 Michael Jonathan
Michael Jonathan Terumasa Ikeda
Terumasa Ikeda- 1Division of Molecular Virology and Genetics, Joint Research Center for Human Retrovirus Infection, Kumamoto University, Kumamoto, Japan
- 2Graduate School of Medical Sciences, Kumamoto University, Kumamoto, Japan
The apolipoprotein B mRNA editing enzyme catalytic polypeptide-like (APOBEC) family consists of cytosine deaminases implicated in diverse and important biological functions. APOBEC3 (A3) proteins belong to the APOBEC/AID family, and they catalyze the deamination of cytosine to uracil in single-stranded DNA and, to a lesser extent, in RNA substrates. In humans, seven A3 genes have been identified (A3A, A3B, A3C, A3D, A3F, A3G, and A3H). The introduction of lethal G-to-A or C-to-U mutations into certain viral genomes leads to virus inactivation. However, the mutagenic capability of A3 proteins could serve as a source of mutations to drive virus evolution. Therefore, recent studies have implied the role of A3 proteins in aiding the evolution of viruses, conferring them with severe manifestations such as drug resistance and/or immune evasion. In this review, we discuss in depth the interactions of A3 proteins with viruses that infect humans and our self-proteins.
1 Introduction
The apolipoprotein B mRNA editing enzyme catalytic polypeptide-like (APOBEC) family comprises cytosine deaminases implicated in diverse and important biological functions. The first member of this group to be characterized was APOBEC1 (A1) (1, 2), which participates in lipid metabolism through the editing of apolipoprotein B mRNA [reviewed in (3–6)]. Later, several A1 proteins in nonhuman species were demonstrated to deaminate single-stranded DNA (ssDNA) (7–9) and exhibit potential to inhibit retroviruses, including human immunodeficiency virus type I (HIV-1), in cell line models (9–13). The second member of this family to be identified was activation-induced cytosine deaminase (AID) (14), known for its importance in the antibody diversification of immunoglobulin genes and class switch recombination [reviewed in (6, 15, 16)]. Other members of this group including APOBEC2 (A2) (17) and APOBEC4 (A4) (18) are less characterized [reviewed in (6)].
APOBEC3 (A3) proteins belong to the APOBEC/AID family. These proteins catalyze the deamination of cytosine to uracil in ssDNA substrates [reviewed in (19–21)]. In humans, seven A3 genes (A3A, A3B, A3C, A3D, A3F, A3G, and A3H) are tandemly arranged into a gene cluster between the flanking genes CBX6 and CBX7 on chromosome 22 [reviewed in (19, 22, 23)] (Figure 1A). A3 genes are the products of arduous evolution over 100 million years (24). The deaminase activity of A3 proteins is attributable to the presence of a conserved zinc-binding motif (Z domain). A3A, A3C, and A3H proteins have a single Z domain, whereas A3B, A3D, A3F, and A3G proteins have double Z domains [reviewed in (19–21, 23, 25)] (Figure 1A). The mechanism of A3 protein-mediated catalysis involves a zinc-mediated hydrolytic reaction (Figure 1B). Histidine and cysteine residues are responsible for the coordination of the zinc ion required for the catalytic activity of these proteins, whereas a glutamate residue facilitates the removal of the amine group from the substrate deoxycytidine [reviewed in (26–28)] (Figure 1B). First, a glutamic acid residue in the Z domain deprotonates water and forms a zinc-stabilized hydroxide ion (Figure 1B). Second, the third nitrogen atom (N3) of the cytosine pyrimidine ring is protonated, leading to the disturbance of a double bond between N3 and the fourth carbon atom (C4) of the cytosine pyrimidine ring (Figure 1B). Third, this causes the zinc-stabilized hydroxide ion to attack C4, forming a double bond and essentially converting cytosine into uracil [reviewed in (26–28)] (Figure 1B). The differences among the A3 proteins also extend to their nucleic acid sequences. The stretch of nucleotides recognized by A3 proteins can be grouped into two types, namely 5′-CC and 5′-TC (the target cytosines are underlined; see Figure 2). The former is recognized by A3G protein, whereas the latter is preferred by other A3 proteins (12, 29–35).
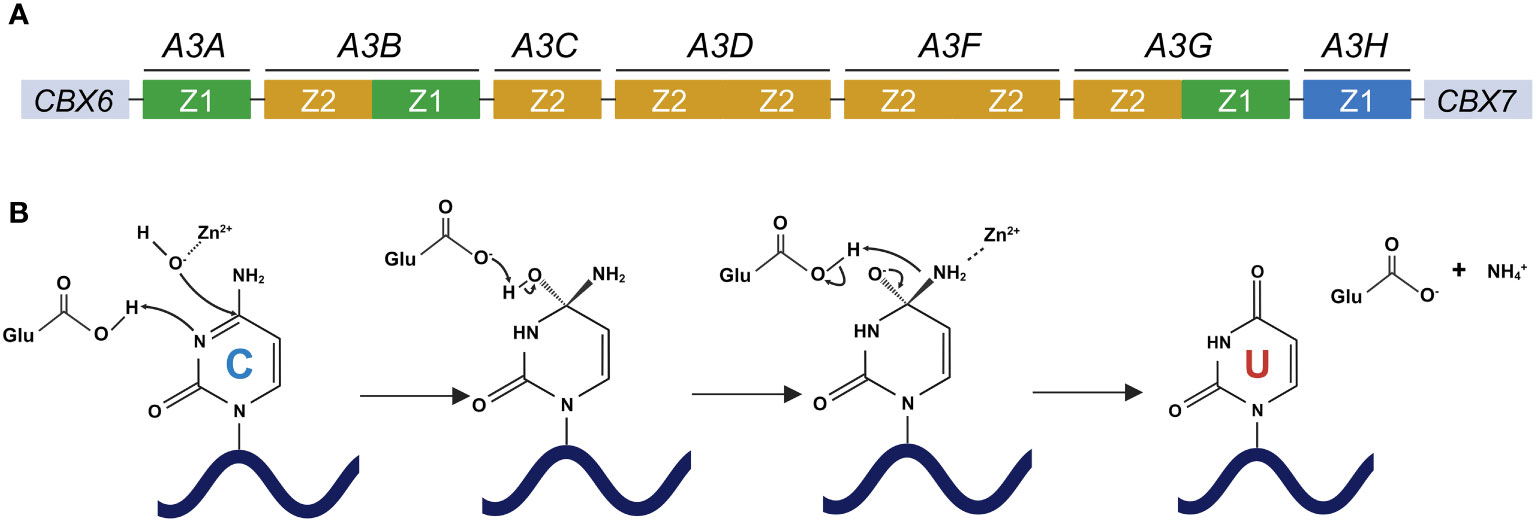
Figure 1 Human A3 gene organization and deamination catalyzed by A3 proteins. (A) Human A3 genes are arranged tandemly between CBX6 and CBX7 on chromosome 22. A3 family proteins include single or double deaminases, which are composed of three phylogenetically different groups, namely Z1 (green), Z2 (orange), and Z3 (blue) domains. (B) The deamination process by which cytosine is converted to uracil by A3 proteins. A glutamic acid residue in the Z domain of A3 proteins deprotonates water and forms a zinc-stabilized hydroxide ion. The third nitrogen atom (N3) of the cytosine pyrimidine ring is protonated, thereby disturbing the double bond between N3 and the fourth carbon atom (C4) of the cytosine pyrimidine ring. This causes the zinc-stabilized hydroxide ion to attack C4, forming a double bond and essentially converting cytosine into uracil. CBX6, chromobox 6; CBX7, chromobox 7; APOBEC3/A3, apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3; A3A, APOBEC3A; A3B, APOBEC3B; A3C, APOBEC3C; A3D, APOBEC3D; A3F, APOBEC3F; A3G, APOBEC3G; A3H, APOBEC3H. Created with BioRender.com.
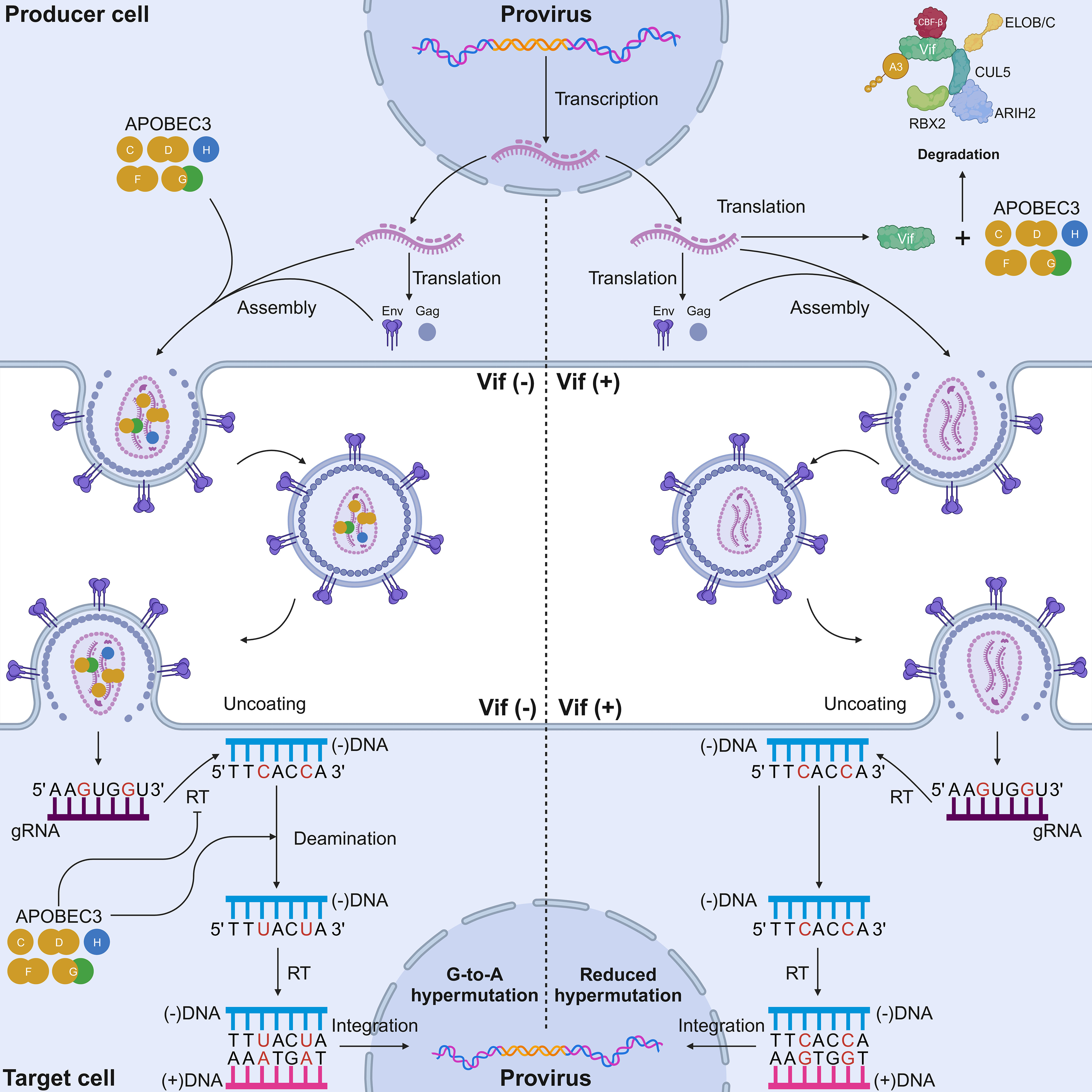
Figure 2 Mechanisms of HIV-1 inhibition by A3 proteins and the counteraction of Vif. In a Vif-deficient virus infection scenario (left), the lack of Vif causes A3 proteins to be packaged into nascent virions. A G-to-A mutation occurs in the single-stranded cDNA intermediate of HIV-1, resulting in deleterious and potentially lethal mutations. A3 proteins have different dinucleotide motifs. A3G prefers 5′-CC-3′ motifs, whereas the other six A3 proteins prefer 5′-TC-3′ motifs (target cytosines are presented in red). A3 proteins also physically inhibit reverse transcription via their deaminase-independent activity to prevent HIV replication. In a Vif-proficient virus infection scenario (right), Vif ubiquitinates A3 proteins for degradation. None or a reduced number of A3 proteins is packaged into nascent virions. Sublethal levels of A3-mediated mutagenesis can occur in the single-stranded cDNA intermediate of HIV-1. RBX2, RING box protein 2; ARIH2, ariadne RBR E3 ubiquitin protein ligase 2; CBF-β, core-binding factor subunit beta; ELOB, elongin B; ELOC, elongin C; Vif, viral infectivity factor; CUL5, cullin-5; A3, apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3; RT, reverse transcriptase; Env, envelope; Gag, group-specific antigen. Created with BioRender.com.
Interestingly, because of their RNA-binding capacity, most A3 proteins are incorporated into ribonucleoprotein complexes in cells immediately after translation. Multiple reports have documented that A3G protein can form higher-order, high molecular mass ribonucleoprotein (HMM RNP) complexes composed of A3G-binding RNAs, A3G-binding proteins, and numerous cellular RNA-binding proteins (36–47). Similarly, the ability to form HMM RNP complexes is conserved in other A3 members (at least in A3B, A3C, A3F, and stable A3H proteins, but not A3A protein) (39, 44, 48, 49). Importantly, this capability inactivates the catalytic activity of A3B, A3G, and A3H proteins, as RNase treatment can restore their enzymatic activity by abrogating HMM complex formation, leading to a shift toward low molecular mass (LMM) complexes (40, 49–52). The shift toward LMM complex formation allows the A3G protein to be packaged into nascent virions for anti-HIV-1 activity (40). Therefore, the formation of A3 HMM RNP complexes (with the exception of A3A) is one of the regulatory mechanisms for the mutagenic activity of A3 proteins, and A3 enzymatic activity is altered by cytokines such as interferons (IFNs) and different cell types (41, 46, 47, 53). Furthermore, differences in each A3 interactor potentially lead to differences in the localization of A3 proteins in cells. A3A and A3C proteins are distributed throughout the cell, whereas A3B protein is localized exclusively to the nucleus (30, 51, 54–62). Notably, endogenous A3A protein in CD14+ primary cells and IFN-stimulated THP-1 cells is localized to the cytoplasm, which is distinct from the overexpressed A3A protein in HEK293 cells (56). This distinct subcellular localization pattern of overexpressed and endogenous A3A protein suggests the existence of regulatory mechanisms for A3A activity and/or provides a potential explanation for its genotoxicity. Meanwhile, A3D, A3F, and A3G proteins are exclusively localized in the cytoplasm (30, 36, 44, 54, 55, 57, 59–61). However, A3G protein was recently reported to localize to the nucleus of bladder cancer cells in vivo (63) and in human T cell lines in small amounts (64, 65). Interestingly, A3H protein has a haplotype-dependent localization in cells: haplotype I is distributed through the cell (55, 66) and haplotype II is present in only distinct subcellular compartments, namely the cytoplasm and nucleolus (30, 50, 66, 67). The distinct subcellular localization of A3H haplotypes may be a regulatory mechanism for the antiviral activity of this protein.
2 A3 proteins as innate antiviral restriction factors
Much of the knowledge about the function of A3 proteins as restriction factors stemmed from studies of retroviruses, with HIV-1 being the most well-documented retrovirus [reviewed in (19–21, 68–70)]. Initially, A3G was identified as a specific inhibitor of viral infectivity factor (Vif)-deficient HIV-1 (71). Thereafter, it was elucidated that this antiviral protein is packaged in nascent virions in virus-producing cells, and it triggers the lethal deamination of cytosine to uracil in the HIV-1 minus-strand cDNA intermediate in target cells (71–73) (Figure 2). Currently, up to five A3 proteins (A3C-I188, A3D, A3F, A3G, and stable A3H haplotypes) contribute to HIV-1 restriction in human CD4+ T cells (30, 32, 34, 74–77).
In addition to their deaminase activity, the antiviral activity of A3 proteins can also be conferred through their deaminase-independent activity (Figure 2). Although the catalytic activity of A3G protein is important for HIV-1 inhibition, the N-terminal domain of this protein has the ability to bind to RNAs, although it lacks any catalytic activity (78–81). Owing to this domain, A3G protein can be packaged in HIV-1 particles, in which it inhibits viral replication through its deaminase-independent activity (78–84). One of the well-known restriction models based on this ability is the “roadblock” model, whereby A3G protein physically blocks the elongation of reverse transcription (82, 83). A3G protein also interacts with HIV-1 reverse transcriptase (RT), thereby preventing reverse transcription (85, 86). Interestingly, the antiviral activity of A3F and A3H proteins has been largely attributed to the deaminase-independent mode (50, 52, 87, 88). An alternative action of A3 proteins is the inhibition of HIV-1 integration. One study highlighted the possibility that A3G protein can interrupt the structural integrity of the HIV-1 pre-integration complex (PIC), as it is reported that A3F and A3G proteins can interact with the integrase protein of HIV-1, thereby inhibiting provirus formation (89). Furthermore, it has been postulated that A3 proteins can interact with host cellular factors through their deaminase-independent activity. Specifically, recent work illustrated that A3B protein can interact with host protein kinase R and RNase L to limit the gene expression of Sendai virus, poliovirus, and Sindbis virus by inactivating host RNA translation in vitro (62). Taken together, the antiviral capability of A3 proteins must be measured through their deaminase and deaminase-independent activities.
3 Arms race between A3 proteins and viruses
Heterogeneity in the number of Z domains carried by A3 genes is predicted to be caused by a gene duplication event (24, 90–93). This event appears to have occurred within a placental mammal ancestor that had already possessed the three types of Z domains (Z1, Z2, and Z3) carried by the present-day human A3 genes (24, 90–93). Each of the human A3 genes is composed of a combination of these domains (Figure 1A). Intriguingly, the copy number of A3 genes varies greatly within the mammalian lineage (24, 90–95), suggesting an arms race between placental mammals and genetic threats including endogenous and exogenous retroviruses [reviewed in (5, 19, 69, 96, 97)]. As up to five A3 proteins (A3C-I188, A3D, A3F, A3G, and stable A3H haplotypes) are involved in HIV-1 restriction in human CD4+ T cells and myeloid cells (30, 32, 34, 35, 74–77, 98–100), A3 enzymes have overlapping functions to protect their hosts from various threats. Therefore, it is plausible that the “fortifications” in genes encoding host restriction factors differ because the interaction between the host and pathogen is independent in each mammal. For instance, the mouse genome encodes at least eight TRIM5α-like proteins (101), whereas the human genome encodes only one (102). By contrast, mice have only one A3 protein (92, 103). Thus, it can be said that the seven copies of A3 genes in humans could be the product of pathogenic pressures imposed on our ancestors.
The same question can also be asked about the pathogens themselves. Because of this “arms race”, if viruses had not coevolved with our ancestors, they would have succumbed to the mutagenic properties of A3 proteins. For example, HIV-1 encodes the Vif protein, which can induce degradation of A3 proteins through a proteasome-mediated pathway (34, 35, 65, 71, 75, 98, 104–110), inhibit A3 mRNA translation (111, 112), and alter the expression of RUNX-regulated genes (113). Vif protein inactivates both deaminase-dependent and deaminase-independent processes. It is hypothesized that HIV-1 Vif evolved to recognize the expansive repertoire of human A3 proteins (114). It is possible that a cross-species jump to primates occurred in an ancestral lentivirus that had originally infected non-primate mammals. As previously explained, mammals have evolved different repertoires of innate restriction factors depending on the individual interactions between themselves and their respective threats. The feline A3 repertoire, containing four genes producing five A3 proteins (114–117), is much simpler than the human A3 repertoire with seven A3 proteins [reviewed in (3, 19, 22, 23)]. The feline lentiviral Vif protein must therefore, have adapted to the A3 repertoire of its new host. This theory is supported by several in vitro reports demonstrating that the simian immunodeficiency virus (SIV) Vif protein is still able to degrade feline A3 proteins (117–119). The mechanism through which lentiviral Vif can adapt to an expanded A3 repertoire is termed the “Wobble model” (120) (Figure 3). A zoonotic event causes a partial interaction between the lentiviral Vif and the A3 repertoire of its new host (Figure 3). Initially, a more expansive A3 repertoire may bind to the same recognition site (Figure 3). Over time, “wobbles” may occur, in which Vif adapts and fortifies its interaction with the A3 proteins of its new host (Figure 3). This adaptation event could result in overlapping interaction surfaces of lentiviral Vif with those of A3 proteins, leading to slight differences arising from this independent adaptation event (Figure 3). Over a longer period, the interaction surface shifts drastically because many independent wobble events occur, leading to the current distinct interaction surface between Vif and various human A3 proteins (Figure 3).
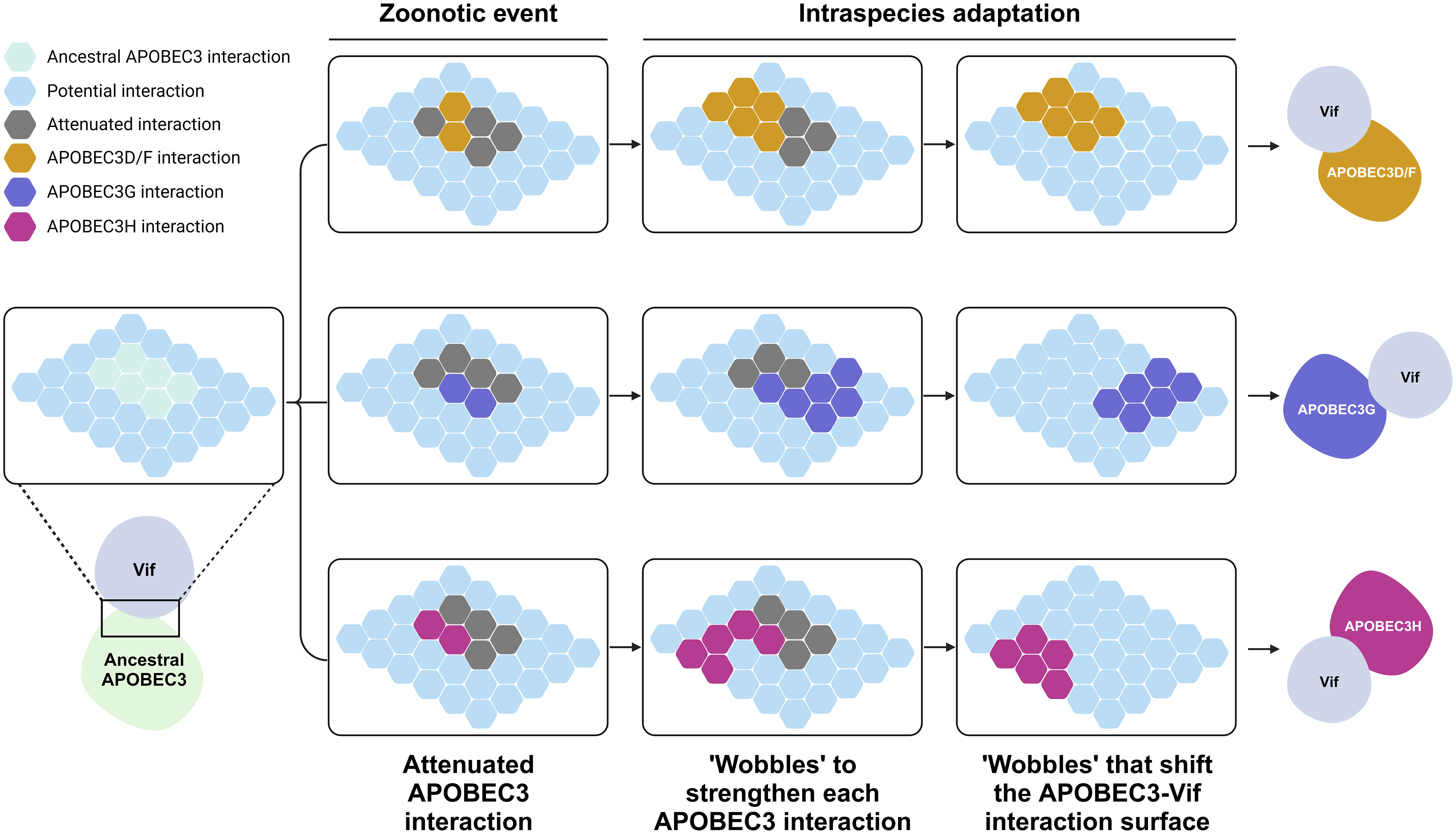
Figure 3 Evolution of the interaction between A3 proteins and lentiviral Vif. This schematic is adopted from (120). Each hexagon tiles represent the binding surface of an A3 protein and Vif. Initially, an ancestral A3 protein bound strongly to an ancient lentiviral Vif protein. A cross-species jump to humans occurred, forcing protein to adapt to the wide repertoire of A3 proteins in humans. This resulted in a diminished yet overlapping interaction between Vif and the new repertoire of A3 proteins. Over an evolutionary period, intraspecies adaptation occurred whereby Vif further adapted to its new host, creating wobbles that shifted the binding interface of Vif, resulting in the nonoverlapping interaction surface presently observed between Vif and human A3 proteins. A3, apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3; Vif, viral infectivity factor. Created with BioRender.com.
The interaction between HIV-1 Vif and the human transcription factor CBF-β (105, 121) is another example of how a virus adapts to a new host. Vif forms an E3 ubiquitin ligase complex consisting of the transcriptional cofactors CBF-β, CUL5, ELOB, ELOC, RBX2, and ARIH2, which allows the viral protein to interact with A3 proteins and induce polyubiquitination, leading to degradation by the 26S proteasome (105, 107, 121–126) (Figure 2). The interaction between Vif and CBF-β was reported to be specific only to HIV-1 and SIVs and not essential for non-primate lentiviruses in vitro (127–131). Generally, CBF-β forms heterodimers with RUNX transcription factors to regulate the expression of genes important for hematopoiesis, T-cell development, osteogenesis, and neurogenesis [reviewed in (132–134)]. It has been reported that this recruitment of CBF-β to the E3 ubiquitin ligase complex impacts the heterodimerization of CBF-β with the transcription factor RUNX1 and consequently represses the transcription of genes regulated by RUNX1, one of which is T-bet, a repressor of IL-2 expression (113).
Vif is also reported to be able to interfere with A3G protein translation. A short conserved upstream open reading frame (uORF) located within two secondary structures of A3G mRNA is reported to be a negative regulator of A3G protein translation and is crucial for inhibition of translation mediated by Vif (111, 112). HIV-1 Vif requires the presence of this uORF in A3G mRNA to inhibit A3G protein translation (presumably by inhibiting the leaky scanning mechanism of A3G mRNAs) and redirect A3G mRNAs to stress granules; both these actions reduce global A3G mRNA translation (112).
Therefore, it can be said that primate lentiviral Vif has evolved multiple ways to adapt to its new host after presumed cross-species transmission from non-primates. First, primate lentivirus hijacks CBF-β to degrade primate A3 proteins and, coincidentally, perturbs the expression of RUNX-stimulated target genes, presumably also for the benefit of the virus. Second, HIV-1 Vif is able to inhibit the translation of A3G mRNAs to prevent (or at least reduce) the packaging of A3 proteins to nascent virions. Collectively, A3 proteins have roles in exerting selection pressure on viruses, thereby facilitating the evolution of the virus in some cases, as will be discussed in the upcoming sections of this review.
4 Implications of A3 proteins as drivers of retrovirus evolution
Retroviruses belong to group VI of the Baltimore classification (135). The genome of retroviruses consists of a homodimer of linear, positive-sense, single-stranded RNA of 7–13 kb in length (136). Unlike most other viruses, retroviruses use an obligate reverse transcription process to convert viral genomic RNA (gRNA) into double-stranded DNA (dsDNA) with long terminal repeats (LTRs). This dsDNA is then integrated into the host genome, resulting in the formation of a provirus, which serves as a template for the transcription of viral RNAs for gRNA and viral protein production (137). During reverse transcription, an ssDNA intermediate is produced. A3 proteins target this ssDNA intermediate as a substrate. In this section, we discuss the implications of A3 proteins as evolutionary drivers of human retroviruses.
4.1 HIV-1
HIV-1 cDNA is the most well-characterized substrate for A3 proteins. A3 proteins induce C-to-U mutations in reverse-transcription ssDNA intermediates, resulting in G-to-A mutations on the genomic strand (Figure 2). These G-to-A mutations can affect more than 10% of all G residues within HIV-1 gRNA, causing an abortive infection attributable to the lethal level of mutations in the HIV-1 genome (73, 138–141). HIV-1 counteracts the antiviral activity of A3 proteins through Vif (Figure 2). Studies examining samples from patients infected with HIV-1 revealed that A3 proteins form a distinct twin gradient of G-to-A hypermutation (142, 143). This gradient is well aligned with the amount of time that the minus-strand DNA remains single-stranded during reverse transcription. The frequency of G-to-A mutations increases away from the primer binding site, reaching a maximum upstream of the 5′-central polypurine tract (cPPT) half-way along the provirus, then reducing significantly, increasing when moving away from the 5′-cPPT and then reaching a maximum upstream of the 3′-PPT of the HIV-1 3′-LTR (142, 143).
An interesting question is whether the mutagenic capability of A3 proteins can aid in virus evolution. Regarding this question, it must first be stressed that G-to-A hypermutation induced by A3 proteins can be beneficial or deleterious for the virus and the host. The inherent error-prone replication of virus genomic material allows viruses to diversify themselves, permitting the existence of variants that are more fit for replication, including those that can evade the immune system, such as what we currently see in the situation regarding severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) (144–151), and those that become resistant to antiviral medications, such as influenza A virus [reviewed in (152)]. This also appears to be the case with A3 proteins. A possible treatment strategy for HIV-1 that focuses on the interplay between A3 and Vif could revolve around the modulation of A3 protein-mediated hypermutation. The theory was predicated on a previous in vitro study that demonstrated a proof-of-concept of targeting the A3G–Vif axis to tip the balance in favor of HIV inhibition (153) (Figures 4A, B). In this model, A3-mediated hypermutation (or hypomutation) can be utilized to combat HIV-1 infection (Figure 4). However, a niche between these two extremities that is beneficial for HIV-1 exists because of the existence of a threshold whereby HIV-1 can garner benefits from the mutagenic property of A3 proteins if the action of these proteins is counterbalanced stably by Vif to achieve a certain level of mutagenesis (154–157) (Figures 4A, B). Conversely, the extremities of the A3 mutagenic axes (hypermutation and hypomutation) are deleterious for HIV-1 [reviewed in (28, 158, 159)] (Figures 4A, C, D). For instance, hypomutagenic therapy might work under the assumption that A3 mutagenesis is important and substantial toward the diversification of HIV-1 genome. The idea is that by eliminating this mutagenic potential, HIV-1 cannot sufficiently diversify, allowing the elimination of the virus through the adaptive immune response and antiviral drugs. The premise of the opposite side of this strategy, i.e., hypermutation, is that by modulating A3 activity to induce high levels of mutagenesis, the lethal consequence would eventually eliminate HIV-1 replication, as demonstrated in vitro by small-molecule drugs that can upregulate the activity of A3 proteins, resulting in HIV-1 inhibition (153, 160–166). Hypermutated viral cDNA would be subjected to degradation before integration (167). Even if the integration is successful, the cDNA would contain large numbers of missense and nonsense mutations. These proviral DNAs are assumed to be nonreproductive because no progenies or noninfectious viruses will be produced (168–171).
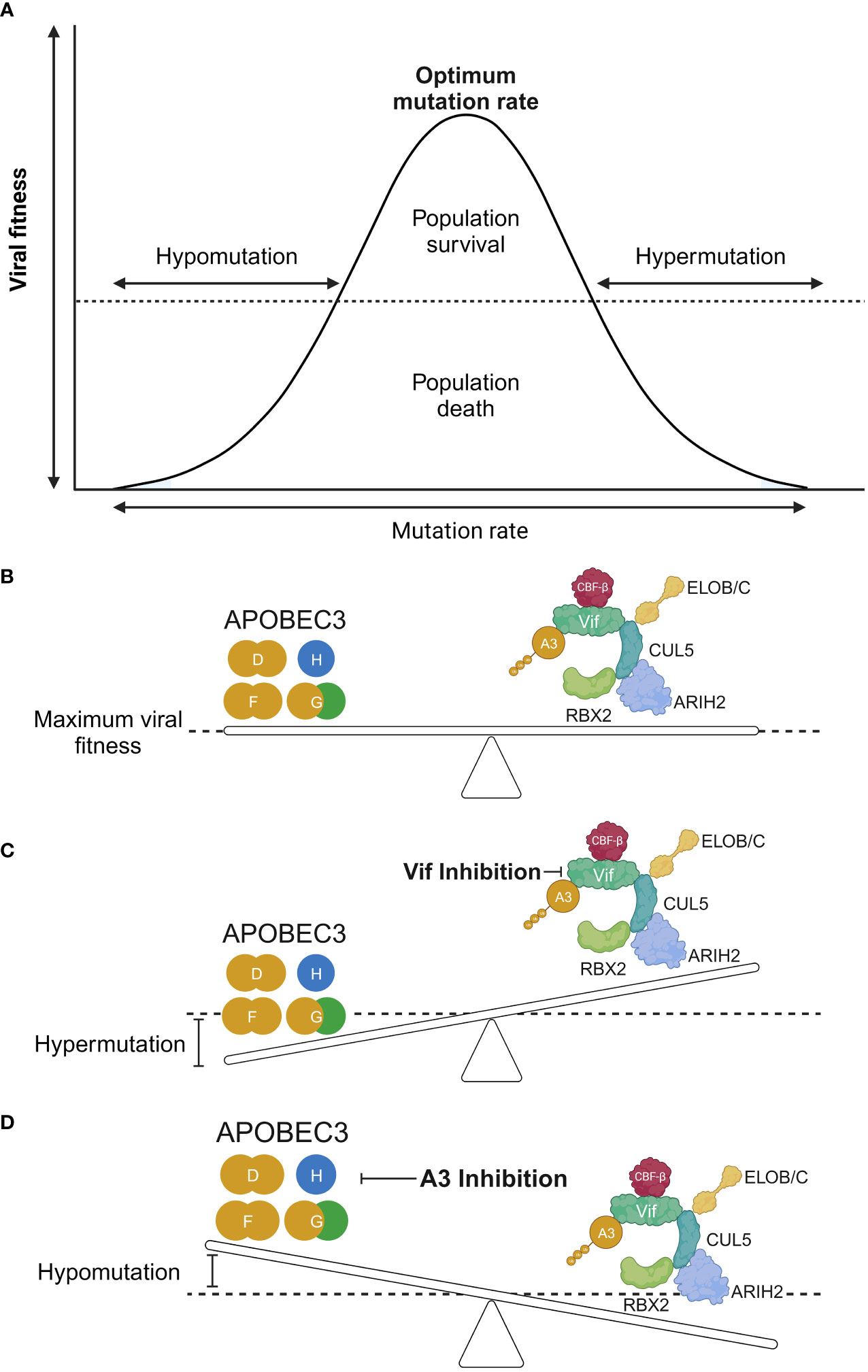
Figure 4 The hypomutation and hypermutation activities of A3 proteins. (A, B) The interplay between HIV-1 Vif and A3 proteins is an interaction between viral fitness and the mutation rate. An equilibrium between these two axes results in an optimal mutation rate for HIV-1 through the use of A3 proteins as additional sources of mutagenesis. This will achieve a maximum viral fitness greatly exceeding the “dead zone” depicted as a horizontal dotted line along the graph. (C) Increased activity of A3 proteins results in hypermutation of the HIV-1 genome, leading to loss of the genome in the population. (D) Conversely, it is argued that reduced A3 protein activity results in a less diversified HIV-1 genome, causing it to be more easily eliminated by the adaptive immune response, provided that A3 proteins are important sources of mutagenesis for HIV-1 diversification. RBX2, RING box protein 2; ARIH2, ariadne RBR E3 ubiquitin protein ligase 2; CBF-β, core-binding factor subunit beta; ELOB, elongin B; ELOC, elongin C; Vif, viral infectivity factor; CUL5, cullin-5; A3, apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3. Created with BioRender.com.
We discussed that A3 can exert mutagenic pressure and that a balance exists between the actions of A3 and Vif (Figure 4). This is because the degradation of A3 proteins by Vif is not absolute, meaning that sublethal mutagenesis can possibly be utilized by HIV-1 to diversify its genome (154, 156, 157, 172, 173). Clinical studies reported that a considerable number of patients infected with HIV-1 at different clinical stages harbored abundant G-to-A hypermutation in proviral DNA (169–171, 174–181), further suggesting that Vif does not eliminate A3 function completely.
Because A3 proteins are mutagens for HIV-1, their potential to confer drug resistance should also be considered. The A3G protein is reported to be a source of genetic variation that confers resistance to 2′,3′-dideoxy-3′-thiacytidine (3TC) in vitro and in vivo (154, 155). This observation is further supported by an ex vivo study using peripheral blood mononuclear cells (PBMCs) obtained from healthy donors (156). Naturally occurring HIV-1 Vif point mutants with suboptimal anti-A3G activity induce the development of proviruses with 3TC resistance and these cytosine deamination mutation events are detected in more than 40% of proviruses with partially defective Vif (156). This partially Vif-defective phenotype can outcompete wild-type virus growth, implying that G-to-A hypermutation alone can alter the phenotype of a viral population. An in vivo study using humanized mice found that an isogenic HIV-1 molecular clone with Vif 45G (HIV-45G) had lower replication over time than the wild-type HIV-1 (NL4-3) (155). However, when these mice were treated with 3TC, treatment failure occurred in 91% of mice infected with HIV-45G, which was in clear contrast to the treatment failure of 36% in mice infected with wild-type HIV-1 (155). The mutations attributed to 3TC resistance are M184I (ATG-to-ATA) and M184V (ATG-to-GTG) (155). Although both of these mutations can result from reverse transcription errors, the M184I mutation contains a dinucleotide motif preferred by A3G (ATGG-to-ATAG) (155). Differentiating G-to-A mutations that are derived from either A3 proteins or RT in this mutation is difficult because both can induce G-to-A mutations. However, it is noteworthy that A3G-mediated mutations significantly increase the incidence of 3TC-resistant mutations in humanized mice (155). This agrees with an in vitro study demonstrating that the mutation rate of A3 proteins is higher than that of HIV-1 RT (182, 183). Specific patterns can be discerned regarding G-to-A mutations derived from A3 proteins and HIV-1 RT. A study of PBMCs from patients infected with HIV revealed that A3D/F/G/H-derived mutations introduced more stop codons than HIV-1 RT-derived mutations (181). In addition, in vitro studies found that HIV-1 RT was biased to the introduction of A-to-G mutations (183, 184), which is fundamentally different to the exclusive G-to-A mutations introduced by A3 proteins. As expected, the result of this difference is that the hypermutation index (A3 protein-mediated G-to-A mutation/100bp) is calculated by subtracting it from the A-to-G mutation rate to correct for background mutation by HIV-1 RT (143).
An in vitro study suggested a role for A3 proteins in modulating, but not necessarily conferring, drug resistance (185). It has been argued that A3 proteins have a role in enhancing resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs) by inducing a V179I substitution, which arises from a G-to-A mutation (185). In and of itself, V179I does not confer drug resistance. However, when present with the NNRTI resistance mutation Y181C/V, NNRTI resistance is increased by 3-to-8 fold. In addition, the frequency of V179I substitution is higher in CD4+ T cells that express A3F and A3G proteins (185).
A clinical study found that A3G-mediated hypermutation was significantly associated with antiretroviral therapy (ART) failure in Indian patients infected with HIV-1 subtype C (186). However, it should be noted that A3G-mediated hypermutation was only observed in the provirus; no hypermutation was recorded in the plasma HIV RNA of the patients tested (186). This agrees with an in vitro study demonstrating that A3G induces purifying selection that will eventually select HIV-1 variants with no hypermutation in their RNA genome that can still produce virions (187). Furthermore, in vitro and in silico studies support the proposition that A3-mediated hypermutation is an “all or nothing” phenomenon because the production of proviruses with A3G-induced sublethal hypermutation (proviruses with low levels of hypermutation yet bearing no stop codons) is statistically very unlikely (188).
Based on the aforementioned clinical study, drug-resistance phenotype derived from A3G-mediated hypermutation was not detected in plasma HIV-1 RNA because it also harbors stop codons due to the deaminating action of A3G in tryptophan codons (TGG-to-TGA/TAA/TAG) (186), further supporting the “all or nothing” phenomenon of A3G-mediated hypermutation. Therefore, it is unlikely that HIV-1 will continue to develop A3-related mutations under effective ART treatment because a clinical study found no G-to-A hypermutation on plasma HIV-1 RNA in patients with successful ART (186).
Another interesting aspect that A3 proteins can influence is the cytotoxic T lymphocyte (CTL) response against HIV-1 (Figure 5). A3 proteins can introduce changes in the HIV-1 provirus, inadvertently changing the amino acid sequence of a given viral protein (Figure 5A). Therefore, one might also assume that this change will eventually accumulate and generate epitopes that can avoid HIV-1–specific CTL responses. This is supported by a previous in vitro report illustrating the inherent trait of A3 proteins as restriction factors that eventually select variants bearing lesser (sublethal) mutations in their genomes because heavily edited genomes would not survive in the population (187). These variants with minor changes in their genomes could result in overall reduced detection by our adaptive immune response. Along these lines, it was reported that A3F/G-induced mutations on common CTL epitopes cause a reduction in HIV-1–specific CTL responses ex vivo (189). Meanwhile, another ex vivo study revealed that hypermutation can facilitate detection by CTLs (190) (Figure 5B). Defective proviruses that were heavily edited by A3G protein could produce truncated proteins that can be sources of antigens to CTLs (190). Newly synthesized and rapidly degraded polypeptides [defective ribosomal products (DRiPs)] are major sources of epitopes for major histocompatibility complex (MHC) class I (191). DRiPs are polypeptides with errors in transcription, translation, or post-translational processes [reviewed in (192)]. A previous study demonstrated that targeting HIV-1 group-specific antigen protein (Gag) to the DRiP pathway boosts MHC class I antigen presentation and CD8+ T-cell activation in vivo (193). This further demonstrates that A3 proteins have a role in stimulating the adaptive immune response by providing another source of viral antigens for recognition.
Figure 5 Effects of A3 proteins on the CTL response against HIV-1 infection. (A) Sublethal mutations mediated by A3F and A3G proteins lead to reduced or even ablated recognition by the CTL because of small changes in the epitope sequence caused by A3F and A3G proteins, depicted by the change of the epitope color and shape. (B) Lethal levels of G-to-A mutations mediated by A3F and A3G proteins improve the MHC class I antigen presentation pathway, providing the adaptive immune system with an additional source of antigen, possibly improving detection by the adaptive immune system, as depicted by the increase of MHC class I expression with varying epitope shapes. A3, apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3; APC, antigen-presenting cell; CTL, cytotoxic T lymphocyte; MHC, major histocompatibility complex; TCR, T-cell receptor. Created with BioRender.com.
Research groups argued that the mutagenicity of A3 proteins is not correlated with the evolutionary pressure from these enzymes (139, 194). If the HIV-1 genome has been changed because of evolutionary pressure from A3 proteins, there should be an imprint left by A3 proteins in the form of an underrepresentation of A3 target motifs and an overrepresentation of A3 product motifs (195). In other words, the nucleotide G must be depleted and A must be enriched in the HIV-1 genome. This is because repetitive exposure to A3 activity can result in nonlethal mutations, leading to the underrepresentation of nucleotide sequences favored by A3 proteins. This underrepresentation is considered a footprint (195). However, this was not found to be the case (194). An explanation based on these two contradicting studies is that G-to-A mutations caused by A3 proteins might not represent the primary mechanism by which they inhibit HIV-1 replication and that they are merely a side product of this inhibition. As previously explained, the antiviral effect of A3 proteins extends beyond their deamination capabilities. Therefore, it is not far-fetched that A3 proteins do not confer any evolutionary pressure on HIV-1. An alternative explanation is that the mutations induced by A3 proteins are highly deleterious, leading to loss of the population. Studies reported that the number of A3G molecules within Vif-deficient HIV-1 virions range from 3–11 (196). This number is greatly reduced to approximately 0.3–0.8 molecules in Vif-proficient virions (197). Furthermore, it was reported that virions packaged with one or two A3G molecules exhibit significantly reduced infectivity (182). This number is close to the number of A3 proteins packaged in virions expressing wild-type Vif. This fuels the hypothesis that the mutagenic property of A3 proteins, as low as it might be, confers a lethal effect on the overall fitness of HIV-1. On average, A3 proteins mutate 2.3 bases for every 1 kb (182). To put things into better perspective, the optimal mutation rate for HIV-1 is agreed to be approximately 0.3 mutations per genome per replication cycle in vivo (198). Therefore, it is tempting to think that the mutational burden levied upon the HIV-1 genome by A3 proteins might be too high to tolerate, such that it immediately produces lethal repercussions, leaving no evolutionary footprints on the genome of the surviving HIV-1 in the population. There could be heterogeneity among A3 molecules, including differences in the amount of A3 molecules incorporated per particle and/or the mutation efficiency of A3 proteins in target cells during infection events. Further investigation elucidating whether A3 proteins contribute to HIV-1 evolution is required.
4.2 HTLV-1
Another member of the retrovirus family is human T-lymphotropic virus type 1 (HTLV-1), the causative agent of adult T-cell leukemia (ATL) (199). HTLV-1 was also reported to be susceptible to restriction by A3 proteins (200, 201). Unlike HIV-1, the HTLV-1 genome does not encode Vif. Instead, a peptide motif in the C-terminal of the HTLV-1 nucleocapsid (NC) domain inhibits the packaging of A3G protein into virions, as mutations in this region lead to increased A3G protein packaging and increased susceptibility to this protein (202). Therefore, it is assumed that the mechanism to counteract the antiviral activity of A3 proteins might not be identical to that of HIV-1 Vif. Although endogenous and overexpressed A3G protein could be packaged into nascent HTLV-1 virions in MT-2 and HEK293T cells, few G-to-A hypermutation were observed in the HTLV-1 genome (200). This observation supports the idea that HTLV-1 is relatively more resistant to the antiviral activity of A3 proteins than HIV-1. Another study reported a relatively lower frequency of G-to-A mutations in the HTLV-1 provirus (203). Interestingly, the base composition of HTLV-1 genome is rich in G-C compared with the HIV-1 genome, which is more A-T rich, as reported decades ago (204–208). Therefore, it appears that the frequency of nucleotide preference for the A3 protein within the HTLV-1 provirus is not a major determining factor, given that the HTLV-1 genome has a higher G-C content than HIV-1, but the former is more resistant to A3 proteins. This supports the idea that the difference in replication strategy between HIV-1 and HTLV-1 may be the biggest determinant in A3 susceptibility of HTLV-1. The primary replication route of HTLV-1 in vivo is through the mitotic division of host cells because the HTLV-1 provirus is also cloned [reviewed in (209)]. Therefore, ssDNA, as a substrate of A3 proteins, would not be present because of the clonal expansion of HTLV-1-infected cells. Furthermore, it has been reported that the HTLV-1 NC domain in the C-terminus is important for inhibiting the packaging of A3G protein into virions (202). The resistance of HTLV-1 to A3G protein is also evident when considering that, although HTLV-1 RT and HIV-1 RT have comparable mutation rates (210), the diversity of HTLV-1 is lower than that of HIV-1 when comparing the variability of their respective env genes (211, 212). These findings provide further emphasis of the greater resistance of HTLV-1 than HIV-1 to the antiviral activity of the A3G protein. Nevertheless, HTLV-1 is still susceptible to A3G protein-mediated deamination during reverse transcription because GG-to-AG mutations preferred by A3G protein were observed in proviruses from HTLV-1 carriers and patients with ATL (203).
An interesting question is whether HTLV-1 can benefit from A3 mutagenesis because A3 proteins leave a footprint on the genome of HTLV-1 (195, 203). HTLV-1 encodes two oncogenic genes, namely tax in the sense (plus) strand and HTLV-1 bZIP factor (HBZ) in the antisense (minus) strand (213). Tax protein is a potent oncoprotein that is essential for viral transcription and cell transformation (214–216). This protein is highly immunogenic because of the strong inducibility of other viral proteins (217, 218). HBZ is also an oncogenic protein, but it has low immunogenicity (219, 220). Importantly, a previous study by Fan et al. reported that nonsense mutations deriving from the A3G protein occur in minus-strand viral cDNA during reverse transcription, resulting in the inactivation of Tax and all viral proteins excluding HBZ (203). Conversely, G-to-A mutations preferred by A3G protein in the plus-strand region were less frequent, whereas the HBZ gene remains intact (203). A nonsense mutation in the tax gene of HTLV-1–infected cells would result in the loss of Tax protein expression, which sounds counterproductive to viral infection. However, considering that Tax protein can halt cell cycle progression, losing expression of this protein could confer a growth advantage. An additional explanation is that the loss of Tax protein expression causes the loss of expression of other viral proteins, resulting in escape from the host immune system.
A plethora of studies have been conducted on the interaction between retroviruses and A3 proteins. Results are indicating the role of A3 proteins as drivers of retrovirus evolution, evident in the fact that A3 proteins confer drug resistance and promote immune escape in HIV-1 and promote uncontrolled cell proliferation and immune evasion in HTLV-1–infected cells. Some studies deny the direct contribution of A3 proteins to the aforementioned phenotypes because of their perceived lack of A3-mediated hypermutation in the retrovirus genome. More research should be conducted to ascertain whether the lack of a footprint found in some studies is because A3 proteins do not exert any evolutionary pressure on HIV-1 or because of the lethal mutagenesis induced by A3 proteins in the HIV-1 genome.
5 Implications of A3 proteins as drivers of DNA virus evolution
It is undisputed that A3 proteins can act as antiviral factors against retroviruses. However, A3 proteins are not only antiviral factors for retroviruses. In essence, A3 proteins are DNA mutators, and their best-characterized substrate is HIV-1. DNA viruses belonging to groups I, II, and VII of the Baltimore classification (135) were reported to be susceptible to the mutagenic ability of A3 proteins, as explained later in this review. This lesser-known interaction might not be far-fetched considering that some A3 proteins were reported to be localized to the nucleus (30, 51, 54–62, 66). Therefore, these proteins can physically access viral genomes during DNA virus infection because replication of the genomes of most DNA viruses occurs in the nucleus [reviewed in (221–223)], although some viruses such as poxvirus can replicate in the cytoplasm (224).
5.1 Parvovirus
Parvoviruses have a linear ssDNA genome [reviewed in (225)], which is a prime candidate for a substrate for A3 deamination. It was reported that A3A protein can inhibit parvoviruses (226, 227). However, this inhibition is entirely deamination-independent, as no A3A-induced mutations were observed (226, 227). A3A protein can induce lethal mutagenesis in the genome and prevent survival of the virus in the population, similar to the previously discussed explanation regarding A3 proteins pushing the HIV genome beyond its genomic stability (Figure 4A). However, it is difficult to ascertain whether this is the case, as no studies have confirmed the mutational rate of human parvoviruses. The closest virus belonging to the same family is canine parvovirus, which has a similar mutation rate as RNA viruses (228, 229), implying that A3 proteins can drive the genomes of parvoviruses beyond their genetic stability. The second explanation is that the nucleic acid-binding capability of A3A protein prevented the replication of the parvovirus genome, leading to inhibition (226). This explanation is highly supported by a study demonstrating that the deamination activity of A3A protein is not required for inhibition of parvovirus replication (227).
However, Poulain et al. recorded different results, demonstrating that in the case of parvoviruses, A3 proteins do leave footprints, and there is a difference in terms of the magnitude and localization of the footprint among the viruses within this family in silico (195). One of the viruses with the largest A3 protein footprints is the B19 erythroparvovirus (195). This virus was strongly depleted of A3-favored motifs, which has been observed to a lesser extent in parvovirus 4 and bocavirus 4 (195). Interestingly, the researchers found no A3 footprints on adeno-associated dependoparvovirus (195), in accordance with previous in vitro reports revealing a lack of A3-induced mutations and the independence of parvovirus replication inhibition from the deaminase activity of A3A protein (226, 227).
If parvoviruses are susceptible to the antiviral activity of A3 proteins, it should be clarified whether they have evolved a mechanism to counteract A3 proteins. However, we found no reports of such an A3 counteraction mechanism by parvoviral proteins akin to Vif protein of HIV-1 or the C-terminal peptide motif of HTLV-1 NC protein. Although it is evident that some viruses belonging to the Parvoviridae family are primarily susceptible to the antiviral activity of A3 proteins, whether A3 proteins impart evolutionary pressure on parvoviruses remains unclear. Furthermore, it is unknown whether A3 proteins can help change the phenotype of parvoviruses by conferring drug resistance and/or immune escape.
5.2 Human herpesvirus
Human herpesviruses (HHVs) possess a linear dsDNA genome of 125–240 kbp in length that encodes 70–165 genes (230). DNA replication in HHVs occurs in the nucleus (231). Hence, it is tempting to think that HHVs are susceptible to the antiviral activity of A3 proteins. A3C protein is reported to be an important restriction factor against human herpesvirus 1 (HSV-1), as indicated by the reduced virus titer and particle/plaque-forming unit ratio upon its overexpression. In addition, A3-edited genomes have been discovered in vitro and in vivo (232). Furthermore, the same A3-mediated editing was detected in the genome of Epstein–Barr virus (EBV) samples (232). Further fueling this theory, recent reports illustrated that the ribonucleotide reductases (RNRs) of HHVs could antagonize A3 proteins by triggering their relocalization and enzymatic inhibition (51, 59, 233, 234), proving that A3 proteins have a sufficient effect on HHV replication to necessitate a counteraction mechanism.
RNRs synthesize deoxyribonucleoside triphosphates, which are required for DNA replication both by viruses and cells (235). EBV has been reported to antagonize A3B protein through the binding of its RNR subunit BORF2 (51, 59, 233, 234). This binding was reported to enzymatically inhibit the cytosine deaminase activity of A3B protein and relocalize A3B protein from the nucleus to perinuclear bodies to maintain genomic integrity (51, 59, 233, 234). This ability of viral RNR to antagonize the antiviral activity of A3B is argued to be the product of evolution for herpesviruses because it is conserved among viruses that infect humans, who naturally express A3B protein, but it is absent in homologous viruses that infect New World monkeys, which naturally lack the A3B gene (233). These results suggest the possibility that A3B protein aided the evolution of HHVs. Furthermore, many of their genes are footprinted by A3 proteins (195).
Another interesting finding is that HHVs that infect lymphocytes, such as varicella zoster virus (VZV), EBV, and Kaposi’s sarcoma-associated herpesvirus, do not have reduced A3G protein-preferred motifs in vitro (236), even though lymphocytes are known to express extremely high levels of A3G protein (237). This makes sense when considering that HHVs replicate solely in the nucleus (231), whereas A3G protein is primarily localized to the cytoplasm (36, 44, 54, 55, 57, 60) and only a very small amount localize to the nucleus (63–65). VZV, but not HSV-1, is depleted of recognition motifs preferred by A3A, A3B, and A3H proteins (238, 239). Taken together, the depletion of A3-preferred motifs might involve multiple factors that can modulate the frequency of these features. A counterpoint to the notion that A3 proteins aid the evolution of HHVs is a study reporting that although G-to-A hypermutation was found in in vivo buccal swabs, they argued that it does not affect HHV genome evolution (232). This connects to the idea mentioned in previous sections of this review in which the highly mutagenic potential of A3 proteins pushed the viral genome beyond its genetic stability, causing the virus to not survive in the population (Figure 4A). Apparently, this might also hold true for HHVs or at least for HSV-1. Although it is true that highly edited genomes are defective, a study monitoring the molecular evolution of herpesviruses found that HSV-1 has 68.3% GC content (240). If A3 proteins contributed to HSV-1 evolution, the percentage should be lower considering the C-to-U mutations that A3 proteins cause on the single-stranded viral DNA.
Therefore, HHVs are demonstrated to be susceptible to the antiviral activity of A3 proteins, and they also possess a counteraction mechanism (51, 59, 233). It can be said that A3 proteins exert evolutionary pressure on HHVs, considering that certain subtypes of HHVs that infect old world monkeys possess a counteraction mechanism against the antiviral activity of A3B protein whereas it is absent in homologous viruses that infect New World monkeys lacking natural A3B protein expression.
5.3 Human papillomavirus
Vartanian et al. sought to clarify whether the human papillomavirus (HPV) genome is susceptible to the antiviral activity of A3 proteins (241). A3H protein is expressed within keratinocytes (target cells of HPV) (241). A3A and A3B proteins are expressed in psoriatic keratinocytes (241), a manifestation that is commonly attributed to HPV infection (242). Prior research focused on the region corresponding to the promoter region of HPV because this region more frequently exists as ssDNA. Hypermutation has been detected in HPV1a plantar wart samples and HPV16 precancerous cervical biopsies, albeit at low cytosine editing frequencies of 11% and 9% for plantar warts and precancerous cervical biopsies, respectively (241). This is supported by a study examining the HPV16, HPV18, and HPV31 genomes (195). However, the researchers found that the footprint left by A3 proteins is genome-wide opposed to being limited to the promoter region (195). This strong presence of an A3 footprint implies that the genome will receive less exposure to A3 proteins, experience less uracil introduction, and consequently less degradation of the viral genome through base excision repair (195). However, an important highlight is the existence of reports of these HPVs stabilizing (243) or even upregulating the expression of A3B protein (244), which is counterproductive for the virus when considering both facts. If the genome of the virus itself is slowly acquiring increased resistance to A3 proteins, why would the proteins encoded in this genome also increase the half-life of A3B protein in an infected cell? Poulain et al. speculated that A3 proteins are required to increase the mutational rate in the HPV genome because the host DNA polymerase hijacked by HPVs is not sufficient to drive viral evolution (195). This might not be far-fetched as a previous publication also suggested a similar narrative (245). Prior research suggested that the TC dinucleotide sequence preferred by several A3 proteins is highly depleted in HPV genomes (195).
HPVs can be roughly categorized into two groups based on tissue tropism. Specifically, beta- and gamma-HPVs infect cutaneous tissues such as the skin, whereas alpha-HPVs infect mucosal tissues such as the cervix (246). Warren et al. found a difference in terms of the basal expression of A3 protein isoforms between cutaneous and mucosal tissues, the latter having extremely high expression of all A3 isoforms excluding A3B (245). They argued that the differential expression of A3 proteins in both types of tissues helps shape the evolution of HPVs, conferring them with tropism toward one tissue over the other by depleting the TC dinucleotide (245). When considering the various findings, it can be said that A3 proteins exert evolutionary pressure on HPVs.
5.4 Human polyomavirus
Poulain et al. reported that several human PyVs, namely BK polyomavirus (BKPyV), JC polyomavirus, and Merkel cell polyomavirus, are susceptible to the antiviral activity of A3 proteins (195). They found that BKPyV exhibits an extreme depletion of TC dinucleotides, as previously reported (247). Interestingly, BKPyV infection upregulated the expression of A3B protein (247). Similarly as HPV, BKPyV also upregulated the expression of A3B protein, which does not improve nor impair BKPyV infection (247). PyVs upregulate A3B protein via their large T antigen (TaG) (248), although the precise mechanism of the process is unclear. TaG has a domain that targets p53 for inactivation and another domain that targets retinoblastoma protein (RB) for inactivation, leading to the inhibition of cellular apoptosis [reviewed in (249, 250)]. Starrett et al. revealed that the RB-interacting domain is important for A3B gene upregulation in vitro (248). They proposed a model whereby disruption of the RB–E2F pathway by TaG resulted in the expression of the antiviral enzyme A3B. Reiterating the speculation by Poulain et al. (195), it is highly plausible that DNA viruses such as HPV and PyV utilize A3B protein as a means to evolve their genomes, modulating pathogenesis and/or allowing immune escape. This is further supported by a clinical study demonstrating that A3A and A3B proteins are major sources of mutations in the BKPyV genome (251), implying that A3 proteins act as drivers for BKPyV evolution.
6 Implications of A3 proteins as drivers of hepadnavirus evolution
The most well-studied hepadnavirus affected by A3 proteins is hepatitis B virus (HBV) (195, 252–259). However, there is a debate on exactly when and how A3 proteins exert their antiviral effects. This is because although HBV possesses RT, the process by which it performs reverse transcription is extremely different than that in retroviruses such as HIV-1. The complete synthesis of the plus-strand DNA of the HBV genome does not occur in the producer cell, meaning that a portion of the minus-strand DNA of HBV is exposed as a single strand, making it a prime target for A3 proteins (253). This largely appears to be the case, as a full genome ultra-deep pyrosequencing study revealed that G-to-A hypermutation largely occurs in single-stranded regions of the HBV genome (253). Furthermore, their results point to the contribution of G-to-A hypermutation, which is characteristic of cytosine deamination mediated by A3 proteins, in the progression of HBV infection to liver cirrhosis because there was a correlation between the increased frequency of G-to-A hypermutation and a higher degree of liver fibrosis (253). Another interesting point from that study was the key difference in the action of A3 between hepadnaviruses and retroviruses, as deamination occurs in mature virions within producer cells before viral egress in hepadnaviruses but cells in retroviruses (253).
The aforementioned G-to-A editing that occurs within the HBV genome contributes to HBeAg seroconversion (253). HBeAg seroconversion is a phenomenon whereby the expression of an accessory nonparticulate protein encoded by preC mRNA is lost during a natural infection, changing the phenotype from a highly replicative, low-inflammatory phenotype of the HBeAg-positive phase to a low-replicative, HbeAg-negative chronic hepatitis phase (257). This loss of HBeAg protein expression can be considered shortsighted evolution because HBV has to establish an infection (high replication) and then persist (low replication and immune response escape). Interestingly, this G-to-A editing is primarily performed by A3 proteins (253), demonstrating that A3 proteins edit the HBV genome and that these mutations naturally help HBV to establish a persistent infection.
Another aspect is the deaminase-independent activity of A3 proteins against hepadnaviruses. The anti-HBV activity of A3G protein is independent of its DNA editing activity (258), implying that this protein physically blocks DNA strand elongation. This is further supported by the fact that A3G protein interferes with reverse transcription by binding to pre-genome RNA (pgRNA) and preventing nucleocapsid formation (256). However, researchers also found that A3C protein, which is smaller than A3G protein, does not prevent nucleocapsid formation and instead deaminates the HBV genome (256). Another group reported similar findings demonstrating that the blockade of HBV DNA accumulation results from the inhibition of pgRNA packaging (260). Therefore, the next question is whether HBVs have a mechanism to counter A3 proteins. An in vitro study demonstrated that the small nonstructural X protein (HBx) of HBV decreases the protein levels of A3G in cells through a process that does not involve proteasomal degradation (255). Interestingly, it was indicated that HBx induces A3G exosome secretion, as the A3G protein concentration was maintained by inhibiting exosome biogenesis (255).
Taken together, the anti-HBV activity of A3 proteins is more of a collective action by several different A3 proteins exerting both deaminase-dependent and deaminase-independent effects to achieve an anti-HBV effect. Considering the evidence that A3 proteins have a role in the evolution of HBV as infection proceeds, it can be said that A3 proteins aid in HBV evolution.
7 Implications of A3 proteins as drivers of RNA virus evolution
The first mention of APOBEC/AID family proteins stems from the discovery of the RNA-editing enzyme A1 (1, 2). A1 protein was later revealed to deaminate DNAs (7). Although A3 proteins generally prefer to deaminate ssDNA, several reports demonstrated that certain A3 proteins induce C-to-U mutation in the ssRNA substrate (261–266). Therefore, it is interesting to speculate that A3 proteins also contribute to RNA virus evolution. Apparently, this has been observed for a few RNA viruses, as discussed in the subsequent sections.
7.1 Coronaviridae
A study revealed that A3C, A3F, and A3H proteins can inhibit the human coronavirus NL63, a nonzoonotic coronavirus (267). Although hypermutation was not found in the tested coronavirus genome (267), cytosine deamination was still found to occur because the study demonstrated that wild-type A3 proteins confer a higher level of antiviral activity than the catalytically inactive A3 mutants (267). There are two possible explanations for the mechanism by which A3 proteins inhibit coronavirus replication: A3 proteins may deaminate a cellular target or the genome of the coronavirus itself; or, the inhibition may be caused by the interaction of A3 with viral RNA and/or proteins. The antiviral activity of A3 proteins against coronaviruses is supported in another study, which demonstrated marked cytosine deamination in all coronavirus genomes (268), and A3 proteins did have an effect on HKU-1, a nonzoonotic human coronavirus (195). Interestingly, despite being in the same family of coronaviruses, zoonotic coronaviruses such as Middle East respiratory syndrome coronavirus (MERS-CoV), severe acute respiratory syndrome coronavirus-1 (SARS-CoV-1), and SARS-CoV-2, do not contain footprints of A3 proteins (195). For the cases of MERS-CoV and SARS-CoV-1, it was speculated that the relatively low number of infected individuals and the short time of viral circulation may have affected the outcome (195). For SARS-CoV-2, it was stated that the additional viral protein encoded in ORF10 might have some Vif-like activity through an interaction with the CUL2 ubiquitin ligase to degrade A3 proteins (269). However, this does not eliminate the possibility of deamination, as several study groups have reported the contribution of A3 proteins to UC-to-UU mutations in the SARS-CoV-2 genome (270, 271). The first report of deamination by A3 proteins in the SARS-CoV-2 genome demonstrated that wild-type A3A protein expression greatly increased the viral titer production, by 100-fold (270). Furthermore, SARS-CoV-2 mutants harboring C-to-U mutations were found to be circulating during the early part of the pandemic (January 2020) and rapidly became a signature of the dominant strains (including Delta and Omicron variants) that spread worldwide (270). This is further supported by several reports mentioning that C-to-U mutations are among the most frequently accumulated mutations in the SARS-CoV-2 genome (271–275). A study of the SARS-CoV-2 transcriptome also detected C-to-U mutations and revealed that the sequence context was compatible with APOBEC-mediated deamination (272). In addition, their study indicated that the inherent error-prone RNA-dependent RNA polymerase (RdRp) of SARS-CoV-2 represented only a fraction of the C-to-U mutations observed (272).
Collectively, A3 proteins may contribute to the evolution of some coronaviruses, notably SARS-CoV-2. However, a correlation between the A3-mediated deamination of coronaviruses and alteration of their phenotype to confer drug resistance and/or immune escape has not yet been identified, because C-to-U mutations were found in the 5′-UTR, which has an important function in the replication of SARS-CoV-2 RNA and viral protein expression (270). Further investigations examining the role of A3 protein-mediated hypermutation in SARS-CoV-2 evolution, including drug resistance and/or immune escape, are needed.
7.2 Rubella virus
It has been reported that the rubella virus belonging to Matonaviridae is susceptible to the RNA-editing activity of A3 proteins (276). The sequence evolution of immunodeficiency-related vaccine-derived rubella virus (iVDRV) revealed a divergence from the genome of the parental rubella virus contained within the vaccine (276). A clear bias toward C-to-U mutations was observed in the iVDRV positive-strand genome, indicating that A3 proteins represent an important cause of the divergence (276). Furthermore, the mutation caused a decrease in nucleotide preference for A3G protein but an increase in nucleotide preference for other A3 proteins and A1 protein, implying that multiple APOBEC family proteins act on iVDRV to generate sequence diversity (276). Similar to a study of coronavirus, the researchers also found that the error-prone RdRp is not the major contributor to this sequence diversity (276). Evidence suggests that rubella virus, at least in immunodeficient patients, is susceptible to the deaminating properties of A3 proteins and that A3 proteins aided the evolution of this virus, as indicated by the changes in its sequence (276). These changes also alter the phenotype of the virus, as different strains of iVDRV have unique replicative and persistence properties (276). Of note, iVDRVs were less cytopathic in cell culture than the parental vaccine virus, but they had better capability to sustain an infection, similar to the characteristics of wild-type rubella virus (276). This strongly supports the idea that A3 proteins can shape the evolution of a virus and even alter its phenotype to improve viral fitness. However, we also note that no other similar studies have confirmed these findings.
The change of iVDRV phenotype allows a higher level of persistence (277). This was later elucidated to primarily stem from A3 protein-derived mutagenesis (276). Therefore, the editing activity of A3 proteins on the rubella virus genome implies that the proteins can shape the evolution of a virus and even alter its phenotype to improve viral fitness.
8 Conclusion
The pathogen–host interaction is a complex “arms race” to outcompete each other for survival. Some viruses have adapted to live with A3 proteins, utilizing their mutagenic ability for their own benefit, whereas some have evolved to negate their action altogether. Therefore, A3 proteins have been established as extremely important restriction factors because of their plethora of interactions with several viruses, helping to shape viral evolution and their own evolution. Modulating A3 proteins for our own benefit remains a difficult task. Because of the deep interactions of A3 proteins with viruses and host proteins, it is difficult to predict the repercussions of modulating these proteins, as it can lead to unwanted outcomes or even promote beneficial mutation in viruses. More research is needed to better elucidate the interactions of A3 proteins with host and pathogens.
Author contributions
MJ: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. TI: Conceptualization, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported in part by AMED Research Program on Emerging and Re-emerging Infectious Diseases (JP22fk0108146, to TI; JP22fk0108511, to TI; JP22fk0108534, to TI); AMED Research Program on HIV/AIDS (JP22fk0410055, to TI); JSPS KAKENHI Grant in-Aid for Scientific Research C (22K07103, to TI); JSPS Leading Initiative for Excellent Young Researchers (LEADER) (to TI); Takeda Science Foundation (to TI); Mochida Memorial Foundation for Medical and Pharmaceutical Research (to TI); The Naito Foundation (to TI); International Joint Research Project of the Institute of Medical Science, the University of Tokyo (to TI).
Acknowledgments
We would like to thank all Ikeda lab members for providing many supports.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science (1993) 260(5115):1816–9. doi: 10.1126/science.8511591
2. Hadjiagapiou C, Giannoni F, Funahashi T, Skarosi SF, Davidson NO. Molecular cloning of a human small intestinal apolipoprotein B mRNA editing protein. Nucleic Acids Res (1994) 22(10):1874–9. doi: 10.1093/nar/22.10.1874
3. Koito A, Ikeda T. Apolipoprotein B mRNA-editing, catalytic polypeptide cytidine deaminases and retroviral restriction. Wiley Interdiscip Rev RNA (2012) 3(4):529–41. doi: 10.1002/wrna.1117
4. Keegan LP, Gallo A, O’Connell MA. The many roles of an RNA editor. Nat Rev Genet (2001) 2(11):869–78. doi: 10.1038/3509858435098584
5. Prohaska KM, Bennett RP, Salter JD, Smith HC. The multifaceted roles of RNA binding in APOBEC cytidine deaminase functions. Wiley Interdiscip Rev RNA (2014) 5(4):493–508. doi: 10.1002/wrna.1226
6. Pecori R, Di Giorgio S, Paulo Lorenzo J, Nina Papavasiliou F. Functions and consequences of AID/APOBEC-mediated DNA and RNA deamination. Nat Rev Genet (2022) 23(8):505–18. doi: 10.1038/s41576-022-00459-8
7. Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell (2002) 10(5):1247–53. doi: 10.1016/S1097-2765(02)00742-6
8. Ikeda T, Abd El Galil KH, Tokunaga K, Maeda K, Sata T, Sakaguchi N, et al. Intrinsic restriction activity by apolipoprotein B mRNA editing enzyme APOBEC1 against the mobility of autonomous retrotransposons. Nucleic Acids Res (2011) 39(13):5538–54. doi: 10.1093/nar/gkr124
9. Ikeda T, Shimoda M, Ebrahimi D, VandeBerg JL, Harris RS, Koito A, et al. Opossum APOBEC1 is a DNA mutator with retrovirus and retroelement restriction activity. Sci Rep (2017) 7:46719. doi: 10.1038/srep46719
10. Ikeda T, Ohsugi T, Kimura T, Matsushita S, Maeda Y, Harada S, et al. The antiretroviral potency of APOBEC1 deaminase from small animal species. Nucleic Acids Res (2008) 36(21):6859–71. doi: 10.1093/nar/gkn802
11. Ikeda T, Ong EB, Watanabe N, Sakaguchi N, Maeda K, Koito A. Creation of chimeric human/rabbit APOBEC1 with HIV-1 restriction and DNA mutation activities. Sci Rep (2016) 6:19035. doi: 10.1038/srep19035
12. Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol (2004) 14(15):1392–6. doi: 10.1016/j.cub.2004.06.057
13. Bishop KN, Holmes RK, Sheehy AM, Malim MH. APOBEC-mediated editing of viral RNA. Science (2004) 305(5684):645. doi: 10.1126/science.1100658.305/5684/645
14. Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem (1999) 274(26):18470–6. doi: 10.1074/jbc.274.26.18470
15. Feng Y, Seija N, Di Noia JM, Martin A. AID in antibody diversification: there and back again. Trends Immunol (2020) 41(7):586–600. doi: 10.1016/j.it.2020.04.009
16. Methot SP, Di Noia JM. Molecular mechanisms of somatic hypermutation and class switch recombination. Adv Immunol (2017) 133:37–87. doi: 10.1016/bs.ai.2016.11.002
17. Anant S, Mukhopadhyay D, Sankaranand V, Kennedy S, Henderson JO, Davidson NO. ARCD-1, an apobec-1-related cytidine deaminase, exerts a dominant negative effect on C to U RNA editing. Am J Physiol Cell Physiol (2001) 281(6):C1904–16. doi: 10.1152/ajpcell.2001.281.6.C1904
18. Rogozin IB, Basu MK, Jordan IK, Pavlov YI, Koonin EV. APOBEC4, a new member of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases predicted by computational analysis. Cell Cycle (2005) 4(9):1281–5. doi: 10.4161/cc.4.9.1994
19. Harris RS, Dudley JP. APOBECs and virus restriction. Virology (2015) 479-480:131–45. doi: 10.1016/j.virol.2015.03.012
20. Desimmie BA, Delviks-Frankenberrry KA, Burdick RC, Qi D, Izumi T, Pathak VK. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J Mol Biol (2014) 426(6):1220–45. doi: 10.1016/j.jmb.2013.10.033
21. Ikeda T, Yue Y, Shimizu R, Nasser H. Potential utilization of APOBEC3-mediated mutagenesis for an HIV-1 functional cure. Front Microbiol (2021) 12:686357. doi: 10.3389/fmicb.2021.686357
22. Cheng AZ, Moraes SN, Shaban NM, Fanunza E, Bierle CJ, Southern PJ, et al. APOBECs and herpesviruses. Viruses (2021) 13(3). doi: 10.3390/v13030390
23. Nakano Y, Aso H, Soper A, Yamada E, Moriwaki M, Juarez-Fernandez G, et al. A conflict of interest: the evolutionary arms race between mammalian APOBEC3 and lentiviral Vif. Retrovirology (2017) 14(1):31. doi: 10.1186/s12977-017-0355-4
24. Münk C, Willemsen A, Bravo IG. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol Biol (2012) 12:71. doi: 10.1186/1471-2148-12-71
25. Kitamura S, Ode H, Iwatani Y. Structural features of antiviral APOBEC3 proteins are linked to their functional activities. Front Microbiol (2011) 2:258. doi: 10.3389/fmicb.2011.00258
26. Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol (2004) 4(11):868–77. doi: 10.1038/nri1489
27. Grillo MJ, Jones KFM, Carpenter MA, Harris RS, Harki DA. The current toolbox for APOBEC drug discovery. Trends Pharmacol Sci (2022) 43(5):362–77. doi: 10.1016/j.tips.2022.02.007
28. Olson ME, Harris RS, Harki DA. APOBEC enzymes as targets for virus and cancer therapy. Cell Chem Biol (2018) 25(1):36–49. doi: 10.1016/j.chembiol.2017.10.007
29. Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, et al. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol (2004) 11(5):435–42. doi: 10.1038/nsmb758
30. Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, et al. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol (2011) 85(21):11220–34. doi: 10.1128/JVI.05238-11
31. Suspene R, Sommer P, Henry M, Ferris S, Guetard D, Pochet S, et al. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res (2004) 32(8):2421–9. doi: 10.1093/nar/gkh554
32. Refsland EW, Hultquist JF, Harris RS. Endogenous origins of HIV-1 G to A hypermutation and restriction in the nonpermissive T cell line CEM2n. PloS Pathog (2012) 8(7):e1002800. doi: 10.1371/journal.ppat.1002800
33. Rathore A, Carpenter MA, Demir O, Ikeda T, Li M, Shaban NM, et al. The local dinucleotide preference of APOBEC3G can be altered from 5’-CC to 5’-TC by a single amino acid substitution. J Mol Biol (2013) 425(22):4442–54. doi: 10.1016/j.jmb.2013.07.040
34. Refsland EW, Hultquist JF, Luengas EM, Ikeda T, Shaban NM, Law EK, et al. Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PloS Genet (2014) 10(11):e1004761. doi: 10.1371/journal.pgen.1004761
35. Ikeda T, Molan AM, Jarvis MC, Carpenter MA, Salamango DJ, Brown WL, et al. HIV-1 restriction by endogenous APOBEC3G in the myeloid cell line THP-1. J Gen Virol (2019) 100(7):1140–52. doi: 10.1099/jgv.0.001276
36. Gallois-Montbrun S, Kramer B, Swanson CM, Byers H, Lynham S, Ward M, et al. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol (2007) 81(5):2165–78. doi: 10.1128/JVI.02287-06
37. Chiu YL, Witkowska HE, Hall SC, Santiago M, Soros VB, Esnault C, et al. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc Natl Acad Sci USA (2006) 103(42):15588–93. doi: 10.1073/pnas.0604524103
38. Kozak SL, Marin M, Rose KM, Bystrom C, Kabat D. The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J Biol Chem (2006) 281(39):29105–19. doi: 10.1074/jbc.M601901200
39. Niewiadomska AM, Tian C, Tan L, Wang T, Sarkis PT, Yu XF. Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J Virol (2007) 81(17):9577–83. doi: 10.1128/JVI.02800-06
40. Soros VB, Yonemoto W, Greene WC. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PloS Pathog (2007) 3(2):e15. doi: 10.1371/journal.ppat.0030015
41. Stopak KS, Chiu YL, Kropp J, Grant RM, Greene WC. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J Biol Chem (2007) 282(6):3539–46. doi: 10.1074/jbc.M610138200
42. Shirakawa K, Takaori-Kondo A, Yokoyama M, Izumi T, Matsui M, Io K, et al. Phosphorylation of APOBEC3G by protein kinase A regulates its interaction with HIV-1 Vif. Nat Struct Mol Biol (2008) 15(11):1184–91. doi: 10.1038/nsmb.1497
43. Sugiyama R, Nishitsuji H, Furukawa A, Katahira M, Habu Y, Takeuchi H, et al. Heat shock protein 70 inhibits HIV-1 Vif-mediated ubiquitination and degradation of APOBEC3G. J Biol Chem (2011) 286(12):10051–7. doi: 10.1074/jbc.M110.166108
44. Gallois-Montbrun S, Holmes RK, Swanson CM, Fernandez-Ocana M, Byers HL, Ward MA, et al. Comparison of cellular ribonucleoprotein complexes associated with the APOBEC3F and APOBEC3G antiviral proteins. J Virol (2008) 82(11):5636–42. doi: 10.1128/JVI.00287-08
45. Maeda K, Almofty SA, Singh SK, Eid MM, Shimoda M, Ikeda T, et al. GANP interacts with APOBEC3G and facilitates its encapsidation into the virions to reduce HIV-1 infectivity. J Immunol (2013) 191(12):6030–9. doi: 10.4049/jimmunol.1302057
46. Pion M, Granelli-Piperno A, Mangeat B, Stalder R, Correa R, Steinman RM, et al. APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J Exp Med (2006) 203(13):2887–93. doi: 10.1084/jem.20061519
47. Ellery PJ, Tippett E, Chiu YL, Paukovics G, Cameron PU, Solomon A, et al. The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol (2007) 178(10):6581–9. doi: 10.4049/jimmunol.178.10.6581
48. Ito F, Yang H, Xiao X, Li SX, Wolfe A, Zirkle B, et al. Understanding the structure, multimerization, subcellular localization and mC selectivity of a genomic mutator and anti-HIV factor APOBEC3H. Sci Rep (2018) 8(1):3763. doi: 10.1038/s41598-018-21955-0
49. Xiao X, Yang H, Arutiunian V, Fang Y, Besse G, Morimoto C, et al. Structural determinants of APOBEC3B non-catalytic domain for molecular assembly and catalytic regulation. Nucleic Acids Res (2017) 45(12):7494–506. doi: 10.1093/nar/gkx362
50. Shaban NM, Shi K, Lauer KV, Carpenter MA, Richards CM, Salamango D, et al. The antiviral and cancer genomic DNA deaminase APOBEC3H is regulated by an RNA-mediated dimerization mechanism. Mol Cell (2018) 69(1):75–86 e9. doi: 10.1016/j.molcel.2017.12.010
51. Cheng AZ, Yockteng-Melgar J, Jarvis MC, Malik-Soni N, Borozan I, Carpenter MA, et al. Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat Microbiol (2019) 4(1):78–88. doi: 10.1038/s41564-018-0284-6
52. Mitra M, Singer D, Mano Y, Hritz J, Nam G, Gorelick RJ, et al. Sequence and structural determinants of human APOBEC3H deaminase and anti-HIV-1 activities. Retrovirology (2015) 12:3. doi: 10.1186/s12977-014-0130-8
53. Kreisberg JF, Yonemoto W, Greene WC. Endogenous factors enhance HIV infection of tissue naive CD4 T cells by stimulating high molecular mass APOBEC3G complex formation. J Exp Med (2006) 203(4):865–70. doi: 10.1084/jem.20051856
54. Lackey L, Law EK, Brown WL, Harris RS. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle (2013) 12(5):762–72. doi: 10.4161/cc.23713
55. Kinomoto M, Kanno T, Shimura M, Ishizaka Y, Kojima A, Kurata T, et al. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res (2007) 35(9):2955–64. doi: 10.1093/nar/gkm181
56. Land AM, Law EK, Carpenter MA, Lackey L, Brown WL, Harris RS. Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic. J Biol Chem (2013) 288(24):17253–60. doi: 10.1074/jbc.M113.458661
57. Stenglein MD, Matsuo H, Harris RS. Two regions within the amino-terminal half of APOBEC3G cooperate to determine cytoplasmic localization. J Virol (2008) 82(19):9591–9. doi: 10.1128/JVI.02471-07
58. Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature (2013) 494(7437):366–70. doi: 10.1038/nature11881
59. Cheng AZ, Moraes SN, Attarian C, Yockteng-Melgar J, Jarvis MC, Biolatti M, et al. A conserved mechanism of APOBEC3 relocalization by herpesviral ribonucleotide reductase large subunits. J Virol (2019) 93(23). doi: 10.1128/JVI.01539-19
60. Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, et al. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem (2006) 281(31):22161–72. doi: 10.1074/jbc.M601716200
61. Salamango DJ, McCann JL, Demir O, Brown WL, Amaro RE, Harris RS. APOBEC3B nuclear localization requires two distinct N-terminal domain surfaces. J Mol Biol (2018) 430(17):2695–708. doi: 10.1016/j.jmb.2018.04.044
62. Manjunath L, Oh S, Ortega P, Bouin A, Bournique E, Sanchez A, et al. APOBEC3B drives PKR-mediated translation shutdown and protects stress granules in response to viral infection. Nat Commun (2023) 14(1):820. doi: 10.1038/s41467-023-36445-9
63. Liu W, Newhall KP, Khani F, Barlow L, Nguyen D, Gu L, et al. The cytidine deaminase APOBEC3G contributes to cancer mutagenesis and clonal evolution in bladder cancer. Cancer Res (2023) 83(4):506–20. doi: 10.1158/0008-5472.CAN-22-2912
64. Nowarski R, Wilner OI, Cheshin O, Shahar OD, Kenig E, Baraz L, et al. APOBEC3G enhances lymphoma cell radioresistance by promoting cytidine deaminase-dependent DNA repair. Blood (2012) 120(2):366–75. doi: 10.1182/blood-2012-01-402123
65. Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell (2003) 12(3):591–601. doi: 10.1016/s1097-2765(03)00353-8
66. Li MM, Emerman M. Polymorphism in human APOBEC3H affects a phenotype dominant for subcellular localization and antiviral activity. J Virol (2011) 85(16):8197–207. doi: 10.1128/JVI.00624-11
67. Salamango DJ, Becker JT, McCann JL, Cheng AZ, Demir O, Amaro RE, et al. APOBEC3H subcellular localization determinants define zipcode for targeting HIV-1 for restriction. Mol Cell Biol (2018) 38(23). doi: 10.1128/MCB.00356-18
68. Simon V, Bloch N, Landau NR. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol (2015) 16(6):546–53. doi: 10.1038/ni.3156
69. Refsland EW, Harris RS. The APOBEC3 family of retroelement restriction factors. Intrinsic Immun (2013) 371:1–27. doi: 10.1007/978-3-642-37765-5_1
70. Sadeghpour S, Khodaee S, Rahnama M, Rahimi H, Ebrahimi D. Human APOBEC3 variations and viral infection. Viruses (2021) 13(7). doi: 10.3390/v13071366
71. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature (2002) 418(6898):646–50. doi: 10.1038/nature00939
72. Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, et al. DNA deamination mediates innate immunity to retroviral infection. Cell (2003) 113(6):803–9. doi: 10.1016/s0092-8674(03)00423-9
73. Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature (2003) 424(6944):99–103. doi: 10.1038/nature01709
74. Nakano Y, Misawa N, Juarez-Fernandez G, Moriwaki M, Nakaoka S, Funo T, et al. HIV-1 competition experiments in humanized mice show that APOBEC3H imposes selective pressure and promotes virus adaptation. PloS Pathog (2017) 13(5):e1006348. doi: 10.1371/journal.ppat.1006348
75. Ooms M, Brayton B, Letko M, Maio SM, Pilcher CD, Hecht FM, et al. HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe (2013) 14(4):411–21. doi: 10.1016/j.chom.2013.09.006
76. Wittkopp CJ, Adolph MB, Wu LI, Chelico L, Emerman M. A single nucleotide polymorphism in human APOBEC3C enhances restriction of lentiviruses. PloS Pathog (2016) 12(10):e1005865. doi: 10.1371/journal.ppat.1005865
77. Anderson BD, Ikeda T, Moghadasi SA, Martin AS, Brown WL, Harris RS. Natural APOBEC3C variants can elicit differential HIV-1 restriction activity. Retrovirology (2018) 15(1):78. doi: 10.1186/s12977-018-0459-5
78. Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, et al. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol (2005) 15(2):166–70. doi: 10.1016/j.cub.2004.12.068
79. Belanger K, Savoie M, Rosales Gerpe MC, Couture JF, Langlois MA. Binding of RNA by APOBEC3G controls deamination-independent restriction of retroviruses. Nucleic Acids Res (2013) 41(15):7438–52. doi: 10.1093/nar/gkt527
80. Huthoff H, Autore F, Gallois-Montbrun S, Fraternali F, Malim MH. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PloS Pathog (2009) 5(3):e1000330. doi: 10.1371/journal.ppat.1000330
81. Navarro F, Bollman B, Chen H, Konig R, Yu Q, Chiles K, et al. Complementary function of the two catalytic domains of APOBEC3G. Virology (2005) 333(2):374–86. doi: 10.1016/j.virol.2005.01.011
82. Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PloS Pathog (2008) 4(12):e1000231. doi: 10.1371/journal.ppat.1000231
83. Iwatani Y, Chan DS, Wang F, Stewart-Maynard K, Sugiura W, Gronenborn AM, et al. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res (2007) 35(21):7096–108. doi: 10.1093/nar/gkm750
84. Li XY, Guo F, Zhang L, Kleiman L, Cen S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem (2007) 282(44):32065–74. doi: 10.1074/jbc.M703423200
85. Pollpeter D, Parsons M, Sobala AE, Coxhead S, Lang RD, Bruns AM, et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat Microbiol (2018) 3(2):220–33. doi: 10.1038/s41564-017-0063-9
86. Wang X, Ao Z, Chen L, Kobinger G, Peng J, Yao X. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J Virol (2012) 86(7):3777–86. doi: 10.1128/JVI.06594-11
87. Kobayashi T, Koizumi Y, Takeuchi JS, Misawa N, Kimura Y, Morita S, et al. Quantification of deaminase activity-dependent and -independent restriction of HIV-1 replication mediated by APOBEC3F and APOBEC3G through experimental-mathematical investigation. J Virol (2014) 88(10):5881–7. doi: 10.1128/JVI.00062-14
88. Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem (2007) 282(4):2587–95. doi: 10.1074/jbc.M607298200
89. Luo K, Wang T, Liu B, Tian C, Xiao Z, Kappes J, et al. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J Virol (2007) 81(13):7238–48. doi: 10.1128/JVI.02584-06
90. LaRue RS, Jonsson SR, Silverstein KA, Lajoie M, Bertrand D, El-Mabrouk N, et al. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol Biol (2008) 9:104. doi: 10.1186/1471-2199-9-104
91. Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol Biol Evol (2005) 22(2):367–77. doi: 10.1093/molbev/msi026
92. Ito J, Gifford RJ, Sato K. Retroviruses drive the rapid evolution of mammalian APOBEC3 genes. Proc Natl Acad Sci USA (2020) 117(1):610–8. doi: 10.1073/pnas.1914183116
93. Larue RS, Lengyel J, Jonsson SR, Andresdottir V, Harris RS. Lentiviral Vif degrades the APOBEC3Z3/APOBEC3H protein of its mammalian host and is capable of cross-species activity. J Virol (2010) 84(16):8193–201. doi: 10.1128/JVI.00685-10
94. Yamada E, Yoshikawa R, Nakano Y, Misawa N, Kobayashi T, Ren F, et al. A naturally occurring bovine APOBEC3 confers resistance to bovine lentiviruses: implication for the co-evolution of bovids and their lentiviruses. Sci Rep (2016) 6:33988. doi: 10.1038/srep33988
95. Hayward JA, Tachedjian M, Cui J, Cheng AZ, Johnson A, Baker ML, et al. Differential evolution of antiretroviral restriction factors in pteropid bats as revealed by APOBEC3 gene complexity. Mol Biol Evol (2018) 35(7):1626–37. doi: 10.1093/molbev/msy048
96. Koito A, Ikeda T. Intrinsic restriction activity by AID/APOBEC family of enzymes against the mobility of retroelements. Mobile Genet Elements (2011) 1(3):197–202. doi: 10.4161/mge.1.3.17430
97. Koito A, Ikeda T. Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front Microbiol (2013) 4:28. doi: 10.3389/fmicb.2013.00028
98. Ikeda T, Shimizu R, Nasser H, Carpenter MA, Cheng AZ, Brown WL, et al. APOBEC3 degradation is the primary function of HIV-1 Vif determining virion infectivity in the myeloid cell line THP-1. mBio (2023) 14(4):e0078223. doi: 10.1128/mbio.00782-23
99. Chaipan C, Smith JL, Hu WS, Pathak VK. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J Virol (2013) 87(1):444–53. doi: 10.1128/JVI.00676-12
100. Ebrahimi D, Richards CM, Carpenter MA, Wang J, Ikeda T, Becker JT, et al. Genetic and mechanistic basis for APOBEC3H alternative splicing, retrovirus restriction, and counteraction by HIV-1 protease. Nat Commun (2018) 9(1):4137. doi: 10.1038/s41467-018-06594-3
101. Tareen SU, Sawyer SL, Malik HS, Emerman M. An expanded clade of rodent Trim5 genes. Virology (2009) 385(2):473–83. doi: 10.1016/j.virol.2008.12.018
102. Sawyer SL, Emerman M, Malik HS. Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PloS Pathog (2007) 3(12):e197. doi: 10.1371/journal.ppat.0030197
103. Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, et al. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell (2003) 114(1):21–31. doi: 10.1016/s0092-8674(03)00515-4
104. Jäger S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature (2012) 481(7381):371–5. doi: 10.1038/nature10693
105. Zhang W, Du J, Evans SL, Yu Y, Yu XF. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature (2012) 481(7381):376–9. doi: 10.1038/nature10718
106. Salamango DJ, Ikeda T, Moghadasi SA, Wang J, McCann JL, Serebrenik AA, et al. HIV-1 Vif triggers cell cycle arrest by degrading cellular PPP2R5 phospho-regulators. Cell Rep (2019) 29(5):1057–65 e4. doi: 10.1016/j.celrep.2019.09.057
107. Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science (2003) 302(5647):1056–60. doi: 10.1126/science.1089591
108. Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol (2003) 13(22):2009–13. doi: 10.1016/j.cub.2003.10.034
109. Kitamura S, Ode H, Nakashima M, Imahashi M, Naganawa Y, Kurosawa T, et al. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat Struct Mol Biol (2012) 19(10):1005–10. doi: 10.1038/nsmb.2378
110. Dang Y, Wang XJ, Esselman WJ, Zheng YH. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol (2006) 80(21):10522–33. doi: 10.1128/Jvi.01123-06
111. Guerrero S, Libre C, Batisse J, Mercenne G, Richer D, Laumond G, et al. Translational regulation of APOBEC3G mRNA by Vif requires its 5’UTR and contributes to restoring HIV-1 infectivity. Sci Rep (2016) 6:39507. doi: 10.1038/srep39507
112. Libre C, Seissler T, Guerrero S, Batisse J, Verriez C, Stupfler B, et al. A conserved uORF regulates APOBEC3G translation and is targeted by HIV-1 Vif protein to repress the antiviral factor. Biomedicines (2021) 10(1). doi: 10.3390/biomedicines10010013
113. Kim DY, Kwon E, Hartley PD, Crosby DC, Mann S, Krogan NJ, et al. CBFbeta stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol Cell (2013) 49(4):632–44. doi: 10.1016/j.molcel.2012.12.012
114. Münk C, Beck T, Zielonka J, Hotz-Wagenblatt A, Chareza S, Battenberg M, et al. Functions, structure, and read-through alternative splicing of feline APOBEC3 genes. Genome Biol (2008) 9(3):R48. doi: 10.1186/gb-2008-9-3-r48
115. Yoshikawa R, Izumi T, Yamada E, Nakano Y, Misawa N, Ren F, et al. A naturally occurring domestic cat APOBEC3 variant confers resistance to feline immunodeficiency virus infection. J Virol (2016) 90(1):474–85. doi: 10.1128/JVI.02612-15
116. Troyer RM, Malmberg JL, Zheng X, Miller C, MacMillan M, Sprague WS, et al. Expression of APOBEC3 lentiviral restriction factors in cats. Viruses (2019) 11(9). doi: 10.3390/v11090831
117. Zhang Z, Gu Q, Jaguva Vasudevan AA, Hain A, Kloke BP, Hasheminasab S, et al. Determinants of FIV and HIV Vif sensitivity of feline APOBEC3 restriction factors. Retrovirology (2016) 13(1):46. doi: 10.1186/s12977-016-0274-9
118. Stern MA, Hu C, Saenz DT, Fadel HJ, Sims O, Peretz M, et al. Productive replication of Vif-chimeric HIV-1 in feline cells. J Virol (2010) 84(14):7378–95. doi: 10.1128/JVI.00584-10
119. Yoshikawa R, Takeuchi JS, Yamada E, Nakano Y, Ren F, Tanaka H, et al. Vif determines the requirement for CBF-beta in APOBEC3 degradation. J Gen Virol (2015) 96(Pt 4):887–92. doi: 10.1099/jgv.0.000027
120. Richards C, Albin JS, Demir Ö, Shaban NM, Luengas EM, Land AM, et al. The binding interface between human APOBEC3F and HIV-1 Vif elucidated by genetic and computational approaches. Cell Rep (2015) 13(9):1781–8. doi: 10.1016/j.celrep.2015.10.067
121. Jäger S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature (2011) 481(7381):371–5. doi: 10.1038/nature10693
122. Li YL, Langley CA, Azumaya CM, Echeverria I, Chesarino NM, Emerman M, et al. The structural basis for HIV-1 Vif antagonism of human APOBEC3G. Nature (2023) 615(7953):728–33. doi: 10.1038/s41586-023-05779-1
123. Hu Y, Desimmie BA, Nguyen HC, Ziegler SJ, Cheng TC, Chen J, et al. Structural basis of antagonism of human APOBEC3F by HIV-1 Vif. Nat Struct Mol Biol (2019) 26(12):1176–83. doi: 10.1038/s41594-019-0343-6
124. Huttenhain R, Xu J, Burton LA, Gordon DE, Hultquist JF, Johnson JR, et al. ARIH2 is a Vif-dependent regulator of CUL5-mediated APOBEC3G degradation in HIV infection. Cell Host Microbe (2019) 26(1):86–99 e7. doi: 10.1016/j.chom.2019.05.008
125. Ito F, Alvarez-Cabrera AL, Kim K, Zhou ZH, Chen XS. Structural basis of HIV-1 Vif-mediated E3 ligase targeting of host APOBEC3H. Nat Commun (2023) 14(1):5241. doi: 10.1038/s41467-023-40955-x
126. Ito F, Alvarez-Cabrera AL, Liu S, Yang H, Shiriaeva A, Zhou ZH, et al. Structural basis for HIV-1 antagonism of host APOBEC3G via Cullin E3 ligase. Sci Adv (2023) 9(1):eade3168. doi: 10.1126/sciadv.ade3168
127. Zhang W, Wang H, Li Z, Liu X, Liu G, Harris RS, et al. Cellular requirements for bovine immunodeficiency virus Vif-mediated inactivation of bovine APOBEC3 proteins. J Virol (2014) 88(21):12528–40. doi: 10.1128/JVI.02072-14
128. Han X, Liang W, Hua D, Zhou X, Du J, Evans SL, et al. Evolutionarily conserved requirement for core binding factor beta in the assembly of the human immunodeficiency virus/simian immunodeficiency virus Vif-cullin 5-RING E3 ubiquitin ligase. J Virol (2014) 88(6):3320–8. doi: 10.1128/JVI.03833-13
129. Ai Y, Zhu D, Wang C, Su C, Ma J, Ma J, et al. Core-binding factor subunit beta is not required for non-primate lentiviral Vif-mediated APOBEC3 degradation. J Virol (2014) 88(20):12112–22. doi: 10.1128/JVI.01924-14
130. Zhang J, Wu J, Wang W, Wu H, Yu B, Wang J, et al. Role of cullin-elonginB-elonginC E3 complex in bovine immunodeficiency virus and maedi-visna virus Vif-mediated degradation of host A3Z2-Z3 proteins. Retrovirology (2014) 11:77. doi: 10.1186/s12977-014-0077-9
131. Hultquist JF, Binka M, LaRue RS, Simon V, Harris RS. Vif proteins of human and simian immunodeficiency viruses require cellular CBFbeta to degrade APOBEC3 restriction factors. J Virol (2012) 86(5):2874–7. doi: 10.1128/JVI.06950-11
132. Collins A, Littman DR, Taniuchi I. RUNX proteins in transcription factor networks that regulate T-cell lineage choice. Nat Rev Immunol (2009) 9(2):106–15. doi: 10.1038/nri2489
133. Stifani S, Ma Q. ‘Runxs and regulations’ of sensory and motor neuron subtype differentiation: implications for hematopoietic development. Blood Cells Mol Dis (2009) 43(1):20–6. doi: 10.1016/j.bcmd.2009.03.001
134. Shimizu H, Yokoyama S, Asahara H. Growth and differentiation of the developing limb bud from the perspective of chondrogenesis. Dev Growth Differ (2007) 49(6):449–54. doi: 10.1111/j.1440-169X.2007.00945.x
135. Baltimore D. Expression of animal virus genomes. Bacteriol Rev (1971) 35(3):235–41. doi: 10.1128/br.35.3.235-241.1971
136. Coffin J, Blomberg J, Fan H, Gifford R, Hatziioannou T, Lindemann D, et al. ICTV virus taxonomy profile: retroviridae 2021. J Gen Virol (2021) 102(12). doi: 10.1099/jgv.0.001712
137. Nilson KA, Price DH. The role of RNA polymerase II elongation control in HIV-1 gene expression, replication, and latency. Genet Res Int (2011) 2011:726901. doi: 10.4061/2011/726901
138. Gillick K, Pollpeter D, Phalora P, Kim EY, Wolinsky SM, Malim MH. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J Virol (2013) 87(3):1508–17. doi: 10.1128/JVI.02587-12
139. Armitage AE, Deforche K, Chang CH, Wee E, Kramer B, Welch JJ, et al. APOBEC3G-induced hypermutation of human immunodeficiency virus type-1 is typically a discrete “all or nothing” phenomenon. PloS Genet (2012) 8(3):e1002550. doi: 10.1371/journal.pgen.1002550
140. Ikeda T, Symeonides M, Albin JS, Li M, Thali M, Harris RS. HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PloS Pathog (2018) 14(4):e1007010. doi: 10.1371/journal.ppat.1007010
141. Haché G, Shindo K, Albin JS, Harris RS. Evolution of HIV-1 isolates that use a novel Vif-independent mechanism to resist restriction by human APOBEC3G. Curr Biol (2008) 18(11):819–24. doi: 10.1016/j.cub.2008.04.073
142. Suspène R, Rusniok C, Vartanian JP, Wain-Hobson S. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res (2006) 34(17):4677–84. doi: 10.1093/nar/gkl555
143. Kijak GH, Janini LM, Tovanabutra S, Sanders-Buell E, Arroyo MA, Robb ML, et al. Variable contexts and levels of hypermutation in HIV-1 proviral genomes recovered from primary peripheral blood mononuclear cells. Virology (2008) 376(1):101–11. doi: 10.1016/j.virol.2008.03.017
144. Kimura I, Yamasoba D, Tamura T, Nao N, Suzuki T, Oda Y, et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 subvariants, including BA.4 and BA.5. Cell (2022) 185(21):3992–4007 e16. doi: 10.1016/j.cell.2022.09.018
145. Saito A, Tamura T, Zahradnik J, Deguchi S, Tabata K, Anraku Y, et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2.75 variant. Cell Host Microbe (2022) 30(11):1540–1555.e15. doi: 10.1016/j.chom.2022.10.003
146. Suzuki R, Yamasoba D, Kimura I, Wang L, Kishimoto M, Ito J, et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature (2022) 603(7902):700–5. doi: 10.1038/s41586-022-04462-1
147. Yamasoba D, Kimura I, Nasser H, Morioka Y, Nao N, Ito J, et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell (2022) 185(12):2103–15 e19. doi: 10.1016/j.cell.2022.04.035
148. Ito J, Suzuki R, Uriu K, Itakura Y, Zahradnik J, Kimura KT, et al. Convergent evolution of SARS-CoV-2 Omicron subvariants leading to the emergence of BQ.1.1 variant. Nat Commun (2023) 14(1):2671. doi: 10.1038/s41467-023-38188-z
149. Tamura T, Ito J, Uriu K, Zahradnik J, Kida I, Anraku Y, et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat Commun (2023) 14(1):2800. doi: 10.1038/s41467-023-38435-3
150. Uriu K, Ito J, Kosugi Y, Tanaka YL, Mugita Y, Guo Z, et al. Transmissibility, infectivity, and immune evasion of the SARS-CoV-2 BA.2.86 variant. Lancet Infect Dis (2023) 23(11):460–461. doi: 10.1016/S1473-3099(23)00575-3
151. Saito A, Irie T, Suzuki R, Maemura T, Nasser H, Uriu K, et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature (2022) 602(7896):300–6. doi: 10.1038/s41586-021-04266-9
152. Lampejo T. Influenza and antiviral resistance: an overview. Eur J Clin Microbiol Infect Dis (2020) 39(7):1201–8. doi: 10.1007/s10096-020-03840-9
153. Nathans R, Cao H, Sharova N, Ali A, Sharkey M, Stranska R, et al. Small-molecule inhibition of HIV-1 Vif. Nat Biotechnol (2008) 26(10):1187–92. doi: 10.1038/nbt.1496
154. Kim EY, Bhattacharya T, Kunstman K, Swantek P, Koning FA, Malim MH, et al. Human APOBEC3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. J Virol (2010) 84(19):10402–5. doi: 10.1128/JVI.01223-10
155. Hernandez MM, Fahrny A, Jayaprakash A, Gers-Huber G, Dillon-White M, Audige A, et al. Impact of suboptimal APOBEC3G neutralization on the emergence of HIV drug resistance in humanized mice. J Virol (2020) 94(5). doi: 10.1128/JVI.01543-19
156. Mulder LC, Harari A, Simon V. Cytidine deamination induced HIV-1 drug resistance. Proc Natl Acad Sci USA (2008) 105(14):5501–6. doi: 10.1073/pnas.0710190105
157. Sato K, Takeuchi JS, Misawa N, Izumi T, Kobayashi T, Kimura Y, et al. APOBEC3D and APOBEC3F potently promote HIV-1 diversification and evolution in humanized mouse model. PloS Pathog (2014) 10(10):e1004453. doi: 10.1371/journal.ppat.1004453
158. Harris RS. Enhancing immunity to HIV through APOBEC. Nat Biotechnol (2008) 26(10):1089–90. doi: 10.1038/nbt1008-1089
159. Hultquist JF, Harris RS. Leveraging APOBEC3 proteins to alter the HIV mutation rate and combat AIDS. Future Virol (2009) 4(6):605. doi: 10.2217/fvl.09.59
160. Sharkey M, Sharova N, Mohammed I, Huff SE, Kummetha IR, Singh G, et al. HIV-1 escape from small-molecule antagonism of Vif. mBio (2019) 10(1). doi: 10.1128/mBio.00144-19
161. Zuo T, Liu D, Lv W, Wang X, Wang J, Lv M, et al. Small-molecule inhibition of human immunodeficiency virus type 1 replication by targeting the interaction between Vif and ElonginC. J Virol (2012) 86(10):5497–507. doi: 10.1128/JVI.06957-11
162. Huang W, Zuo T, Luo X, Jin H, Liu Z, Yang Z, et al. Indolizine derivatives as HIV-1 VIF-ElonginC interaction inhibitors. Chem Biol Drug Des (2013) 81(6):730–41. doi: 10.1111/cbdd.12119
163. Mohammed I, Parai MK, Jiang X, Sharova N, Singh G, Stevenson M, et al. SAR and lead optimization of an HIV-1 Vif-APOBEC3G axis inhibitor. ACS Med Chem Lett (2012) 3(6):465–9. doi: 10.1021/ml300037k
164. Pery E, Sheehy A, Nebane NM, Brazier AJ, Misra V, Rajendran KS, et al. Identification of a novel HIV-1 inhibitor targeting Vif-dependent degradation of human APOBEC3G protein. J Biol Chem (2015) 290(16):10504–17. doi: 10.1074/jbc.M114.626903
165. Pan T, He X, Chen B, Chen H, Geng G, Luo H, et al. Development of benzimidazole derivatives to inhibit HIV-1 replication through protecting APOBEC3G protein. Eur J Med Chem (2015) 95:500–13. doi: 10.1016/j.ejmech.2015.03.050
166. Matsui M, Shindo K, Izumi T, Io K, Shinohara M, Komano J, et al. Small molecules that inhibit Vif-induced degradation of APOBEC3G. Virol J (2014) 11:122. doi: 10.1186/1743-422X-11-122
167. Weil AF, Ghosh D, Zhou Y, Seiple L, McMahon MA, Spivak AM, et al. Uracil DNA glycosylase initiates degradation of HIV-1 cDNA containing misincorporated dUTP and prevents viral integration. Proc Natl Acad Sci USA (2013) 110(6):E448–57. doi: 10.1073/pnas.1219702110
168. Pollack RA, Jones RB, Pertea M, Bruner KM, Martin AR, Thomas AS, et al. Defective HIV-1 proviruses are expressed and can be recognized by cytotoxic T lymphocytes, which shape the proviral landscape. Cell Host Microbe (2017) 21(4):494–+. doi: 10.1016/j.chom.2017.03.008
169. Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med (2016) 22(9):1043–+. doi: 10.1038/nm.4156
170. Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DIS, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell (2013) 155(3):540–51. doi: 10.1016/j.cell.2013.09.020
171. Bruner KM, Wang Z, Simonetti FR, Bender AM, Kwon KJ, Sengupta S, et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature (2019) 566(7742):120–5. doi: 10.1038/s41586-019-0898-8
172. Sadler HA, Stenglein MD, Harris RS, Mansky LM. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J Virol (2010) 84(14):7396–404. doi: 10.1128/JVI.00056-10
173. Armitage AE, Deforche K, Welch JJ, Van Laethem K, Camacho R, Rambaut A, et al. Possible footprints of APOBEC3F and/or other APOBEC3 deaminases, but not APOBEC3G, on HIV-1 from patients with acute/early and chronic infections. J Virol (2014) 88(21):12882–94. doi: 10.1128/JVI.01460-14
174. Janini M, Rogers M, Birx DR, McCutchan FE. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4(+) T cells. J Virol (2001) 75(17):7973–86. doi: 10.1128/jvi.75.17.7973-7986.2001
175. Kieffer TL, Kwon P, Nettles RE, Han Y, Ray SC, Siliciano RF. G–>A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J Virol (2005) 79(3):1975–80. doi: 10.1128/JVI.79.3.1975-1980.2005
176. Pace C, Keller J, Nolan D, James I, Gaudieri S, Moore C, et al. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J Virol (2006) 80(18):9259–69. doi: 10.1128/JVI.00888-06
177. Land AM, Ball TB, Luo M, Pilon R, Sandstrom P, Embree JE, et al. Human immunodeficiency virus (HIV) type 1 proviral hypermutation correlates with CD4 count in HIV-infected women from Kenya. J Virol (2008) 82(16):8172–82. doi: 10.1128/JVI.01115-08
178. Kourteva Y, De Pasquale M, Allos T, McMunn C, D’Aquila RT. APOBEC3G expression and hypermutation are inversely associated with human immunodeficiency virus type 1 (HIV-1) burden in vivo. Virology (2012) 430(1):1–9. doi: 10.1016/j.virol.2012.03.018
179. Eyzaguirre LM, Charurat M, Redfield RR, Blattner WA, Carr JK, Sajadi MM. Elevated hypermutation levels in HIV-1 natural viral suppressors. Virology (2013) 443(2):306–12. doi: 10.1016/j.virol.2013.05.019
180. Imamichi H, Natarajan V, Adelsberger JW, Rehm CA, Lempicki RA, Das B, et al. Lifespan of effector memory CD4+ T cells determined by replication-incompetent integrated HIV-1 provirus. AIDS (2014) 28(8):1091–9. doi: 10.1097/QAD.0000000000000223
181. Cuevas JM, Geller R, Garijo R, Lopez-Aldeguer J, Sanjuan R. Extremely high mutation rate of HIV-1 in vivo. PloS Biol (2015) 13(9):e1002251. doi: 10.1371/journal.pbio.1002251
182. Browne EP, Allers C, Landau NR. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology (2009) 387(2):313–21. doi: 10.1016/j.virol.2009.02.026
183. Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J Virol (2010) 84(19):9864–78. doi: 10.1128/Jvi.00915-10
184. Yeo JY, Koh DWS, Yap P, Goh GR, Gan SKE. Spontaneous mutations in HIV-1 gag, protease, RT p66 in the first replication cycle and how they appear: insights from an in vitro assay on mutation rates and types. Int J Mol Sci (2021) 22(1). doi: 10.3390/ijms22010370
185. Dwivedi R, Wang Y, Kline C, Fischer DK, Ambrose Z. APOBEC3 selects V179I in HIV-1 reverse transcriptase to provide selective advantage for non-nucleoside reverse transcriptase inhibitor-resistant mutants. Front Virol (2022) 2:919825. doi: 10.3389/fviro.2022.919825
186. Neogi U, Shet A, Sahoo PN, Bontell I, Ekstrand ML, Banerjea AC, et al. Human APOBEC3G-mediated hypermutation is associated with antiretroviral therapy failure in HIV-1 subtype C-infected individuals. J Int AIDS Soc (2013) 16(1):18472. doi: 10.7448/IAS.16.1.18472
187. Russell RA, Moore MD, Hu WS, Pathak VK. APOBEC3G induces a hypermutation gradient: purifying selection at multiple steps during HIV-1 replication results in levels of G-to-A mutations that are high in DNA, intermediate in cellular viral RNA, and low in virion RNA. Retrovirology (2009) 6:16. doi: 10.1186/1742-4690-6-16
188. Delviks-Frankenberry KA, Nikolaitchik OA, Burdick RC, Gorelick RJ, Keele BF, Hu WS, et al. Minimal contribution of APOBEC3-induced G-to-A hypermutation to HIV-1 recombination and genetic variation. PloS Pathog (2016) 12(5):e1005646. doi: 10.1371/journal.ppat.1005646
189. Monajemi M, Woodworth CF, Zipperlen K, Gallant M, Grant MD, Larijani M. Positioning of APOBEC3G/F mutational hotspots in the human immunodeficiency virus genome favors reduced recognition by CD8 T cells. PloS One (2014) 9(4). doi: 10.1371/journal.pone.0093428
190. Casartelli N, Guivel-Benhassine F, Bouziat R, Brandler S, Schwartz O, Moris A. The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells. J Exp Med (2010) 207(1):39–49. doi: 10.1084/jem.20091933
191. Schubert U, Antón LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature (2000) 404(6779):770–4. doi: 10.1038/35008096
192. Yewdell JW, Nicchitta CV. The DRiP hypothesis decennial: support, controversy, refinement and extension. Trends Immunol (2006) 27(8):368–73. doi: 10.1016/j.it.2006.06.008
193. Goldwich A, Hahn SS, Schreiber S, Meier S, Kampgen E, Wagner R, et al. Targeting HIV-1 Gag into the defective ribosomal product pathway enhances MHC class I antigen presentation and CD8+ T cell activation. J Immunol (2008) 180(1):372–82. doi: 10.4049/jimmunol.180.1.372
194. Ebrahimi D, Anwar F, Davenport MP. APOBEC3 has not left an evolutionary footprint on the HIV-1 genome. J Virol (2011) 85(17):9139–46. doi: 10.1128/JVI.00658-11
195. Poulain F, Lejeune N, Willemart K, Gillet NA. Footprint of the host restriction factors APOBEC3 on the genome of human viruses. PloS Pathog (2020) 16(8):e1008718. doi: 10.1371/journal.ppat.1008718
196. Xu H, Chertova E, Chen J, Ott DE, Roser JD, Hu WS, et al. Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology (2007) 360(2):247–56. doi: 10.1016/j.virol.2006.10.036
197. Nowarski R, Britan-Rosich E, Shiloach T, Kotler M. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat Struct Mol Biol (2008) 15(10):1059–66. doi: 10.1038/nsmb.1495
198. Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol (1995) 69(8):5087–94. doi: 10.1128/JVI.69.8.5087-5094.1995
199. Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood (1977) 50(3):481–92. doi: 10.1182/blood.V50.3.481.481
200. Sasada A, Takaori-Kondo A, Shirakawa K, Kobayashi M, Abudu A, Hishizawa M, et al. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology (2005) 2:32. doi: 10.1186/1742-4690-2-32
201. Ooms M, Krikoni A, Kress AK, Simon V, Munk C. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J Virol (2012) 86(11):6097–108. doi: 10.1128/JVI.06570-11
202. Derse D, Hill SA, Princler G, Lloyd P, Heidecker G. Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. P Natl Acad Sci USA (2007) 104(8):2915–20. doi: 10.1073/pnas.0609444104
203. Fan J, Ma GY, Nosaka K, Tanabe J, Satou Y, Koito A, et al. APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J Virol (2010) 84(14):7278–87. doi: 10.1128/Jvi.02239-09
204. Kypr J, Mrazek J. Unusual codon usage of HIV. Nature (1987) 327(6117):20. doi: 10.1038/327020a0
205. Kypr J, Mrazek J, Reich J. Nucleotide composition bias and CpG dinucleotide content in the genomes of HIV and HTLV 1/2. Biochim Biophys Acta (1989) 1009(3):280–2. doi: 10.1016/0167-4781(89)90114-0
206. Berkhout B, van Hemert FJ. The unusual nucleotide content of the HIV RNA genome results in a biased amino acid composition of HIV proteins. Nucleic Acids Res (1994) 22(9):1705–11. doi: 10.1093/nar/22.9.1705
207. Bronson EC, Anderson JN. Nucleotide composition as a driving force in the evolution of retroviruses. J Mol Evol (1994) 38(5):506–32. doi: 10.1007/BF00178851
208. van Hemert FJ, Berkhout B. The tendency of lentiviral open reading frames to become A-rich: constraints imposed by viral genome organization and cellular tRNA availability. J Mol Evol (1995) 41(2):132–40. doi: 10.1007/BF00170664
209. Martin JL, Maldonado JO, Mueller JD, Zhang W, Mansky LM. Molecular studies of HTLV-1 replication: an update. Viruses-Basel (2016) 8(2). doi: 10.3390/v8020031
210. Mansky LM. In vivo analysis of human T-cell leukemia virus type 1 reverse transcription accuracy. J Virol (2000) 74(20):9525–31. doi: 10.1128/Jvi.74.20.9525-9531.2000
211. Geller R, Domingo-Calap P, Cuevas JM, Rossolillo P, Negroni M, Sanjuan R. The external domains of the HIV-1 envelope are a mutational cold spot. Nat Commun (2015) 6:8571. doi: 10.1038/ncomms9571
212. Pique C, Tursz T, Dokhelar MC. Mutations introduced along the HTLV-I envelope gene result in a non-functional protein: a basis for envelope conservation? EMBO J (1990) 9(13):4243–8. doi: 10.1002/j.1460-2075.1990.tb07872.x
213. Satou Y, Matsuoka M. HTLV-1 and the host immune system: how the virus disrupts immune regulation, leading to HTLV-1 associated diseases. J Clin Exp Hematop (2010) 50(1):1–8. doi: 10.3960/jslrt.50.1
214. Matsuoka M, Jeang KT. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer (2007) 7(4):270–80. doi: 10.1038/nrc2111
215. Boxus M, Twizere JC, Legros S, Dewulf JF, Kettmann R, Willems L. The HTLV-1 tax interactome. Retrovirology (2008) 5:76. doi: 10.1186/1742-4690-5-76
216. Matsuoka M, Yasunaga J. Human T-cell leukemia virus type 1: replication, proliferation and propagation by Tax and HTLV-1 bZIP factor. Curr Opin Virol (2013) 3(6):684–91. doi: 10.1016/j.coviro.2013.08.010
217. Nomura M, Ohashi T, Nishikawa K, Nishitsuji H, Kurihara K, Hasegawa A, et al. Repression of tax expression is associated both with resistance of human T-cell leukemia virus type 1-infected T cells to killing by tax-specific cytotoxic T lymphocytes and with impaired tumorigenicity in a rat model. J Virol (2004) 78(8):3827–36. doi: 10.1128/jvi.78.8.3827-3836.2004
218. Kurihara K, Harashima N, Hanabuchi S, Masuda M, Utsunomiya A, Tanosaki R, et al. Potential immunogenicity of adult T cell leukemia cells in vivo. Int J Cancer (2005) 114(2):257–67. doi: 10.1002/ijc.20737
219. Bangham CRM, Matsuoka M. Human T-cell leukaemia virus type 1: parasitism and pathogenesis. Philos Trans R Soc Lond B Biol Sci (2017) 372(1732). doi: 10.1098/rstb.2016.0272
220. Ma G, Yasunaga J, Matsuoka M. Multifaceted functions and roles of HBZ in HTLV-1 pathogenesis. Retrovirology (2016) 13:16. doi: 10.1186/s12977-016-0249-x
221. Knipe DM, Prichard A, Sharma S, Pogliano J. Replication compartments of eukaryotic and bacterial DNA viruses: common themes between different domains of host cells. Annu Rev Virol (2022) 9:307–27. doi: 10.1146/annurev-virology-012822-125828
222. Weitzman MD, Fradet-Turcotte A. Virus DNA replication and the host DNA damage response. Annu Rev Virol (2018) 5:141–64. doi: 10.1146/annurev-virology-092917-043534
223. Charman M, Weitzman MD. Replication compartments of DNA viruses in the nucleus: location, location, location. Viruses-Basel (2020) 12(2). doi: 10.3390/v12020151
224. De Silva FS, Lewis W, Berglund P, Koonin EV, Moss B. Poxvirus DNA primase. Proc Natl Acad Sci USA (2007) 104(47):18724–9. doi: 10.1073/pnas.0709276104
225. Cotmore SF, Tattersall P. Parvovirus diversity and DNA damage responses. Cold Spring Harb Perspect Biol (2013) 5(2). doi: 10.1101/cshperspect.a012989
226. Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, et al. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol (2006) 16(5):480–5. doi: 10.1016/j.cub.2006.01.031
227. Narvaiza I, Linfesty DC, Greener BN, Hakata Y, Pintel DJ, Logue E, et al. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PloS Pathog (2009) 5(5):e1000439. doi: 10.1371/journal.ppat.1000439
228. Shackelton LA, Parrish CR, Truyen U, Holmes EC. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc Natl Acad Sci USA (2005) 102(2):379–84. doi: 10.1073/pnas.0406765102
229. Jenkins GM, Rambaut A, Pybus OG, Holmes EC. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J Mol Evol (2002) 54(2):156–65. doi: 10.1007/s00239-001-0064-3
230. Gatherer D, Depledge DP, Hartley CA, Szpara ML, Vaz PK, Benko M, et al. ICTV virus taxonomy profile: 2021. J Gen Virol (2021) 102(10). doi: 10.1099/jgv.0.001673
231. Jr. ESM. Comparative analysis of herpesvirus-common proteins. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, et al., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press (2007).
232. Suspene R, Aynaud MM, Koch S, Pasdeloup D, Labetoulle M, Gaertner B, et al. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J Virol (2011) 85(15):7594–602. doi: 10.1128/JVI.00290-11
233. Moraes SN, Becker JT, Moghadasi SA, Shaban NM, Auerbach AA, Cheng AZ, et al. Evidence linking APOBEC3B genesis and evolution of innate immune antagonism by gamma-herpesvirus ribonucleotide reductases. Elife (2022) 11. doi: 10.7554/eLife.83893
234. Shaban NM, Yan R, Shi K, Moraes SN, Cheng AZ, Carpenter MA, et al. Cryo-EM structure of the EBV ribonucleotide reductase BORF2 and mechanism of APOBEC3B inhibition. Sci Adv (2022) 8(17). doi: 10.1126/sciadv.abm2827
235. Dutia BM, Frame MC, Subaksharpe JH, Clark WN, Marsden HS. Specific-inhibition of herpesvirus ribonucleotide reductase by synthetic peptides. Nature (1986) 321(6068):439–41. doi: 10.1038/321439a0
236. Minkah N, Chavez K, Shah P, Maccarthy T, Chen H, Landau N, et al. Host restriction of murine gammaherpesvirus 68 replication by human APOBEC3 cytidine deaminases but not murine APOBEC3. Virology (2014) 454-455:215–26. doi: 10.1016/j.virol.2014.02.022
237. Koning FA, Newman ENC, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol (2009) 83(18):9474–85. doi: 10.1128/Jvi.01089-09
238. Schelhaas M, Jansen M, Haase I, Knebel-Morsdorf D. Herpes simplex virus type 1 exhibits a tropism for basal entry in polarized epithelial cells. J Gen Virol (2003) 84:2473–84. doi: 10.1099/vir.0.19226-0
239. Arvin AM, Moffat JF, Sommer M, Oliver S, Che XB, Vleck S, et al. Varicella-zoster virus T cell tropism and the pathogenesis of skin infection. Curr Top Microbiol (2010) 342:189–209. doi: 10.1007/82_2010_29
240. Karlin S, Mocarski ES, Schachtel GA. Molecular evolution of herpesviruses - genomic and protein-sequence comparisons. J Virol (1994) 68(3):1886–902. doi: 10.1128/Jvi.68.3.1886-1902.1994
241. Vartanian JP, Guetard D, Henry M, Wain-Hobson S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science (2008) 320(5873):230–3. doi: 10.1126/science.1153201
242. Chen ML, Kao WM, Huang JY, Hung YM, Wei JC. Human papillomavirus infection associated with increased risk of new-onset psoriasis: a nationwide population-based cohort study. Int J Epidemiol (2020) 49(3):786–97. doi: 10.1093/ije/dyaa027
243. Westrich JA, Warren CJ, Klausner MJ, Guo K, Liu CW, Santiago ML, et al. Human papillomavirus 16 E7 stabilizes APOBEC3A protein by inhibiting cullin 2-dependent protein degradation. J Virol (2018) 92(7). doi: 10.1128/JVI.01318-17
244. Mori S, Takeuchi T, Ishii Y, Yugawa T, Kiyono T, Nishina H, et al. Human papillomavirus 16 E6 upregulates APOBEC3B via the TEAD transcription factor. J Virol (2017) 91(6):e02413–16. doi: 10.1128/JVI.02413-16
245. Warren CJ, Van Doorslaer K, Pandey A, Espinosa JM, Pyeon D. Role of the host restriction factor APOBEC3 on papillomavirus evolution. Virus Evol (2015) 1(1):vev015. doi: 10.1093/ve/vev015
246. de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology (2004) 324(1):17–27. doi: 10.1016/j.virol.2004.03.033
247. Verhalen B, Starrett GJ, Harris RS, Jiang M. Functional upregulation of the DNA cytosine deaminase APOBEC3B by polyomaviruses. J Virol (2016) 90(14):6379–86. doi: 10.1128/JVI.00771-16
248. Starrett GJ, Serebrenik AA, Roelofs PA, McCann JL, Verhalen B, Jarvis MC, et al. Polyomavirus T antigen induces APOBEC3B expression using an LXCXE-dependent and TP53-independent mechanism. mBio (2019) 10(1):e02690–18. doi: 10.1128/mBio.02690-18
249. Marei HE, Althani A, Afifi N, Hasan A, Caceci T, Pozzoli G, et al. p53 signaling in cancer progression and therapy. Cancer Cell Int (2021) 21(1):703. doi: 10.1186/s12935-021-02396-8
250. Ahlander J, Bosco G. The RB/E2F pathway and regulation of RNA processing. Biochem Biophys Res Commun (2009) 384(3):280–3. doi: 10.1016/j.bbrc.2009.04.107
251. McIlroy D, Peltier C, Nguyen ML, Manceau L, Mobuchon L, Le Baut N, et al. Quantification of APOBEC3 mutation rates affecting the gene of BK polyomavirus in vivo. Viruses-Basel (2022) 14(9). doi: 10.3390/v14092077
252. Baumert TF, Rosler C, Malim MH, von Weizsacker F. Hepatitis B virus DNA is subject to extensive editing by the human deaminase APOBEC3C. Hepatology (2007) 46(3):682–9. doi: 10.1002/hep.21733
253. Beggel B, Munk C, Daumer M, Hauck K, Haussinger D, Lengauer T, et al. Full genome ultra-deep pyrosequencing associates G-to-A hypermutation of the hepatitis B virus genome with the natural progression of hepatitis B. J Viral Hepat (2013) 20(12):882–9. doi: 10.1111/jvh.12110
254. Bonvin M, Achermann F, Greeve I, Stroka D, Keogh A, Inderbitzin D, et al. Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology (2006) 43(6):1364–74. doi: 10.1002/hep.21187
255. Chen R, Zhao X, Wang Y, Xie Y, Liu J. Hepatitis B virus X protein is capable of down-regulating protein level of host antiviral protein APOBEC3G. Sci Rep (2017) 7:40783. doi: 10.1038/srep40783
256. Chen Z, Eggerman TL, Bocharov AV, Baranova IN, Vishnyakova TG, Patterson AP. APOBEC3-induced mutation of the hepatitis virus B DNA genome occurs during its viral RNA reverse transcription into (-)-DNA. J Biol Chem (2021) 297(2):100889. doi: 10.1016/j.jbc.2021.100889
257. Kramvis A, Kostaki EG, Hatzakis A, Paraskevis D. Immunomodulatory function of HBeAg related to short-sighted evolution, transmissibility, and clinical manifestation of hepatitis B virus. Front Microbiol (2018) 9:2521. doi: 10.3389/fmicb.2018.02521
258. Nguyen DH, Gummuluru S, Hu J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J Virol (2007) 81(9):4465–72. doi: 10.1128/JVI.02510-06
259. Suspene R, Guetard D, Henry M, Sommer P, Wain-Hobson S, Vartanian JP. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci USA (2005) 102(23):8321–6. doi: 10.1073/pnas.0408223102
260. Turelli P, Mangeat B, Jost S, Vianin S, Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science (2004) 303(5665):1829. doi: 10.1126/science.1092066
261. Sharma S, Patnaik SK, Taggart RT, Kannisto ED, Enriquez SM, Gollnick P, et al. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat Commun (2015) 6:6881. doi: 10.1038/ncomms7881
262. Sharma S, Patnaik SK, Taggart RT, Baysal BE. The double-domain cytidine deaminase APOBEC3G is a cellular site-specific RNA editing enzyme. Sci Rep (2016) 6:39100. doi: 10.1038/srep39100
263. Sharma S, Baysal BE. Stem-loop structure preference for site-specific RNA editing by APOBEC3A and APOBEC3G. PeerJ (2017) 5:e4136. doi: 10.7717/peerj.4136
264. Sharma S, Patnaik SK, Kemer Z, Baysal BE. Transient overexpression of exogenous APOBEC3A causes C-to-U RNA editing of thousands of genes. RNA Biol (2017) 14(5):603–10. doi: 10.1080/15476286.2016.1184387
265. Sharma S, Wang J, Alqassim E, Portwood S, Cortes Gomez E, Maguire O, et al. Mitochondrial hypoxic stress induces widespread RNA editing by APOBEC3G in natural killer cells. Genome Biol (2019) 20(1):37. doi: 10.1186/s13059-019-1651-1
266. Alqassim EY, Sharma S, Khan A, Emmons TR, Cortes Gomez E, Alahmari A, et al. RNA editing enzyme APOBEC3A promotes pro-inflammatory M1 macrophage polarization. Commun Biol (2021) 4(1):102. doi: 10.1038/s42003-020-01620-x
267. Milewska A, Kindler E, Vkovski P, Zeglen S, Ochman M, Thiel V, et al. APOBEC3-mediated restriction of RNA virus replication. Sci Rep (2018) 8(1):5960. doi: 10.1038/s41598-018-24448-2
268. Woo PC, Wong BH, Huang Y, Lau SK, Yuen KY. Cytosine deamination and selection of CpG suppressed clones are the two major independent biological forces that shape codon usage bias in coronaviruses. Virology (2007) 369(2):431–42. doi: 10.1016/j.virol.2007.08.010
269. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature (2020) 583(7816):459–68. doi: 10.1038/s41586-020-2286-9
270. Kim K, Calabrese P, Wang S, Qin C, Rao Y, Feng P, et al. The roles of APOBEC-mediated RNA editing in SARS-CoV-2 mutations, replication and fitness. Sci Rep (2022) 12(1):14972. doi: 10.1038/s41598-022-19067-x
271. Nakata Y, Ode H, Kubota M, Kasahara T, Matsuoka K, Sugimoto A, et al. Cellular APOBEC3A deaminase drives mutations in the SARS-CoV-2 genome. Nucleic Acids Res (2023) 51(2):783–95. doi: 10.1093/nar/gkac1238
272. Di Giorgio S, Martignano F, Torcia MG, Mattiuz G, Conticello SG. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci Adv (2020) 6(25):eabb5813. doi: 10.1126/sciadv.abb5813
273. Simmonds P. Rampant C–>U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses: causes and consequences for their short- and long-term evolutionary trajectories. mSphere (2020) 5(3). doi: 10.1128/mSphere.00408-20
274. Siqueira JD, Goes LR, Alves BM, de Carvalho PS, Cicala C, Arthos J, et al. SARS-CoV-2 genomic analyses in cancer patients reveal elevated intrahost genetic diversity. Virus Evol (2021) 7(1):veab013. doi: 10.1093/ve/veab013
275. Kosuge M, Furusawa-Nishii E, Ito K, Saito Y, Ogasawara K. Point mutation bias in SARS-CoV-2 variants results in increased ability to stimulate inflammatory responses. Sci Rep (2020) 10(1):17766. doi: 10.1038/s41598-020-74843-x
276. Perelygina L, Chen MH, Suppiah S, Adebayo A, Abernathy E, Dorsey M, et al. Infectious vaccine-derived rubella viruses emerge, persist, and evolve in cutaneous granulomas of children with primary immunodeficiencies. PloS Pathog (2019) 15(10):e1008080. doi: 10.1371/journal.ppat.1008080
Keywords: APOBEC3, C-to-U/G-to-A mutations, deamination, retroviruses, DNA viruses, RNA viruses, virus evolution
Citation: Jonathan M and Ikeda T (2023) APOBEC3 family proteins as drivers of virus evolution. Front. Virol. 3:1332010. doi: 10.3389/fviro.2023.1332010
Received: 02 November 2023; Accepted: 04 December 2023;
Published: 18 December 2023.
Edited by:
Yasumasa Iwatani, National Hospital Organization (NHO), JapanReviewed by:
Hang Su, Albert Einstein College of Medicine, United StatesJean-Christophe Paillart, Université de Strasbourg, France
Xiaojiang Chen, University of Southern California, United States
Copyright © 2023 Jonathan and Ikeda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Terumasa Ikeda, aWtlZGF0QGt1bWFtb3RvLXUuYWMuanA=