"Corrigendum: Enterovirus-D68 – A Reemerging Non-Polio Enterovirus that Causes Severe Respiratory and Neurological Disease in Children"
 Cassandra S. Grizer
Cassandra S. Grizer Kevin Messacar
Kevin Messacar Joseph J. Mattapallil
Joseph J. Mattapallil- 1Department of Microbiology & Immunology, The Henry M. Jackson Foundation for Military Medicine, Uniformed Services University, Bethesda, MD, United States
- 2Department of Hospital Medicine, The Children’s Hospital Colorado and University of Colorado School of Medicine, Aurora, CO, United States
- 3Department of Microbiology and Immunology, Uniformed Services University, Bethesda, MD, United States
The past decade has seen the global reemergence and rapid spread of enterovirus D68 (EV-D68), a respiratory pathogen that causes severe respiratory illness and paralysis in children. EV-D68 was first isolated in 1962 from children with pneumonia. Sporadic cases and small outbreaks have been reported since then with a major respiratory disease outbreak in 2014 associated with an increased number of children diagnosed with polio-like paralysis. From 2014-2018, major outbreaks were reported every other year in a biennial pattern with > 90% of the cases occurring in children under the age of 16. With the outbreak of SARS-CoV-2 and the subsequent COVID-19 pandemic, there was a significant decrease in the prevalence EV-D68 cases along with other respiratory diseases. However, since the relaxation of pandemic social distancing protocols and masking mandates the number of EV-D68 cases have begun to rise again-culminating in another outbreak in 2022. Here we review the virology, pathogenesis, and the immune response to EV-D68, and discuss the epidemiology of EV-D68 infections and the divergence of contemporary strains from historical strains. Finally, we highlight some of the key challenges in the field that remain to be addressed.
Introduction
Enterovirus D68 (EV-D68) has re-emerged as a major public health concern in the last decade. Children <16 years of age account for >90% of infections worldwide. EV-D68 infection leads to severe acute respiratory distress in children below 5 years of age with clinical symptoms of hypoxia and wheezing associated with a significant increase in pediatric hospitalizations (1). A subset of children infected with EV-D68 develop polio-like acute flaccid myelitis (AFM) that affects lower motor neurons in the spinal cord gray matter, and is characterized by sudden onset of muscle weakness, particularly in the arms or legs, with decreased muscle tone and compromised reflexes, sometimes associated with difficulty in swallowing, drooping of eyelids, and in serious cases respiratory failure (2).
The genus Enterovirus of the family Picornaviridae consists of some of the most common viral pathogens. Enteroviruses are classified into 4 groups namely, polioviruses, Coxsackie A viruses, Coxsackie B viruses, and echoviruses and 15 species comprising 4 human enteroviruses, 8 animal enteroviruses, and 3 rhinoviruses. Most enterovirus infections are asymptomatic, but they can cause a wide range of illnesses from febrile illness to severe neurological diseases in humans. The most well-studied neuropathogenic enteroviruses are the polioviruses which are known to cause poliomyelitis (3). However, poliomyelitis has been largely eliminated throughout the world due to decades of mass vaccination efforts. While reported cases of poliomyelitis have waned, the recognition of neurologic complications associated with non-polio enteroviruses, including EV-A71, coxsackievirus A16, EV-D70, and EV-D68, has globally increased in recent years (4). Among these enteroviruses, a surge in the number of reported EV-D68 infections in children has raised significant public health concern as they have tended to coincide with the large outbreaks of respiratory disease in children.
EV-D68 was first isolated from children with pneumonia and bronchiolitis in 1962 and since then 26 cases were reported from 1970 – 2005, and 699 cases from 2005 – 2012. Large outbreaks of EV-D68 infections were reported worldwide in 2014 with about 1,395 confirmed cases in the United States (US) from Aug 2014 – Jan 2015. Based on syndromic surveillance, however, the number of cases worldwide may be a gross underestimate of the actual number of infections due to limited availability of clinical testing for EV-D68 (5).
In temperate climates, EV-D68 circulation predominantly occurs in the summer-fall season between Aug – Oct, which has temporally and geographically correlated with spikes in AFM cases since 2014 (6). A recent study (7) for the first time demonstrated the presence of EV-D68 nucleic acid and antigen in motor neurons in the spinal cord grey matter of a child who died following an AFM-like illness. Interestingly, EV-D68 outbreaks have displayed a biennial pattern with a significant increase in the number of reported cases in 2014, 2016, and 2018. Recent studies have reported the re-emergence and rapid increase in EV-D68 infections in Europe and the US (8).
Virus structure, receptors, and replication
Enterovirus D68 is an RNA virus with a positive-sense, single-stranded RNA genome (9–11). The genome is ~7.2 kb and consists of a 5’ untranslated region (UTR) with an internal ribosome entry site (IRES), an open reading frame (ORF) that encodes for a single precursor polypeptide, and a 3’ UTR with a poly-A tail (12). The precursor polypeptide is post-translationally processed, yielding 4 structural proteins namely, VP 1 - 4 that form the non-enveloped icosahedral capsid and 7 non-structural proteins that include 2A protease, 2B and 2C ATPase and 3A, 3B, 3C protease, and 3D polymerase. The VP 1, 2, and 3 create the outer shell of the capsid, whereas VP4 lines the interior (Figure 1). The vertices of VP 1-3 come together and alternate between threefold and fivefold symmetry. A “canyon,” considered important for binding, surrounds the fivefold vertex (13–15). A hydrophobic pocket in the VP1 subunit at the base of each canyon contains a host-derived lipid-like “pocket factor.” This structure is mostly conserved in all picornaviruses and confirmed to be present in EV-D68 by crystallography (16, 17).
Figure 1
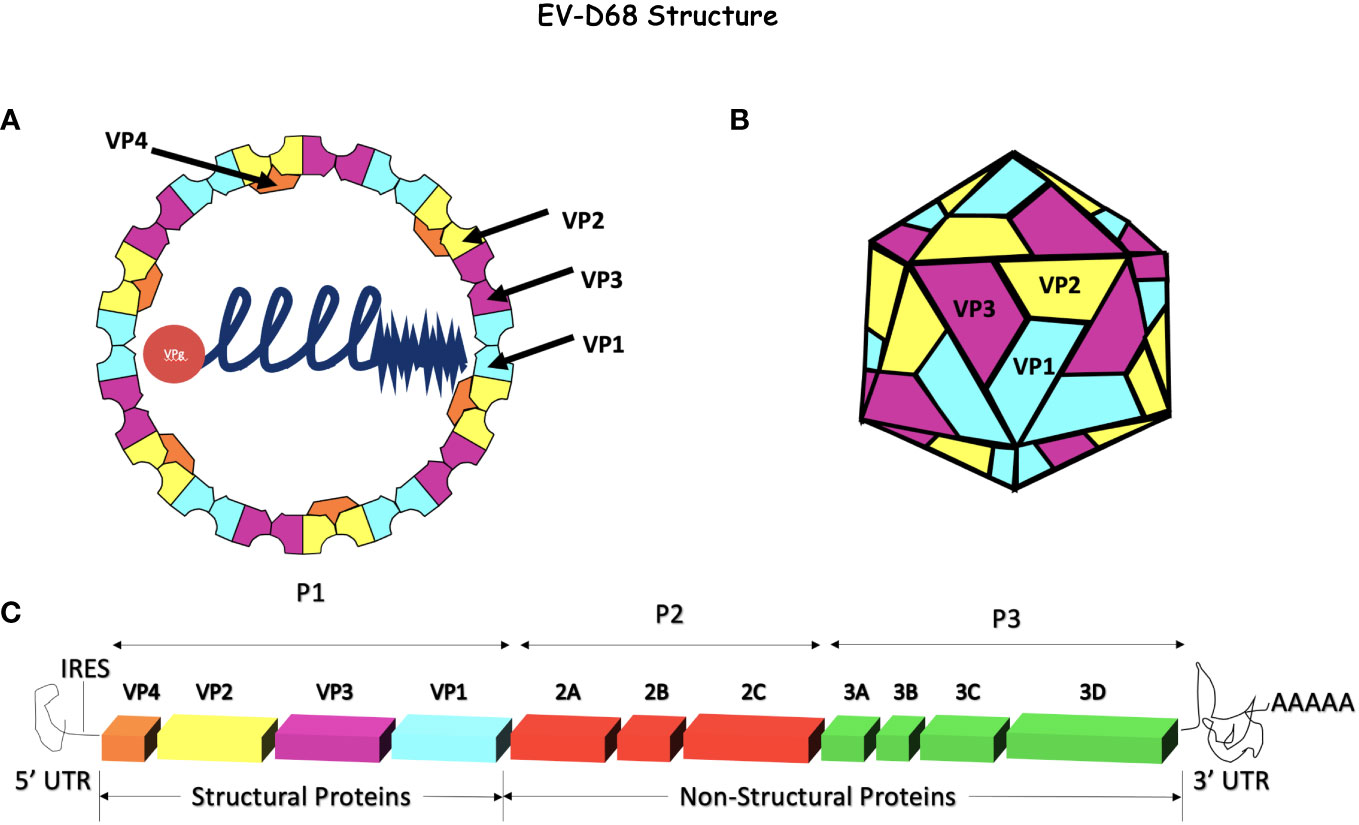
Figure 1 Enterovirus-D68 Genome and capsid. (A, B) EV-D68 has an icosahedral, non-enveloped capsid consisting of 4 structural proteins, VP1, VP2, and VP3 on the external side of the capsid and VP4 on the internal side. This capsid surrounds the +ssRNA naked genome attached to VPg. (C) The EV-D68 genome encodes for 4 structural proteins (VP1 - 4) and 7 non-structural proteins (2A - C and 3A - D). The internal ribosome entry site (IRES) is at the 5’ end and a poly-A tail terminates the 3’ end. The genome is ~7.2 kb in size and is composed of a single open reading frame (ORF). Initially, the polyprotein is processed into 3 precursor proteins, P1-P3. P1 is later proteolytically cleaved into the 4 structural proteins (VP1 - 4) while P2 and P3 are processed into replicase, VPg, proteases (2A and 3C), a polymerase (3D), and other non-structural proteins.
EV-D68 replication is initiated when the viral capsid binds to its receptor on the host cells. The exact receptor that EV-D68 uses to infect its target cells is still under investigation. Treatment of cells with neuraminidase, an enzyme that removes sialic acid, was associated with a marked decrease in EV-D68 binding suggesting that sialic acid could be the entry receptor (18). Others have shown that EV-D68 Fermon strain along with other historical strains bind to α2, 6- and α2, 3-linked sialic acids, displacing the pocket factor (16, 19). More contemporary strains of EV-D68 have been shown to bind to cells independently of sialic acid (20) unlike the historical Fermon strain that requires sialic acid (21). Recent studies have suggested that EV-D68 could use sulfated glycosaminoglycans or other receptors in the absence of sialic acid (22). The role of intercellular adhesion molecule-5 (ICAM-5) as a possible EV-D68 receptor has been studied, given that ICAM-5 is expressed on neurons (23). In cell culture, enhanced viral replication was observed with both sialic acid - independent and -dependent strains in the presence of ICAM-5 (24). However, little or no expression of ICAM-5 has been found in the human respiratory tract or spinal cord, hence the relevance of these receptors is less clear (21). Uncapher et al., and others have suggested that EV-D68 likely uses sialic acid, ICAM-5, or sulfated glycosaminoglycans as either co-receptors or attachment factors (18). In picornaviruses such as poliovirus, after internalization and uncoating, the viral capsid undergoes a structural reorganization step within the endosomes to become an enlarged intermediate structure known as the “A particle” (25, 26). Studies have shown that the transformation of mature EV-D68 to A particle occurs after binding to ICAM-5 (24, 27). To exit the endosome, the capsid reorganizes its structure to form a pore in the lipid bilayer and releases the viral genome into the cytoplasm (28–30). For enteroviruses in general, this uncoating is thought to be acid sensitive; EV-D68 requires a pH of 6.0 to reorganize from the A particle to the pore-containing particle in vitro (31, 32). Some picornaviruses such as CV-B1, CV-A7, and EV-A71 including EV-D68 also require the pan-enterovirus host factor, Adipose-specific phospholipase A2 (PLA2G16), for uncoating (22, 33, 34).
In most enteroviruses, translation is initiated immediately following the release of the enteroviral genome into the cytosol. Translation overcomes a 5’ cap requirement by directly utilizing the IRES found within the 5’ UTR (35). Host poly-A binding protein 1 (PABP-1) interacts with the genome’s poly-A tail and the eukaryotic initiation factor 4G (eIF4G), which is also bound to the IRES; this forms a circular structure of the mRNA that mirrors host cell RNA (36). Translation requires many other host factors including eIF1A, eIF2, eIF3, eIF4A and B, and IRES transactivation factors. Together, these factors recruit the translation initiation complex to the IRES (37, 38). Translation of the enteroviral genome results in a single polypeptide that is then proteolytically processed to yield 4 structural (VP 1 - 4) and 7 non-structural (2 A - C and 3 A - D) proteins. The first cleavage is performed by the viral protease 2Apro which cleaves the structural proteins from the non-structural proteins. The viral proteases 3Cpro and 3CDpro then cleave the structural proteins into VP0, VP1, and VP3, along with other intermediate and mature non-structural peptides. Lastly, during maturation of the viral capsid, a self-cleavage event occurs, whereas VP2 and VP4 are generated from VP0 (39). When 3Cpro reaches a sufficiently high concentration, a “switch” ends the predominant translation phase. This switch occurs when 3Cpro cleaves polypyrimidine tract-binding protein 1 (PTBP-1), poly(rC)binding protein 2 (PCBP-2), and PABP-1, which are all required for translation (40–43). After the switch, the viral RNA is synthesized.
The enteroviral RNA-dependent RNA polymerase, 3Dpol, is responsible for generating the viral RNA. Negative sense RNA (-ssRNA) is first synthesized and acts as a template to generate copies of genomic RNA (+ssRNA). The -ssRNA is circularized by the binding of the host factor heterogeneous ribonucleoprotein (hnRNP) C1/C2 (44). Translation and synthesis of antisense RNA cannot occur simultaneously, possibly due to ribosomes and polymerase being unable to travel along viral RNA in opposite directions at the same time (45). Synthesis of the viral genome occurs on replication organelles, which are single and double membrane-bound vesicles. These vesicles are thought to be derived from autophagosomes and fatty acid droplets or the endoplasmic reticulum/Golgi apparatus (46–48). Replication organelles are thought to promote colocalization of RNA-binding proteins and likely allow the viral RNA to evade host RNases (49–51).
Once the viral genome is synthesized, the virus is assembled in the cytoplasm. The structural proteins VP0, VP1, and VP3 form a trimeric structure and then self-assemble into a pentameric unit. Twelve pentameric units self-assemble to form the icosahedral enterovirus capsid (14). The newly synthesized viral RNA (+ssRNA) is covalently linked to VPg (viral protein genome-linked), a protein derived from the non-structural protein 3B, at the 5’ end before being packed into the viral capsid (52). The capsid is then enclosed in an autophagosome-derived vesicle. Maturation of the viral particle containing autophagosomes is prevented by 3Cpro, which cleaves host snap receptor (SNARE) protein, blocking the fusion of the endosome/lysosome to the autophagosome (53). Capsid maturation is triggered in the vesicle and leads to cleavage of VP0 into VP2 and VP4. Viral particles are then released by either lysis or exocytosis of the autophagosome-derived vesicles containing multiple viral particles (Figure 2). Studies using poliovirus have shown that similar vesicles contain mature virions, negative- and positive-sense nonencapsulated viral RNA, several host proteins, and non-structural viral proteins. This mechanism may allow for increased cell-to-cell spread efficiency as these particles initiate viral replication faster than non-membrane-enclosed viruses. Vesicles may also help with immune evasion by shielding the antigenic viral capsid (54).
Figure 2
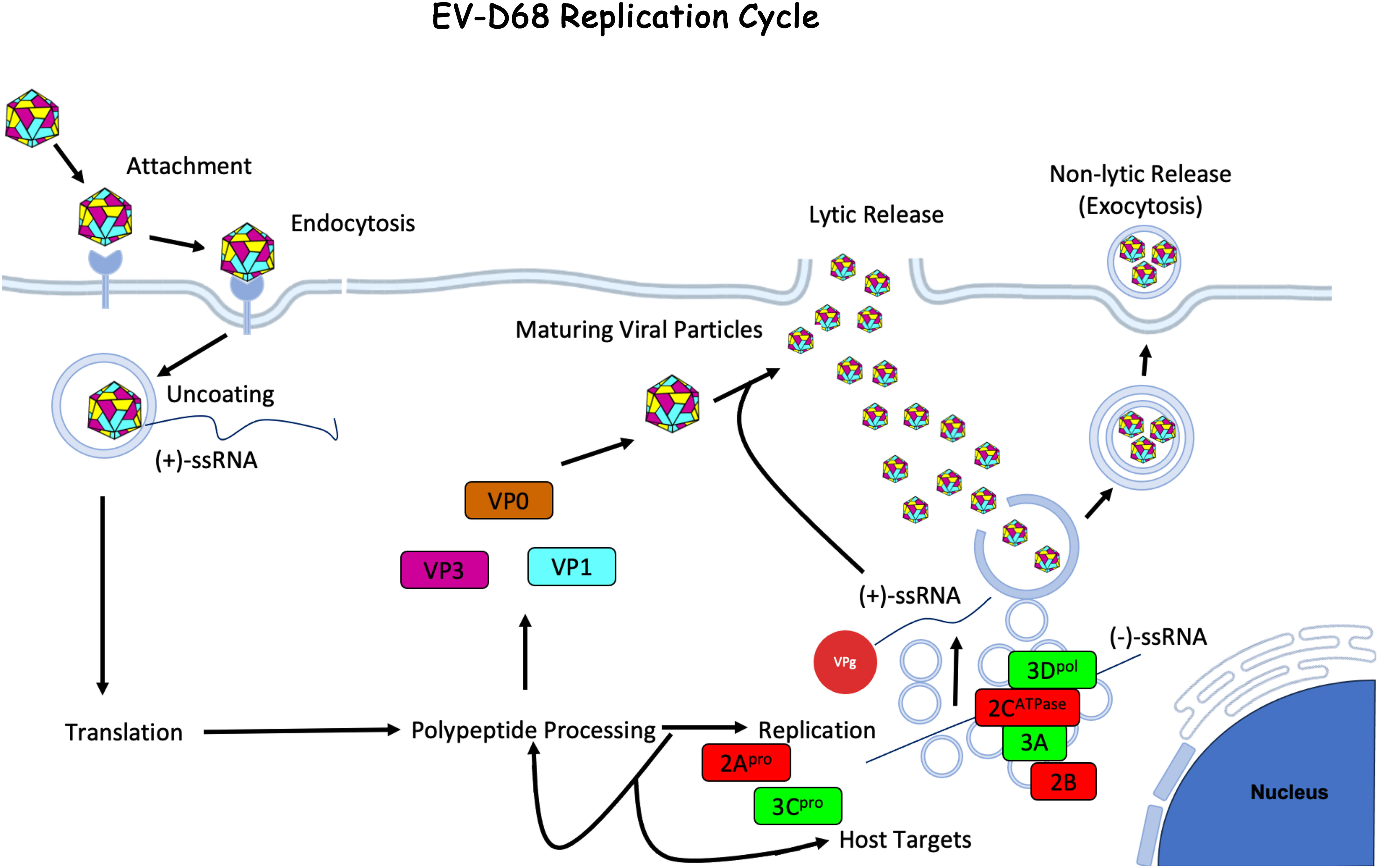
Figure 2 Enterovirus-D68 Life Cycle. EV-D68 attaches to the host cell membrane and is internalized through receptor-mediated endocytosis. The viral capsid undergoes uncoating and creates a pore through the endosomal membrane and the +ssRNA viral genome is released into the cytoplasm. The +ssRNA is translated into a polyprotein that is cleaved by host and viral proteases to generate structural (VP1 - 4) and non-structural (2A - C and 3A - D) proteins. During viral replication, -ssRNA is created and used as a template for new viral genome copies, which occurs on vesicular structures known as replication organelles. Progeny virions are assembled with structural proteins and VPg-linked RNA. The immature viral particles are then mostly taken up by autophagosomes where the acidic environment triggers viral capsid maturation. Virus is then released by either exocytosis of the autophagic vesicles (53) or by cell lysis. The figure was made using BioRender.com under license to CG.
Enterovirus replication is both temperature and pH sensitive. Classic enteroviruses, species A to C, are known to replicate best at temperatures near mammalian body temperature, 37°C, and can replicate in acidic conditions, both of which allow these viruses to replicate in the gastrointestinal tract. This is in contrast to rhinoviruses that typically replicate in the nasopharynx and replicate more efficiently at lower temperatures. Rhinoviruses are also sensitive to acid (55). EV-D68 is unique; it is genetically and antigenically similar to other enteroviruses but is known to replicate most efficiently at 33°C and is also acid labile, making EV-D68 phenotypically similar to rhinoviruses, including transmission in respiratory droplets and primarily causing respiratory disease. Some contemporary strains of EV-D68 from the 2014 outbreak are known to efficiently replicate at both 33°C and 37°C; this may contribute to the increased ability of more contemporary EV-D68 strains to cause systemic infection and neuroinvasion (56, 57).
Epidemiology
Children are at the greatest risk of severe EV-D68 infection. Most cases occur in children under the age of 16, with the greatest proportion of cases in children under the age of 5. Children with a history of asthma are more likely to have severe respiratory disease, including requiring intensive care support (58–60). Healthy adults may be infected with EV-D68 but are more likely to have mild or asymptomatic cases (61, 62). The elderly or adults with co-morbidities have been shown to experience severe respiratory disease, similar to children. EV-D68 infections are widespread, with a high rate of mild cases found in prospective surveillance studies. In temperate regions of the United States, enterovirus infections peak in the late summer and early fall unlike other respiratory viruses that peak during winter months (63–65). In addition to seasonality, EV-D68 outbreaks appear to follow a biennial cycle with reported outbreaks occurring every two years from 2014 to 2018 (Figure 3). This biennial cycle was broken in 2020, likely due to control measures initiated by the COVID-19 pandemic. Since the relaxation of control measures, EV-D68 has started to reemerge, with increased circulation in Europe in 2021, and a large outbreak of respiratory illness in children occurring in the fall of 2022 in the US (66, 67). Currently, very little is known about reinfection with EV-D68, though the current serological data suggests reinfection of older, previously exposed individuals could potentially drive antigenic evolution of EV-D68 (61).
Figure 3
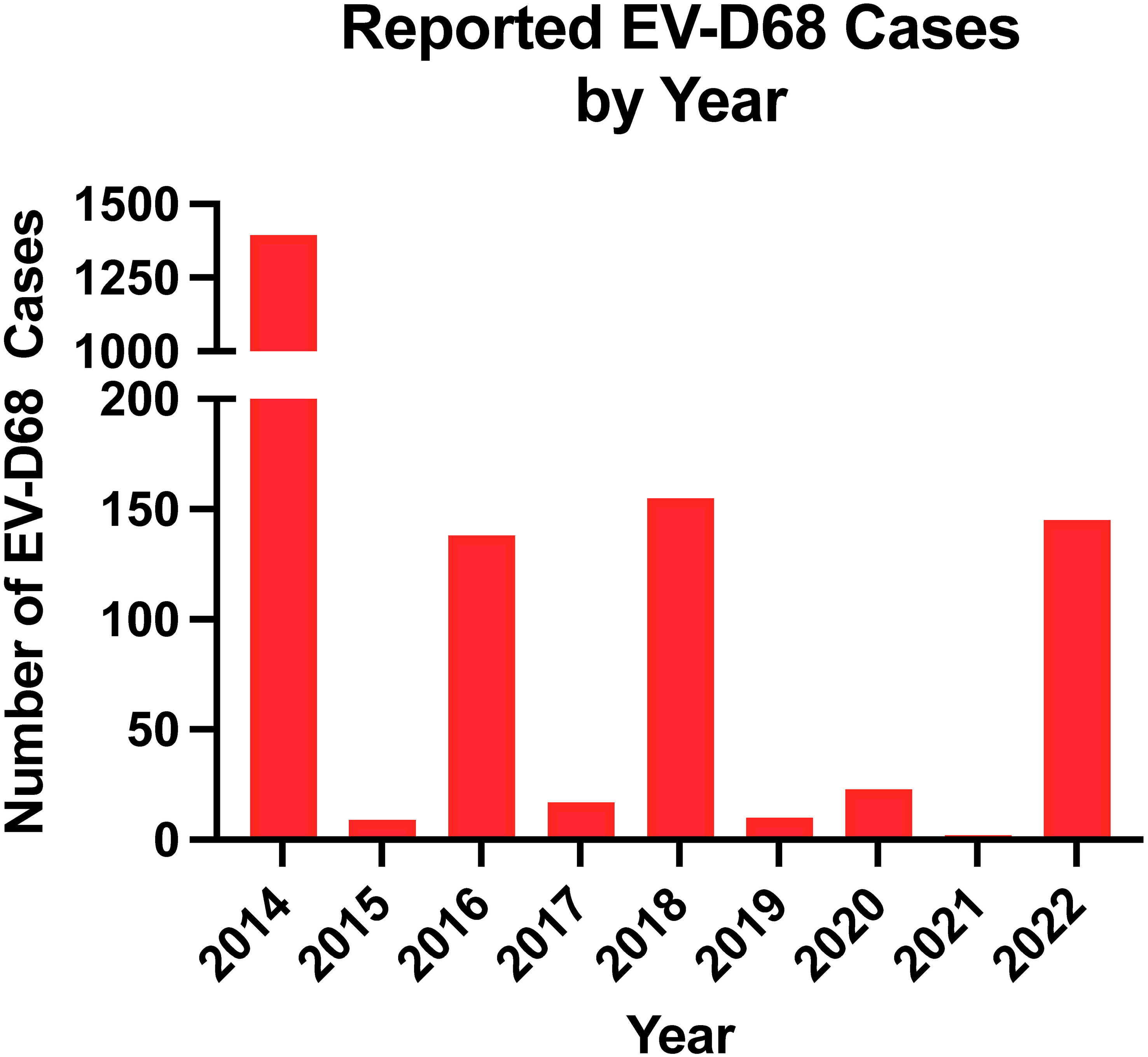
Figure 3 Number of EV-D68 reported by year. Data reported by the National Enterovirus Surveillance System was used.
There are 15 species in the Enterovirus genus (Figures 4, 5). These include four human enteroviruses, A to D, three human rhinoviruses, A to C, and enteroviruses, E to L, which infect nonhuman hosts. Enterovirus genus members have recently been reclassified based on genomic taxonomy, but initially, they were classified based on clinical manifestations and subtyped using serology. This led to the traditional names in the Enterovirus genus, including poliovirus and coxsackievirus. After genetic reclassification, all three poliovirus subtypes are placed in Enterovirus C, while coxsackieviruses are spread across Enterovirus A, B, and C. The remaining viruses in this genus maintain the enterovirus name. In 1970, newly discovered enterovirus subtypes were numbered in order of discovery, starting with enterovirus 68. After genetic reclassification, the numbers were attached to the species letter name (e.g., enterovirus 68 was renamed as EV-D68) (73).
Figure 4
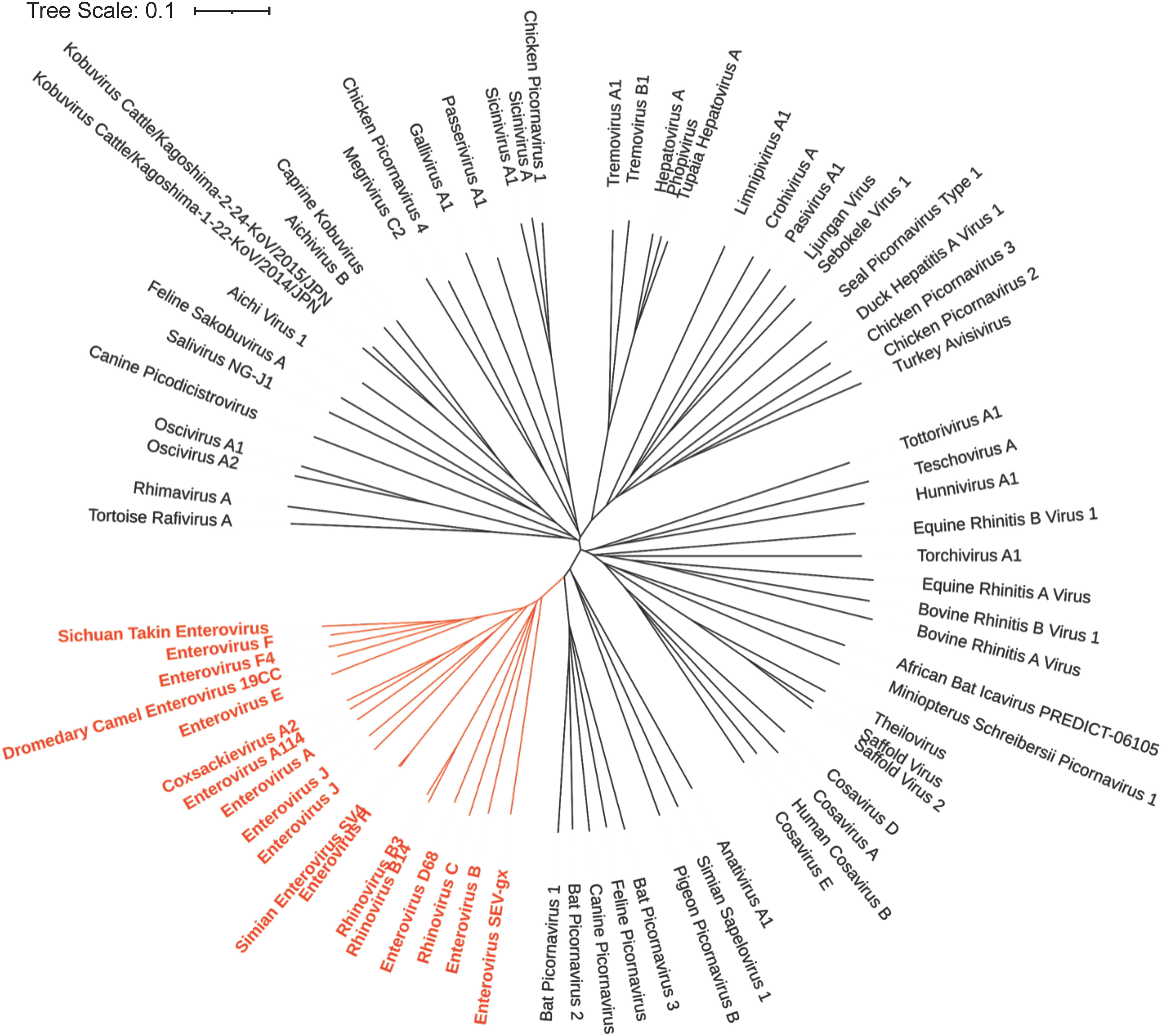
Figure 4 Phylogenetic Tree based on Picornavirus VP1 sequence. The NCBI Virus database was used to download all available Picornavirus VP1 capsid protein reference sequences (68). The sequences were then aligned using the online Clustal Omega Multiple Sequence Alignment Tool (69). The output from Clustal Omega was then transferred to Simple Phylogeny to create the phylogenetic tree (70). The results were then uploaded to iTOL for annotation (71). The genus enterovirus (shown in red) is genetically similar and descended from a common ancestor based on their VP1 capsid sequence.
Figure 5
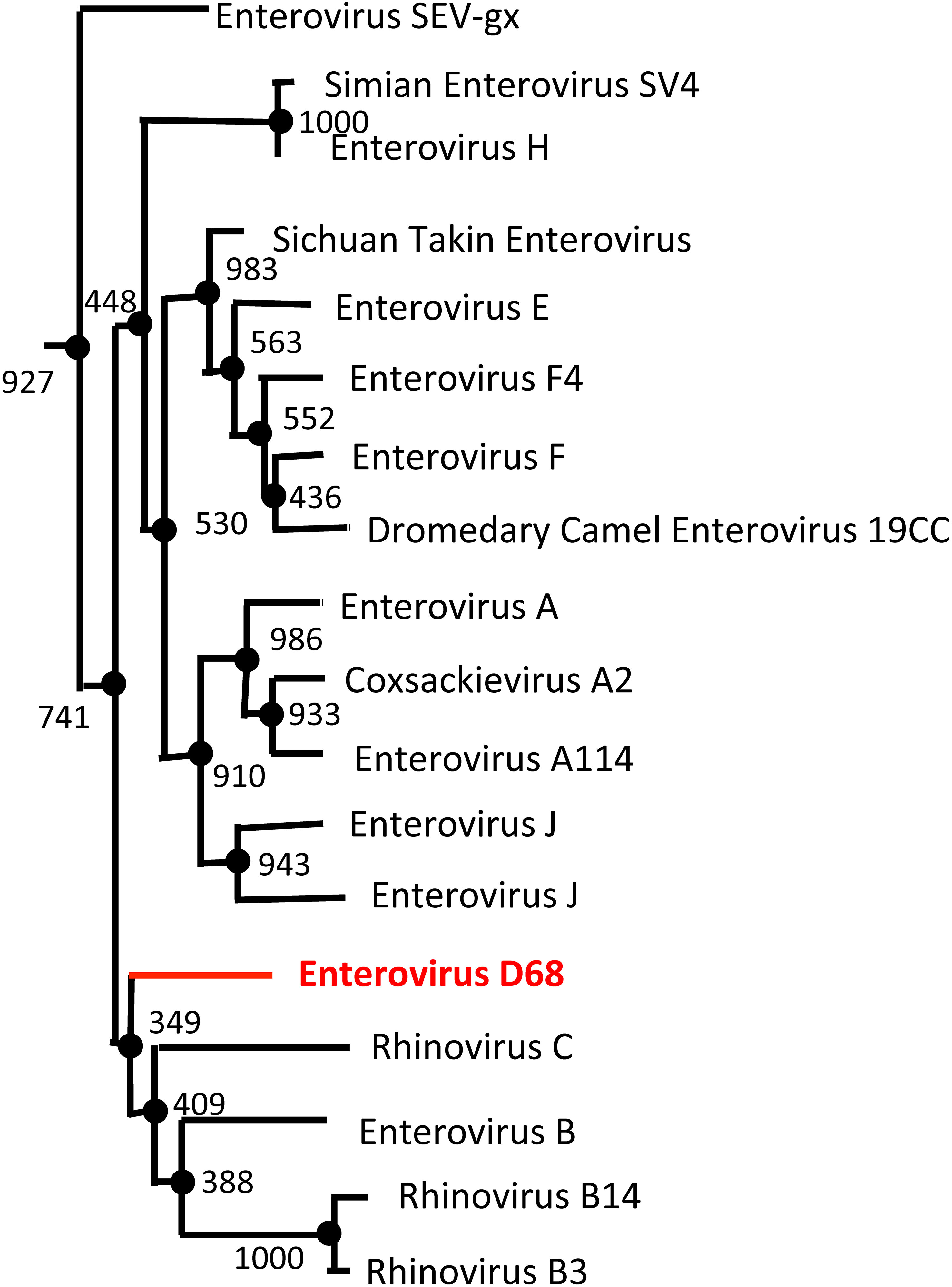
Figure 5 Enterovirus Phylogenetic Tree based on VP1 sequence. The NCBI Virus database was used to download all available enterovirus VP1 capsid protein reference sequences (68). The sequences were then aligned using the Clustal Omega Multiple Sequence Alignment Tool (69). The resulting output from Clustal Omega was then transferred to the online tool, ATGCPhyl, to create the phylogenetic tree with bootstrap values. One thousand Bootstrap replicates were computed to estimate the accuracy of the phylogenetic tree (72). The results were then annotated in PowerPoint. EV-D68 (shown in red) is more genetically similar to rhinoviruses (Rhinovirus B and C) than other enterovirus species (Enterovirus C and D).
In 1962, EV-D68 was first isolated from the pharyngeal swabs of children with acute lower respiratory tract illness. These isolated strains were named the Fermon, Franklin, Rhyne, and Robinson strains, with Fermon being considered, to this day, the prototypic EV-D68 strain (56). In 1963, a similar strain was classified as rhinovirus 87 due to its strong phenotypic resemblance to other rhinoviruses, but genomic data revealed that it was an EV-D68 strain (74, 75). There are currently four identified clades of EV-D68, A-D (Figure 6). These clades are separated primarily based on their VP1 sequence, the gene that encodes a significant component of the viral capsid (Figure 7). Two clades, A and B, are pervasive globally in Europe, Asia, and the United States. Clade D split from clade A and shares a common ancestor. Clades C and D are also prevalent globally but in much lower frequencies. Clade B has been split further into newly emergent subclades, B1-B3 (77–81).
Figure 6
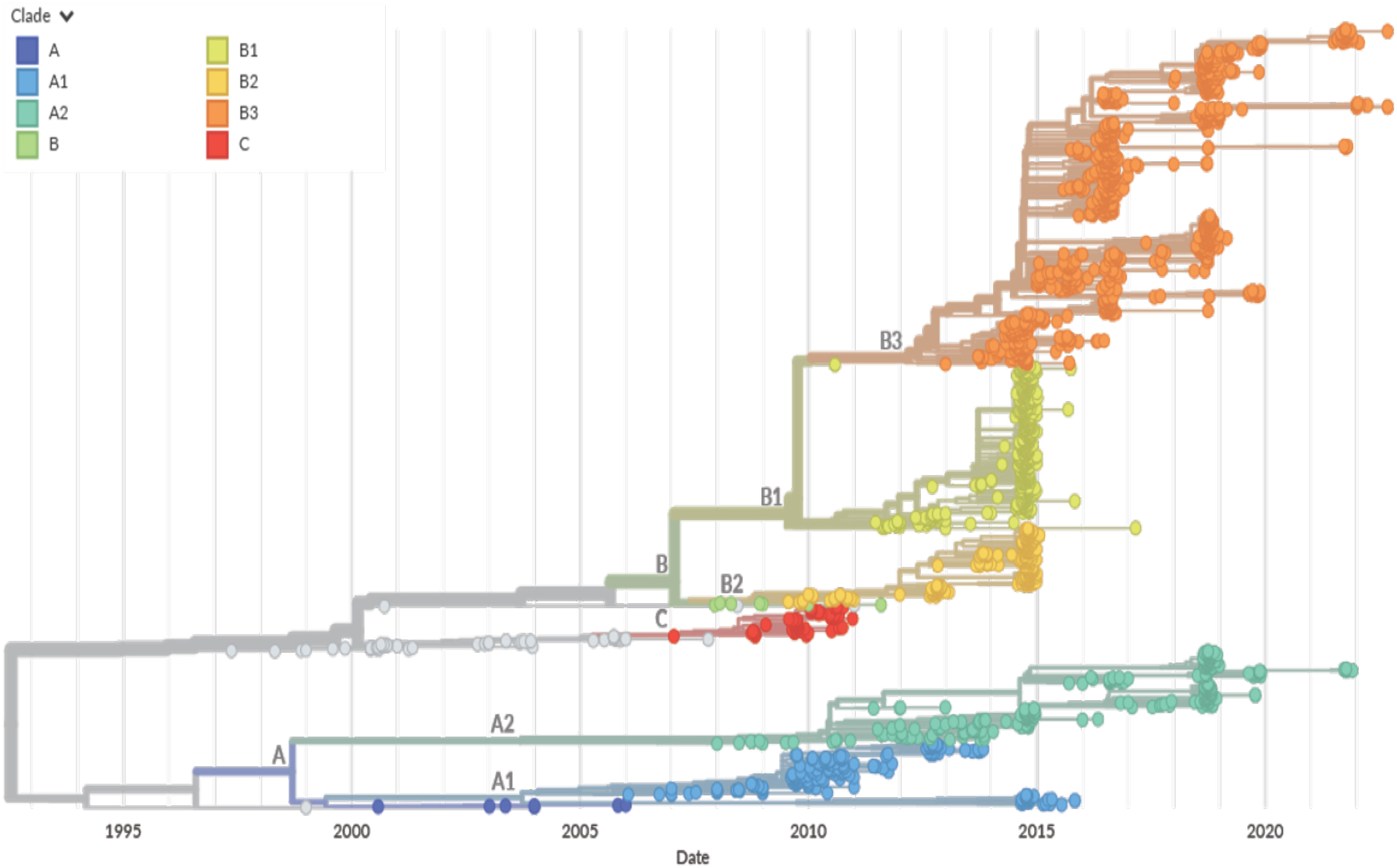
Figure 6 NextStrain based EV-D68 Phylogenetic Tree. A time-scaled phylogenetic tree was visualized using NextStrain based on the VP1 capsid protein sequence from 1992 to 2022 (76). The year that the EV-D68 isolate was detected is represented on the x-axis. The clade for each isolate is denoted by color per the legend (Clade A in blue, Clade B in green, yellow, and orange, and Clade C in red. Clade D is not represented). Major clade branches are labeled at the branch points. EV-D68 clade B1 was the major clade for strains identified in the 2014 outbreak, but a shift has been seen with clades A2 and B3 currently circulating.
Figure 7
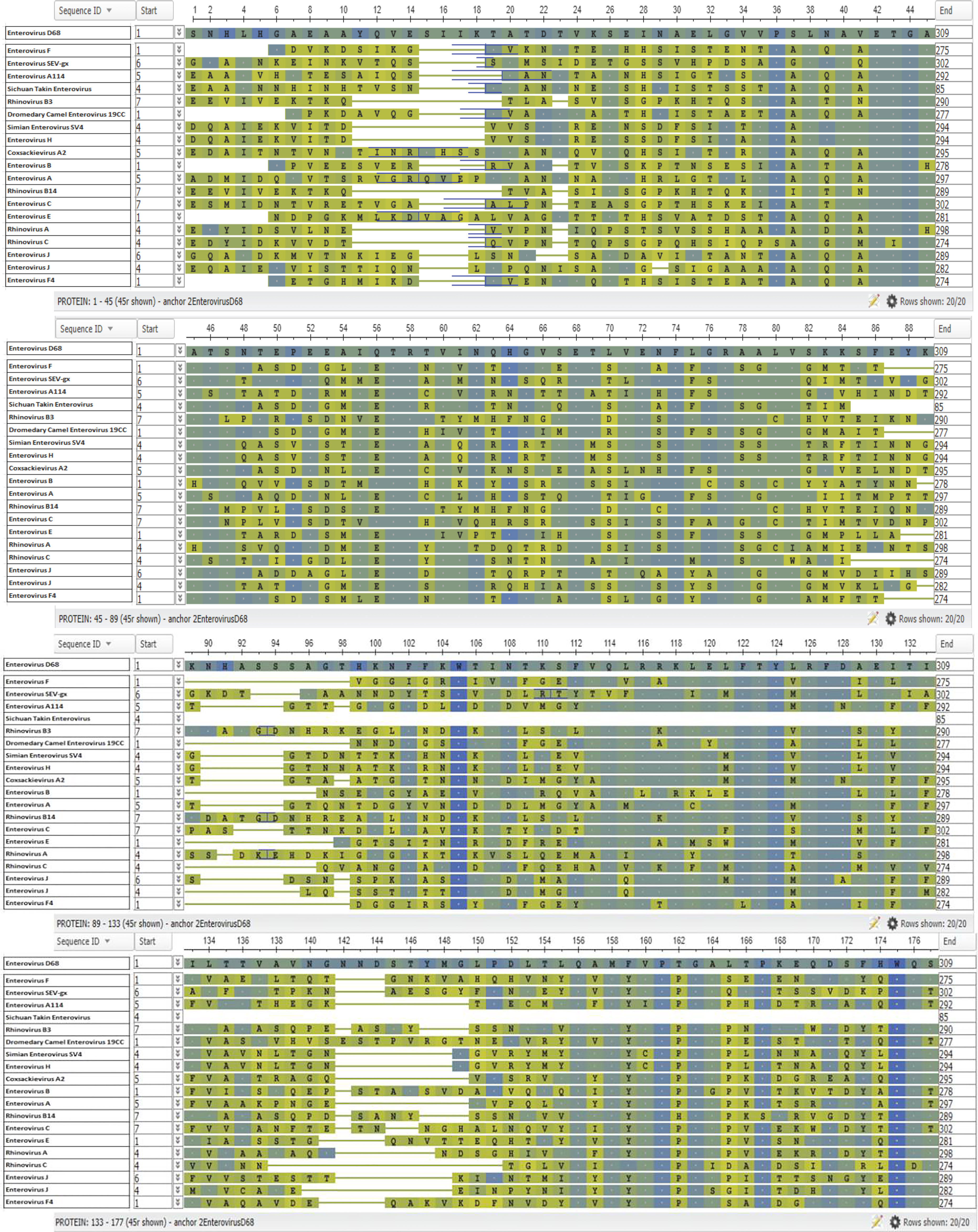
Figure 7 Enterovirus VP1 Protein Alignment. The NCBI Virus database was used to download all available enterovirus VP1 capsid protein reference sequences (68). The sequences were then aligned using the Clustal Omega Multiple Sequence Alignment Tool (69). The resulting output was then transferred to the online tool, NCBI Multiple Alignment. EV-D68 was selected as the anchor sequence, the top row, and BLOSUM62 was used for analysis and coloring (68). Amino acids identical to the anchor sequence (EV-D68) are represented by a dot and shown in blue, while amino acids not identical to the anchor sequence are shown in green a, with blue/green coloring indicating that the substitution is conservative.
The contemporary strains have diverged from the prototypic strains and have accumulated significant genetic changes. The region with the most variation appears to be the VP1 capsid protein, which plays a major role in antigenicity, determines serotype, and is the site of receptor binding (14, 15, 82), and may be a contributing factor to the shifting epidemiology of EV-D68 (57). A high level of variation was first seen in Netherlands surveillance data from 1994 - 2010, which showed that an increase in EV-D68 circulation coincided with an increase in VP1 diversity (83). Samples taken in Colorado from 2012 - 2014 contained multiple polymorphisms within the ORF; some were similar to other neurotropic enteroviruses, poliovirus, and EV-D70 sequences. The strains with these polymorphisms belonged to the B1 subclade (84). In Missouri, 2014 strains were identified to be multiple independent lineages, all belonging to the B1 clade that were co-circulating at the same time (85). Others have shown that most patients diagnosed with AFM in 2014 carried EV-D68 strains of the B1 clade (86–88). Since 2014, many other clades of EV-D68 have been linked to AFM (81, 89).
The B3 subclade was first identified in Taiwan and China in 2014 and shares a common ancestor with B1. The B3 subclade had multiple polymorphisms in the ORF, but these did not match the sequences on neurotropic viruses, whereas this sequence in the B1 subclade is conserved (90). The B3 subclade was still the predominant subclade in the 2016 and 2018 outbreaks worldwide, in both respiratory cases and cases associated with AFM. In the US, all isolates from the 2018 outbreak belonged to the B3 subclade (8, 91–94). The predominant mechanism driving these polymorphisms in the circulating clades appear to be single nucleotide mutations, most likely occurring randomly during viral replication. However, more extensive rearrangements have occurred, and even one incidence of recombination between subclades, though it is unknown if these changes have led to changes in pathogenicity between the clades (85, 95).
Since its discovery in 1962, sporadic cases of EV-D68 infections were reported with infections remaining largely benign. Only 26 cases were identified in the US National Surveillance System from 1970 to 2005 (only 0.1% of total reported enterovirus cases) (65). Between 2008 and 2010, sporadic outbreaks of EV-D68 were reported worldwide. These outbreaks occurred in the US (Georgia, Pennsylvania, Arizona, and New York), Asia (The Philippines, Japan, and Cambodia), and Europe (The Netherlands, Italy, and France) (96–102). The frequency of outbreaks increased between 2010 and 2014, with reports of outbreaks in Africa (Gambia, Kenya, Senegal, and South Africa), Asia (China, Japan, the Philippines and Thailand), Europe (England, Finland, France, Italy, and The Netherlands), and Oceania (Australia and New Zealand) (83, 99, 103–110).
The largest documented outbreak of EV-D68 to date occurred in August - October 2014. Pediatricians at Children’s Mercy Hospital in Kansas City, MO, reported an unexpected increase in severe respiratory disease in children along with a surge of positive PCR tests for enterovirus/rhinovirus in respiratory specimens (111, 112). The Centers for Disease Control and Prevention (CDC) identified EV-D68 as the primary virus by sequencing these specimens. Likewise, University of Chicago Comer Children’s Hospital and Children’s Hospital in Colorado reported a significant increase in severe respiratory disease in children around the same time with the CDC identifying EV-D68 in a majority of respiratory specimens from these hospitals (58, 113). Shortly thereafter, a number of other hospitals across the US reported an increase in pediatric patient volumes with respiratory disease suggesting a rapid spread of infections (114, 115).
Between August 2014 and January 2015, the CDC reported 1,395 confirmed EV-D68 cases across the US (49 states and the District of Columbia) (116). This number is most likely a gross underestimate of the true number of cases, as the CDC report only included cases that were confirmed by genome sequencing, as clinical testing for EV-D68 was not widely available. Compared to the previous two years before 2014, hospitals saw a major increase in emergency department visits, hospitalizations, and pediatric intensive care admissions though only some of those cases were tested for EV-D68. The CDC estimated that millions of milder EV-D68 cases likely occurred during the summer and fall of 2014 in the US (113, 116, 117). The United States was not the only country with an outbreak of EV-D68 in 2014; outbreaks also occurred in other North American countries (Canada), South America (Brazil and Chile), and Europe (Denmark, France, Germany, Italy, Spain, Sweden, and The Netherlands) (106, 118–129).
Unlike the earlier EV-D68 outbreaks, there was a significant increase in the number of children with AFM during the 2014 outbreak; 120 cases of AFM were reported that year (1) with respiratory specimens from about 43% of these cases testing positive for enterovirus/rhinovirus (130). EV-D68 was found in cerebrospinal fluid (CSF) from a young adult patient and from a child who died of paralysis and respiratory failure in 2005 and 2008, respectively (65, 131). Localized clusters of AFM were reported in California in 2012 (132). As in 2014, significant numbers of AFM cases coincided with EV-D68 outbreaks again in 2016 (160 cases) and 2018 (238 cases). However, EV-D68 was isolated only from 20% to 40% of these cases (133) likely due to limitations in sample collection, given that respiratory sampling is necessary to detect viral shedding, and samples taken closer to the onset of respiratory illness are more likely to test positive (134). Unlike some of the earlier outbreaks, the 2022 outbreak of EV-D68 respiratory disease after the relaxation of lockdown measures did not show a high association with AFM. The decoupling of EV-D68 circulation and AFM cases merits further investigation to determine the virologic, immunologic, and/or epidemiologic factors that may have played a role (135). EV-D68 mouse models have been shown to replicate key symptoms of AFM, with one mouse model fulfilling Koch’s postulates for causation (136–142). Koch’s postulates are a list of four criteria that needs to be met to establish a causal relationship between the microbe and the disease namely, a microbe should be found in all organisms with the disease and not found in healthy organisms, it should be isolated from an infected organism and grown in pure culture, the cultured organism should cause disease when introduced to a healthy organism, lastly the microbe should be isolated from the inoculated host and identified as the original microbe (143). Using a neonatal mouse model, Hixon et al. demonstrated that mice infected with EV-D68 exhibited significant signs of paralysis, with lysates from the spinal cords of paralyzed mice showing significant CPE in cell culture. Koch’s postulates were fulfilled when the mice newly infected with the cultured EV-D68 went on to develop paralytic disease themselves, with the same EV-D68 strain isolated from their spinal cords (136–142).
Though no direct causal link had been established between EV-D68 infection and AFM, studies have reported that CSF from AFM patients showed a strong enrichment of enterovirus-associated antibodies compared to controls (144, 145). Furthermore, in 2022, a study by Vogt et al. reported the presence of EV-D68 RNA and protein in a preserved spinal cord specimen from a young child who died from AFM in 2008 (7). Other enteroviruses and rhinoviruses, mainly EV-A71 and coxsackievirus A16, have been associated with AFM, though at lower rates (134). EV-D68 is not commonly found in CSF at the time of clinical presentation of AFM, and the brain and spinal cord tissue has rarely been available for testing. Delay between prodromal symptom onset of respiratory disease to the development of neurologic disease (median 5-7 days), delayed recognition and diagnosis, and incomplete or late biologic specimen collection has likely impeded the ability to detect virus in AFM cases. Virus has been more readily identified from nonsterile sites, particularly respiratory specimens, though these specimens still need to be collected in a timely manner to be detected while still within the shedding period (median 12 days from symptom onset) (62).
Respiratory tract and EV-D68 pathogenesis
Children below the age of 5 years are at the highest risk for EV-D68 infection. Most infections are mild with respiratory symptoms such as runny nose, coughing, sneezing, and body and muscle aches. A subset of children experience severe symptoms such as respiratory distress, coryza, wheezing and shortness of breath requiring supplemental oxygen and hospitalization especially in those who are prone to asthma (146).
Pathogenic changes in the respiratory tract of patients who are infected with EV-D68 have been difficult to study largely due to the transient nature of infection. EV-D68 RNA has been readily detected by RT-PCR in samples from both the upper and lower respiratory tract, suggesting that EV-D68 can infect cells throughout the respiratory tract (147, 148). More detailed understanding of the pathogenic events following EV-D68 infection has come from experimental infection of animal models. EV-D68 RNA was detected in nasal washes and lungs of ferrets following intranasal infection that was accompanied by an acute inflammatory response with histological evidence of mild interstitial pneumonia in the lungs. EV-D68 VP1 was detected in the connective tissue surrounding the alveoli with detectable EV-D68 in cells that expressed α2,6-linked sialic acids in ferrets by confocal microscopy (149, 150). Studies using in vitro nasal airway cultures have reported that EV-D68 infects ciliated epithelial cells that was associated with lysis and induction of numerous pro-inflammatory cytokines such as IL-8, IP-10, IL-1β, IL-6, and GM-CSF (151). In IFN- α/β/ɣ receptor deficient mice, EV-D68 infection was associated with histopathological changes characterized by moderate interstitial pneumonia and mononuclear cell infiltration with detectable VP2 in alveolar epithelial cells. Others have reported an increase of neutrophils suggesting that EV-D68 infection induced neutrophilic airway inflammation that is likely mediated by IL-17A suggesting a potential role for this pathway in the aggravation of asthma in patients infected with EV-D68 (152, 153). The mechanism by which children and immunocompromised adults infected with EV-D68 develop severe disease is not clear though some have suggested that the subdued or the lack of effective immune responses likely plays a role in this process (148, 154–156).
Though the primary route of EV-D68 infection is the respiratory tract, EV-D68 RNA has less commonly been detected in blood, sera, and stool samples, as well as in CSF of EV-D68-associated AFM patients suggesting that the virus has the potential to disseminate systemically following respiratory infection (84, 89, 131, 157–159). The exact mechanism of extra-respiratory spread is currently unknown. Viral RNA has been detected in EV-D68 patient blood and sera of children (158) suggesting that EV-D68 could disseminate systemically to other tissues in the body as some animal studies have shown; EV-D68 has been isolated from blood, liver, lungs, kidneys, muscle, spinal cord, and spleen of mice infected with EV-D68 (137, 141, 152).
Studies have reported the presence of EV-D68 in patient stool samples, though acute gastroenteritis is less commonly reported with EV-D68 infection (132, 157, 160, 161). Human intestinal epithelial cell lines can be infected by EV-D68 in vitro though not very efficiently (162). EV-D68 RNA has been detected in stool samples of both infected ferrets and macaques (150, 163). Other extra-respiratory symptoms have been associated with EV-D68 infection, including cardiac problems (e.g., myocarditis, pericarditis, and acute cardiac failure) and skin rashes (93, 164–166) though the exact mechanism for these sequelae are not clear. In mice, infection with EV-D68 has led to muscle disease and infection of muscle tissue, though its relevance to human infections is not clear (138, 152).
Central nervous system and EV-D68 pathogenesis
EV-D68 infection has been associated with various neurological complications that include AFM, cranial nerve dysfunction, encephalitis, and meningoencephalitis. These complications usually arise after febrile illness and respiratory (coughing, rhinorrhea, and pharyngitis) and gastrointestinal (vomiting and diarrhea) symptoms. Direct detection of EV-D8 in the CSF to document infection of the CNS has been rarely demonstrated, likely due to the delay in the onset of CNS related symptoms following infection and presentation in the clinic. EV-D68 can, however, be detected in nasopharyngeal samples, usually within the first week of CNS disease onset (1, 13, 62, 130, 132). Given a median duration of RNA shedding of 12 days, prompt recognition and early respiratory sampling is key to EV-D68 detection in suspected AFM cases.
Acute flaccid myelitis associated with EV-D68 can progress quickly, within days or even hours after the onset of symptoms (132, 167). Early symptoms include pain or stiffness in the affected limb or back and neck, headache, followed by absent or reduced reflexes (1, 132). Upper limbs are more likely to be affected by AFM, and anywhere from one to all limbs can be affected, usually with asymmetric distribution (1, 2, 130, 132). In severe cases, intubation and ventilator support may be necessary due to bulbar paralysis and inability to protect the airway or paralysis of the diaphragmatic muscles controlling breathing (132, 167). Lesions in the anterior horn of the spinal cord usually occur in the cervical and upper thoracic regions that can be detected by Magnetic Resonance Imaging (MRI) (2). The majority of patients with persistent weakness will develop noticeable muscle atrophy in the affected limb and many report muscle aches and pain that have lasted months after onset (1, 132).
Cranial nerve dysfunction has been reported in patients with and without AFM (1, 2, 130, 132, 167, 168), including facial weakness, double vision, soft speech, difficulty speaking, and difficulty swallowing (2, 130). Sensory deficits in paralyzed limbs and autonomic deficits associated with bowel and bladder dysfunction have also been less commonly reported (132, 167). Cases of brainstem encephalitis (that led to cardiopulmonary failure), fatal meningoencephalitis, and non-fatal aseptic meningitis have all been reported to be associated with EV-D68 infection, but appear rare compared to AFM (84, 89, 125, 131, 169–171). The lesions in the anterior horn of the spinal cord and lesions in the cranial motor nuclei of the medulla, midbrain, and pons suggest that the motor neuron function and cranial nerves are significantly compromised during EV-D68 associated AFM (1, 2, 168, 172). It is not clear if these lesions are a direct result of infection, even though EV-D68 viral RNA has been detected in CSF samples, or due to bystander damage caused by immune responses (84, 131, 169, 170, 172, 173).
Though the exact mechanisms of how EV-D68 gains entry to the spinal cord and, spreads within the CNS in vivo is not clear, one theory is that EV-D68 infects peripheral nerves in the respiratory tract and transmits by retrograde axonal transport to reach the spinal cord. EV-D68 was shown to use axonal microtubules for retrograde transport across the neurons in vitro in hiPSCs derived motor neurons (20). In mice, after intramuscular infection, EV-D68 was detected in the spinal cord, which may have entered the motor neurons either via the neuromuscular junctions or through hematogenous spread (136, 138, 140, 141).
Cranial nerve dysfunction has been observed in patients without limb paralysis. Cranial nerve motor nuclei innervate the muscles of the face, oral cavity, respiratory tract, and tongue. It is possible that EV-D68 uses these neurons to enter the CNS or it is possible that these motor neurons are infected similarly to those in the anterior horn of the spinal cord due to viral tropism (1). On the other hand, the presence of EV-D68 in the CSF suggests that EV-D68 may enter via breaching the blood-brain barrier (BBB). Whether crossing the BBB into the CSF can lead to direct infection of the spinal cord gray matter remains to be determined. EV-D68 may also invade the CNS by infecting lymphocytes and other immune cells that enter the CNS (174, 175) though there is little or no evidence to support this hypothesis.
In mice that were infected with EV-D68 either intracranially or intraperitoneally, viral antigens and RNA have been detected in the spinal cord (136, 140, 141). Infection led to efficient replication in the motor neurons in the anterior horn of the spinal cord, but viral replication was low to absent in the cerebellum and cerebrum (136). In vitro, human and mouse neuroblastoma cell lines and neurons derived from hiPSCs (astrocytes, cortical neurons, and motor neurons) are all permissive to EV-D68 infection. In hiPSCs, viral replication was sustained for 72 hours without cytopathic effect, contrasting in vivo data. In mouse models, viral replication in the CNS is restricted to the spinal cord motor neurons, while hiPSCs show the permissiveness of astrocytes and human cortical neurons (21, 77, 136, 139, 176). In a patient with fatal meningomyeloencephalitis, widespread lymphocytic meningomyelitis, and encephalitis were associated with neuronal destruction in motor nuclei in the cerebellum, cervical cord, medulla, midbrain, and pons. Infiltrating CD3+ T cells were observed in the spinal cord around motor nuclei, and CD20+ infiltrating B cells were observed in perivascular areas (131).
The exact receptor that EV-D68 uses for viral entry and infecting the CNS is still under investigation. Although both sialic acid and ICAM-5 are expressed in the CNS, ICAM-5 is not expressed on motor neuronal cells (21, 24). Likewise, heparan sulfate is abundantly expressed in the CNS cells and used by EV-A71 for entry though it is not known if EV-D68 uses heparan sulfate as an entry receptor to infect the CNS (177).
Immune response to EV-D68 infection
EV-D68 infects and replicates in the respiratory tract. In most cases, infection is rapidly cleared from the upper respiratory tract suggesting that EV-D68 infection induces a robust innate immune response (137). In others who experience symptomatic disease or remain asymptomatic, infection is associated with the induction of EV-D68 specific adaptive B and T cell responses (7). Evidence of EV-D68 specific B cell responses has largely come from serological studies. Though T cell responses have been examined in some cases, a detailed characterization of T cell responses remains an understudied area of research.
Innate immune responses have been shown to play an important role in early protection from viral infections. Type 1 interferon (IFNα and β) produced in response to infection induces over 100 interferon stimulated genes (ISGs) that contribute to an antiviral state among the neighboring cells thereby limiting and containing the spread of infection. How exactly EV-D68 drives innate immune responses is still under investigation though the interplay between the innate immune system and other enteroviruses have been extensively examined. During Coxsackievirus B3 (CV-B3) infection, TLR7 and TLR8 have been reported to recognize ssRNA, leading to the recruitment of MyD88 and a downstream innate immune response (178). Studies using TLR3 deficient mice have implicated TLR3 signaling in macrophages as a driver of host innate responses to CV-B4 (179), CV-B3 (180, 181), and poliovirus (PV) (182) with significantly higher levels of enterovirus viral replication in TLR3 deficient mice. On the other hand, Hsiao et al. demonstrated that TLR9 plays an important role in the induction of the innate immune response against EV-A71 (183). Overexpression of MDA5 and RIG-I during EV-A71 infection was shown to enhance the production of type I IFNs (184) and dsRNA from CV-B3, PV, and EV-A71 induced IFNβ through activation of MDA5 (185). Lastly, the activation of the STING pathway has been shown to inhibit viral replication for multiple viruses including EV-D68 (186) and EV-A71 (187).
Why innate immune responses fail to control infection in some children who go on to experience severe disease is not clear. Numerous studies suggest that EV-D68 like other enteroviruses interferes with the induction of Type I IFN responses. Multiple viral proteins, both structural and non-structural, have been implicated in inhibiting the innate immune response by suppressing Type I IFN signaling. Kang et al. showed that the capsid protein VP3 suppresses the phosphorylation and nuclear translocation of IRF7 as well as ubiquitination of IRF7 by TRAF6 by competitive inhibition, leading to the repression of interferon transcription (154). The nonstructural protein, 3D polymerase, is an important viral protein during viral replication that has also been implicated in mitochondrial dynamics and suppressing expression of type I interferons. Yang et al. showed that the 3D polymerase interacted with PGAM5 and upregulated the mitofusin 2 protein, leading to a change in mitochondrial morphology and impairing activation of the RIG-I receptor pathway that leads to IFNβ production (188). EV-D68 was shown to upregulate suppressor of cytokine signaling 3 (SOCS3) that inhibits the phosphorylation of STAT3 thereby suppressing the expression of downstream ISGs (189).
Two major EV-D68 proteases namely, the 2A and 3C proteases have been implicated in the evasion of innate immune responses. Kang et al. demonstrated that 2A protease cleaves TRAF3, a key protein in the induction of Type I IFN, during EV-D68 infection leading to a suppression of interferon production (154). Others have shown that 2A protease inhibits stress granule formation that contributes to IFN signaling (190). Xiao et al. demonstrated that 3C protease cleaves both the RIG-I and TRIM25, which ubiquitinates RIG-I, essential for the activation of the receptor (191). EV-D68 3C protease has been shown to bind MDA5 and inhibit its interaction between MDA5 and MAVS (155). Lastly, 3C protease has been shown to cleave IRF7 and TRIF, as well as prevent the activation of IRF3 to inhibit IFN production and to cleave the proteins TAK1 and TRIF, preventing NF-κB signaling (156, 192). Taken together, these studies suggest that innate IFN responses are induced early in infection, but evasion of these innate responses likely enables EV-D68 to establish productive infection in some patients leading to disease progression.
Evidence of adaptive immune responses induced by EV-D68 comes from serological analysis of patient samples. A screen for antibodies in CSF of AFM patients showed an enrichment of Picornaviridae specific antibody responses, compared to control patients, which was dominated by the Enterovirus genus; EV-D68 specific VP1 capsid protein specific antibodies were confirmed by ELISA in 85% of AFM patients. Schubert et al. reported detectable levels of EV-D68 specific binding antibodies in the CSF of AFM patients by ELISA (145). Harrison et al. demonstrated a high prevalence of EV-D68 seropositivity in the US using serum samples that were collected between 2012 - 2013 before the first major EV-D68 outbreak suggesting that EV-D68 infection may have been prevalent in the US prior to the outbreak of 2014 (193). In a cohort in Taiwan, EV-D68 specific neutralizing antibody responses were found to increase with age suggesting that exposure over time; ~18% of children between 1 and 2 years of age were seropositive where 100% 16-49-year-olds were seropositive (194). Whether these responses were specific to EV-D68 is not clear as numerous studies have reported significant cross-reactivity between related enteroviruses. Rosenfeld et al. demonstrated that antibodies induced against recent isolates of EV-D68 (2009, 2014, and 2018) in mice and guinea pigs show significant cross reactivity with the poliovirus type 1/Mahoney strain (195). Interestingly, healthy human sera from adults were found to have neutralization activity against EV-D68, EV-A71, and Poliovirus P1/Mahoney suggesting potential cross-neutralization across different enterovirus species. This was confirmed in the mouse model, where antibodies specifically raised against poliovirus were found to neutralize EV-D68 and vice versa (195). Most neutralizing antibodies appear to target EV-D68 VP1 (17, 61, 196, 197) though some of these antibodies have been shown to cross- neutralize across different clades (197). Vogt et al. (197) isolated and characterized a number of monoclonal antibodies (mAb) against EV-D68 VP1. Sixty mAb were isolated from 12 subjects previously infected with EV-D68 during the 2014 outbreak (11 subjects were infected with EV-D68 from clade B1 and 1 subject was infected with EV-D68 from clade A1) and tested them for neutralizing and binding capabilities. Seven of the sixty mAb were shown, by Western blot, to bind to EV-D68 VP1 and three of those seven mAb (EV-48, EV-46, and EV-40) were demonstrated to have cross clade neutralization. mAb EV-48 and EV-46 protected cells from infection with an isolate from Clade D (US/KY/14-18953), and EV-40 neutralized the prototypic Fermon strain, exhibiting cross-clade neutralization. Thirteen other monoclonal antibodies tested, not confirmed as anti-VP1 antibodies, were also able to cross-neutralize the clade D (4 mAbs) and Fermon (9 mAbs) strains (197). Anti-EV-D68 neutralizing antibodies have been shown to neutralize poliovirus infection in cell culture suggesting that heterotypic neutralization does occur, but likely at a lower efficacy than type-specific neutralizing antibodies (195).
There is a paucity of information regarding the nature and type of EV-D68 specific T cell responses induced during infection. Grifoni et al. using in silico approaches demonstrated that CD4+ T cell epitopes appear to be primarily focused within VP1 and other structural proteins (198). Rajput et al. reported that EV-D68 infected mice produced IL-17 at levels higher than mice similarly infected with RV-A, suggesting that infection induces EV-D68 specific Th17 and γδT cells (153). Kreuter et al. found abundant T cells in the spinal cord and brain of a child who died from meningomyeloencephalitis caused by EV-D68. Others have reported that perforin, a marker of CD8 T cell activity, was detectable in the tissues suggesting that infection likely induces EV-D68 specific CD8 T cell responses (131). Additional studies are needed to understand the kinetics and nature of EV-D68 specific CD4 and CD8 T cells induced during infection and the role these responses play in protection.
EV-D68 treatments and vaccines
Currently, there are no approved antiviral therapies, monoclonal antibodies, or vaccines for EV-D68. The current guidelines for EV-D68 treatment are supportive care with asthma management and treatment of bronchoreactivity with bronchodilators and steroids, if necessary. Commercial intravenous immunoglobulin (IVIG) has also been used in EV-D68-associated AFM cases as it has been found to have high levels of anti-EV-D68 antibodies that may confer passive immunity, and also has immunomodulatory properties. IVIG has been shown to be protective against paralysis when given early after EV-D68 infection in a neonatal mouse model (199). Vogt et al. generated two monoclonal antibody candidates that have been shown to neutralize EV-D68 with high potency (197). Wide screening of compounds for activity against EV-D68 has identified several potential antiviral candidates (200). Fluoxetine, an FDA approved selective serotonin reuptake inhibitor, was found to have in vitro antiviral activity against circulating strains of EV-D68, however a retrospective analysis of non-randomized use in human AFM cases in 2018 failed to demonstrate any signal of efficacy (201). The small molecule guanidine has shown promise in mouse models, but the mechanism is currently unknown. Telaprevir, an FDA approved protease inhibitor has been shown to prevent EV-D68 replication by irreversibly binding to EV-D68 2A protease. In a mouse model, telaprevir improved paralysis outcomes. While several antivirals designed against enteroviruses, including pocapavir and pleconaril, failed to demonstrate activity against EV-D68, other candidates such as the protease inhibitors, rupintrivir and V7404, that were shown to have EC50s as low as 0.0015 to 0.0051 μM, warrant further study as potential antiviral candidates with in vitro EV-D68 activity (202). Lastly, EV-D68 vaccine candidates are being developed as a preventive strategy against future waves of EV-D68 respiratory disease and AFM if cases continue to rise. Zhang et al. used an EV-D68 VLP vaccine with Pichia pastoris co-expressing the precursor P1 protein and 3CD protease that protected both neonatal mice born from vaccinated dams from lethal EV-D68 infection and mice who passively received anti-VLP sera (203, 204). Lin et al. developed a bivalent EV-D68/EV-A71 mucosal vaccine that was shown to induce high neutralizing antibody titers of EV-D68 and EV-A71 specific IgG and IgA that cross neutralized multiple EV-D868 and EV-A71 subtypes, and protected neonatal mice against lethal EV-A71 and EV-D68 infection (203, 204). Krug et al. (205) demonstrated that an EV-D68 VLP candidate vaccine elicited high levels of neutralizing antibodies against multiple EV-D68 clades in both mouse and non-human primate models. Additional trials and studies will be required to fully elucidate antivirals and vaccines that will prevent EV-D68 infection and severe symptoms.
Conclusions
EV-D68 causes severe respiratory disease in young children and has been temporally associated with spinal cord pathology and paralysis. There has been a significant increase in EV-D68 infections worldwide since the major outbreak of 2014. Infections appear to follow a biennial pattern until 2018. Epidemiological data suggests that the likelihood of future EV-D68 outbreaks remain quite high. The exact reason for this increase in the incidence of infections is not clear and remains an active area of research. Though progress continue to be made in developing better surveillance and diagnostic tools, a number of challenges remain to be addressed. Why children below the age of 16 are more susceptible to EV-D68 infection is not clear. Likewise, the exact mechanism of how respiratory infection with EV-D68 leads to the destruction of spinal cord gray matter is not known. Better understanding of the nature and kinetics of immune responses that contribute to protection is key to the development of better vaccines and therapeutic approaches to control future outbreaks.
Author contributions
CG: Data curation, Formal Analysis, Visualization, Writing – original draft, Writing – review & editing. KM: Writing – review & editing. JM: Conceptualization, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by a grant (R21AI152076) from the National Institutes of Health to JJM.
Acknowledgments
We would like to thank Dr. Andrew Frank and Dr. Kenneth Elliott at the Uniformed Services University for their expert assistance with the analysis of the data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The opinions or assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of Defense, the Uniformed Services University of the Health Sciences or any other agency of the U.S. Government.
References
1. Messacar K, Schreiner TL, Maloney JA, Wallace A, Ludke J, Oberste MS, et al. A cluster of acute flaccid paralysis and cranial nerve dysfunction temporally associated with an outbreak of enterovirus D68 in children in Colorado, USA. Lancet (2015) 385(9978):1662–71. doi: 10.1016/S0140-6736(14)62457-0
2. Messacar K, Schreiner TL, Van Haren K, Yang M, Glaser CA, Tyler KL, et al. Acute flaccid myelitis: A clinical review of US cases 2012-2015. Ann Neurol (2016) 80(3):326–38. doi: 10.1002/ana.24730
3. Racaniello VR. One hundred years of poliovirus pathogenesis. Virology (2006) 344(1):9–16. doi: 10.1016/j.virol.2005.09.015
4. Pons-Salort M, Parker EP, Grassly NC. The epidemiology of non-polio enteroviruses: recent advances and outstanding questions. Curr Opin Infect Dis (2015) 28(5):479–87. doi: 10.1097/QCO.0000000000000187
5. Holm-Hansen CC, Midgley SE, Fischer TK. Global emergence of enterovirus D68: a systematic review. Lancet Infect Dis (2016) 16(5):e64–75. doi: 10.1016/S1473-3099(15)00543-5
6. Park SW, Pons-Salort M, Messacar K, Cook C, Meyers L, Farrar J, et al. Epidemiological dynamics of enterovirus D68 in the United States and implications for acute flaccid myelitis. Sci Transl Med (2021) 13(584):eabd2400. doi: 10.1126/scitranslmed.abd2400
7. Vogt MR, Wright PF, Hickey WF, De Buysscher T, Boyd KL, Crowe JE Jr. Enterovirus D68 in the anterior horn cells of a child with acute flaccid myelitis. N Engl J Med (2022) 386(21):2059–60. doi: 10.1056/NEJMc2118155
8. Kujawski SA, Midgley CM, Rha B, Lively JY, Nix WA, Curns AT, et al. Enterovirus D68-associated acute respiratory illness - new vaccine surveillance network, United States, July-October, 2017 and 2018. MMWR Morb Mortal Wkly Rep (2019) 68(12):277–80. doi: 10.15585/mmwr.mm6812a1
9. Baggen J, Thibaut HJ, Strating J, van Kuppeveld FJM. Publisher Correction: The life cycle of non-polio enteroviruses and how to target it. Nat Rev Microbiol (2018) 16(6):391. doi: 10.1038/s41579-018-0022-3
10. Lulla V, Dinan AM, Hosmillo M, Chaudhry Y, Sherry L, Irigoyen N, et al. An upstream protein-coding region in enteroviruses modulates virus infection in gut epithelial cells. Nat Microbiol (2019) 4(2):280–92. doi: 10.1038/s41564-018-0297-1
11. Oberste MS, Maher K, Schnurr D, Flemister MR, Lovchik JC, Peters H, et al. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J Gen Virol (2004) 85(Pt 9):2577–84. doi: 10.1099/vir.0.79925-0
12. Huang W, Wang G, Zhuge J, Nolan SM, Dimitrova N, Fallon JT. Whole-genome sequence analysis reveals the enterovirus D68 isolates during the United States 2014 outbreak mainly belong to a novel clade. Sci Rep (2015) 5:15223. doi: 10.1038/srep15223
13. Hogle JM, Chow M, Filman DJ. Three-dimensional structure of poliovirus at 2.9 A resolution. Science (1985) 229(4720):1358–65. doi: 10.1126/science.2994218
14. Rossmann MG, Arnold E, Erickson JW, Frankenberger EA, Griffith JP, Hecht HJ, et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature (1985) 317(6033):145–53. doi: 10.1038/317145a0
15. Sherry B, Mosser AG, Colonno RJ, Rueckert RR. Use of monoclonal antibodies to identify four neutralization immunogens on a common cold picornavirus, human rhinovirus 14. J Virol (1986) 57(1):246–57. doi: 10.1128/jvi.57.1.246-257.1986
16. Liu Y, Sheng J, Baggen J, Meng G, Xiao C, Thibaut HJ, et al. Sialic acid-dependent cell entry of human enterovirus D68. Nat Commun (2015) 6:8865. doi: 10.1038/ncomms9865
17. Liu Y, Sheng J, Fokine A, Meng G, Shin WH, Long F, et al. Structure and inhibition of EV-D68, a virus that causes respiratory illness in children. Science (2015) 347(6217):71–4. doi: 10.1126/science.1261962
18. Uncapher CR, DeWitt CM, Colonno RJ. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology (1991) 180(2):814–7. doi: 10.1016/0042-6822(91)90098-V
19. Imamura T, Okamoto M, Nakakita S, Suzuki A, Saito M, Tamaki R, et al. Antigenic and receptor binding properties of enterovirus 68. J Virol (2014) 88(5):2374–84. doi: 10.1128/JVI.03070-13
20. Baggen J, Thibaut HJ, Staring J, Jae LT, Liu Y, Guo H, et al. Enterovirus D68 receptor requirements unveiled by haploid genetics. Proc Natl Acad Sci U S A (2016) 113(5):1399–404. doi: 10.1073/pnas.1524498113
21. Hixon AM, Clarke P, Tyler KL. Contemporary circulating enterovirus D68 strains infect and undergo retrograde axonal transport in spinal motor neurons independent of sialic acid. J Virol (2019) 93(16):e00578-19. doi: 10.1128/JVI.00578-19
22. Baggen J, Liu Y, Lyoo H, van Vliet ALW, Wahedi M, de Bruin JW, et al. Bypassing pan-enterovirus host factor PLA2G16. Nat Commun (2019) 10(1):3171. doi: 10.1038/s41467-019-11256-z
23. Yoshihara Y, Oka S, Nemoto Y, Watanabe Y, Nagata S, Kagamiyama H, et al. An ICAM-related neuronal glycoprotein, telencephalin, with brain segment-specific expression. Neuron (1994) 12(3):541–53. doi: 10.1016/0896-6273(94)90211-9
24. Wei W, Guo H, Chang J, Yu Y, Liu G, Zhang N, et al. ICAM-5/telencephalin is a functional entry receptor for enterovirus D68. Cell Host Microbe (2016) 20(5):631–41. doi: 10.1016/j.chom.2016.09.013
25. Belnap DM, Filman DJ, Trus BL, Cheng N, Booy FP, Conway JF, et al. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J Virol (2000) 74(3):1342–54. doi: 10.1128/JVI.74.3.1342-1354.2000
26. Huang Y, Hogle JM, Chow M. Is the 135S poliovirus particle an intermediate during cell entry? J Virol (2000) 74(18):8757–61. doi: 10.1128/JVI.74.18.8757-8761.2000
27. Zheng Q, Zhu R, Xu L, He M, Yan X, Liu D, et al. Atomic structures of enterovirus D68 in complex with two monoclonal antibodies define distinct mechanisms of viral neutralization. Nat Microbiol (2019) 4(1):124–33. doi: 10.1038/s41564-018-0275-7
28. Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM, et al. Imaging poliovirus entry in live cells. PLoS Biol (2007) 5(7):e183. doi: 10.1371/journal.pbio.0050183
29. Panjwani A, Strauss M, Gold S, Wenham H, Jackson T, Chou JJ, et al. Capsid protein VP4 of human rhinovirus induces membrane permeability by the formation of a size-selective multimeric pore. PLoS Pathog (2014) 10(8):e1004294. doi: 10.1371/journal.ppat.1004294
30. Strauss M, Levy HC, Bostina M, Filman DJ, Hogle JM. RNA transfer from poliovirus 135S particles across membranes is mediated by long umbilical connectors. J Virol (2013) 87(7):3903–14. doi: 10.1128/JVI.03209-12
31. Hussain KM, Leong KL, Ng MM, Chu JJ. The essential role of clathrin-mediated endocytosis in the infectious entry of human enterovirus 71. J Biol Chem (2011) 286(1):309–21. doi: 10.1074/jbc.M110.168468
32. Liu Y, Sheng J, van Vliet ALW, Buda G, van Kuppeveld FJM, Rossmann MG. Molecular basis for the acid-initiated uncoating of human enterovirus D68. Proc Natl Acad Sci U S A (2018) 115(52):E12209–17. doi: 10.1073/pnas.1803347115
33. Elling U, Wimmer RA, Leibbrandt A, Burkard T, Michlits G, Leopoldi A, et al. A reversible haploid mouse embryonic stem cell biobank resource for functional genomics. Nature (2017) 550(7674):114–8. doi: 10.1038/nature24027
34. Staring J, von Castelmur E, Blomen VA, van den Hengel LG, Brockmann M, Baggen J, et al. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature (2017) 541(7637):412–6. doi: 10.1038/nature21032
35. Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature (1988) 334(6180):320–5. doi: 10.1038/334320a0
36. Herold J, Andino R. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol Cell (2001) 7(3):581–91. doi: 10.1016/S1097-2765(01)00205-2
37. Flather D, Semler BL. Picornaviruses and nuclear functions: targeting a cellular compartment distinct from the replication site of a positive-strand RNA virus. Front Microbiol (2015) 6:594. doi: 10.3389/fmicb.2015.00594
38. Sweeney TR, Abaeva IS, Pestova TV, Hellen CU. The mechanism of translation initiation on Type 1 picornavirus IRESs. EMBO J (2014) 33(1):76–92. doi: 10.1002/embj.201386124
39. Pallansch M, Roos R. . Enteroviruses: Polioviruses, Coxsackieviruses, Echoviruses, and Newer Enteroviruses. U: Knipe DM, Howley PM, ur. Fields virology. 2007, Philadelphia: Lippincott Williams & Wilkins.
40. Bonderoff JM, Larey JL, Lloyd RE. Cleavage of poly(A)-binding protein by poliovirus 3C proteinase inhibits viral internal ribosome entry site-mediated translation. J Virol (2008) 82(19):9389–99. doi: 10.1128/JVI.00006-08
41. Kanda T, Gauss-Muller V, Cordes S, Tamura R, Okitsu K, Shuang W, et al. Hepatitis A virus (HAV) proteinase 3C inhibits HAV IRES-dependent translation and cleaves the polypyrimidine tract-binding protein. J Viral Hepat (2010) 17(9):618–23. doi: 10.1111/j.1365-2893.2009.01221.x
42. Perera R, Daijogo S, Walter BL, Nguyen JH, Semler BL. Cellular protein modification by poliovirus: the two faces of poly(rC)-binding protein. J Virol (2007) 81(17):8919–32. doi: 10.1128/JVI.01013-07
43. Zhang B, Seitz S, Kusov Y, Zell R, Gauss-Muller V. RNA interaction and cleavage of poly(C)-binding protein 2 by hepatitis A virus protease. Biochem Biophys Res Commun (2007) 364(4):725–30. doi: 10.1016/j.bbrc.2007.09.133
44. Ertel KJ, Brunner JE, Semler BL. Mechanistic consequences of hnRNP C binding to both RNA termini of poliovirus negative-strand RNA intermediates. J Virol (2010) 84(9):4229–42. doi: 10.1128/JVI.02198-09
45. Barton DJ, Morasco BJ, Flanegan JB. Translating ribosomes inhibit poliovirus negative-strand RNA synthesis. J Virol (1999) 73(12):10104–12. doi: 10.1128/JVI.73.12.10104-10112.1999
46. Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. Complex dynamic development of poliovirus membranous replication complexes. J Virol (2012) 86(1):302–12. doi: 10.1128/JVI.05937-11
47. Laufman O, Perrino J, Andino R. Viral generated inter-organelle contacts redirect lipid flux for genome replication. Cell (2019) 178(2):275–89.e16. doi: 10.1016/j.cell.2019.05.030
48. Limpens RW, van der Schaar HM, Kumar D, Koster AJ, Snijder EJ, van Kuppeveld FJ, et al. The transformation of enterovirus replication structures: a three-dimensional study of single- and double-membrane compartments. mBio (2011) 2(5):e00166-11. doi: 10.1128/mBio.00166-11
49. den Boon JA, Ahlquist P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol (2010) 64:241–56. doi: 10.1146/annurev.micro.112408.134012
50. Miller S, Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol (2008) 6(5):363–74. doi: 10.1038/nrmicro1890
51. Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog (2012) 8(12):e1003056. doi: 10.1371/journal.ppat.1003056
52. Goodfellow I. The genome-linked protein VPg of vertebrate viruses - a multifaceted protein. Curr Opin Virol (2011) 1(5):355–62. doi: 10.1016/j.coviro.2011.09.003
53. Corona AK, Saulsbery HM, Corona Velazquez AF, Jackson WT. Enteroviruses remodel autophagic trafficking through regulation of host SNARE proteins to promote virus replication and cell exit. Cell Rep (2018) 22(12):3304–14. doi: 10.1016/j.celrep.2018.03.003
54. Yang JE, Rossignol ED, Chang D, Zaia J, Forrester I, Raja K, et al. Complexity and ultrastructure of infectious extracellular vesicles from cells infected by non-enveloped virus. Sci Rep (2020) 10(1):7939. doi: 10.1038/s41598-020-64531-1
55. Lennette EH, Lennette DA, Lennette ET. Diagnostic procedures for viral, rickettsial, and chlamydial infections. 7th ed. Washington, DC: American Public Health Association (1995).
56. Schieble JH, Fox VL, Lennette EH. A probable new human picornavirus associated with respiratory diseases. Am J Epidemiol (1967) 85(2):297–310. doi: 10.1093/oxfordjournals.aje.a120693
57. Smith BD, Pekosz A. Contemporary Enterovirus D68 strains show enhanced replication and translation at 37°C. bioRxiv (2020), 2020.03.31.019380. doi: 10.1101/2020.03.31.019380.
58. Midgley CM, Jackson MA, Selvarangan R, Turabelidze G, Obringer E, Johnson D, et al. Severe respiratory illness associated with enterovirus D68 - Missouri and Illinois, 2014. MMWR Morb Mortal Wkly Rep (2014) 63(36):798–9. doi: 10.1111/ajt.13035
59. Rao S, Messacar K, Torok MR, Rick AM, Holzberg J, Montano A, et al. Enterovirus D68 in critically ill children: A comparison with pandemic H1N1 influenza. Pediatr Crit Care Med (2016) 17(11):1023–31. doi: 10.1097/PCC.0000000000000922
60. Schuster JE, Miller JO, Selvarangan R, Weddle G, Thompson MT, Hassan F, et al. Severe enterovirus 68 respiratory illness in children requiring intensive care management. J Clin Virol (2015) 70:77–82. doi: 10.1016/j.jcv.2015.07.298
61. Hodcroft EB, Dyrdak R, Andres C, Egli A, Reist J, Garcia Martinez de Artola D, et al. Evolution, geographic spreading, and demographic distribution of Enterovirus D68. PLoS Pathog (2022) 18(5):e1010515. doi: 10.1371/journal.ppat.1010515
62. Nguyen-Tran H, Thompson C, Butler M, Miller KR, Pyle L, Jung S, et al. Duration of enterovirus D68 RNA shedding in the upper respiratory tract and transmission among household contacts, Colorado, USA. Emerg Infect Dis (2023) 29(11):2315–24. doi: 10.3201/eid2911.230947
63. Abedi GR, Watson JT, Nix WA, Oberste MS, Gerber SI. Enterovirus and parechovirus surveillance - United States, 2014-2016. MMWR Morb Mortal Wkly Rep (2018) 67(18):515–8. doi: 85910.15585/mmwr.mm6718a2
64. Abedi GR, Watson JT, Nix WA, Oberste MS, Gerber SI. Enterovirus and human parechovirus surveillance - United States, 2009-2013. MMWR Morb Mortal Wkly Rep (2015) 64(34):940–3. doi: 10.15585/mmwr.mm6434a3
65. Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA. Enterovirus surveillance–United States, 1970-2005. MMWR Surveill Summ (2006) 55(8):1–20.
66. Benschop KS, Albert J, Anton A, Andres C, Aranzamendi M, Armannsdottir B, et al. Re-emergence of enterovirus D68 in Europe after easing the COVID-19 lockdown, September 2021. Euro Surveill (2021) 26(45):e2100998. doi: 10.2807/1560-7917.ES.2021.26.45.2100998
67. Ma KC, Winn A, Moline HL, Scobie HM, Midgley CM, Kirking HL, et al. Increase in acute respiratory illnesses among children and adolescents associated with rhinoviruses and enteroviruses, including enterovirus D68 - United States, July-September 2022. MMWR Morb Mortal Wkly Rep (2022) 71(40):1265–70. doi: 10.15585/mmwr.mm7140e1
68. Sayers EW, Bolton EE, Brister JR, Canese K, Chan J, Comeau DC, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res (2022) 50(D1):D20–6. doi: 10.1093/nar/gkab1112
69. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol (2011) 7:539. doi: 10.1038/msb.2011.75
70. Madeira F, Pearce M, Tivey ARN, Basutkar P, Lee J, Edbali O, et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res (2022) 50(W1):W276–9. doi: 10.1093/nar/gkac240
71. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res (2021) 49(W1):W293–6. doi: 10.1093/nar/gkab301
72. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol (2010) 59(3):307–21. doi: 10.1093/sysbio/syq010
73. Simmonds P, Gorbalenya AE, Harvala H, Hovi T, Knowles NJ, Lindberg AM, et al. Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch Virol (2020) 165(3):793–7. doi: 10.1007/s00705-019-04520-6
74. Blomqvist S, Savolainen C, Raman L, Roivainen M, Hovi T. Human rhinovirus 87 and enterovirus 68 represent a unique serotype with rhinovirus and enterovirus features. J Clin Microbiol (2002) 40(11):4218–23. doi: 10.1128/JCM.40.11.4218-4223.2002
75. Ishiko H, Miura R, Shimada Y, Hayashi A, Nakajima H, Yamazaki S, et al. Human rhinovirus 87 identified as human enterovirus 68 by VP4-based molecular diagnosis. Intervirology (2002) 45(3):136–41. doi: 10.1159/000065866
76. Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics (2018) 34(23):4121–3. doi: 10.1093/bioinformatics/bty407
77. Brown DM, Hixon AM, Oldfield LM, Zhang Y, Novotny M, Wang W, et al. Contemporary circulating enterovirus D68 strains have acquired the capacity for viral entry and replication in human neuronal cells. mBio (2018) 9(5):e01954-18. doi: 10.1128/mBio.01954-18
78. Du J, Zheng B, Zheng W, Li P, Kang J, Hou J, et al. Analysis of enterovirus 68 strains from the 2014 North American outbreak reveals a new clade, indicating viral evolution. PLoS One (2015) 10(12):e0144208. doi: 10.1371/journal.pone.0144208
79. Eshaghi A, Duvvuri VR, Isabel S, Banh P, Li A, Peci A, et al. Global distribution and evolutionary history of enterovirus D68, with emphasis on the 2014 outbreak in Ontario, Canada. Front Microbiol (2017) 8:257. doi: 10.3389/fmicb.2017.00257
80. Shen L, Gong C, Xiang Z, Zhang T, Li M, Li A, et al. Upsurge of enterovirus D68 and circulation of the new subclade D3 and subclade B3 in Beijing, China, 2016. Sci Rep (2019) 9(1):6073. doi: 10.1038/s41598-019-42651-7
81. Sun J, Hu XY, Yu XF. Current understanding of human enterovirus D68. Viruses (2019) 11(6):e490. doi: 10.3390/v11060490
82. Oberste MS, Maher K, Kilpatrick DR, Pallansch MA. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol (1999) 73(3):1941–8. doi: 10.1128/JVI.73.3.1941-1948.1999
83. Meijer A, van der Sanden S, Snijders BE, Jaramillo-Gutierrez G, Bont L, van der Ent CK, et al. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in The Netherlands in 2010. Virology (2012) 423(1):49–57. doi: 10.1016/j.virol.2011.11.021
84. Greninger AL, Naccache SN, Messacar K, Clayton A, Yu G, Somasekar S, et al. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012-14): a retrospective cohort study. Lancet Infect Dis (2015) 15(6):671–82. doi: 10.1016/S1473-3099(15)70093-9
85. Tan Y, Hassan F, Schuster JE, Simenauer A, Selvarangan R, Halpin RA, et al. Molecular evolution and intraclade recombination of enterovirus D68 during the 2014 outbreak in the United States. J Virol (2016) 90(4):1997–2007. doi: 10.1128/JVI.02418-15
86. Dyda A, Stelzer-Braid S, Adam D, Chughtai AA, MacIntyre CR. The association between acute flaccid myelitis (AFM) and Enterovirus D68 (EV-D68) - what is the evidence for causation? Euro Surveill (2018) 23(3):e17-00310. doi: 10.2807/1560-7917.ES.2018.23.3.17-00310
87. Hixon AM, Frost J, Rudy MJ, Messacar K, Clarke P, Tyler KL. Understanding enterovirus D68-induced neurologic disease: A basic science review. Viruses (2019) 11(9):e821. doi: 10.3390/v11090821
88. Zhang Y, Cao J, Zhang S, Lee AJ, Sun G, Larsen CN, et al. Genetic changes found in a distinct clade of Enterovirus D68 associated with paralysis during the 2014 outbreak. Virus Evol (2016) 2(1):vew015. doi: 10.1093/ve/vew015
89. Esposito S, Chidini G, Cinnante C, Napolitano L, Giannini A, Terranova L, et al. Acute flaccid myelitis associated with enterovirus-D68 infection in an otherwise healthy child. Virol J (2017) 14(1):4. doi: 10.1186/s12985-016-0678-0
90. Gong YN, Yang SL, Shih SR, Huang YC, Chang PY, Huang CG, et al. Molecular evolution and the global reemergence of enterovirus D68 by genome-wide analysis. Med (Baltimore) (2016) 95(31):e4416. doi: 10.1097/MD.0000000000004416
91. Dyrdak R, Grabbe M, Hammas B, Ekwall J, Hansson KE, Luthander J, et al. Outbreak of enterovirus D68 of the new B3 lineage in Stockholm, Sweden, August to September 2016. Euro Surveill (2016) 21(46):e30403. doi: 10.2807/1560-7917.ES.2016.21.46.30403
92. Knoester M, Scholvinck EH, Poelman R, Smit S, Vermont CL, Niesters HG, et al. Upsurge of enterovirus D68, the Netherlands, 2016. Emerg Infect Dis (2017) 23(1):140–3. doi: 10.3201/eid2301.161313
93. Midgley SE, Benschop K, Dyrdak R, Mirand A, Bailly JL, Bierbaum S, et al. Co-circulation of multiple enterovirus D68 subclades, including a novel B3 cluster, across Europe in a season of expected low prevalence, 2019/20. Euro Surveill (2020) 25(2):e1900749. doi: 10.2807/1560-7917.ES.2020.25.2.1900749
94. Wang G, Zhuge J, Huang W, Nolan SM, Gilrane VL, Yin C, et al. Enterovirus D68 subclade B3 strain circulating and causing an outbreak in the United States in 2016. Sci Rep (2017) 7(1):1242. doi: 10.1038/s41598-017-01349-4
95. Sooksawasdi Na Ayudhya S, Laksono BM, van Riel D. The pathogenesis and virulence of enterovirus-D68 infection. Virulence (2021) 12(1):2060–72. doi: 10.1080/21505594.2021.1960106
96. Centers for Disease, C. and Prevention. Clusters of acute respiratory illness associated with human enterovirus 68–Asia, Europe, and United States, 2008-2010. MMWR Morb Mortal Wkly Rep (2011) 60(38):1301–4.
97. Hasegawa S, Hirano R, Okamoto-Nakagawa R, Ichiyama T, Shirabe K. Enterovirus 68 infection in children with asthma attacks: virus-induced asthma in Japanese children. Allergy (2011) 66(12):1618–20. doi: 10.1111/j.1398-9995.2011.02725.x
98. Ikeda T, Mizuta K, Abiko C, Aoki Y, Itagaki T, Katsushima F, et al. Acute respiratory infections due to enterovirus 68 in Yamagata, Japan between 2005 and 2010. Microbiol Immunol (2012) 56(2):139–43. doi: 10.1111/j.1348-0421.2012.00411.x
99. Imamura T, Fuji N, Suzuki A, Tamaki R, Saito M, Aniceto R, et al. Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg Infect Dis (2011) 17(8):1430–5. doi: 10.3201/eid1708.101328
100. Kaida A, Kubo H, Sekiguchi J, Kohdera U, Togawa M, Shiomi M, et al. Enterovirus 68 in children with acute respiratory tract infections, Osaka, Japan. Emerg Infect Dis (2011) 17(8):1494–7. doi: 10.3201/eid1708.110028
101. Rahamat-Langendoen J, Riezebos-Brilman A, Borger R, van der Heide R, Brandenburg A, Scholvinck E, et al. Upsurge of human enterovirus 68 infections in patients with severe respiratory tract infections. J Clin Virol (2011) 52(2):103–6. doi: 10.1016/j.jcv.2011.06.019
102. Renois F, Bouin A, Andreoletti L. Enterovirus 68 in pediatric patients hospitalized for acute airway diseases. J Clin Microbiol (2013) 51(2):640–3. doi: 10.1128/JCM.02640-12
103. Furuse Y, Chaimongkol N, Okamoto M, Imamura T, Saito M, Tamaki R, et al. Molecular epidemiology of enterovirus D68 from 2013 to 2014 in Philippines. J Clin Microbiol (2015) 53(3):1015–8. doi: 10.1128/JCM.03362-14
104. Levy A, Roberts J, Lang J, Tempone S, Kesson A, Dofai A, et al. Enterovirus D68 disease and molecular epidemiology in Australia. J Clin Virol (2015) 69:117–21. doi: 10.1016/j.jcv.2015.06.079
105. Lu QB, Wo Y, Wang HY, Wei MT, Zhang L, Yang H, et al. Detection of enterovirus 68 as one of the commonest types of enterovirus found in patients with acute respiratory tract infection in China. J Med Microbiol (2014) 63(Pt 3):408–14. doi: 10.1099/jmm.0.068247-0
106. Meijer A, Benschop KS, Donker GA, van der Avoort HG. Continued seasonal circulation of enterovirus D68 in the Netherlands, 2011-2014. Euro Surveill (2014) 19(42):e20935. doi: 10.2807/1560-7917.ES2014.19.42.20935
107. Opanda SM, Wamunyokoli F, Khamadi S, Coldren R, Bulimo WD. Genetic diversity of human enterovirus 68 strains isolated in Kenya using the hypervariable 3'-end of VP1 gene. PloS One (2014) 9(7):e102866. doi: 10.1371/journal.pone.0102866
108. Piralla A, Girello A, Grignani M, Gozalo-Marguello M, Marchi A, Marseglia G, et al. Phylogenetic characterization of enterovirus 68 strains in patients with respiratory syndromes in Italy. J Med Virol (2014) 86(9):1590–3. doi: 10.1002/jmv.23821
109. Todd AK, Hall RJ, Wang J, Peacey M, McTavish S, Rand CJ, et al. Detection and whole genome sequence analysis of an enterovirus 68 cluster. Virol J (2013) 10:103. doi: 10.1186/1743-422X-10-103
110. Tokarz R, Firth C, Madhi SA, Howie SRC, Wu W, Sall AA, et al. Worldwide emergence of multiple clades of enterovirus 68. J Gen Virol (2012) 93(Pt 9):1952–8. doi: 10.1099/vir.0.043935-0
111. Oermann CM, Schuster JE, Conners GP, Newland JG, Selvarangan R, Jackson MA, et al. Enterovirus d68. A focused review and clinical highlights from the 2014 U.S. Outbreak. Ann Am Thorac Soc (2015) 12(5):775–81.doi: 10.1513/AnnalsATS.201412-592FR
112. Schuster JE, Newland JG. Management of the 2014 enterovirus 68 outbreak at a pediatric tertiary care center. Clin Ther (2015) 37(11):2411–8. doi: 10.1016/j.clinthera.2015.06.015
113. Midgley CM, Watson JT, Nix WA, Curns AT, Rogers SL, Brown BA, et al. Severe respiratory illness associated with a nationwide outbreak of enterovirus D68 in the USA (2014): a descriptive epidemiological investigation. Lancet Respir Med (2015) 3(11):879–87. doi: 10.1016/S2213-2600(15)00335-5
114. Influx of patients with asthma-like symptoms strains resources in many pediatric EDs. ED Manag (2014) 26(11):121–4. doi: 10.2807/1560-7917.Es.2016.21.4.30119
115. Shaw J, Welch TR, Milstone AM. The role of syndromic surveillance in directing the public health response to the enterovirus D68 epidemic. JAMA Pediatr (2014) 168(11):981–2. doi: 10.1001/jamapediatrics.2014.2628
116. Centers for Disease Control and Prevention (2023). Available at: https://www.cdc.gov/non-polio-enterovirus/about/EV-D68.html.
117. Messacar K, Hawkins SM, Baker J, Pearce K, Tong S, Dominguez SR, et al. Resource burden during the 2014 enterovirus D68 respiratory disease outbreak at children's hospital Colorado: an unexpected strain. JAMA Pediatr (2016) 170(3):294–7. doi: 10.1001/jamapediatrics.2015.3879
118. Bal A, Schuffenecker I, Casalegno JS, Josset L, Valette M, Armand N, et al. Enterovirus D68 nosocomial outbreak in elderly people, France, 2014. Clin Microbiol Infect (2015) 21(8):e61–2. doi: 10.1016/j.cmi.2015.05.008
119. Carney S, Brown D, Siqueira MM, Dias JP, da Silva EE. Enterovirus D68 detected in children with severe acute respiratory illness in Brazil. Emerg Microbes Infect (2015) 4(10):e66. doi: 10.1038/emi.2015.66
120. Drews SJ, Simmonds K, Usman HR, Yee K, Fathima S, Tipples G, et al. Characterization of enterovirus activity, including that of enterovirus D68, in pediatric patients in Alberta, Canada, in 2014. J Clin Microbiol (2015) 53(3):1042–5. doi: 10.1128/JCM.02982-14
121. Dyrdak R, Rotzen-Ostlund M, Samuelson A, Eriksson M, Albert J. Coexistence of two clades of enterovirus D68 in pediatric Swedish patients in the summer and fall of 2014. Infect Dis (Lond) (2015) 47(10):734–8. doi: 10.3109/23744235.2015.1047402
122. Edwin JJ, Domingo FR, Booth TF, Mersereau T, Skowronski DM, Chambers C, et al. Surveillance summary of hospitalized pediatric enterovirus D68 cases in Canada, September 2014. Can Commun Dis Rep (2015) 41(Suppl 1):2–8. doi: 10.14745/ccdr.v41is1a01
123. Esposito S, Zampiero A, Ruggiero L, Madini B, Niesters H, Principi N. Enterovirus D68-associated community-acquired pneumonia in children living in Milan, Italy. J Clin Virol (2015) 68:94–6. doi: 10.1016/j.jcv.2015.05.017
124. Gimferrer L, Campins M, Codina MG, Esperalba J, Martin Mdel C, Fuentes F, et al. First Enterovirus D68 (EV-D68) cases detected in hospitalised patients in a tertiary care university hospital in Spain, October 2014. Enferm Infecc Microbiol Clin (2015) 33(9):585–9. doi: 10.1016/j.eimc.2015.01.008
125. Lang M, Mirand A, Savy N, Henquell C, Maridet S, Perignon R, et al. Acute flaccid paralysis following enterovirus D68 associated pneumonia, France, 2014. Euro Surveill (2014) 19(44):e20952. doi: 10.2807/1560-7917.ES2014.19.44.20952
126. Midgley SE, Christiansen CB, Poulsen MW, Hansen CH, Fischer TK. Emergence of enterovirus D68 in Denmark, June 2014 to February 2015. Euro Surveill (2015) 20(17):e21105. doi: 10.2807/1560-7917.ES2015.20.17.21105
127. Poelman R, Scholvinck EH, Borger R, Niesters HG, van Leer-Buter C. The emergence of enterovirus D68 in a Dutch University Medical Center and the necessity for routinely screening for respiratory viruses. J Clin Virol (2015) 62:1–5. doi: 10.1016/j.jcv.2014.11.011
128. Reiche J, Bottcher S, Diedrich S, Buchholz U, Buda S, Haas W, et al. Low-level circulation of enterovirus D68-associated acute respiratory infections, Germany, 2014. Emerg Infect Dis (2015) 21(5):837–41. doi: 10.3201/eid2105.141900
129. Torres JP, Farfan MJ, Izquierdo G, Piemonte P, Henriquez J, O'Ryan ML, et al. Enterovirus D68 infection, Chile, spring 2014. Emerg Infect Dis (2015) 21(4):728–9. doi: 10.3201/eid2104.141766
130. Sejvar JJ, Lopez AS, Cortese MM, Leshem E, Pastula DM, Miller L, et al. Acute flaccid myelitis in the United States, August-December 2014: results of nationwide surveillance. Clin Infect Dis (2016) 63(6):737–45. doi: 10.1093/cid/ciw372
131. Kreuter JD, Barnes A, McCarthy JE, Schwartzman JD, Oberste MS, Rhodes CH, et al. A fatal central nervous system enterovirus 68 infection. Arch Pathol Lab Med (2011) 135(6):793–6. doi: 10.5858/2010-0174-CR.1
132. Van Haren K, Ayscue P, Waubant E, Clayton A, Sheriff H, Yagi S, et al. Acute flaccid myelitis of unknown etiology in California, 2012-2015. JAMA (2015) 314(24):2663–71. doi: 10.1001/jama.2015.17275
133. Helfferich J, Knoester M, Van Leer-Buter CC, Neuteboom RF, Meiners LC, Niesters HG, et al. Acute flaccid myelitis and enterovirus D68: lessons from the past and present. Eur J Pediatr (2019) 178(9):1305–15. doi: 10.1007/s00431-019-03435-3
134. Ayers T, Lopez A, Lee A, Kambhampati A, Nix WA, Henderson E, et al. Acute flaccid myelitis in the United States: 2015-2017. Pediatrics (2019) 144(5):e20191619. doi: 10.1542/peds.2019-1619
135. Forero EL, Knoester M, Gard L, Ott A, Brandenburg AH, McCall MBB, et al. Changes in enterovirus epidemiology after easing of lockdown measures. J Clin Virol (2023) 169:105617. doi: 10.1016/j.jcv.2023.105617
136. Hixon AM, Yu G, Leser JS, Yagi S, Clarke P, Chiu CY, et al. A mouse model of paralytic myelitis caused by enterovirus D68. PloS Pathog (2017) 13(2):e1006199. doi: 10.1371/journal.ppat.1006199
137. Hurst BL, Evans WJ, Smee DF, Van Wettere AJ, Tarbet EB, et al. Evaluation of antiviral therapies in respiratory and neurological disease models of Enterovirus D68 infection in mice. Virology (2019) 526:146–54. doi: 10.1016/j.virol.2018.10.014
138. Morrey JD, Wang H, Hurst BL, Zukor K, Siddharthan V, Van Wettere AJ, et al. Causation of acute flaccid paralysis by myelitis and myositis in enterovirus-D68 infected mice deficient in interferon alphabeta/gamma receptor deficient mice. Viruses (2018) 10(1):e33. doi: 10.3390/v10010033
139. Rosenfeld AB, Warren AL, Racaniello VR. Neurotropism of enterovirus D68 isolates is independent of sialic acid and is not a recently acquired phenotype. mBio (2019) 10(5):e02370-19. doi: 10.1128/mBio.02370-19
140. Sun S, Bian L, Gao F, Du R, Hu Y, Fu Y, et al. A neonatal mouse model of Enterovirus D68 infection induces both interstitial pneumonia and acute flaccid myelitis. Antiviral Res (2019) 161:108–15. doi: 10.1016/j.antiviral.2018.11.013
141. Zhang C, Zhang X, Dai W, Liu Q, Xiong P, Wang S, et al. A mouse model of enterovirus D68 infection for assessment of the efficacy of inactivated vaccine. Viruses (2018) 10(2):e58. doi: 10.3390/v10020058
142. Messacar K, Asturias EJ, Hixon AM, Van Leer-Buter C, Niesters HGM, Tyler KL, et al. Enterovirus D68 and acute flaccid myelitis-evaluating the evidence for causality. Lancet Infect Dis (2018) 18(8):e239–47. doi: 10.1016/S1473-3099(18)30094-X
143. Bednarski Z, Bednarska H. [First research work by Robert Koch on etiology of anthrax-in cooperation with Jozef Knechtel, Polish apothecary]. Arch Hist Filoz Med (2003) 66(2):161–8.
144. Mishra N, Ng TFF, Marine RL, Jain K, Ng J, Thakkar R, et al. Antibodies to enteroviruses in cerebrospinal fluid of patients with acute flaccid myelitis. mBio (2019) 10(4):e01903-19. doi: 10.1128/mBio.01903-19
145. Schubert RD, Hawes IA, Ramachandran PS, Ramesh A, Crawford ED, Pak JE, et al. Pan-viral serology implicates enteroviruses in acute flaccid myelitis. Nat Med (2019) 25(11):1748–52. doi: 10.1038/s41591-019-0613-1
146. Biggs HM, McNeal M, Nix WA, Kercsmar C, Curns AT, Connelly B, et al. Enterovirus D68 infection among children with medically attended acute respiratory illness, Cincinnati, Ohio, July-October 2014. Clin Infect Dis (2017) 65(2):315–23. doi: 10.1093/cid/cix314
147. Schuster JE, Selvarangan R, Hassan F, Briggs KB, Hays L, Miller JO, et al. Clinical course of enterovirus D68 in hospitalized children. Pediatr Infect Dis J (2017) 36(3):290–5. doi: 10.1097/INF.0000000000001421
148. Waghmare A, Pergam SA, Jerome KR, Englund JA, Boeckh M, Kuypers J. Clinical disease due to enterovirus D68 in adult hematologic Malignancy patients and hematopoietic cell transplant recipients. Blood J Am Soc Hematol (2015) 125(11):1724–9. doi: 10.1182/blood-2014-12-616516
149. Patel MC, Wang W, Pletneva LM, Rajagopala SV, Tan Y, Hartert TV, et al. Enterovirus D-68 infection, prophylaxis, and vaccination in a novel permissive animal model, the cotton rat (Sigmodon hispidus). PLoS One (2016) 11(11):e0166336. doi: 10.1371/journal.pone.0166336
150. Zheng HW, Sun M, Guo L, Wang JJ, Song J, Li JQ, et al. Nasal Infection of Enterovirus D68 Leading to Lower Respiratory Tract Pathogenesis in Ferrets (Mustela putorius furo). Viruses (2017) 9(5). doi: 10.3390/v9050104
151. Essaidi-Laziosi M, Brito F, Benaoudia S, Royston L, Cagno V, Fernandes-Rocha M, et al. Propagation of respiratory viruses in human airway epithelia reveals persistent virus-specific signatures. J Allergy Clin Immunol (2018) 141(6):2074–84. doi: 10.1016/j.jaci.2017.07.018
152. Evans WJ, Hurst BL, Peterson CJ, Van Wettere AJ, Day CW, Smee DF, et al. Development of a respiratory disease model for enterovirus D68 in 4-week-old mice for evaluation of antiviral therapies. Antiviral Res (2019) 162:61–70. doi: 10.1016/j.antiviral.2018.11.012
153. Rajput C, Han M, Bentley JK, Lei J, Ishikawa T, Wu Q, et al. Enterovirus D68 infection induces IL-17-dependent neutrophilic airway inflammation and hyperresponsiveness. JCI Insight (2018) 3(16):e121882. doi: 10.1172/jci.insight.121882
154. Kang J, Pang Z, Zhou Z, Li X, Liu S, Cheng J, et al. Enterovirus D68 protease 2A(pro) targets TRAF3 to subvert host innate immune responses. J Virol (2021) 95(3):e01856-20. doi: 10.1128/JVI.01856-20
155. Rui Y, Su J, Wang H, Chang J, Wang S, Zheng W, et al. Disruption of MDA5-mediated innate immune responses by the 3C proteins of coxsackievirus A16, coxsackievirus A6, and enterovirus D68. J Virol (2017) 91(13):e00546-17. doi: 10.1128/JVI.00546-17
156. Xiang Z, Liu L, Lei X, Zhou Z, He B, Wang J, et al. 3C protease of enterovirus D68 inhibits cellular defense mediated by interferon regulatory factor 7. J Virol (2016) 90(3):1613–21. doi: 10.1128/JVI.02395-15
157. Chong PF, Kira R, Mori H, Okumura A, Torisu H, Yasumoto S, et al. Clinical features of acute flaccid myelitis temporally associated with an enterovirus D68 outbreak: results of a nationwide survey of acute flaccid paralysis in Japan, August–December 2015. Clin Infect Dis (2018) 66(5):653–64. doi: 10.1093/cid/cix860
158. Imamura T, Suzuki A, Lupisan S, Kamigaki T, Okamoto M, Roy CN, et al. Detection of enterovirus 68 in serum from pediatric patients with pneumonia and their clinical outcomes. Influenza other Respir viruses (2014) 8(1):21–4. doi: 10.1111/irv.12206
159. Kusabe Y, Takeshima A, Seino A, Nishida M, Takahashi M, Yamada S, et al. An adult case of enterovirus D68 encephalomyelitis presenting as bilateral facial nerve palsy and dysphagia. Brain Nerve= Shinkei Kenkyu no Shinpo (2017) 69(8):957–61. doi: 10.11477/mf.1416200848
160. Fall A, Ndiaye N, Jallow MM, Barry MA, Toure CSB, Kebe O, et al. Enterovirus D68 subclade B3 circulation in Senegal, 2016: detection from influenza-like illness and acute flaccid paralysis surveillance. Sci Rep (2019) 9(1):13881. doi: 10.1038/s41598-019-50470-z
161. Williams CJ, Thomas RH, Pickersgill TP, Lyons M, Lowe G, Stiff RE, et al. Cluster of atypical adult Guillain-Barré syndrome temporally associated with neurological illness due to EV-D68 in children, South Wales, United Kingdom, October 2015 to January 2016. Eurosurveillance (2016) 21(4):30119.
162. Guo H, Li Y, Liu G, Jiang Y, Shen S, Bi R, et al. A second open reading frame in human enterovirus determines viral replication in intestinal epithelial cells. Nat Commun (2019) 10(1):4066. doi: 10.1038/s41467-019-12040-9
163. Zheng H, Wang J, Li B, Guo L, Li H, Song J, et al. A novel neutralizing antibody specific to the DE loop of VP1 can inhibit EV-D68 infection in mice. J Immunol (2018) 201(9):2557–69. doi: 10.4049/jimmunol.1800655
164. Antona D, Kossorotoff M, Schuffenecker I, Mirand A, Leruez-Ville M, Bassi C, et al. Severe paediatric conditions linked with EV-A71 and EV-D68, France, May to October 2016. Eurosurveillance (2016) 21(46):30402. doi: 10.2807/1560-7917.ES.2016.21.46.30402
165. Barnadas C, Midgley SE, Skov MN, Jensen L, Poulsen MW, Fischer TK. An enhanced Enterovirus surveillance system allows identification and characterization of rare and emerging respiratory enteroviruses in Denmark, 2015-16. J Clin Virol (2017) 93:40–4. doi: 10.1016/j.jcv.2017.05.017
166. Chang TH, Yang TI, Hsu WY, Huang LM, Chang LY, Lu CY. Case report: painful exanthems caused by enterovirus D68 in an adolescent. Med (Baltimore) (2019) 98(33):e16493. doi: 10.1097/MD.0000000000016493
167. Nelson GR, Bonkowsky JL, Doll E, Green M, Hedlund GL, Moore KR, et al. Recognition and management of acute flaccid myelitis in children. Pediatr Neurol (2016) 55:17–21. doi: 10.1016/j.pediatrneurol.2015.10.007
168. Maloney JA, Mirsky DM, Messacar K, Dominguez SR, Schreiner T, Stence NV. MRI findings in children with acute flaccid paralysis and cranial nerve dysfunction occurring during the 2014 enterovirus D68 outbreak. AJNR Am J Neuroradiol (2015) 36(2):245–50. doi: 10.3174/ajnr.A4188
169. Esposito S, Lunghi G, Zampiero A, Tagliabue C, Orlandi A, Torresani E, et al. Enterovirus-D68 in the cerebrospinal fluid of two children with aseptic meningitis. Pediatr Infect Dis J (2016) 35(5):589–91. doi: 10.1097/INF.0000000000001085
170. Giombini E, Rueca M, Barberi W, Iori AP, Castilletti C, Scognamiglio P, et al. Enterovirus D68-associated acute flaccid myelitis in immunocompromised woman, Italy. Emerg Infect Dis (2017) 23(10):1690–3. doi: 10.3201/eid2310.170792
171. Wali RK, Lee AH, Kam JC, Jonsson J, Thatcher A, Poretz D, et al. Acute neurological illness in a kidney transplant recipient following infection with enterovirus-D68: an emerging infection? Am J Transplant (2015) 15(12):3224–8. doi: 10.1111/ajt.13398
172. Ruggieri V, Paz MI, Peretti MG, Rugilo C, Bologna R, Freire C, et al. Enterovirus D68 infection in a cluster of children with acute flaccid myelitis, Buenos Aires, Argentina, 2016. Eur J Paediatr Neurol (2017) 21(6):884–90. doi: 10.1016/j.ejpn.2017.07.008
173. Greenberg B, Plumb P, Cutter G, Dean J, Desena A, Hopkins S, et al. Acute flaccid myelitis: long-term outcomes recorded in the CAPTURE study compared with paediatric transverse myelitis. BMJ Neurol Open (2021) 3(1):e000127. doi: 10.1136/bmjno-2021-000127
174. Coyne CB, Kim KS, Bergelson JM. Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. EMBO J (2007) 26(17):4016–28. doi: 10.1038/sj.emboj.7601831
175. Ohka S, Nihei C, Yamazaki M, Nomoto A. Poliovirus trafficking toward central nervous system via human poliovirus receptor-dependent and -independent pathway. Front Microbiol (2012) 3:147. doi: 10.3389/fmicb.2012.00147
176. Sooksawasdi Na Ayudhya S, Meijer A, Bauer L, Oude Munnink B, Embregts C, Leijten L, et al. Enhanced enterovirus D68 replication in neuroblastoma cells is associated with a cell culture-adaptive amino acid substitution in VP1. mSphere (2020) 5(6):e00941-20. doi: 10.1128/mSphere.01147-20
177. Tseligka ED, Sobo K, Stoppini L, Cagno V, Abdul F, Piuz I, et al. A VP1 mutation acquired during an enterovirus 71 disseminated infection confers heparan sulfate binding ability and modulates ex vivo tropism. PLoS Pathog (2018) 14(8):e1007190. doi: 10.1371/journal.ppat.1007190
178. Triantafilou K, Orthopoulos G, Vakakis E, Ahmed MA, Golenbock DT, Lepper PM, et al. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell Microbiol (2005) 7(8):1117–26. doi: 10.1111/j.1462-5822.2005.00537.x
179. Richer MJ, Lavallee DJ, Shanina I, Horwitz MS. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PLoS One (2009) 4(1):e4127. doi: 10.1371/journal.pone.0004127
180. Negishi H, Osawa T, Ogami K, Ouyang X, Sakaguchi S, Koshiba R, et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc Natl Acad Sci U S A (2008) 105(51):20446–51. doi: 10.1073/pnas.0810372105
181. Abston ED, Coronado MJ, Bucek A, Onyimba JA, Brandt JE, Frisancho JA, et al. TLR3 deficiency induces chronic inflammatory cardiomyopathy in resistant mice following coxsackievirus B3 infection: role for IL-4. Am J Physiol Regul Integr Comp Physiol (2013) 304(4):R267–77. doi: 10.1152/ajpregu.00516.2011
182. Abe Y, Fujii K, Nagata N, Takeuchi O, Akira S, Oshiumi H, et al. The toll-like receptor 3-mediated antiviral response is important for protection against poliovirus infection in poliovirus receptor transgenic mice. J Virol (2012) 86(1):185–94. doi: 10.1128/JVI.05245-11
183. Hsiao HB, Chou AH, Lin SI, Chen IH, Lien SP, Liu CC, et al. Toll-like receptor 9-mediated protection of enterovirus 71 infection in mice is due to the release of danger-associated molecular patterns. J Virol (2014) 88(20):11658–70. doi: 10.1128/JVI.00867-14
184. Kuo RL, Kao LT, Lin SJ, Wang RY, Shih SR. MDA5 plays a crucial role in enterovirus 71 RNA-mediated IRF3 activation. PLoS One (2013) 8(5):e63431. doi: 10.1371/journal.pone.0063431
185. Feng Q, Hato SV, Langereis MA, Zoll J, Virgen-Slane R, Peisley A. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep (2012) 2(5):1187–96. doi: 10.1016/j.celrep.2012.10.005
186. Garcia G Jr., Irudayam JI, Jeyachandran AV, Dubey S, Chang C, Castillo Cario S, et al. Innate immune pathway modulator screen identifies STING pathway activation as a strategy to inhibit multiple families of arbo and respiratory viruses. Cell Rep Med (2023) 4(5):101024. doi: 10.1016/j.xcrm.2023.101024
187. Zheng W, Zhou Z, Rui Y, Ye R, Xia F, Guo F, et al. TRAF3 activates STING-mediated suppression of EV-A71 and target of viral evasion. Signal Transduct Target Ther (2023) 8(1):79. doi: 10.1038/s41392-022-01287-2
188. Yang Z, Zheng H, Li H, Chen Y, Hou D, Fan Q, et al. The expression of IFN-beta is suppressed by the viral 3D polymerase via its impact on PGAM5 expression during enterovirus D68 infection. Virus Res (2021) 304:198549. doi: 10.1016/j.virusres.2021.198549
189. Zhang Y, Xu L, Zhang Z, Su X, Wang Z, Wang T, et al. Enterovirus D68 infection upregulates SOCS3 expression to inhibit JAK-STAT3 signaling and antagonize the innate interferon response of the host. Virol Sin (2023) 38(5):755–66. doi: 10.1016/j.virs.2023.08.007
190. Visser LJ, Langereis MA, Rabouw HH, Wahedi M, Muntjewerff EM, de Groot RJ, et al. Essential role of enterovirus 2A protease in counteracting stress granule formation and the induction of type I interferon. J Virol (2019) 93(10):e00222-19. doi: 10.1128/JVI.00222-19
191. Xiao H, Li J, Yang X, Li Z, Wang Y, Rui Y, et al. Ectopic expression of TRIM25 restores RIG-I expression and IFN production reduced by multiple enteroviruses 3C(pro). Virol Sin (2021) 36(6):1363–74. doi: 10.1007/s12250-021-00410-x
192. Xiang Z, Li L, Lei X, Zhou H, Zhou Z, He B, et al. Enterovirus 68 3C protease cleaves TRIF to attenuate antiviral responses mediated by Toll-like receptor 3. J Virol (2014) 88(12):6650–9. doi: 10.1128/JVI.03138-13
193. Harrison CJ, Weldon WC, Pahud BA, Jackson MA, Oberste MS, Selvarangan R, et al. Neutralizing Antibody against Enterovirus D68 in Children and Adults before 2014 Outbreak, Kansas City, Missouri, USA(1). Emerg Infect Dis (2019) 25(3):585–8. doi: 10.3201/eid2503.180960
194. Lee JT, Shih WL, Yen TY, Cheng AL, Lu CY, Chang LY, et al. Enterovirus D68 seroepidemiology in Taiwan, a cross sectional study from 2017. PLoS One (2020) 15(3):e0230180. doi: 10.1371/journal.pone.0230180
195. Rosenfeld AB, Shen EQL, Melendez M, Mishra N, Lipkin WI, Racaniello VR. Cross-reactive antibody responses against nonpoliovirus enteroviruses. mBio (2022) 13(1):e0366021. doi: 10.1128/mbio.03660-21
196. Fang Y, Chen Q, Wang H, Wang L, Rong H, Liao Q, et al. The role of conformational epitopes in the evolutionary divergence of enterovirus D68 clades: A bioinformatics-based study. Infect Genet Evol (2021) 93:104992. doi: 10.1016/j.meegid.2021.104992
197. Vogt MR, Fu J, Kose N, Williamson LE, Bombardi R, Setliff I, et al. Human antibodies neutralize enterovirus D68 and protect against infection and paralytic disease. Sci Immunol (2020) 5(49):eaba4902. doi: 10.1126/sciimmunol.aba4902
198. Grifoni A, Mahajan S, Sidney J, Martini S, Scheuermann RH, Peters B, et al. A survey of known immune epitopes in the enteroviruses strains associated with acute flaccid myelitis. Hum Immunol (2019) 80(11):923–9. doi: 10.1016/j.humimm.2019.08.004
199. Hixon AM, Clarke P, Tyler KL. Evaluating treatment efficacy in a mouse model of enterovirus D68-associated paralytic myelitis. J Infect Dis (2017) 216(10):1245–53. doi: 10.1093/infdis/jix468
200. Rhoden E, Zhang M, Nix WA, Oberste MS. In vitro efficacy of antiviral compounds against enterovirus D68. Antimicrob Agents Chemother (2015) 59(12):7779–81. doi: 10.1128/AAC.00766-15
201. Messacar K, Sillau S, Hopkins SE, Otten C, Wilson-Murphy M, Wong B, et al. Safety, tolerability, and efficacy of fluoxetine as an antiviral for acute flaccid myelitis. Neurology (2019) 92(18):e2118–26. doi: 10.1212/WNL.0000000000006670
202. Hu Y, Musharrafieh R, Zheng M, Wang J. Enterovirus D68 antivirals: past, present, and future. ACS Infect Dis (2020) 6(7):1572–86. doi: 10.1021/acsinfecdis.0c00120
203. Lin YL, Cheng PY, Chin CL, Chuang KT, Lin JY, Chang N, et al. A novel mucosal bivalent vaccine of EV-A71/EV-D68 adjuvanted with polysaccharides from Ganoderma lucidum protects mice against EV-A71 and EV-D68 lethal challenge. J BioMed Sci (2023) 30(1):96. doi: 10.1186/s12929-023-00987-3
204. Zhang C, Zhang X, Zhang W, Dai W, Xie J, Ye L, et al. Enterovirus D68 virus-like particles expressed in Pichia pastoris potently induce neutralizing antibody responses and confer protection against lethal viral infection in mice. Emerg Microbes Infect (2018) 7(1):3. doi: 10.1038/s41426-017-0005-x
Keywords: enterovirus, EV-D68, acute flaccid myelitis, respiratory distress, children
Citation: Grizer CS, Messacar K and Mattapallil JJ (2024) Enterovirus-D68 – a reemerging non-polio enterovirus that causes severe respiratory and neurological disease in children. Front. Virol. 4:1328457. doi: 10.3389/fviro.2024.1328457
Received: 19 November 2023; Accepted: 26 January 2024;
Published: 15 February 2024.
Edited by:
Yoon-Seok Chung, Korea Disease Control and Prevention Agencyorea, Republic of KoreaReviewed by:
Pedro A. Piedra, Baylor College of Medicine, United StatesJun Wang, Rutgers, The State University of New Jersey, United States
Copyright © 2024 Grizer, Messacar and Mattapallil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph J. Mattapallil, am9zZXBoLm1hdHRhcGFsbGlsQHVzdWhzLmVkdQ==