
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Virol. , 25 July 2023
Sec. Viral Diversification and Evolution
Volume 3 - 2023 | https://doi.org/10.3389/fviro.2023.1221156
 Wendy G. Marchant1†
Wendy G. Marchant1† Habibu Mugerwa1†
Habibu Mugerwa1† Saurabh Gautam1Hamed Al-Aqeel2,3Jane E. Polston3Gabriel Rennberger4,5,6Hugh Smith4Bill Turechek5Scott Adkins5
Saurabh Gautam1Hamed Al-Aqeel2,3Jane E. Polston3Gabriel Rennberger4,5,6Hugh Smith4Bill Turechek5Scott Adkins5 Judith K. Brown7
Judith K. Brown7 Rajagopalbabu Srinivasan1*
Rajagopalbabu Srinivasan1*Tomato yellow leaf curl virus (TYLCV) is a monopartite DNA virus with a genome size of ~ 2,800 base pairs. The virus belongs to the genus Begomovirus within the family Geminiviridae. Extant TYLCV strains are differentiated based on an established threshold of 94% genome-wide pairwise nucleotide identity. The phylogenetic relationships, diversification mechanisms, including recombination, and extent of spread within and from the center of origin for TYLCV have been reported in previous studies. However, the evolutionary relationships among strains, strains’ distribution and genomic diversification, and genetic mechanisms shaping TYLCV strains’ evolution have not been re-evaluated to consider globally representative genome sequences in publicly available sequence database, including herein newly sequenced genomes from the U.S. and Middle East, respectively. In this study, full-length genome sequences for the extant strains and isolates of TYLCV (n=818) were downloaded from the GenBank database. All previously published genome sequences, and newly sequenced TYLCV genomes of TYLCV isolates from Kuwait and USA, determined herein (n=834), were subjected to recombination analysis. To remove the ‘phylogenetic noise’ imparted by interspecific recombination, the recombinant genomes were removed from the data set, and the remaining non-recombinant genome sequences (n=423) were subjected to population genetics and Bayesian analyses. Results of the phylogeographical analysis indicated that the type strain, TYLCV-Israel, and TYLCV-Mild strain, were globally distributed, spanning Africa, America, Asia, Australia/Oceania, Europe, and New Caledonia, while the other TYLCV strains were prevalent only throughout the Middle East. The results of Bayesian evolutionary (ancestral) analysis predicted that TYLCV-Israel represents the oldest, most recent common ancestor (MRCA) (41,795 years), followed by TYLCV-Mild at 39,808 years. These were closely followed by two Iranian strains viz., TYLCV-Kerman and TYLCV-Iran at 37,529 and 36,420 years, respectively. In contrast, the most recently evolving strains were TYLCV-Kuwait and TYLCV-Kahnooj at 12,445 and 298 years, respectively. Results of the neutrality test indicated that TYLCV-Israel and TYLCV-Mild populations are undergoing purifying selection and/or population expansion, although statistically significant selection was documented for only TYLCV-Israel, based on positive selection acting on five codons.
Viruses in the family Geminiviridae are pathogens of plants that are transmitted by hemipteran insects. Members of this plant virus group cause diseases resulting in yield loss and reduced quality of fiber, ornamental, and vegetable crops (1, 2). Begomoviruses have a circular, single-stranded DNA genome that can be bipartite, referred to as the DNA-A (~2.6 kb) and DNA-B (~2.5–2.6 kb) components, or monopartite, with one DNA molecule of approximately 2.8 kb in size. Each DNA fragment is encapsidated within a virion that has a twinned icosohedral or ‘geminate’ structure (3).
The genus, Begomovirus is the largest of nine genera within the Geminiviridae (4). The viruses in this genus are transmitted in a persistent, circulative, and non-propagative manner by the whitefly Bemisia tabaci (Gennadius) cryptic species (family Aleyrodidae) and infect primarily dicotyledonous plants (5–7). Among begomovirus species, Tomato yellow leaf curl virus, has been the most extensively studied because of its economic importance (8). The abundance of full-length genome sequences available in public databases for tomato yellow leaf curl virus (TYLCV) has provided a valuable resource for investigating begomoviral intraspecific and interspecific evolutionary relationships (2, 9). Also, the small genome size has facilitated molecular cloning and sequencing of many, globally-representative, begomovirus genomes (8). Different strains of TYLCV causes leaf curl disease of tomato and infect other crop species, with several strains being particularly widely distributed from the zone(s) of endemism primarily within the last 50 years, by human-mediated transport of infected plants and/or the viruliferous whitefly vector (2, 10–14).
The genome of TYLCV is approximately 2,800 base-pairs in size and encodes six genes. Two genes, V1 and V2, are encoded on the virus sense strand. The V1 open reading frame (ORF) encodes the capsid protein, while the V2 ORF encodes a protein involved in modulating host symptom expression and influences virus accumulation (15). Four genes, designated as C1–C4, are encoded on the viral complementary strand (16). The C1 ORF encodes the viral replication-associated protein, while C2 encodes the transcriptional activator protein involved in the suppression of host plant post-transcriptional gene silencing. The C3 and C4 ORFs encode the replication enhancer protein and a determinant of host symptom development and severity, host range, and systemic spread in the host plant, respectively (16–19). The highly variable intergenic region (IR) of approximately 200 nucleotides in length, contains the promoters for transcription of the viral-sense genes, V1 and V2, and complementary sense C1 and C4 genes (15).
Disease symptoms in tomato plants caused by TYLCV include stunted growth, leaf chlorosis, and curling. Reduced fruit yield was observed for the first time in Israel and Jordan during the 1930s and 1940s, but the causal agent was not identified until the 1960s when the genome sequence was determined, earning notoriety as the first monopartite begomovirus (20, 21). The initial spread of two strains of TYLCV-Mild and TYLCV-Israel from Israel and Jordan occurred during the 1990s (9, 22) through international trade of ornamentals and vegetable seedlings, either virus-infected or harboring the viruliferous B. tabaci vector (23–25).
The vector of TYLCV, B. tabaci, is a complex of cryptic species that are morphologically indistinguishable but possess distinct biological and behavioral characteristics that have facilitated adaptation to different environments (26–29). They can be differentiated phylogeographically based on divergence of the rapidly evolving cytochrome oxidase subunit I gene (30–32). Recently, six cryptic species groups have been recognized based on analysis of 2,184 nuclear orthologs (7, 32, 33). The TYLCV vector native to the Middle East and adjacent areas is the B mitotype [North Africa-Middle East (NAF-ME) cryptic species (previously, B biotype)] (34), which has co-evolved with the TYLCV begomovirus species (26). Several studies have shown that the Q mitotype, or the North Africa-Mediterranean (NAF-MED) cryptic species, also transmits TYLCV (35–37), an observation that is consistent with the predicted close evolutionary relationship among members of the NAF-MED-ME cryptic species group (7, 33). The cryptic species of the NAF-MED-ME group harbor invasive haplotypes, and they have displaced certain native B. tabaci species and established in vegetable and fiber cropping systems throughout the world (26, 37–44). The dispersal of certain B. tabaci cryptic species and its co-evolved strains of TYLCV also has been documented on six continents, including Africa (45, 46), Asia (47, 48), Australia (49), Europe (35, 50–53), North America and the Caribbean region (11, 54, 55), and South America (56, 57).
The species TYLCV as a tomato pathogen has been studied at the phylogenetic, population genetics, and biological levels, to elucidate virus origins and diversification patterns and to facilitate resistance breeding efforts (14, 22, 58–63). Interestingly, TYLCV accumulates mutations nearly as rapidly as single-stranded RNA viruses, at a rate of 2.88 × 10−4 substitutions/site/year (9). Additionally, mutations were found to accumulate at a higher rate in the viral intergenic compared with coding regions (62). Such a high substitution rate is consistent with the ‘quasispecies’ concept, recognized for single-stranded RNA plant and animal viruses (25, 64). This concept envisions quasi-species as having no ‘specific members’ that exists within the host as closely related variants of a population. In addition, population dynamics of a virus quasi-species is hypothesized to facilitate virus adaptation to the environment through the exploration of landscape fitness (65). The high rates of mutation and genetic recombination as powerful forces in the evolution of begomoviruses, including TYLCV, are well-established (9, 25).
The dispersal of TYLCV-infected plant material and/or of TYLCV-viruliferous whiteflies has contributed to the global spread of two of the several other well-characterized strains, TYLCV-Israel and TYLCV-Mild (59, 66); however, the basis for the restricted distribution of the other extant strains endemic to the Middle East and adjacent vicinities where the NAF-ME-MED cryptic species co-occur, is not understood. The objective of this study was to use all TYLCV genome sequences available (as of 2022), for a comprehensive analysis of the genomic evolution of extant TYLCV strains. A curated genome sequence set consisting of recently determined genome sequences from U.S. and Middle East isolates, and globally- and temporally-representative reference sequences from the GenBank database were examined with respect to phylogenomics, population genetics signatures reminiscent of gene flow and genetic differentiation, evidence for neutral or positive selection within viral coding regions, and fixed mutations. The goal was to reveal potentially static and dynamic diversification scenarios that could shape new population structures and invasion paths or hotspots of TYLCV strains and variants that could emerge. The analyses considered all TYLCV genomes available in the NIH-NBCI-GenBank database and recently sequenced TYLCV genomes from US TYLCV-hotspots (Georgia and Florida) and Kuwait. The TYLCV reference genomes represented all recognized strains of TYLCV (67). Recombinant genomes were not included in the population genetics analyses because of their potential for inferring unreliable evolutionary history.
Representative genome sequences were downloaded from the GenBank database, representing the following seven TYLCV strains, TYLCV-Israel: X15656, TYLCV-Boushehr: GU076454, TYLCV-Iran: AJ132711, TYLCV-Kahnooj: EU635776, TYLCV-Kerman: GU076442, TYLCV-Mild: X76319 and TYLCV-Oman: FJ956700. The genome sequences were queried using the Basic Local Alignment Search Tool (BLAST) available at the National Center for Biotechnology Information (NCBI) database. Among the obtained genome sequences, a 94% pairwise nucleotide identity threshold was applied as the strain cut-off, and the requirement for at least 90% coverage to the queried sequence (67). The initial number of downloaded genomes for each strain available in GenBank were: TYLCV-Israel = 849, TYLCV-Boushehr = 3, TYLCV-Iran = 33, TYLCV-Kahnooj = 37, TYLCV-Kerman = 11, TYLCV-Mild = 215 and TYLCV-Oman = 33. To enhance computing efficiency, highly similar genome sequences and therefore redundant at the isolate level, were uniformly removed across all strains with a filter query coverage, at 100% and 99%, respectively. Twenty-two previously unpublished sequences from the Middle East (Kuwait), determined in the Florida laboratories were also included in the initial data set. In these instances, the DNA samples were subjected to rolling circle amplification to obtain complete sequences following the protocols described by Inoue-Nagata et al. (68) and modifications by Marchant et al. (69). The total number of complete genome sequences for each TYLCV strain was: TYLCV-Israel (n = 709), TYLCV-Boushehr (n = 2), TYLCV-Iran (n = 11), TYLCV-Kahnooj (n = 4), TYLCV-Kerman (n = 11), TYLCV-Mild (n = 48), TYLCV- Kuwait (16), and TYLCV-Oman (n = 33).
The TYLCV genome sequences were subjected to recombination analysis using the Recombination Detection Program (RDP4) v4.100 (70). A genome was considered to represent a recombinant when a significant result was identified for three or more of the seven methods available in the RDP4 software, namely, RDP, GENECONV, Bootscan, Maxchi, Chimaera, SiSscan, and 3Seq. A predicted event was confirmed by a shift in tree position based on phylogenetic analysis of the genome sequence, minus the predicted recombined region(s) (Table 1). The predicted recombinant genomes were removed from the data set, leaving 422 remaining TYLCV genomes. The genome sequences were labeled to indicate strain, GenBank accession number, and country of collection.
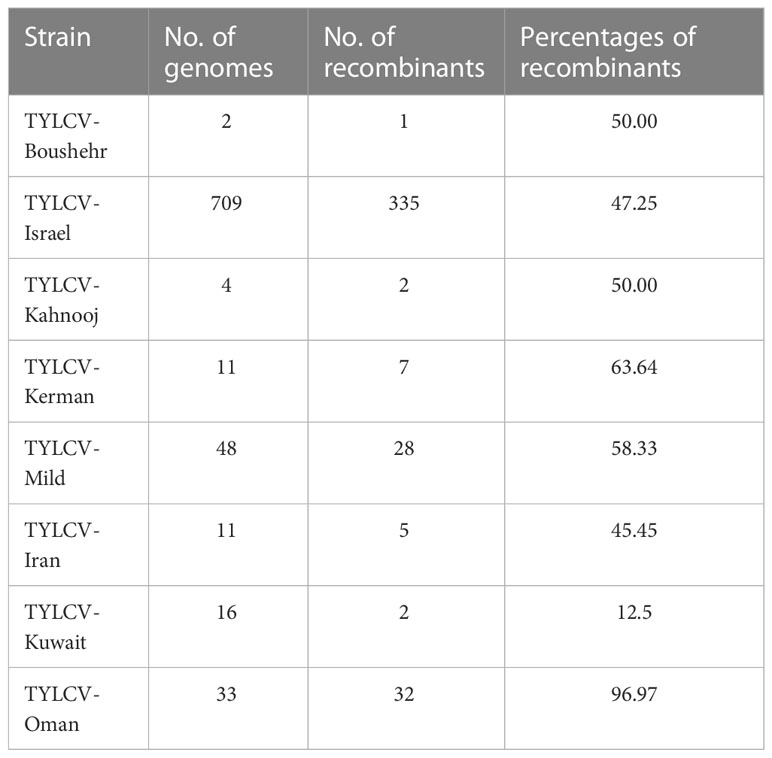
Table 1 Number and percentage of TYLCV recombinant genomes for each strain.
The evolutionary analysis was carried out using the Bayesian Evolutionary Analyses Sampling Trees algorithm (BEAST v2.7.4) (71). The optimal nucleotide substitution model for coding and untranslatable regions was identified using PartitionFinder2 and selecting the BEAST suitability option (72). PartitionFinder2 suggested models were approximated to the closest umbrella models available in BEAST. The Generalized Time Reversible (GTR) was identified as the best fitting model based on the lowest Bayesian Information Criterion (BIC) Scores for all the coding and untranslatable genomic regions selected. The genome sequences and nucleotide substitution model parameters were exported in BEAUti2: Standard. Also, a gamma site heterogeneity model was selected to permit an analysis that considered the between-site-variation. The sequence data were partitioned into six taxa, each representing a strain of TYLCV. TYLCV-Boushehr and TYLCV-Oman strains were not partitioned because they were represented by a single sequence. A Strict clock model was selected, and the coalescent constant population model was set as the tree prior, under the default assumption of uniform distribution of taxon sets. The calibration time limits for estimating the Most Recent Common Ancestor (MRCA) per species were set to 290 and 40211 years ago, based on previously published calibrators (58). The Monte Carlo Markov Chain was set to run at 75,000,000 with trace log generated for every 1000 trees. The subsequent Xml file was used as an input file to BEAST with which two runs were carried out. The combined tracer (log) file was produced with LogCombiner, and the MCMC output file was visualized using Tracer v1.7.1 (73). TREEANNOTATOR was used to select a tree from among all trees sampled and available in the BEAST output files. The maximum clade credibility tree was reconstructed. The tree was drawn using the interactive tree of life software (iTOL v6- https://itol.embl.de).
Polymorphic traits associated with the TYLCV genomes were characterized in DnaSP v6.12.03 using the DNA Polymorphism tool (74). Six TYLCV strains were subject to this analysis, excluding TYLCV-Boushehr and TYLCV-Oman, the only genome representing this strain. Polymorphic traits examined included the number of polymorphic sites and haplotypes, haplotype diversity, nucleotide diversity (Pi), and average number of nucleotide differences (k). Polymorphism analysis was conducted with all the samples in each strain group as well as with only two randomly selected samples (genome sequences from each strain grouping) to offset the influence of vastly differing sample sizes.
The extent of genomic differentiation was analyzed for six TYLCV strains, both within and between strains. The statistical analyses, Ks, Kst, Snn, Z, and Fst (75) were carried out using DnaSP and the Gene Flow and Genetic Differentiation tool. A permutation test with 1000 replications was conducted to test for levels of significance, with a p-value of < 0.05 considered significant.
To carry out a reliable population neutrality test required a minimum of four genome sequences, a criterion that limited the analysis to only five strains, TYLCV-Iran, TYLCV-Israel, TYLCV-Mild, TYLCV-kerman, and TYLCV-Kuwait. Tajima’s D test (76) estimated the average number of pairwise nucleotide differences, and the number of segregating sites among the genome sequences to calculate a value on a strain-by-strain basis. The Fu and Li’s D and F statistics (77) were computed for the TYLCV strains using the Fu and Li’s Tests tool in DnaSP. The D statistic was based on the number of mutations occurring only once, and the total number of mutations. The F statistic was calculated based on the number of mutations occurring once and the pairwise distances between sequences.
The six TYLCV coding regions for the selected TYLCV genomes were analyzed for potential positive selection. The HyPhy tool (78) in MEGA X (79, 80) was used to identify viral codons evolving under positive selection, based on the number of non-synonymous (dN) and synonymous (dS) substitutions per codon. The Tamura-Nei model (81) was identified as the most parsimonious model for calculating the rate substitution, based on codons with a dN greater than dS, and a p-value less than 0.05, which are considered evidence that a coding region is evolving under positive selection.
The geographic collection site for each TYLCV genome sequence analyzed in this study was provided and used to draw the distribution map with the ArcGIS online tool (https://usg.maps.arcgis.com/home/index.html).
Overall, 59 separate recombination events were observed (Supplementary Table 1). Of which, 44 were significant in at least three of the tests conducted using RDP4 (Table 1). The TYLCV-Oman genome sequences showed evidence of the highest percentage of recombination events, followed by, TYLCV-Kerman, TYLCV-Mild, TYLCV- Boushehr, TYLCV-Kahnooj, and TYLCV-Iran. TYLCV-Kuwait had the least number of recombinants of all strains/isolates (Table 1). The events, though occurring in the same regions, in most instances were unique to each strain data set evaluated. The events did not follow a pattern and were distributed across all six genes on the complementary as well as virus strands and on the untranslatable intergenic regions (UTRs). All are detailed in an Excel spread sheet (Supplementary Tab1e 1). Based on the RDP analysis an example is illustrated below, the genome of MH507499.1 Iran was predicted to have resulted from recombination between the major parent AJ132711.1 Iran and secondary (minor) parent JQ928349.1 Iran. The MH507499.1 Iran sequence shared its high nucleotide identity with AJ132711.1 Iran at bases of the predicted recombination site comprising the 1–130 and 913–2808 nucleotides. In addition, it shared high nucleotide identity with JQ928349.1 Iran at the predicted recombination site consisting of a fragment located at nucleotides 131 to 912 within the genome. This recombination event is supported by the results of phylogenetic analyses (Supplementary Figure 1).
The Markov Chain Monte Carlo (MCMC) analysis yielded an effective sample size (ESS) of >200 relative to the posterior parameters, indicating convergence (Table 2). The summary statistics for the most recent common ancestor (MRCA) per strain are presented in the Tracer panel (Table 2; Figure 1). The MRCA was calculated for each TYLCV strain except for TYLCV-Boushehr and TYLCV-Oman, which were represented by only one non-recombinant genome sequence. The TYLCV-Israel strain was the most ancient at 41,795 years, followed by TYLCV-Mild at 39,808 years. These strains were then closely ensued by TYLCV-Kerman and TYLCV-Iran at 37,529 and 36,420 years, respectively. TYLCV-Kuwait and TYLCV-Kahnooj strain were identified as the most recently diversifying isolates, at 12,445 and 298 years, respectively.
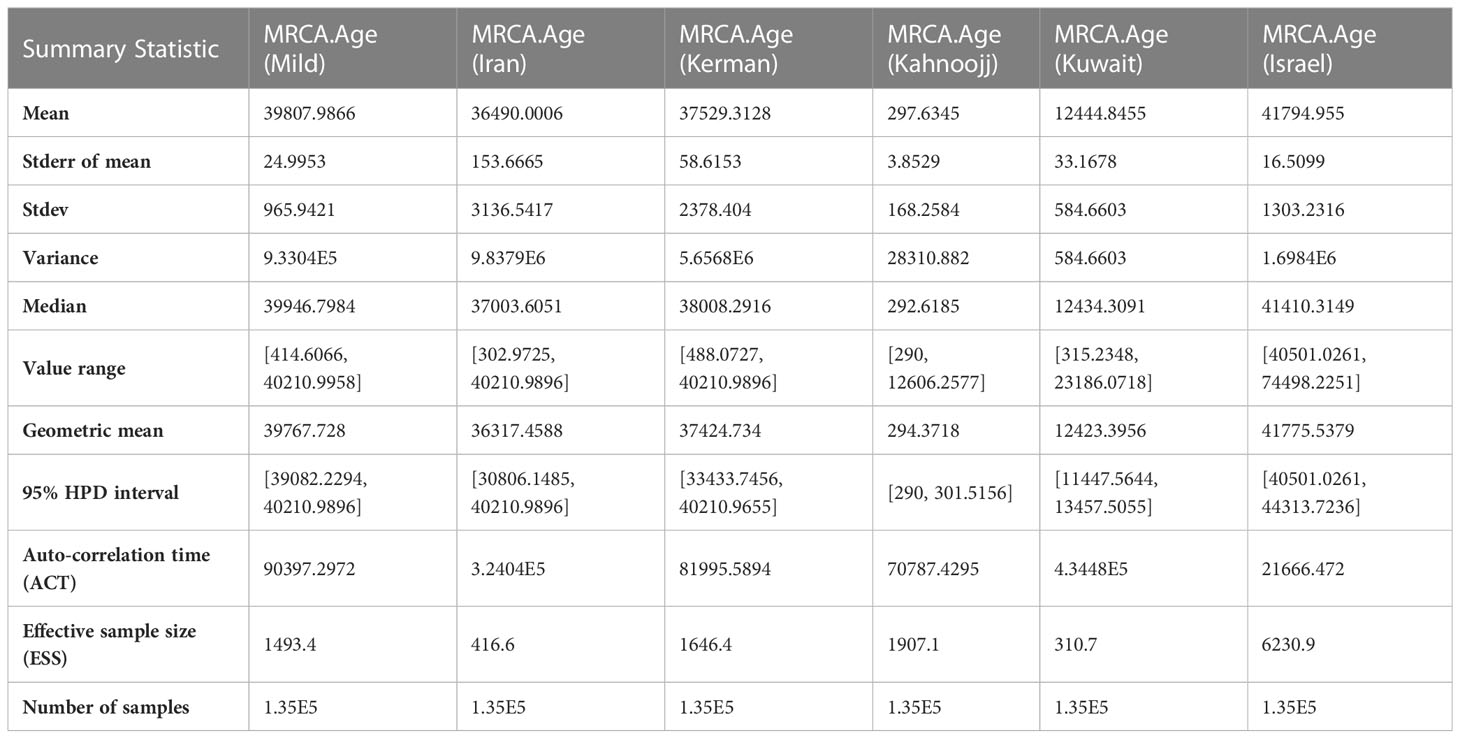
Table 2 Summary Statistics for the time of most recent common ancestor (MCRA) among six strains, as determined by BEAST analysis.
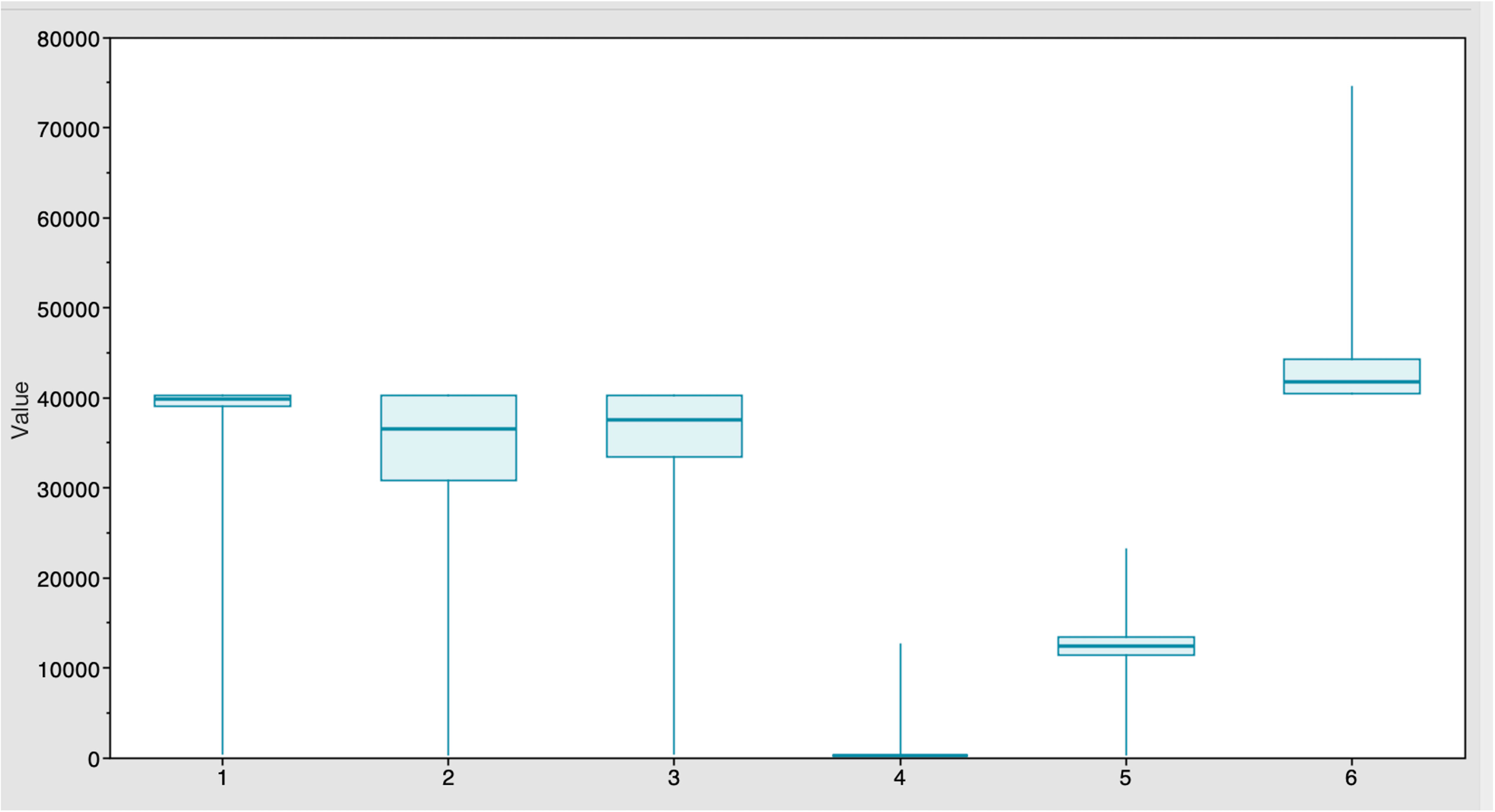
Figure 1 Estimates of time of the most recent common ancestor (MRCA) among six strains after BEAST runs. The TYLCV-Israel strain was the most ancient, at 41,795 years, followed by TYLCV-Mild at 39,808 years. These strains were followed by TYLCV-Kerman and TYLCV-Iran at 37,529 and 36,420 years, respectively. TYLCV-Kuwait and TYLCV-Kahnooj strains were identified as the most recently diversifying strains, at 12,445 and 298 years, respectively.
The Tracer marginal density panel kernel density estimate (kda) plots (Figure 2) showed that probability mass distribution for each strain occurred at 290 and 40211 years ago. The Tracer joint-marginal panel (Figure 3) between the six TYLCV strains illustrate the strength and direction of each correlation. The ellipticals indicate the extent of reinforced strength of the correlations, with no correlation appearing as a circle and perfect correlation as a line (Figure 3). Finally, the Trace panel showing line plots connecting sequential samples of six TYLCV strains against character (Figure 4) was suggestive of admixture between Israel and Mild strains based on the MRCA values, which were similar. In addition, admixtures were also observed among the Kerman and Iran strains. The Kuwait strain rarely showed admixture with the other strains. The mean substitution rate for all TYLCV genomes analyzed here was 2.7899 × 10−4 substitutions/site/year (95% HPD with a range of 3.7339 × 10−5 to 4.7489 × 10−5).
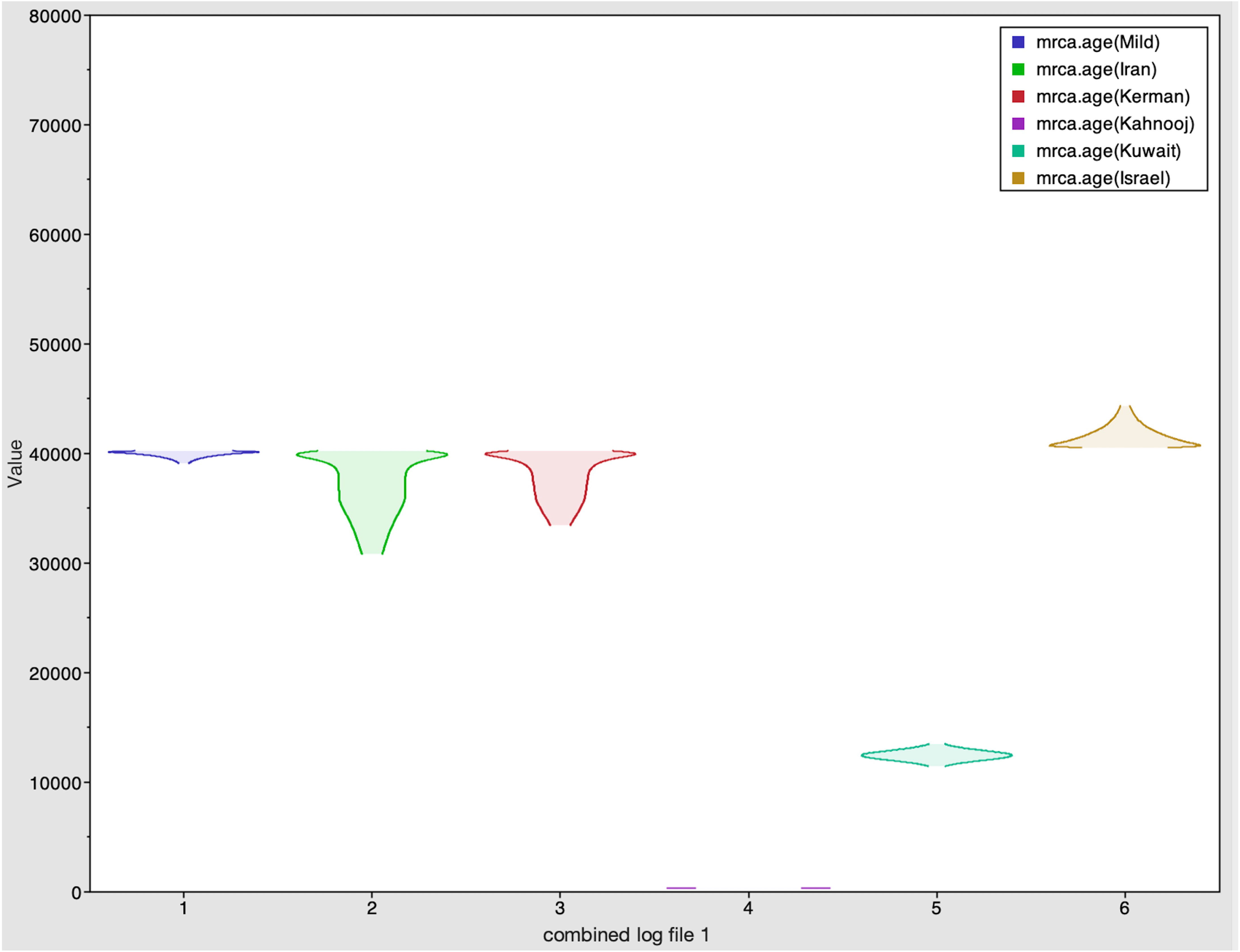
Figure 2 Marginal density plot showing kernel density estimates for the six strains. These plots illustrate each strain's probability mass distribution between 290 and 40211 years ago.
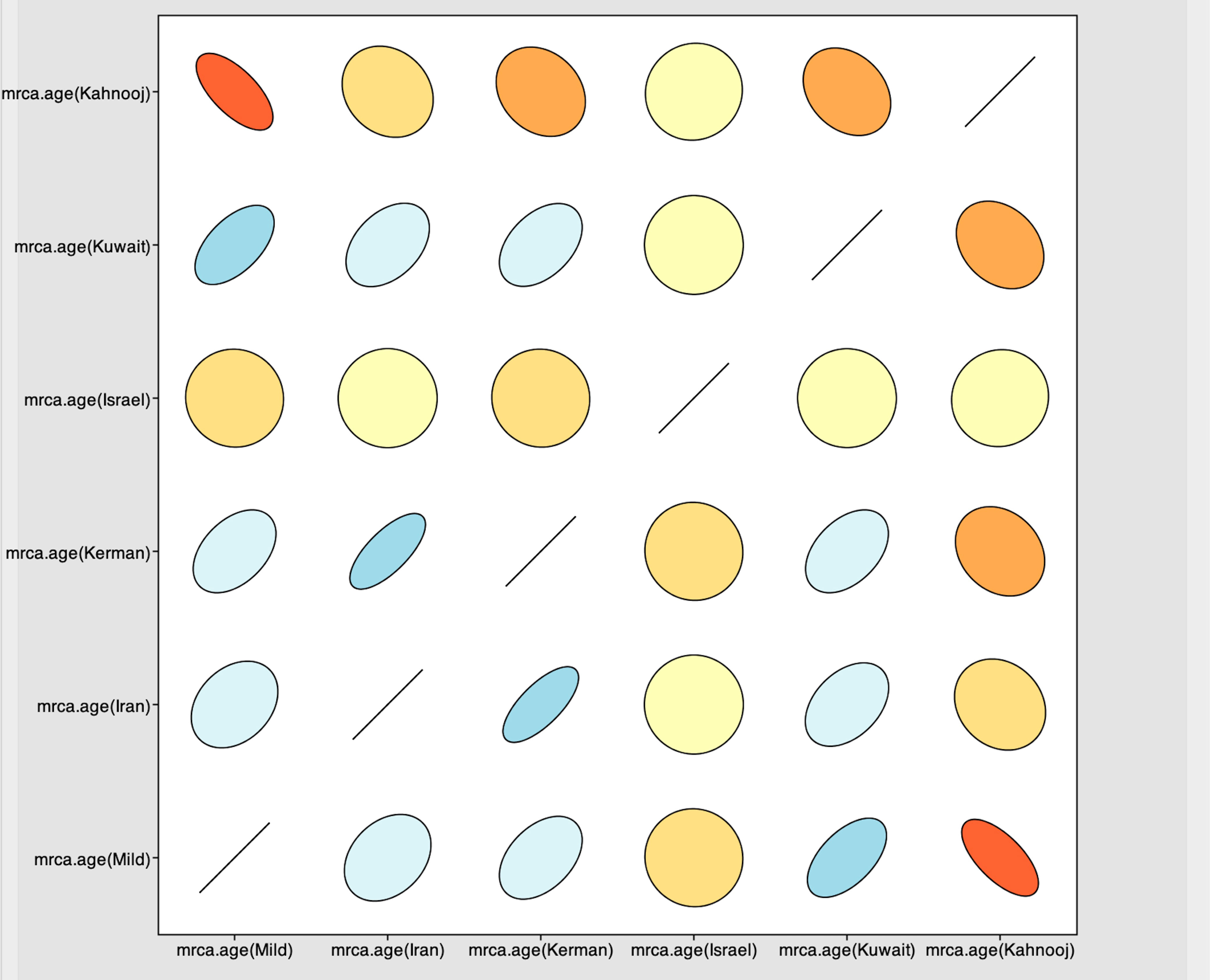
Figure 3 Joint-marginal plot showing extension for correlations between strains – color gradients indicate the strength and direction of the correlation with red and blue indicating strong positive correlation compared with yellow and beige. The ellipse shapes reinforce the strength of correlation, with no correlation appearing as a circle and perfect correlation as a line.
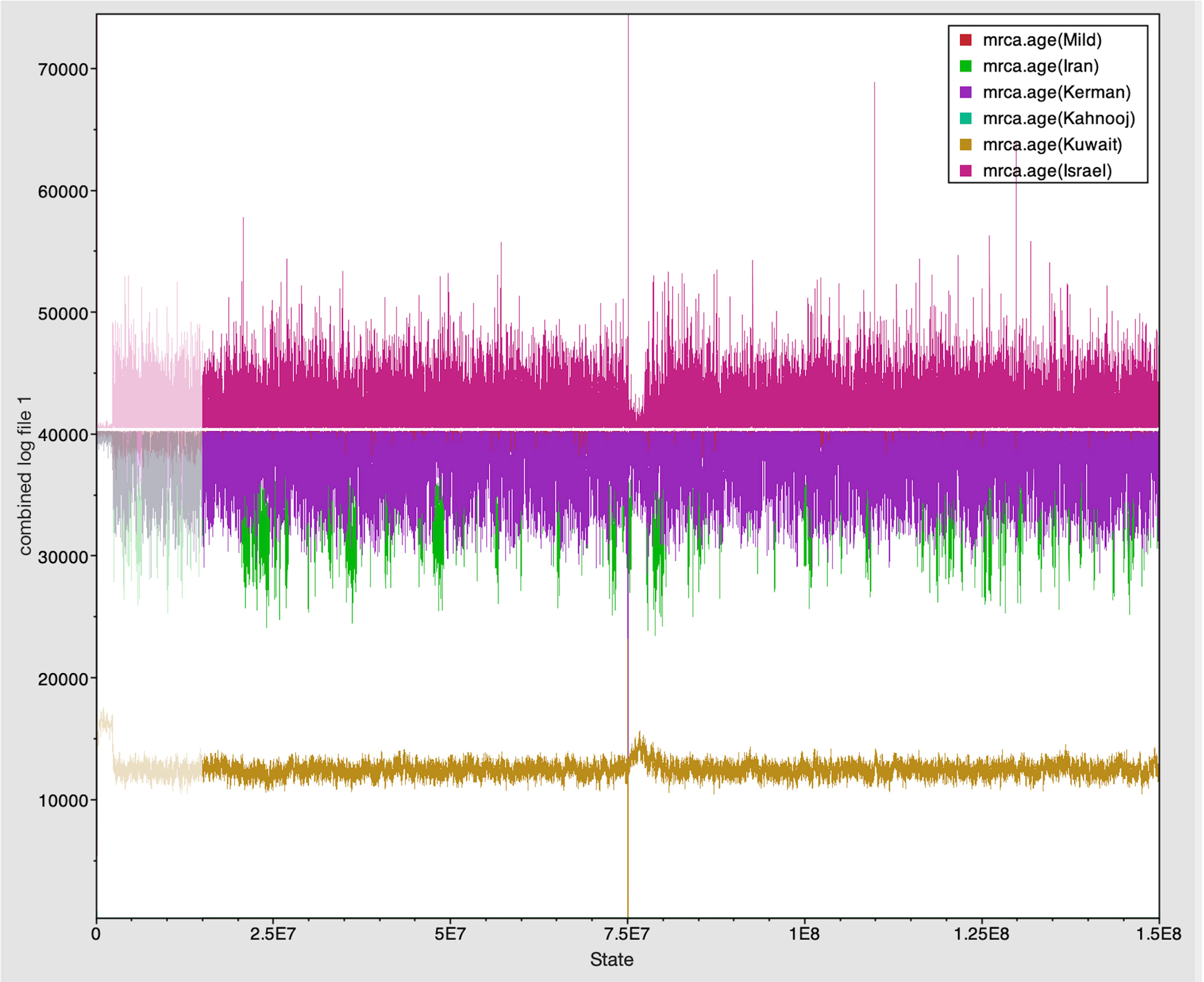
Figure 4 Trace panel displayed line plots connecting the sequential samples of the six TYLCV strains against state. The plot shows mixing between strains with similar MRCA.
In the Bayesian tree, TYLCV genomes grouped with the genome sequences determined in this study as multiple clades (Figure 5). The TYLCV-Israel strain was represented by the greatest number of sequences and formed a monophyletic clade, and consisted of isolates from Africa (Egypt, Tunisia, Morocco, Mauritius, Reunion), Asia (India, Japan, South Korea, China), Australia, the Caribbean region (Cuba, Dominican Republic, and Puerto Rico), Eastern Mediterranean (Jordan, Turkey, Lebanon), Europe (Estonia, Italy, Spain, UK), New Caledonia, and North America (Mexico and USA). However, a few TYLCV-Israel sequences were present in clades with other strain groups such as TYLCV-Mild and TYLCV-Kuwait. A majority of the TYLCV-Mild (70%) sequences were in multiple sister clades within a single node. The TYLCV-Mild isolates formed a monophyletic group together with sequences for isolates from Africa (Reunion), Asia (Japan), Europe (Portugal, Spain, and Sweden), the Eastern Mediterranean (Israel, Jordan and Lebanon), the Caribbean region (Dominican Republic), and South America (Venezuela). The remaining TYLCV-Mild sequences were clustered in sister clades with TYLCV-Israel. Two Iranian isolate sequences (MF536415.1 and KY825714.1) were in a sister clade with other TYLCV isolates from viz., Boushehr, Kerman, and Kahnooj thereby forming a monphyletic clade. Whereas a few other Iranian isolate sequences were found in a sister clade with TYLCV-Israel and TYLCV-Kuwait. TYLCV-Kuwait and TYLCV Oman sequences did not form a monphyletic group and were often found in clades with TYLCV-Israel. The results of this analysis are supported by previous studies that also reported the global distribution of TYLCV-Israel and TYLCV-Mild, compared with TYLCV-Oman, TYLCV-Iran, TYLCV-Kahnooj, TYLCV-Kerman and TYLCV-Boushehr strains, which are restricted to the Middle East (Figure 6) (14, 22, 60).
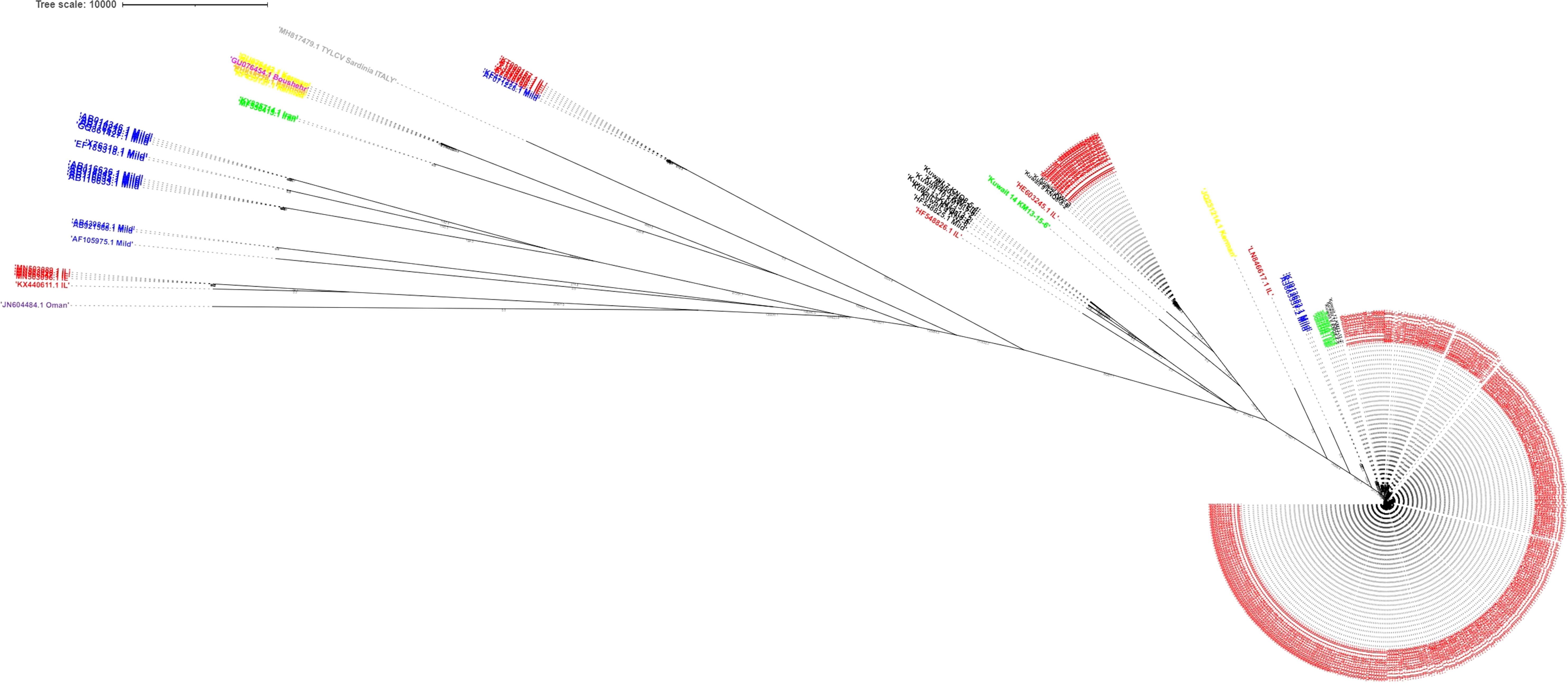
Figure 5 Bayesian tree constructed for tomato yellow leaf curl virus (TYLCV) strains. Genomes are color-coded by strain group: red indicates TYLCV(-Israel), blue indicates TYLCV-mild, green indicates TYLCV-Iran, orange indicates TYLCV-Kahnooj, pink indicates TYLCV-Boushehr, yellow indicates TYLCV-Kerman, and black purple indicates TYLCV-Kuwait. TYLCV Sardinia virus is in Grey. The scale represents that the tree branches are drawn to scale with respect to time.
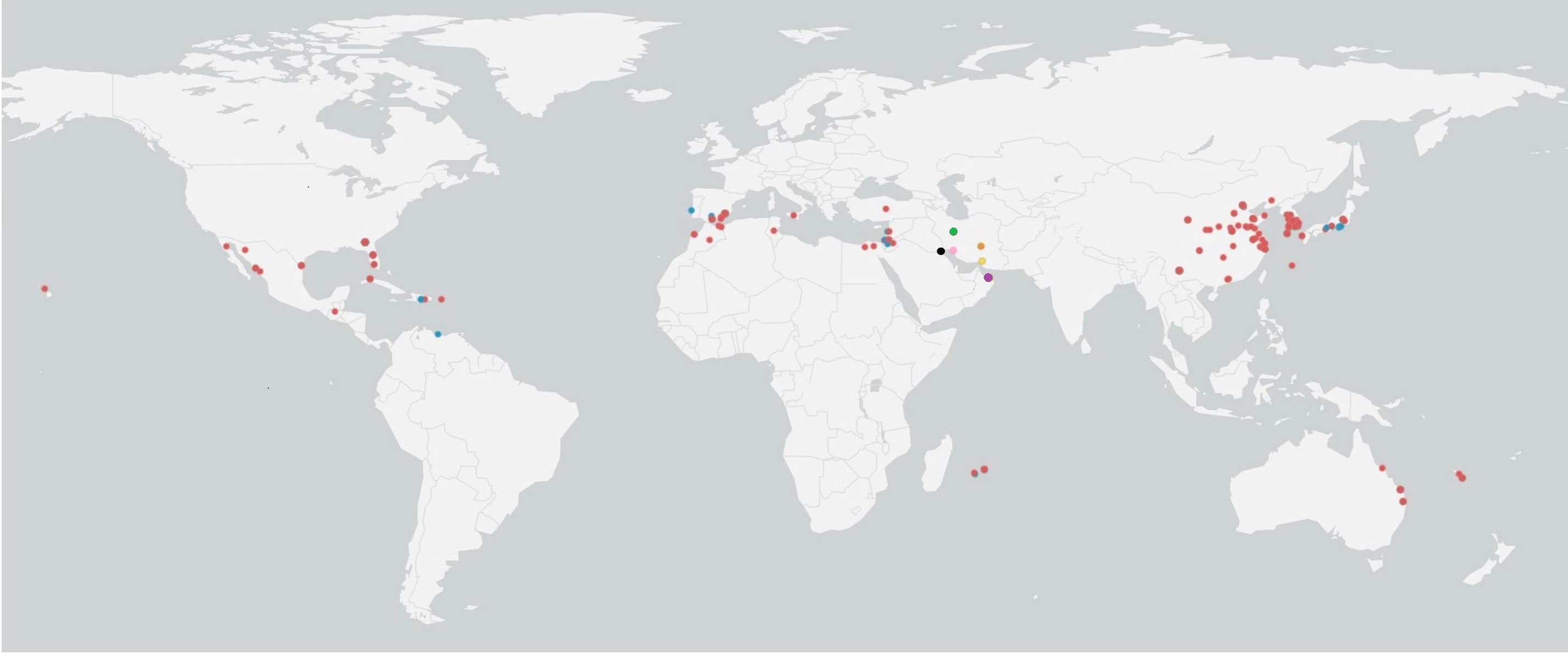
Figure 6 Global distribution of tomato yellow leaf curl virus (TYLCV) strains. Colored dots represent TYLCV strains: red dots indicate TYLCV-Israel, blue dots indicates TYLCV-Mild, green dots indicate TYLCV-Iran, orange dots indicate TYLCV-Kahnooj, pink dots indicate TYLCV-Boushehr, yellow dots indicate TYLCV-Kerman, and black dots indicate TYLCV-Kuwait.
An analysis of genomes for number of polymorphic sites was carried out for all TYLCV strains, except TYLCV-Boushehr and TYLCV-Oman (Table 3). High haplotype diversity was noticed in the case of all TYLCV strains regardless of sample size. The number of polymorphic sites were the highest in TYLCV-Mild followed by TYLCV-Israel and TYLCV-Kuwait. TYLCV-Kahnooj had the fewest polymorphic sites. The highest average number of nucleotide differences (k) – was observed in the case of TYLCV-Mild followed by TYLCV-Kuwait and two Iranian strains TYLCV-Kerman and TYLCV-Iran (Table 3). The results of the analyses may in some instances could have resulted from bias related to too few sequences per group. To overcome the above-stated bias, the polymorphism test was conducted with the same number of randomly selected sequences for all isolates (n = 2) (Table 4). In this scenario, the same haplotype diversity was reflected across all strains (Table 4). Also, the highest average number of nucleotide differences (k) was observed in the case of TYLCV-Iran followed by TYLCV-Israel, and two Iranian strains (Kahnooj and Kerman).
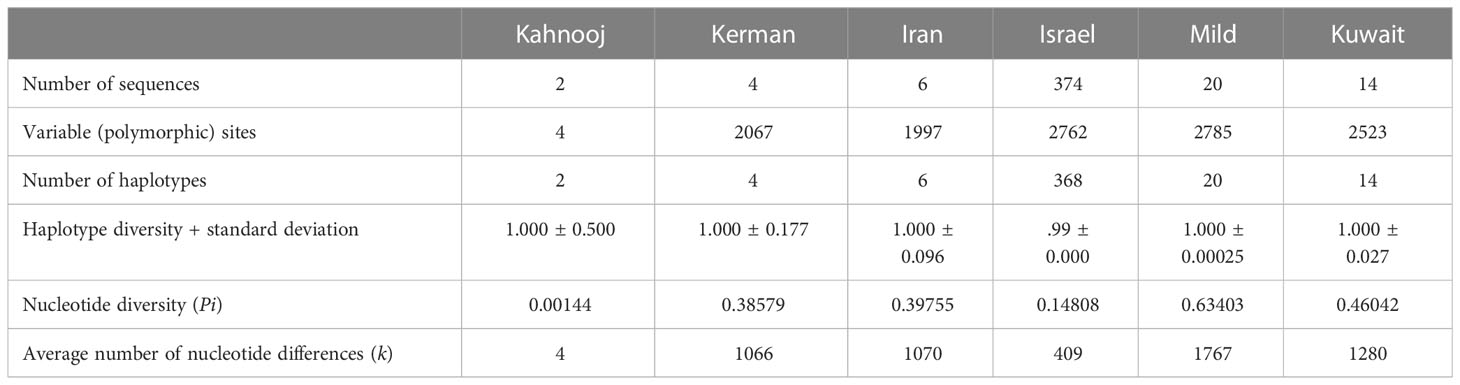
Table 3 Nucleotide polymorphisms among strains of TYLCV worldwide.
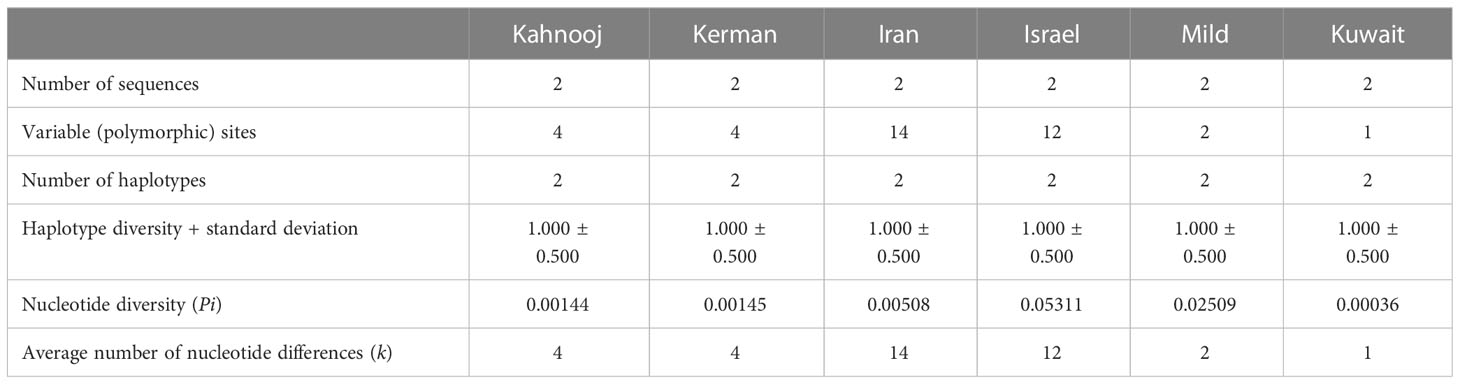
Table 4 Nucleotide polymorphisms among strains of TYLCV worldwide with selected sequences.
The between and within strain nucleotide statistics were calculated for each strain group to estimate the extent of differentiation. The p-values for Ks, Kst, Snn, and Z indicated most TYLCV strains were significantly differentiated from one another, (Table 5). The Snn values were all close to one, which is again indicative of differentiation. The Fst values for between strain comparisons were high (>0.50), indicating substantial differences between TYLCV-Israel and TYLCV-Mild as well as between TYLCV-Israel and the Iranian isolates and TYLCV-Mild and Iranian isolates, especially TYLCV-Kerman and TYLCV-Kahnooj. These patterns were indicative of extensive genetic differentiation with low intra-strain gene flow.
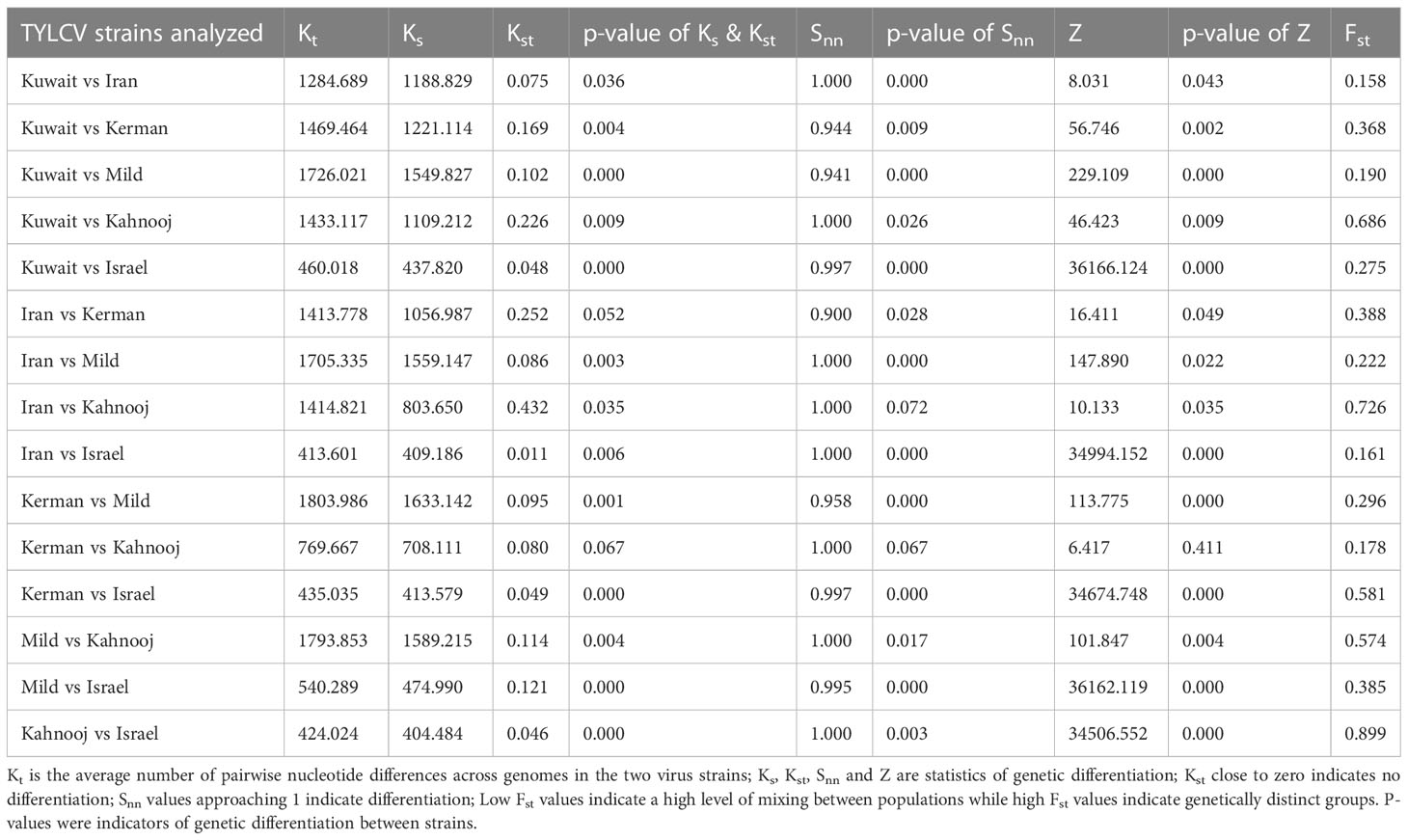
Table 5 Genetic differentiation analyses.
The Fu and Li’s D and F statistics and Tajima’s D statistics were calculated for five TYLCV strains for which sufficient number of strains/populations were available to derive reliable estimates (Table 6). Negative values for all three statistical tests are indicative either of purifying selection or population expansion. The negative values could reflect a large number of mutations occurring rarely or only once within a population. All except for TYLCV-Iran and TYLCV-Kuwait strains exhibited negative Tajima’s D values and only TYLCV-Kerman for all three statistical tests; however, results were significant only for TYLCV-Israel and TYLCV-Kerman strains.
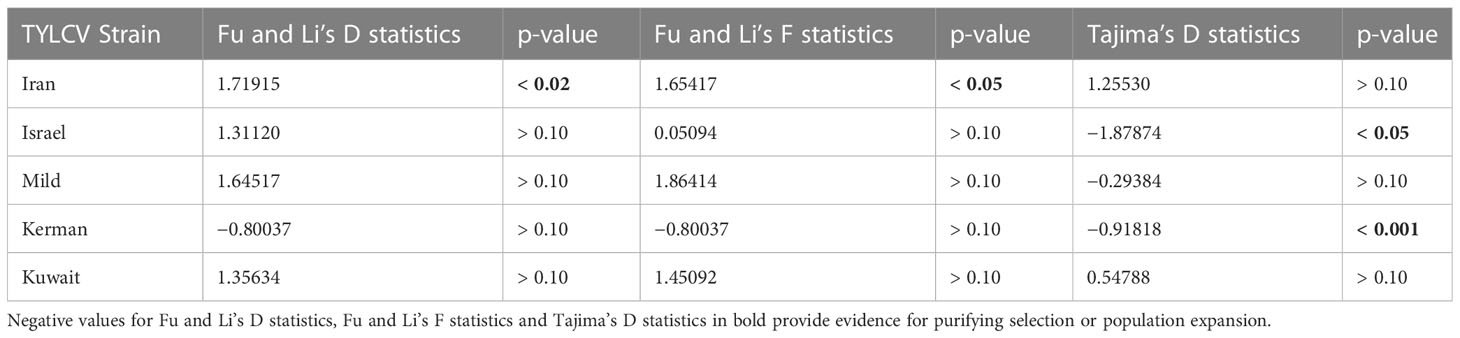
Table 6 Population neutrality analysis.
The six ORFs encoded by all seven TYLCV strains were analyzed for selection to determine if the coding regions were evolving under positive selection. Among the seven strains, positive selection was identified for only TYLCV-Israel, while no signals of positive selection were predicted for the other strains. Positive selection was predicted for the TYLCV-Israel at codons 7, 13, 27, 196, and 319.
The phylogenetic relationships and genetic differentiation of the TYLCV species genomes were analyzed for the extant strains of TYLCV. The results underscore the ancestral position of TYLCV-Israel strain to all the other TYLCV strains based on the most ancient MRCA. However, MRCAs for TYLCV-Mild, TYLCV-Iran, and TYLCV Kerman were not very far apart from TYLCV-Israel. TYLCV-Israel sequences were collected from five continents across the globe. Similarly, TYLCV-Mild sequences also were collected from multiple continents. Interestingly, all the TYLCV-Iran and Kuwait genomes analyzed in the data set were collected in their respective countries alone. Multiple strains were observed in Iran than in any other region. The results reported here are consistent with those reported in a previous study (14), wherein Iran could be the center of divergence.
Although TYLCV-like symptoms were first observed in tomato in the Jordan Valley during 1929 (82), the first economic damage to the tomato crop was documented in Israel during 1931 and shortly after in Jordan during 1935. However, the TYLCV-Israel strain etiology of leaf curl disease was not demonstrated until the 1960s (20, 21). Since then, several TYLCV strains have been identified in Iran, where the TYLCV isolates possess the greatest genetic diversity, an observation that is consistent with the hypotheses that Iran is the center of divergence of TYLCV (22, 60).
Ancestral reconstruction for the TYLCV species indicated an estimated mean MRCA of ~28,000 years ago, which is consistent with the previously reported the 95% highest probability density (HPD) values ranging between 290 and 40,211 years ago (9). Here, using the estimated HPD values, a more recent mean MRCA of approximately 290 years was estimated for TYLCV-Kahnooj and 12445 years for TYLCV-Kuwait, and between 36,490 to 41,795 years for the other strains. These results provide support for the hypothesis that TYLCV diverged from a common ancestor long before tomato yellow leaf curl disease outbreaks were documented in tomato crops (9). Although a lower MRCA was predicted for the TYLCV-Kahnooj from Iran, as shown by the Trace panel line plots, the Joint marginal distribution plot indicated good correlation between TYLCV-Kahnooj and other Iranian strains. TYLCV-Kahnooj also had good correlation with TYLCV-Mild. Interestingly, joint marginal distribution of TYLCV-Israel was not substantial (as indicated by circles than ellipticals) with other TYLCV-strains.
The estimated TYLCV genome substitution rate in the current study was 2.7899 × 10−4 substitutions/site/year (95% HPD with a range of 3.7339 × 10−5 to 4.7489 × 10−5). Estimates from previous studies have suggested a similar substitution rate for TYLCV, at 2.88 × 10−4 substitutions/site/year (95%, HPD with a range of 1.34 × 10−6 to 6.06 × 10−4) and 4.5 × 10−4 substitutions/site/year (95% HPD ranging from 2.4 × 10−4 to 6.8 × 10−4) (9, 22). All three studies were carried out with similar parameters, including tree prior (constant population size). However, this analysis included 423 full-length genome sequences compared with 56 and 82 full genome sequences, respectively, available in two previous studies (9, 22). All three studies found the TYLCV genome substitution rate was similar to that of the ssDNA bacteriophage PhiX174 (~7 × 10−6 nt/generation), and greater than double-stranded DNA viruses (~10−8 nt/generation) (83, 84).
Accelerated substitution rates such as those observed for TYLCV, are expected to contribute to relatively rapid genome diversification. The genome sequences of the TYLCV strains/isolates in this study represented diverse variants or haplotypes. In contrast with viral populations consisting of relatively homogeneous genome sequences, multiple haplotypes among the different TYLCV strains are consistent with the quasispecies concept (65). Previous studies have enumerated the quasispecies-like characteristics of begomoviruses (25, 64). Given the relatively rapid substitution rate of TYLCV, emergence of the most fit or better adapted than previous isolates would be expected.
The TYLCV strains [-Iran (including Boushehr, Kerman, and Kahnooj), -Kuwait, and -Oman] that prevail nearly exclusively in the Middle East, including the Arabian Peninsula and in Iran exhibited the highest average number of nucleotide differences (k) and comprised the highest proportion of recombinant genomes. High, significant inter-strain genetic diversity may have facilitated accelerated recombination. In contrast, TYLCV-Israel isolates, despite the high proportion of recombinant genomes of 46.4%, harbored a lower average number of nucleotide differences compared with TYLCV-Mild and TYLCV-Iranian strains. Despite initial reports of tomato yellow leaf curl disease outbreaks in Israel and the Jordan Valley from 1929 to 1944 (82, 85), the genomic diversity among TYLCV-Israel isolates appears to be surprisingly low. This suggests that one or a few founder events led to first and regular-occurring annual outbreaks, a scenario in which the crop serves as the source of virus for each subsequent infection cycle, creating a potential for a species bottleneck. Notably, among all the TYLCV strains, TYLCV-Israel was the only strain undergoing positive selection. The relatively low average number of nucleotide differences, and the five mutations in the codon positions strongly suggest that these fixed mutations co-exist in populations as the result of a population bottleneck resulting from the nearly continuous cropping of tomato. In contrast, the TYLCV-Kahnooj strains harbored the fewest nucleotide differences. However, this observation may represent an artifact resulting from the very small sample size of only two genomes.
Analysis of the full-length genome and coat protein sequences revealed that the Middle East constitute the zones of TYLCV endemism (22), making them the epicenter of the most ancient, and potentially, the most recent common extant ancestors of TYLCV. The extensive diversity among the genomes of TYLCV-Iranian strains, native to the Middle East, compared with the much lower diversity associated with TYLCV-Israel, can best be explained by differences in endemism and invasion history (22).
Based on the signals from the population neutrality tests only TYLCV-Israel and TYLCV-Kerman were identified as undergoing either purifying selection or population expansion, with evidence favoring TYLCV-Israel expansion. Of course, the inclusion of a substantially high number of TYLCV-Israel sequences could have influenced the results. Nevertheless, records show that TYLCV-Israel was introduced into China in 2006, and by 2009 was prevalent in 11 provinces (37). By 2014, the virus had been identified in 13 provinces or autonomous regions in China (62). The TYLCV-Israel predicted ‘recombinant free’ reference sequences available in GenBank (as of 2022) contained genomes from 12 provinces, two municipalities (Beijing and Shanghai), and 1 autonomous region (Xinjiang Uyghur) of China. Rapid spread of the introduced Q mitotype of B. tabaci (cryptic species NAF-MED) throughout China at about this same time was strongly correlated with the rapid spread of TYLCV (37, 63). Evidence further suggests the Q mitotype was a more fit and/competent B. tabaci vector of TYLCV in China than either of the two B haplotypes introduced to China (32, 37, 63). Thus, the predicted genomic signals of population expansion support the observed rapid TYLCV-Israel population sweep in China.
The TYLCV genomes in this analysis represent all known TYLCV strains, of which only two, TYLCV-Israel and TYLCV-Mild, have spread globally. Although it seems likely that naturally occurring mixed TYLCV-Mild (mild symptoms) and TYLCV-Israel (severe symptoms) infections would be common, the two strains are usually not reported to occur in mixtures, post-introduction. Even so, experiments carried out during the 1960’s, showed that mixed infections of the two strains could be established (58, 86). Recent studies have shown that primarily TYLCV-Israel has been introduced into Africa, the Americas, Asia, and the Mediterranean region on infected plants originating in the Middle East (10, 45–47, 87). The predominant vector involved in establishing the primary infections was B. tabaci B mitotype that infested ornamentals and other TYLCV host plants including tomato seedlings (13, 25, 36, 37, 88) Thus, a combination of factors likely facilitated global spread of TYLCV-Israel and TYLCV-Mild. Although international trade is one of the main drivers for the global spread of TYLCV-Israel and TYLCV-Mild strains, why the other TYLCV strains have not spread through these activities and routes is not clear. For example, dispersal of TYLCV strains native to Iran, from there to other countries/locales that may be expected to occur, may have been lessened by the minimized trade between Iran and other countries due to political reasons. In contrast, movement of TYLCV-Oman strain to other countries is expected because extensive trade of many horticultural crops occurs between Oman and other countries. Similar scenarios of the spread of TYLCV strains endemic to Iran or Oman have not been documented to date.
Among the 834 TYLCV genomes analyzed, 412 exhibited evidence of one or more recombination events, or a frequency of 49.40%. Recombination is a well-known mechanism among begomoviruses that has contributed to genetic diversity among begomovirus populations, including TYLCV. The TYLCV species, as a whole, has undergone frequent intraspecific recombination events, as well as recombination with other begomovirus species. In one study, 18 recombination events among species infecting tomato in the Middle East and Western Mediterranean was identified, with 16 of those events involving two virus species (22). Indeed, in another study, predicted recombination events were identified between TYLCV and tomato leaf curl Iran virus, tomato leaf curl Sardinia virus, tomato leaf curl Karnataka virus from India, and cotton leaf curl Gezira virus. Other analyses have discovered TYLCV interspecific recombination with tomato yellow leaf curl Sardinia virus (89), tomato leaf curl virus (90), tomato leaf curl Comoros virus (91), tomato leaf curl Sudan virus (92), tomato leaf curl Iran virus (93), tobacco leaf curl virus (94), and tomato yellow leaf curl Thailand virus (95). In the current study >40 intra-strain recombination events were identified. The most fit recombinants may be expected to exhibit a selective advantage over parental lineages, potentially manifest as host range shifts or host range expansion, variability in symptom severity, and/or increased transmission frequency (96–98). For example, a recombinant between TYLCV-Israel and tomato yellow leaf curl Sardinia virus was found to have broadened its host range while symptom severity was lessened (99). The unusually high prevalence of TYLCV recombinants may in part be explained by the widespread and overlapping distributions of extant TYLCV strains, and the potential for intra- and inter-specific recombination that has favored their survival over apparently less competitive recombinants.
Despite the additional sequences analyzed in this study, the topography of the Bayesian phylogenetic tree was overall consistent with those reconstructed in previous studies that have also differentiated TYLCV strains (22, 60). Among the TYLCV strains, two have spread rapidly aided by human-facilitated transport into non-endemic areas (10, 12, 22). For example, TYLCV-Israel isolates prevalent in the Americas are extant there as the result of multiple introductions over time, perhaps, six or more. Among the introductions are (i) a large group of U.S. isolates and several from Cuba, Dominican Republic, Guatemala, and Puerto Rico that are most closely related to isolates from Jordan, (ii) a group of isolates extant in both China and Mexico, (iii) one U.S. isolate that grouped with isolates introduced into Japan, (iv) one U.S. isolate that clustered with TYLCV isolates also found in Australia, (v) a single isolate found in the U.S. and Jordan, (vi) one isolate from Lebanon that clustered with a group of U.S. isolates, and (vii) TYLCV variants introduced into Japan apparently before they further spread to South Korea. Based on a previous analysis of fewer TYLCV genome sequences (58), two TYLCV-Israel variants were introduced separately into North America from Asia and the Middle East. Similarly, (22) reported the same two separate introduction events into North America. Later, (14) identified three introductions into mainland North America, and three that had dispersed throughout the Caribbean. The mainland North America lineages apparently originating from introductions to Australia, East Asia (Japan), and the Caribbean, while the Caribbean isolates were most recently of Mediterranean origin. Notably, the Caribbean region has also experienced introductions from the Eastern and Western Mediterranean, and East Asia, making this not only a hot spot for diversification, but also a major pathway for introductions of TYLCV in the long term. In summary, TYLCV in the U.S. have originated from Asia, the Middle East, the Eastern Mediterranean, as well as Australia. Finally, although much less widespread, TYLCV-Mild has been introduced into Japan in two separate events, stemming from prior introductions to Portugal and Reunion Island, respectively, while isolates extant in the Dominican Republic, Jordan, and Sweden were identified for the first time in a unique, previously unreported association. These results may portend a new invasion route for TYLCV-Mild to spread from the latter to new and/or already TYLCV affected locales.
The geographic distribution of TYLCV, once endemic only to the Middle East, has expanded dramatically over the last half century and more of international trade and human travel that have aided in the dispersal of at least three invasive haplotypes of the B. tabaci cryptic species, and virus-infected plant reservoirs throughout much of the world. Multiple introductions, intra- and inter-specific recombination, selection, and the accumulation of fixed mutations have and continue to shape the genomic diversification and evolution of the TYLCV species and its diverse populations. The results of this study offer a recent snapshot of TYLCV population distribution and of the prospective trends involving the spread of new variants and/or strains, and of the potential impact of new and continued TYLCV outbreaks on crop production, in relation to global human demographics
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
WM, HM, JB, and RS conceptualized and designed the study. WM, HM, SA, JP, GR, HS, BT, SA, and HA-A collected samples and/or performed the experiments. HM, SG, WM, JB, and RS analyzed the data sets and/or the experimental results. WM and HM drafted the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by funds from by the USDA Hatch Grant awarded to RS # GEO 00671, Georgia Commodity Commission for vegetables, the USDA-UGA Cooperative Agreement #6080-22000-027-18-S, and USDA-Specialty Crops Research Initiative (USDA NIFA SCRI grant 2018-51181-28420.
The authors thank Ms. Gabrielle LaTora and Sarah Bragg as well as Mr. Michael Catto and Sudeep Pandey for technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors JB and RS declared that they were editorial board members of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2023.1221156/full#supplementary-material
1. Thottappilly G. Plant virus diseases of importance to African agriculture. J Phytopathol (1992) 134:265–88. doi: 10.1111/j.1439-0434.1992.tb01236.x
2. Picó B, Díez MJ, Nuez F. Viral diseases causing the greatest economic losses to the tomato crop. II. the tomato yellow leaf curl virus - a review. Sci Hortic (1996) 67:151–196. doi: 10.1016/S0304-4238(96)00945-4
3. Böttcher B, Unseld S, Ceulemans H, Russell RB, Jeske H. Geminate structures of African cassava mosaic virus. J Virol (2004) 78:6758–65. doi: 10.1128/jvi.78.13.6758-6765.2004
4. Varsani A, Roumagnac P, Fuchs M, Navas-Castillo J, Moriones E, Idris A, et al. Capulavirus and grablovirus: two new genera in the family Geminiviridae. Arch Virol (2017) 162:1819–31. doi: 10.1007/s00705-017-3268-6
5. Rey MEC, Ndunguru J, Berrie LC, Paximadis M, Berry S, Cossa N, et al. Diversity of dicotyledenous-infecting geminiviruses and their associated DNA molecules in Southern Africa, including the South-West Indian ocean islands. Viruses (2012) 4:1753–91. doi: 10.3390/v4091753
6. Czosnek H, Hariton-Shalev A, Sobol I, Gorovits R, Ghanim M. The incredible journey of begomoviruses in their whitefly vector. Viruses (2017) 9:273. doi: 10.3390/v9100273
7. de Moya RS, Brown JK, Sweet AD, Kimberly KK, Paredes-Montero JR, Waterhouse RM, et al. Nuclear orthologs derived from whole genome sequencing indicate cryptic diversity in the Bemisia tabaci (Insecta: Aleyrodidae) complex of whiteflies. Diversity (Basel) (2019) 11:1–19. doi: 10.3390/d11090151
8. Yan Z, Anne-Marie W, Jesus N-C, Yuling B. The global dimension of tomato yellow leaf curl disease: current status and breeding perspectives. Microorganisms (2021) 9:740. doi: 10.3390/MICROORGANISMS9040740
9. Duffy S, Holmes EC. Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded dna begomovirus tomato yellow leaf curl virus. J Virol (2008) 82:957–965. doi: 10.1128/jvi.01929-07
10. Czosnek H, Laterrot H. A worldwide survey of tomato yellow leaf curl viruses. Arch Virol (1997) 142:1391–406. doi: 10.1007/S007050050168
11. Polston JE, McGovern RJ, Brown LG. Introduction of tomato yellow leaf curl virus in Florida and implications for the spread of this and other geminiviruses of tomato. Plant Dis (1999) 83:984–8. doi: 10.1094/PDIS.1999.83.11.984
12. Moriones E, Navas-Castillo J. Tomato yellow leaf curl virus, an emerging virus complex causing epidemics worldwide. Virus Res (2000) 71:123–34. doi: 10.1016/S0168-1702(00)00193-3
13. Brown JK, Czosnek H. Whitefly transmission of plant viruses. In Advances in Botanical Research (pp. 65-76,IN1-IN2,77-100). (Advances in Botanical Research; Vol. 36). Academic Press Inc.. Cambridge, MA. United States. doi: 10.1016/s0065-2296(02)36059-2
14. Mabvakure B, Martin DP, Kraberger S, Cloete L, van Brunschot S, Geering ADW, et al. Ongoing geographical spread of tomato yellow leaf curl virus. Virology (2016) 498:257–264. doi: 10.1016/j.virol.2016.08.033
15. Gronenborn B. The tomato yellow leaf curl virus genome and function of its proteins. In: Tomato yellow leaf curl virus disease: management, molecular biology, breeding for resistance. (Heidelberg, Germany: Springer Netherlands) (2007). p. 67–84. doi: 10.1007/978-1-4020-4769-5_5
16. Czosnek H. Tomato yellow leaf curl virus. In: Encyclopedia of virology. Elsevier Ltd, Amsterdam, Netherlands (2008). p. 138–45. doi: 10.1016/B978-012374410-4.00717-2
17. Jupin I, De Kouchkovsky F, Jouanneau F, Gronenborn B. Movement of tomato yellow leaf curl geminivirus (TYLCV): involvement of the protein encoded by ORF C4. Virology (1994) 204:82–90. doi: 10.1006/viro.1994.1512
18. Rojas MR, Jiang H, Salati R, Xoconostle-Cázares B, Sudarshana MR, Lucas WJ, et al. Functional analysis of proteins involved in movement of the monopartite begomovirus, tomato yellow leaf curl virus. Virology (2001) 291:110–25. doi: 10.1006/viro.2001.1194
19. Van Wezel R, Dong X, Liu H, Tien P, Stanley J, Hong Y. Mutation of three cysteine residues in tomato yellow leaf curl virus-China C2 protein causes dysfunction in pathogenesis and posttranscriptional gene-silencing suppression. Mol Plant-Microbe Interact (2002) 15:203–8. doi: 10.1094/MPMI.2002.15.3.203
20. Navot N, Pichersky E, Zeidan M, Zamir D, Czosnek H. Tomato yellow leaf curl virus: a whitefly-transmitted geminivirus with a single genomic component. Virology (1991) 185:151–61. doi: 10.1016/0042-6822(91)90763-2
21. Cohen S, Antignus Y. Tomato yellow leaf curl virus, a whitefly-borne geminivirus of tomatoes. In: Harris KF, editor. Advances in disease vector research. advances in disease vector research, vol. 10 . New York, NY: Springer (1994). doi: 10.1007/978-1-4612-2590-4_10
22. Lefeuvre P, Martin DP, Harkins G, Lemey P, Gray AJA, Meredith S, et al. The spread of tomato yellow leaf curl virus from the middle East to the world. PLoS Pathog (2010) 6:e1001164. doi: 10.1371/journal.ppat.1001164
23. Brown JK. Whitefly-transmitted geminiviruses and associated disorders in the americas and the Caribbean basin. Plant Dis (1992) 76:220. doi: 10.1094/PD-76-0220
24. Brown JK. Current status of Bemisia tabaci as a plant pest and virus vector in agroecosystems worldwide Vol. 42. FAO (Food and Agriculture Organization of the United Nations) Plant Protection Bulletin, Rome, Italy (1994) p. 3–32.
25. Seal SE, VandenBosch F, Jeger MJ. Factors influencing begomovirus evolution and their increasing global significance: implications for sustainable control. CRC Crit Rev Plant Sci (2006) 25:23–46. doi: 10.1080/07352680500365257
26. Brown JK, Frohlich DR, Rosell RC. The sweetpotato or silverleaf whiteflies: biotypes of Bemisia tabaci or species complex? Annu Rev Entomol (1995) 40:511–34. doi: 10.1146/annurev.en.40.010195.002455
27. Chu D, Zhang YJ, Brown JK, Cong B, Xu BY, Wu QJ, et al. The introduction of the exotic q biotype of Bemisia tabaci from the Mediterranean region into China on ornamental crops. Fla. Entomol (2006) 89:168–74. doi: 10.1653/0015-4040(2006)89[168:tioteq]2.0.co;2
28. Brown JK. Phylogenetic biology of the Bemisia tabaci sibling species group. Bemisia: Bionomics and Management of a Global Pest (2010) p. 31–67. doi: 10.1007/978-90-481-2460-2_2/FIGURES/9
29. Gill RJ, Brown JK. Systematics of Bemisia and Bemisia relatives: can molecular techniques solve the Bemisia tabaci complex conundrum-a taxonomist’s viewpoint. Bemisia: Bionomics Manage Global Pest. Springer Netherlands, Heidelberg, Germany (2010), 5–29. doi: 10.1007/978-90-481-2460-2_1
30. Dinsdale A, Cook L, Riginos C, Buckley YM, De Barro P. Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase 1 to identify species level genetic boundaries. Ann Entomol Soc Am (2010) 103:196–208. doi: 10.1603/AN09061
31. Mugerwa H, Colvin J, Alicai T, Omongo CA, Kabaalu R, Visendi P, et al. Genetic diversity of whitefly (Bemisia spp.) on crop and uncultivated plants in Uganda: implications for the control of this devastating pest species complex in Africa. J Pest Sci (2021) 2004(1):3. doi: 10.1007/s10340-021-01355-6
32. Paredes-Montero JR, Haq QMI, Mohamed AA, Brown JK. Phylogeographic and SNPs analyses of Bemisia tabaci B mitotype populations reveal only two of eight haplotypes are invasive. Biol (Basel) (2021) 10:1048. doi: 10.3390/biology10101048
33. Brown JK, Paredes-Montero JR, Stocks IC. The Bemisia tabaci cryptic (sibling) species group [[/amp]]mdash; imperative for a taxonomic reassessment. Curr Opin Insect Science. (2023) 57:101032. doi: 10.1016/j.cois.2023.101032. ISSN 2214-5745.
34. Costa HS, Brown JK. Variation in biological characteristics and esterase patterns among populations of Bemisia tabaci, and the association of one population with silverleaf symptom induction. Insect Sci (1991) 61:211–9. doi: 10.1111/j.1570-7458.1991.tb01553.x
35. Moriones E. First report of tomato yellow leaf curl virus in Spain. Plant Dis (1993) 77:953. doi: 10.1094/pd-77-0953b
36. Li M, Hu J, Xu F-C, Liu S-S. Transmission of tomato yellow leaf curl virus by two invasive biotypes and a Chinese indigenous biotype of the whitefly Bemisia tabaci. Int J Pest Manag (2010) 56:275–80. doi: 10.1080/09670871003743428
37. Pan H, Chu D, Yan W, Su Q, Liu B, Wang S, et al. Rapid spread of tomato yellow leaf curl virus in china is aided differentially by two invasive whiteflies. PLoS One (2012) 7:e34817. doi: 10.1371/journal.pone.0034817
38. Lima LHC, Campos L, Moretzsohn MC, Navia D, de Oliveira MRV. Genetic diversity of Bemisia tabaci (Genn.) populations in Brazil revealed by RAPD markers. Genet Mol Biol (2002) 25:217–23. doi: 10.1590/s1415-47572002000200016
39. Zang L-S, Chen W-Q, Liu S-S. Comparison of performance on different host plants between the b biotype and a non-b biotype of Bemisia tabaci from zhejiang, China. Entomol Exp Appl (2006) 121:221–7. doi: 10.1111/j.1570-8703.2006.00482.x
40. Brown JK. Phylogenetic biology of the bemisia tabaci sibling species group. In: Bemisia: bionomics and management of a global pest. Dordrecht: Springer Netherlands (2009). p. 31–67. doi: 10.1007/978-90-481-2460-2_2
41. De Barro P, Ahmed M. Genetic networking of the Bemisia tabaci cryptic species complex reveals pattern of biological invasions. PLoS One (2011) 6:e25579. doi: 10.1371/journal.pone.0025579
42. Muñiz Y, Granier M, Caruth C, Umaharan P, Marchal C, Pavis C, et al. Extensive settlement of the invasive MEAM1 population of Bemisia tabaci (Hemiptera: Aleyrodidae) in the caribbean and rare detection of indigenous populations. Environ Entomol (2011) 40:989–998. doi: 10.1603/EN11129
43. Srinivasan R, Riley D, Diffie S, Sparks A, Adkins S. Whitefly population dynamics and evaluation of whitefly-transmitted tomato yellow leaf curl virus (TYLCV)-resistant tomato genotypes as whitefly and TYLCV reservoirs. J Econ Entomol (2012) 105:1447–56. doi: 10.1603/EC11402
44. Esterhuizen LL, Mabasa KG, van Heerden SW, Czosnek H, Brown JK, van Heerden H, et al. Genetic identification of members of the Bemisia tabaci cryptic species complex from South Africa reveals native and introduced haplotypes. J Appl Entomology (2013) 137:1–14. doi: 10.1111/j.1439-0418.2012.01720.x
45. Peterschmitt M, Granier M, Aboulama S. First report of tomato yellow leaf curl geminivirus in Morocco. Plant Dis (1999) 83:1074. doi: 10.1094/pdis.1999.83.11.1074c
46. Peterschmitt M, Granier M, Mekdoud R, Dalmon A, Gambin O, Vayssieres JF, et al. First report of tomato yellow leaf curl virus in réunion island. Plant Dis (1999) 83:303. doi: 10.1094/pdis.1999.83.3.303b
47. Kato K, Onuki M, Fuji S, Hanada K. The first occurrence of tomato yellow leaf curl virus in tomato (Lycopersicon esculentum mill.) in Japan. Japanese J Phytopathol (1998) 64:552–9. doi: 10.3186/jjphytopath.64.552
48. Wu JB, Dai FM, Zhou XP. First report of tomato yellow leaf curl virus in China. Plant Dis (2006) 90:1359. doi: 10.1094/pd-90-1359c
49. Van Brunschot SL, Persley DM, Geering ADW, Campbell PR, Thomas JE. Tomato yellow leaf curl virus in Australia: distribution, detection and discovery of naturally occurring defective DNA molecules. Australas Plant Pathol (2010) 39:412–23. doi: 10.1071/AP10083
50. Louro D. First report of tomato yellow leaf curl virus in Portugal. Plant Dis (1996) 80:1079. doi: 10.1094/pd-80-1079d
51. Avgelis AD, Roditakis N, Dovas CI, Katis NI, Varveri C, Vassilakos N, et al. First report of tomato yellow leaf curl virus on tomato crops in Greece. Plant Dis (2001) 85:678. doi: 10.1094/pdis.2001.85.6.678c
52. Accotto GP, Bragaloni M, Luison D, Davino S, Davino M. First report of tomato yellow leaf curl virus (TYLCV) in Italy. Plant Pathol (2003) 52:799. doi: 10.1111/j.1365-3059.2003.00908.x
53. Botermans M, Verhoeven J, Th. J, Jansen CCC, Roenhorst JW, Stijger CCMM, et al. First report of tomato yellow leaf curl virus in tomato in the Netherlands. Plant Dis (2009) 93:1073. doi: 10.1094/pdis-93-10-1073c
54. Ascencio-Ibáñez JT, Diaz-Plaza R, Méndez-Lozano J, Monsalve-Fonnegra ZI, Argüello-Astorga GR, Rivera-Bustamante RF. First report of tomato yellow leaf curl geminivirus in yucatán, méxico. Plant Dis (1999) 83:1178. doi: 10.1094/pdis.1999.83.12.1178a
55. Brown JK, Idris AM. Introduction of the exotic monopartite tomato yellow leaf curl virus into West coast Mexico. Plant Dis (2006) 90:1360. doi: 10.1094/pd-90-1360a
56. Bird J, Idris AM, Rogan D, Brown JK. Introduction of the exotic tomato yellow leaf curl virus -Israel in tomato to Puerto Rico. Plant Dis (2001) 85:1028. doi: 10.1094/pdis.2001.85.9.1028b
57. Zambrano K, Carballo O, Geraud F, Chirinos D, Fernández C, Marys E. First report of tomato yellow leaf curl virus in Venezuela. Plant Dis (2007) 91:768. doi: 10.1094/pdis-91-6-0768a
58. Duffy S, Holmes EC. Multiple introductions of the old world begomovirus tomato yellow leaf curl virus into the new world. Appl Environ Microbiol (2007) 73:7114–7. doi: 10.1128/AEM.01150-07
59. Lee H, Song W, Kwak HR, Kim JD, Park J, Auh CK, et al. Phylogenetic analysis and inflow route of tomato yellow leaf curl virus (TYLCV) and Bemisia tabaci in Korea. Mol Cells (2010) 30:467–476. doi: 10.1007/s10059-010-0143-7
60. Hosseinzadeh MR, Shams-Bakhsh M, Osaloo SK, Brown JK. Phylogenetic relationships, recombination analysis, and genetic variability among diverse variants of tomato yellow leaf curl virus in Iran and the Arabian peninsula: further support for a TYLCV center of diversity. Arch Virol (2014) 159:485–97. doi: 10.1007/s00705-013-1851-z
61. Romay G, Chirinos DT, Geraud-Pouey F, Gillis A. Full-length genome sequencing of the mild strain of tomato yellow leaf curl virus in Venezuela reveals a third introduction event of this virus in new world. Australas Plant Dis Notes (2014) 9:1–3. doi: 10.1007/s13314-014-0123-4
62. Yang XL, Zhou MN, Qian YJ, Xie Y, Zhou XP. Molecular variability and evolution of a natural population of tomato yellow leaf curl virus in shanghai, China. J Zhejiang Univ Sci B (2014) 15:133–42. doi: 10.1631/jzus.B1300110
63. Zhang WM, Fu H, Wang WH, Piao CS, Tao YL, Guo D, et al. Rapid spread of a recently introduced virus (Tomato yellow leaf curl virus) and its vector Bemisia tabaci (Hemiptera: Aleyrodidae) in liaoning province, China. J Econ Entomol (2014) 107:98–104. doi: 10.1603/EC13348
64. Roossinck MJ. Mechanisms of plant virus evolution. Annu Rev Phytopathol (1997) 35:191–209. doi: 10.1146/annurev.phyto.35.1.191
65. Domingo E, Sheldon J, Perales C. Viral quasispecies evolution. Microbiol Mol Biol Rev (2012) 76:159–216. doi: 10.1128/mmbr.05023-11
66. Polston JE, De Barro P, Boykin LM. Transmission specificities of plant viruses with the newly identified species of the Bemisia tabaci species complex. Pest Manag Sci (2014) 70:1547–52. doi: 10.1002/ps.3738
67. Brown JK, Zerbini FM, Navas-Castillo J, Moriones E, Ramos-Sobrinho R, Silva JCF, et al. Revision of begomovirus taxonomy based on pairwise sequence comparisons. Arch Virol (2015) 160. doi: 10.1007/s00705-015-2398-y
68. Inoue-Nagata AK, Albuquerque LC, Rocha WB, Nagata T. A simple method for cloning the complete begomovirus genome using the bacteriophage ϕ29 DNA polymerase. J Virol Methods (2004) 116:209–11. doi: 10.1016/j.jviromet.2003.11.015
69. Marchant WG, Gautam S, Hutton SF, Srinivasan R. Tomato yellow leaf curl virus-resistant and -susceptible tomato genotypes similarly impact the virus population genetics. Front Plant Sci (2020) 11:599697. doi: 10.3389/fpls.2020.599697
70. Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol (2015) 1:vev003. doi: 10.1093/ve/vev003
71. Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PloS Comput Biol (2019) 15. doi: 10.1371/journal.pcbi.1006650
72. Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol Biol Evol. (2017) 34:772–773. doi: 10.1093/molbev/msw260
73. Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst Biol (2018) 67:901–4. doi: 10.1093/sysbio/syy032
74. Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol (2017) 34:3299–302. doi: 10.1093/molbev/msx248
75. Hudson RR, Slatkin M, Maddison WP. Estimation of levels of gene flow from DNA sequence data. Genetics (1992) 132:583–9. doi: 10.1093/genetics/132.2.583
76. Tajima F. The effect of change in population size on DNA polymorphism. Genetics (1989) 123:597–601. doi: 10.1093/genetics/123.3.597
77. Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics (1993) 133:693–709. doi: 10.1093/genetics/133.3.693
78. Pond K,SL, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics (2005) 21:676–9. doi: 10.1093/bioinformatics/bti079
79. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol (2018) 35:1547–9. doi: 10.1093/molbev/msy096
80. Stecher G, Tamura K, Kumar S. Molecular evolutionary genetics analysis (MEGA) for macOS. Mol Biol Evol (2020) 37:1237–9. doi: 10.1093/molbev/msz312
81. Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol (1993) 10:512–26. doi: 10.1093/oxfordjournals.molbev.a040023
82. Avidov HZ. Tobacco whitefly in israel. tel Aviv: hassadeh (in Hebrew). In: Baery I, Kapoller B, editors. Growing vegetables–part b. summer squash. tel Aviv: hassadeh (in Hebrew) (19441963). p. 1–33.
83. Raney JL, Delongchamp RR, Valentine CR. Spontaneous mutant frequency and mutation spectrum for gene a of phiX174 grown in e. coli. Environ Mol Mutagen (2004) 44:119–27. doi: 10.1002/EM.20041
84. Peck KM, Lauring AS. Complexities of viral mutation rates. J Virol (2018) 92:e01031-17. doi: 10.1128/JVI.01031-17
85. Cohen S, Lapidot M. Appearance and expansion of TYLCV: a historical point of view. In: Tomato yellow leaf curl virus disease: management, molecular biology, breeding for resistance Springer, New York, NY. United States. (2007). doi: 10.1007/978-1-4020-4769-5_1
86. Antignus Y, Cohen S. Complete nucleotide sequence of an infectious clone of a mild isolate of tomato yellow leaf curl virus (TYLCV). Phytopathology (1994) 84:707–12. doi: 10.1094/PHYTO-84-707
87. Polston JE, Anderson PK. The emergence of whitefly-transmitted geminiviruses in tomato in the Western hemisphere. Plant Dis (2007) 81:1358–69. doi: 10.1094/PDIS.1997.81.12.1358
88. Brown JK. The Bemisia tabaci complex: genetic and phenotypic variation and relevance to TYLCV-vector interactions. In: Czosnek H, editor. Tomato yellow leaf curl virus disease. New York and Tokyo: Springer (2007). p. 25–55.
89. Belabess Z, Peterschmitt M, Granier M, Tahiri A, Blenzar A, Urbino C. The non-canonical tomato yellow leaf curl virus recombinant that displaced its parental viruses in Southern Morocco exhibits a high selective advantage in experimental conditions. J Gen Virol (2016) 97:3433–45. doi: 10.1099/jgv.0.000633
90. Navas-Castillo J, Sanchez-Campos S, Noris E, Louro D, Accotto GP, Moriones E. Natural recombination between tomato yellow leaf curl virus-ls and tomato leaf curl virus. J Gen Virol (2000) 81:2797–801. doi: 10.1099/0022-1317-81-11-2797
91. Urbino C, Gutiérrez S, Antolik A, Bouazza N, Doumayrou J, Granier M, et al. Within-host dynamics of the emergence of tomato yellow leaf curl virus recombinants. PloS One (2013) 8:e58375. doi: 10.1371/journal.pone.0058375
92. Idris AM, Brown JK. Evidence for interspecific-recombination for three monopartite begomoviral genomes associated with the tomato leaf curl disease from central Sudan. Arch Virol (2005) 150:1003–12. doi: 10.1007/s00705-004-0484-7
93. Bananej K, Kheyr-Pour A, Hosseini Salekdeh G, Ahoonmanesh A. Complete nucleotide sequence of Iranian tomato yellow leaf curl virus isolate: further evidence for natural recombination amongst begomoviruses. Arch Virol (2004) 149:1435–1443. doi: 10.1007/s00705-004-0308-9
94. Park J, Lee H, Kim MK, Kwak HR, Auh CK, Lee K, et al. Phylogenetic lineage of tobacco leaf curl virus in Korea and estimation of recombination events implicated in their sequence variation. Virus Res (2011) 159:124–31. doi: 10.1016/j.virusres.2011.04.017
95. Kim SH, Oh S, Oh TK, Park JS, Kim SC, Kim SH, et al. Genetic diversity of tomato-infecting tomato yellow leaf curl virus (TYLCV) isolates in Korea. Virus Genes (2011) 42:117–27. doi: 10.1007/s11262-010-0541-0
96. Stenger DC, Davis KR, Bisaro DM. Recombinant beet curly top virus genomes exhibit both parental and novel pathogenic phenotypes. Virology (1994) 200. doi: 10.1006/viro.1994.1231
97. Zhou X, Liu Y, Calvert L, Munoz C, Otim-Nape GW, Robinson DJ, et al. Evidence that DNA-a of a geminivirus associated with severe cassava mosaic disease in Uganda has arisen by interspecific recombination. J Gen Virol (1997) 78(Pt 8):2101–11. doi: 10.1099/0022-1317-78-8-2101
98. Martin DP, Lefeuvre P, Varsani A, Hoareau M, Semegni JY, Dijoux B, et al. Complex recombination patterns arising during geminivirus coinfections preserve and demarcate biologically important intra-genome interaction networks. PloS Pathog (2011) 7:e1002203. doi: 10.1371/journal.ppat.1002203
99. Monci F, Sánchez-Campos S, Navas-Castillo J, Moriones E. A natural recombinant between the geminiviruses tomato yellow leaf curl Sardinia virus and tomato yellow leaf curl virus exhibits a novel pathogenic phenotype and is becoming prevalent in Spanish populations. Virology (2002) 303:317–26. doi: 10.1006/viro.2002.1633
Keywords: Geminiviridae, tomato-infecting begomovirus, TYLCV, strains, ancestral reconstruction
Citation: Marchant WG, Mugerwa H, Gautam S, Al-Aqeel H, Polston JE, Rennberger G, Smith H, Turechek B, Adkins S, Brown JK and Srinivasan R (2023) Phylogenomic and population genetics analyses of extant tomato yellow leaf curl virus strains on a global scale. Front. Virol. 3:1221156. doi: 10.3389/fviro.2023.1221156
Received: 15 May 2023; Accepted: 30 June 2023;
Published: 25 July 2023.
Edited by:
Mark Zwart, Netherlands Institute of Ecology (NIOO-KNAW), NetherlandsReviewed by:
James M. Wainaina, The Ohio State University, United StatesCopyright © 2023 Marchant, Mugerwa, Gautam, Al-Aqeel, Polston, Rennberger, Smith, Turechek, Adkins, Brown and Srinivasan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajagopalbabu Srinivasan, YmFidXNyaUB1Z2EuZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.