected with HIV in the absence of (A) Vif (-Vif) and presence of (B) Vif (+Vif) when A3F-V5 and A3D-HA were expressed alone or together in 239T cells. (A, B) Error bars represent the standard deviation of the mean from (A) three or (B) two independent experiments. Designations for significant differences between values were determined using a t-test and are shown as: p ≤ 0.001 (***), p ≤ 0.01 (**), or p ≤ 0.05 (*). (C, D) Immunoblots show steady-state protein levels of A3F-V5, A3D-HA and co-expressed A3F-V5/A3D-HA in (C) cells and virus in absence of Vif (-Vif) and (D) cells and virus in presence of Vif (+Vif). Two transfection conditions with either a low or high amount of A3 plasmid was used (see Materials and methods). The α-tubulin and p24 were used as loading controls for cell lysates and virions, respectively.
Both A3F and A3D alone could restrict HIV -Vif, but A3D was approximately 2-fold more efficient at the low and high plasmid transfection amounts (Figure 2A). When A3D and A3F were co-expressed, we observed restriction of HIV -Vif, but the restriction amount was only 1.5-fold greater (low) or equal to (high) A3D alone (Figure 2A). To investigate why there was no cooperative effect, we used immunoblotting to determine the A3D and A3F protein levels in virus producing cells and virions. Using different tags, we could detect A3F-V5 and A3D-HA when expressed together. We observed that the expression levels in cells for each, A3F or A3D, were similar when they were expressed alone or together (Figure 2C, Cells). However, the immunoblot showed that encapsidation of A3F and A3D was different (Figure 2C, Virus). In particular, when A3D and A3F were co-expressed, we could not detect any A3F encapsidation by immunoblotting and the A3D encapsidation levels were similar to when A3D was expressed alone (Figure 2C, Virus). These data suggest a competitive encapsidation process, where A3D excludes A3F from encapsidation into virions.
We also examined if this occurred during an HIV +Vif infection. The A3F and A3D alone could not efficiently restrict HIV +Vif, consistent with each A3 being sensitive to Vif-mediated degradation and only detectable on immunoblots at the high transfection level (Figures 2B, D). Accordingly, when A3D and A3F were each expressed alone we could detect encapsidation at the higher transfection level (Figure 2D). However, when A3D and A3F were co-expressed, both A3s were less sensitive to Vif-mediated degradation and could be detected in cell lysates at low and high transfection amounts (Figure 2D). In particular, there was 23-fold more A3D in cells when co-expressed with A3F, than in its absence (Figure 2D, Cells, High). This correlated with 1.5-fold more encapsidation of A3D in virions in the when co-expressed with A3F, than in its absence (Figure 2D, Virus, High). This potentially explains how A3D was able to partially restrict virus replication in the presence of A3F, but not its absence (Figure 2B, High). Notably, we could not detect A3F in virions when co-expressed with A3D, despite more 3-fold more A3F in cell lysates when expressed with A3D compared to A3F expression alone (Figure 2D, High). However, we cannot exclude that A3F becomes encapsidated into virions, but at a level below the detection limit of the immunoblotting. Overall, these data support the HIV -Vif results and further suggest that A3D competes with A3F for encapsidation into virions, even in the presence of Vif. This hypothesis would require that A3D bound more tightly to the HIV genomic RNA than A3F.
Deamination-independent mechanisms of A3s typically rely on RNA binding (34). A3 enzymes can bind HIV genomic RNA and inhibit the progression of reverse transcriptase, which results in less proviral DNA integration. We observed that A3D inhibited proviral integration 13.5-fold more than A3F (Figure 3A). The A3F/A3D co-expression inhibited proviral integration similar to A3D alone (Figure 3A). To exclude another mechanism of inhibiting proviral DNA integration through host-mediated degradation of proviral genomes after excision of uracils, we also determined the level of A3-induced mutations in a 582 bp region of the pol gene (Figure 3B). The A3-induced mutations are analyzed in the (+) DNA where uracils cause G→A mutations, primarily in a GA→AA context for A3F and A3D has a more broad mutation potential that includes GA→AA, GG→AG, and GC→AC (12, 41). Consistent with past studies, we observed that A3D was barely able to cause mutations above the reverse transcriptase background (no A3) (Figure 3B, Supplementary Figure 1). In contrast, A3F was able to cause 26 GA→AA mutations per kb (Figure 3B, Supplementary Figure 2). The A3F/A3D condition had approximately 10-fold fewer mutations than A3F alone, consistent with a decrease in, but not complete exclusion of A3F from virion encapsidation (Figures 2C, 3B; Supplementary Figure 3). Collectively, these data suggest that uracil-based degradation of proviral DNA and RNA binding cause the A3F-induced decrease of proviral DNA integration, whereas A3D decrease of proviral DNA integration is through only RNA binding (Figures 3A, B). This is consistent with a previous study that identified human A3D having lowered catalytic activity due to a Y320C change from chimpanzee A3D (17). These data support that A3D binds RNA more tightly than A3F and this could be a reason why A3D competes A3F off HIV RNA during encapsidation.
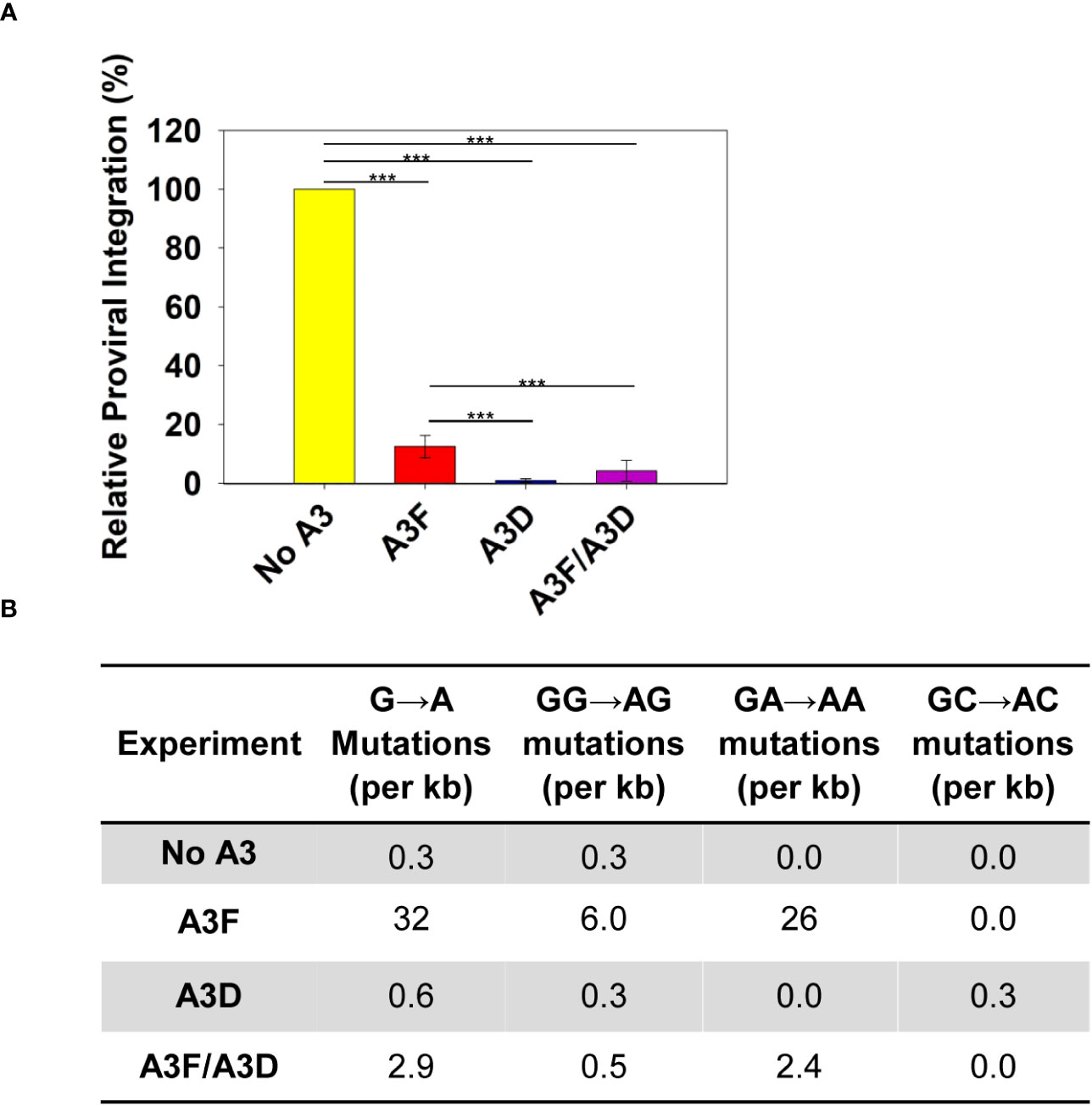
Figure 3 Measurement of A3D and A3F deamination -independent and -dependent mechanisms of HIV restriction. (A) Quantification using qPCR of relative amounts of integrated HIV -Vif proviral DNA. The error bars represent the standard deviations of the mean from three independent experiments. Designations for significant differences between values were determined using a t-test and are shown as: p ≤ 0.001 (***), p ≤ 0.01 (**), or p ≤ 0.05 (*). (B) Analysis of A3-induced mutagenesis in a 582 bp region of pol proviral DNA from an HIV -Vif infection. Clones were sequenced, aligned with Clustal Omega, and analyzed using Hypermut. The total base pairs sequenced for each condition were: No A3: 2910; A3F: 3492; A3D: 2910; and A3F/A3D: 4074. Sequence alignments for each conditions are shown in Supplementary Figures 1-4.
To determine if in addition or alternatively A3D directly interacts with A3F we co-expressed A3D and A3F and used co-immunoprecipitation (co-IP) in the absence or presence of RNAse A to determine if they interacted through a protein-protein interaction, such as hetero-oligomerization, or through an RNA intermediate. The co-IP showed that A3F most strongly immunoprecipitated when RNAse A was not added, demonstrating that their interaction is primarily through an RNA intermediate (Figure 4A). To determine if this was a common interaction mechanism, we also tested A3G. During the co-IP with A3D, A3G was more sensitive to the RNAse A treatment than A3F, and we observed no co-IP of A3G with A3D unless the cellular RNA was intact (Figure 4B). These data demonstrate that the interaction of A3D with A3F and A3G is RNA mediated.
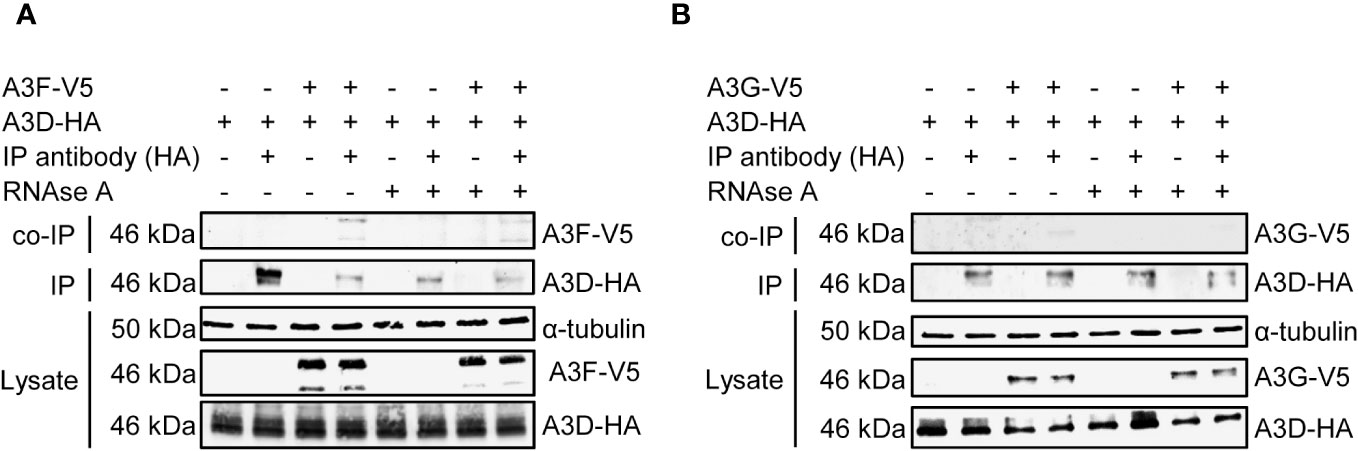
Figure 4 A3F and A3G interact with A3D in a RNA dependent manner. Immunoprecipitation of A3D-HA followed by immunoblotting for (A) A3F-V5 or (B) A3G-V5 in the presence or absence of RNase (A). (A) The experimental and mock samples are prepared from the same lysate. The lysate blot demonstrates the cellular expression of levels of the HA- and V5- tagged proteins. The α-tubulin was used as a loading control.
Since the co-IP is performed in the presence of an abundance of cellular RNAs with different sequence specificities, we performed an additional experiment to directly assess if A3D is competitively excluding A3F from HIV RNA during encapsidation into virions. A3F and A3G have been shown to bind multiple diverse RNAs during encapsidation into HIV, including the HIV genomic RNA (42, 43). We used in vitro transcription to produce two regions of the HIV genomic RNA, the HIV 5’UTR (497 nt) and protease (120 nt), and labeled the RNAs with biotin to enable immobilization onto streptavidin-agarose resin. Then we added cell lysates that had A3D-HA, A3F-V5, A3G-V5, A3D-HA and A3F-V5, or A3D-HA and A3G-V5 (Figure 5). This streptavidin capture experiment showed that both A3D and A3F alone bound to the HIV 5’UTR RNA, but when the A3D and A3F were present together, the A3D maintained binding to the HIV 5’UTR RNA and A3F was prevented from binding (Figure 5). Similar results were found with A3G, however, A3G bound less well to the HIV 5’UTR than A3F and A3D alone (Figure 5). To determine if this was specific to the HIV 5’UTR RNA, we also tested a portion of the HIV protease (Figure 5) We observed similar results, although A3D was not able to completely exclude A3F and A3G from binding (Figure 5). These data in combination with the co-IP of A3D and A3F as an RNA-mediated complex from cell lysates (Figure 4A) support the hypothesis that A3D excludes A3F from encapsidation into virions by competitive inhibition of A3F RNA binding.
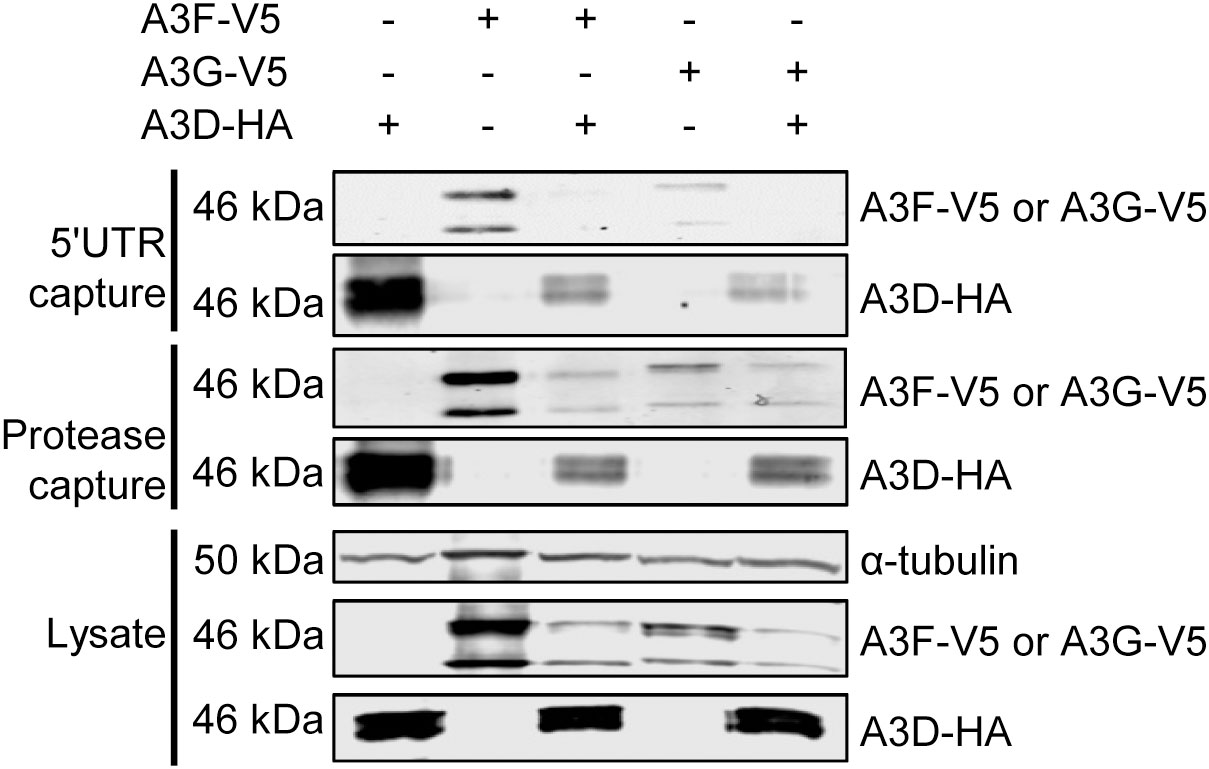
Figure 5 A3F and A3G are competitively excluded from RNA in the presence of A3D. Streptavidin and biotinylated RNA based capture of A3 enzymes from cell lysates on HIV 5’UTR or protease RNA. Immunoblotting for A3D-HA, A3F-V5, or A3G-V5 demonstrated their ability to bind to the RNA alone or in combination with A3D-HA. The lysate blot demonstrates the cellular expression of levels of the HA- and V5- tagged proteins. The α-tubulin was used as a loading control.
The contribution of A3D as a host restriction factor required reassessment due to reports of A3D not being catalytically active and being antagonistic to A3F and A3G during Hepatitis B infection (12, 18, 23, 25). Despite A3D not having catalytic activity it has evolved under positive selection, consistent with a role as host defense factor (23). A3D mRNA levels in some donors were higher than A3F mRNA suggesting that A3D is present at high enough levels to influence A3F restriction in vivo (Figure 1). However, due to the expression of multiple A3 enzymes in primary cells, we had to use a transfection-based system to analyze the relationship between A3F and A3D. While this has potential caveats, we have attempted to mitigate these by using multiple transfection conditions and multiple experimental systems to test the same hypothesis. The data presented here shows that A3D can restrict HIV through a deamination-independent mechanism and to a similar extent as A3F, that uses deamination -independent and -dependent mechanisms (Figure 2). However, the co-encapsidation of A3D and A3F did not occur due to A3D having a higher affinity for HIV RNA than A3F (Figure 5).
Importantly, the data obtained in this study are in line with other data that show co-regulation of A3 enzymes. Our lab previously found that A3F could enhance proteasomal degradation of A3H Haplotype I (44). However, during HIV infection, the increased A3F-induced A3H Haplotype I degradation enabled A3F to be encapsidated at a higher level into HIV virions (44). This suggests that all A3s are competitively binding the HIV RNA for encapsidation. We observed that the RNA interactions between A3D and A3F or A3G were more competitive in the 5’UTR (Figure 5). The limited ssDNA in this highly structured RNA region may create a competitive environment with few A3 binding sites (45). Overall, the data support a model where A3D binds to RNA with higher affinity than A3F. This results in competitive exclusion of A3F from virions (Figure 6A). However, some A3F is likely encapsidated with A3D that is beyond the detection limit of immunoblotting (Figure 2D), but can be detected by sequencing of proviral DNA and suggest approximately 10-fold less A3F encapsidation based on mutation frequencies (Figure 3B, 26 vs. 2.4 GA→AA mutations/kb). It remains to be determined how a partial resistance to Vif is achieved when A3D and A3F are co-expressed, but we propose that A3F and A3D co-binding to cellular or viral RNA, even if transient may prevent Vif-mediated degradation (Figure 6B). For A3D, this resulted in significant restriction of HIV Vif+ (Figure 2B). Altogether, these data suggest a mechanism of co-regulation of virion encapsidation based on RNA binding (Figure 6).
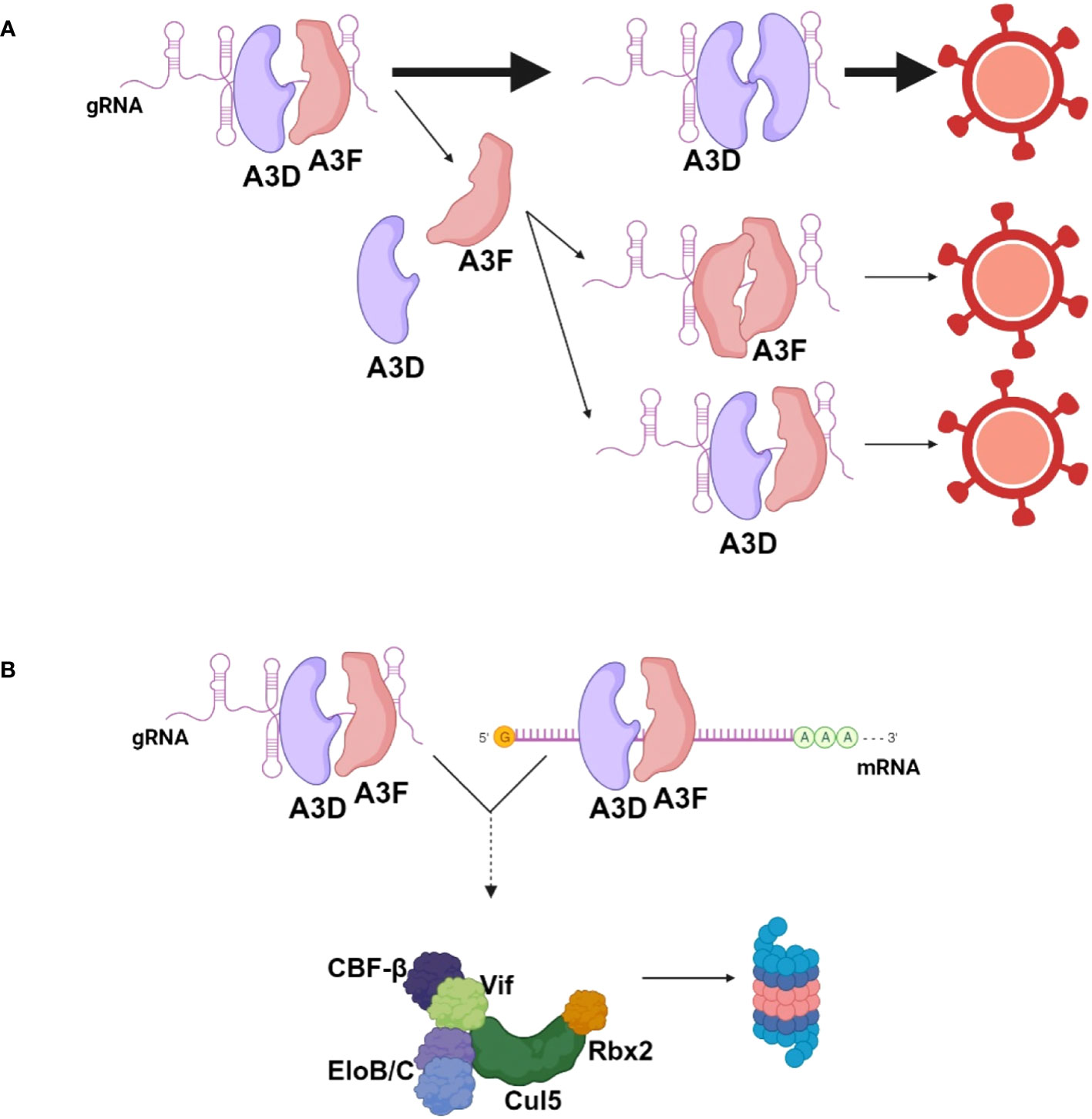
Figure 6 Model for encapsidation of A3D and A3F into HIV during co-expression. (A) When A3D and A3F are co-expressed in HIV infected cells, they both bind HIV genomic RNA (gRNA) to enable encapsidation. A3D binds the gRNA with higher affinity than A3F and competitively excludes A3F from virions. A3F can co-encapsidate with A3D or encapsidate independent of A3D, but this occurs less than A3D encapsidation alone. (B) A3F and A3D co-binding to RNA may prevent Vif-mediated degradation, resulting in more apparent A3F and A3D in cells in the presence of Vif when co-expressed together in comparison to individually. Image created with biorender.com.
Even apart from HIV restriction, studies have found co-regulation of A3 enzymes. Petljak et al. observed that deletion of A3B from cancer cell lines resulted in increased deamination activity of A3A (46). These data suggest that there have been evolutionary pressures to decrease concurrent A3 enzyme activity. While for A3A and A3B that have been implicated in cancer mutagenesis and evolution, this suggests a genome protection mechanism, it is unclear why multiple A3s would be negative for HIV restriction (47). One possibility is that virion exclusion promotes variability in which A3s are packaged over time. Our experimental system was done under single cycle conditions, but under multiple cycles of infections, it is possible that the A3D levels in cells would be decreased upon encapsidation from initial virus particles produced, leaving other A3s to be encapsidated in subsequent viral particles. This cycle could repeat depending on the temporal regulation of mRNA and protein production. Our lab has found that A3F and A3G co-encapsidate and Desimmie et al. found that A3G could co-encapsidate with A3F, A3D, and A3H, but this study is the first to look at A3D and A3F co-encapsidation into HIV virions (26, 48). The co-encapsidation of A3G and A3D observed by Desimmie et al. is in agreement with our study that found less competitive inhibition by A3D of A3G RNA binding in comparison to A3F RNA binding (Figure 5) (48). A co-regulated exclusion mechanism would combat differences in A3 restriction as a result of reverse transcriptase processivity and efficiency (26, 49). The reverse transcriptase efficiency would vary between HIV isolates and possibly over the course of an infection. We previously showed that a combination of A3F and A3G can better restrict HIV than each alone since an enzyme that has a deamination-independent mechanism, such as A3F, could slow down the reverse transcriptase enough to enhance the ssDNA deamination activity of A3G, an enzyme with less deamination-independent restriction (26). Our data suggest that A3D would not require co-encapsidation to efficiently restrict HIV. Multiple cycle infection studies would be needed to test this hypothesis, however, there are caveats to testing this in PBMCs such as inability to parse out effects of multiple A3s and a lack of specific and sensitive native antibodies for A3 enzymes.
A3D-mediated exclusion of A3F enzymes from virions is common between Hepatitis B and HIV (25). In our work, we observed a stronger A3D-mediated exclusion from HIV RNA of A3F compared to A3G, consistent with the Hepatitis B study results (25). This may be due to A3D and A3F having a partial protein-protein interaction whereas none was observed between A3D and A3G (Figure 4). Nonetheless, the protein-protein interaction even for A3D and A3F was much weaker than the interaction mediated by RNA (Figure 4). We did observe A3F interaction with A3D on cellular RNA and did not observe competitive exclusion in this situation, compared to HIV 5’UTR or protease RNA (Figures 4, 5). This collectively suggests that the competitive exclusion is sequence specific.
Altogether, these data demonstrate the importance of studying multiple co-expressed A3 proteins. Here we only studied how co-expression of A3D and A3F affected HIV restriction, but how these data fit in with A3F-mediated instability of A3H Hap I or A3F and A3G hetero-oligomerization still needs to be assessed (26, 27, 44). However, our study supports that A3D can bind HIV RNA with high affinity resulting in exclusion of A3F from virions, similar to what has been observed in Hepatitis B virus (25), and that A3D has an ability to restrict HIV through deamination-independent mechanisms. Development of cellular systems to effectively study more than two A3 enzymes concurrently would provide a framework to assess how the multiple family members coordinate restriction activity of HIV.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving humans were approved by University of Saskatchewan Biomedical Ethics Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
SB: Conceptualization, Formal Analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. AG: Investigation, Methodology, Supervision, Writing – review & editing. LC: Conceptualization, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The project was supported by a Canadian Institutes of Health Research Grant to LC (PJT-162407), a College of Medicine Graduate Scholarship to SB, and a Saskatchewan Health Research Foundation Postdoctoral Scholarship to AG.
We thank members of the Chelico Lab for critical discussion.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2024.1343037/full#supplementary-material
1. Gaba A, Flath B, Chelico L. Examination of the APOBEC3 barrier to cross species transmission of primate lentiviruses. Viruses (2021) 13(6):1084. doi: 10.3390/v13061084
2. Delviks-Frankenberry KA, Desimmie BA, Pathak VK. Structural insights into APOBEC3-mediated lentiviral restriction. Viruses (2020) 12(6):587. doi: 10.3390/v12060587
3. Pollpeter D, Parsons M, Sobala AE, Coxhead S, Lang RD, Bruns AM, et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat Microbiol (2018) 3(2):220–33. doi: 10.1038/s41564-017-0063-9
4. Uriu K, Kosugi Y, Suzuki N, Ito J, Sato K. Elucidation of the complicated scenario of primate APOBEC3 gene evolution. J Virol (2021) 95(12):e00144-21. doi: 10.1128/JVI.00144-21
5. Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, et al. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics (2002) 79(3):285–96. doi: 10.1006/geno.2002.6718
6. Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol Biol Evol (2005) 22(2):367–77. doi: 10.1093/molbev/msi026
7. Uriu K, Kosugi Y, Ito J, Sato K. The battle between retroviruses and APOBEC3 genes: its past and present. Viruses (2021) 13(1):124. doi: 10.3390/v13010124
8. Nakata Y, Ode H, Kubota M, Kasahara T, Matsuoka K, Sugimoto A, et al. Cellular APOBEC3A deaminase drives mutations in the SARS-CoV-2 genome. Nucleic Acids Res (2023) 51(2):783–95. doi: 10.1093/nar/gkac1238
9. Kim K, Calabrese P, Wang S, Qin C, Rao Y, Feng P, et al. The roles of APOBEC-mediated RNA editing in SARS-CoV-2 mutations, replication and fitness. Sci Rep (2022) 12(1):14972. doi: 10.1038/s41598-022-19067-x
10. Cheng AZ, Yockteng-Melgar J, Jarvis MC, Malik-Soni N, Borozan I, Carpenter MA, et al. Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat Microbiol (2019) 4(1):78–88. doi: 10.1038/s41564-018-0284-6
11. Salter JD, Bennett RP, Smith HC. The APOBEC protein family: united by structure, divergent in function. Trends Biochem Sci (2016) 41(7):578–94. doi: 10.1016/j.tibs.2016.05.001
12. Dang Y, Wang X, Esselman WJ, Zheng YH. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol (2006) 80(21):10522–33. doi: 10.1128/JVI.01123-06
13. Pak V, Heidecker G, Pathak VK, Derse D. The role of amino-terminal sequences in cellular localization and antiviral activity of APOBEC3B. J Virol (2011) 85(17):8538–47. doi: 10.1128/JVI.02645-10
14. Lackey L, Demorest ZL, Land AM, Hultquist JF, Brown WL, Harris RS. APOBEC3B and AID have similar nuclear import mechanisms. J Mol Biol (2012) 419(5):301–14. doi: 10.1016/j.jmb.2012.03.011
15. Lackey L, Law EK, Brown WL, Harris RS. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle (2013) 12(5):762–72. doi: 10.4161/cc.23713
16. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature (2002) 418(6898):646–50. doi: 10.1038/nature00939
17. Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, et al. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol (2011) 85(21):11220–34. doi: 10.1128/JVI.05238-11
18. Chaipan C, Smith JL, Hu WS, Pathak VK. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J Virol (2013) 87(1):444–53. doi: 10.1128/JVI.00676-12
19. Harari A, Ooms M, Mulder LC, Simon V. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J Virol (2009) 83(1):295–303. doi: 10.1128/JVI.01665-08
20. OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe (2008) 4(3):249–59. doi: 10.1016/j.chom.2008.07.005
21. Wittkopp CJ, Adolph MB, Wu LI, Chelico L, Emerman M. A single nucleotide polymorphism in human APOBEC3C enhances restriction of lentiviruses. PloS Pathog (2016) 12(10):e1005865. doi: 10.1371/journal.ppat.1005865
22. Adolph MB, Ara A, Feng Y, Wittkopp CJ, Emerman M, Fraser JS, et al. Cytidine deaminase efficiency of the lentiviral viral restriction factor APOBEC3C correlates with dimerization. Nucleic Acids Res (2017) 45(6):3378–94. doi: 10.1093/nar/gkx066
23. Duggal NK, Malik HS, Emerman M. The breadth of antiviral activity of Apobec3DE in chimpanzees has been driven by positive selection. J Virol (2011) 85(21):11361–71. doi: 10.1128/JVI.05046-11
24. Li J, Chen Y, Li M, Carpenter MA, McDougle RM, Luengas EM, et al. APOBEC3 multimerization correlates with HIV-1 packaging and restriction activity in living cells. J Mol Biol (2014) 426(6):1296–307. doi: 10.1016/j.jmb.2013.12.014
25. Bouzidi MS, Caval V, Suspene R, Hallez C, Pineau P, Wain-Hobson S, et al. APOBEC3DE antagonizes hepatitis B virus restriction factors APOBEC3F and APOBEC3G. J Mol Biol (2016) 428(17):3514–28. doi: 10.1016/j.jmb.2016.05.022
26. Ara A, Love RP, Follack TB, Ahmed KA, Adolph MB, Chelico L. Mechanism of enhanced HIV restriction by virion coencapsidated cytidine deaminases APOBEC3F and APOBEC3G. J Virol (2017) 91(3):e02230-16. doi: 10.1128/JVI.02230-16
27. Mohammadzadeh N, Love RP, Gibson R, Arts EJ, Poon AFY, Chelico L. Role of co-expressed APOBEC3F and APOBEC3G in inducing HIV-1 drug resistance. Heliyon (2019) 5(4):e01498. doi: 10.1016/j.heliyon.2019.e01498
28. Feng Y, Love RP, Ara A, Baig TT, Adolph MB, Chelico L. Natural polymorphisms and oligomerization of human APOBEC3H contribute to single-stranded DNA scanning ability. J Biol Chem (2015) 290(45):27188–203. doi: 10.1074/jbc.M115.666065
29. Mohammadzadeh N, Follack TB, Love RP, Stewart K, Sanche S, Chelico L. Polymorphisms of the cytidine deaminase APOBEC3F have different HIV-1 restriction efficiencies. Virology (2019) 527:21–31. doi: 10.1016/j.virol.2018.11.004
30. Love RP, Xu H, Chelico L. Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J Biol Chem (2012) 287(36):30812–22. doi: 10.1074/jbc.M112.393181
31. Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res (2010) 38(13):4274–84. doi: 10.1093/nar/gkq174
32. Gaba A, Hix MA, Suhail S, Flath B, Boysan B, Williams DR, et al. Divergence in dimerization and activity of primate APOBEC3C. J Mol Biol (2021) 433(24):167306. doi: 10.1016/j.jmb.2021.167306
33. Yang H, Ito F, Wolfe AD, Li S, Mohammadzadeh N, Love RP, et al. Understanding the structural basis of HIV-1 restriction by the full length double-domain APOBEC3G. Nat Commun (2020) 11(1):632. doi: 10.1038/s41467-020-14377-y
34. Belanger K, Savoie M, Rosales Gerpe MC, Couture JF, Langlois MA. Binding of RNA by APOBEC3G controls deamination-independent restriction of retroviruses. Nucleic Acids Res (2013) 41(15):7438–52. doi: 10.1093/nar/gkt527
35. Ara A, Love RP, Chelico L. Different mutagenic potential of HIV-1 restriction factors APOBEC3G and APOBEC3F is determined by distinct single-stranded DNA scanning mechanisms. PloS Pathog (2014) 10(3):e1004024. doi: 10.1371/journal.ppat.1004024
36. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol (2011) 7:539. doi: 10.1038/msb.2011.75
37. Rose PP, Korber BT. Detecting hypermutations in viral sequences with an emphasis on G –> A hypermutation. Bioinformatics (2000) 16(4):400–1. doi: 10.1093/bioinformatics/16.4.400
38. Duggal NK, Fu W, Akey JM, Emerman M. Identification and antiviral activity of common polymorphisms in the APOBEC3 locus in human populations. Virology (2013) 443(2):329–37. doi: 10.1016/j.virol.2013.05.016
39. Anderson BD, Harris RS. Transcriptional regulation of APOBEC3 antiviral immunity through the CBF-beta/RUNX axis. Sci Adv (2015) 1(8):e1500296. doi: 10.1126/sciadv.1500296
40. LaRue RS, Andresdottir V, Blanchard Y, Conticello SG, Derse D, Emerman M, et al. Guidelines for naming nonprimate APOBEC3 genes and proteins. J Virol (2009) 83(2):494–7. doi: 10.1128/JVI.01976-08
41. Liddament MT, Brown WL, Schumacher AJ, Harris RS. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol (2004) 14(15):1385–91. doi: 10.1016/j.cub.2004.06.050
42. Apolonia L, Schulz R, Curk T, Rocha P, Swanson CM, Schaller T, et al. Promiscuous RNA binding ensures effective encapsidation of APOBEC3 proteins by HIV-1. PloS Pathog (2015) 11(1):e1004609. doi: 10.1371/journal.ppat.1004609
43. York A, Kutluay SB, Errando M, Bieniasz PD. The RNA binding specificity of human APOBEC3 proteins resembles that of HIV-1 nucleocapsid. PloS Pathog (2016) 12(8):e1005833. doi: 10.1371/journal.ppat.1005833
44. Yousefi M, Annan Sudarsan AK, Gaba A, Chelico L. Stability of APOBEC3F in the presence of the APOBEC3 antagonist HIV-1 vif increases at the expense of co-expressed APOBEC3H haplotype I. Viruses (2023) 15(2):463. doi: 10.3390/v15020463
45. Ye L, Gribling-Burrer AS, Bohn P, Kibe A, Bortlein C, Ambi UB, et al. Short- and long-range interactions in the HIV-1 5’ UTR regulate genome dimerization and packaging. Nat Struct Mol Biol (2022) 29(4):306–19. doi: 10.1038/s41594-022-00746-2
46. Petljak M, Dananberg A, Chu K, Bergstrom EN, Striepen J, von Morgen P, et al. Mechanisms of APOBEC3 mutagenesis in human cancer cells. Nature (2022) 607(7920):799–807. doi: 10.1038/s41586-022-04972-y
47. Petljak M, Green AM, Maciejowski J, Weitzman MD. Addressing the benefits of inhibiting APOBEC3-dependent mutagenesis in cancer. Nat Genet (2022) 54(11):1599–608. doi: 10.1038/s41588-022-01196-8
48. Desimmie BA, Burdick RC, Izumi T, Doi H, Shao W, Alvord WG, et al. APOBEC3 proteins can copackage and comutate HIV-1 genomes. Nucleic Acids Res (2016) 44(16):7848–65. doi: 10.1093/nar/gkw653
Keywords: restriction factor, HIV, APOBEC3, virion encapsidation, mutagenesis
Citation: Bhattacharjee S, Gaba A and Chelico L (2024) APOBEC3D excludes APOBEC3F from HIV-1 virions by competitive binding of RNA. Front. Virol. 4:1343037. doi: 10.3389/fviro.2024.1343037
Received: 22 November 2023; Accepted: 04 January 2024;
Published: 24 January 2024.
Edited by:
Yasumasa Iwatani, National Hospital Organization (NHO), JapanReviewed by:
Jason D. Fernandes, Scribe Therapeutics Inc., United StatesCopyright © 2024 Bhattacharjee, Gaba and Chelico. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda Chelico, bGluZGEuY2hlbGljb0B1c2Fzay5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.