
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Virol., 25 May 2023
Sec. Emerging and Reemerging Viruses
Volume 3 - 2023 | https://doi.org/10.3389/fviro.2023.1181017
 Litty Paul1
Litty Paul1 Jocelynn Morgan1Gloria Pulley1Tirth Uprety2
Jocelynn Morgan1Gloria Pulley1Tirth Uprety2 Ben M. Hause3
Ben M. Hause3 Emma Adam2
Emma Adam2 Feng Li2
Feng Li2 Craig N. Carter1Douglas Marthaler4
Craig N. Carter1Douglas Marthaler4 Erdal Erol1*
Erdal Erol1*Rotaviruses (RVs) are significant enteric pathogens of humans and animals. In March 2021, the University of Kentucky Veterinary Diagnostic Laboratory (UKVDL) received a fecal sample from a 1-week-old goat kid with diarrhea from a farm with 5 additional diarrheic kids. The fecal sample was tested negative for Bovine coronavirus, Bovine rotavirus Group A, E. coli K99+, Cryptosporidium parvum and Salmonella spp by multiplex real-time PCR assays. Interestingly, a novel Equine Rotavirus B (ERVB) in Kentucky was identified from neonatal foals also with watery diarrhea in the Spring of 2021. Once the ERVB-specific real-time PCR assay became available, the fecal sample from the goat kid was tested and found positive for RVB. Genome sequence of the caprine RVB from fecal sample was obtained using shotgun metagenomic sequencing by Illumina MiSeq. All of the eleven viral segments of caprine RVB were sequenced either completely or partially. Genetic and phylogenetic analysis of VP7, VP4, VP6, VP1–VP3, and NSP1- NSP5 genes indicated the caprine RVB strain had the genotype constellation of G3-P[3]-I3- R3-C3-M3-A4-N3-T3-E3-H3. In addition, the caprine RVB sequence showed the highest nucleotide identity and evolutionary relationship to the ERVB and previously sequenced caprine RVB strains. Given the similar geographical location of the equine and caprine strains from our study, these findings suggest a possible common source of infection.
Rotaviruses (RVs) are icosahedral, non-enveloped, double-stranded RNA viruses in the Reoviridae family and cause enteric diseases in humans and animals (1). The RV genome contains 11 RNA segments encoding six structural proteins (VP1–VP4, VP6, and VP7) and five nonstructural proteins (NSP1–NSP5/6). Reassortment of gene segments between hosts are common in RVs due to the segmented nature of the viral genome (2–6). The outer capsid protein VP7 and spike protein VP4 are the major viral antigens in stimulation of RV-neutralizing antibodies and form the dual classification system for RV genotypes, which are further classified into G and P genotypes, respectively. RVs are classified into nine groups or species (A–I) based on the antigenic properties and sequence of the viral protein 6 (VP6) (7, 8). Rotavirus A (RVA) is the most common group causing gastroenteritis and severe diarrhea in children, piglets, calves, foals, goat kids and other young animals. Spillover of RVA of animal origins to humans associated with diarrhea in children is well documented, indicating a zoonotic potential of animal rotaviruses (2, 4, 9).
Rotavirus B (RVB) is genetically and antigenically distinct from RVA and was first detected in adult humans as causing severe gastroenteritis and diarrhea (10). Later, sporadic or epidemic outbreaks of diarrhea associated with RVB were reported in calves, adult cows, lambs, goat kids, and piglets (11–13). Two earlier studies on phylogenetic analyses of bovine RVB from calves in India and adult cattle in Japan revealed that bovine RVB strains represented a distant cluster from other RVBs (14, 15). In a recent report, a RVB strain was identified in intestinal samples from a few day-old goat kids and shown to be genetically related to bovine RVB strains (16).
Despite frequent detection of RVB in humans and animals, reports in horses are very rare. Although a single horse tested positive (out of total 37 samples) for ERVB in Germany some years ago, the disease status and genome analysis were not investigated (17). In the Spring of 2021, the full genome of a novel equine RVB (ERVB) from newborn foals with diarrhea in Kentucky, USA was reported (18). Genomic analysis demonstrated that the ERVB exhibited more than 96% overall amino acid identity to previously sequenced cattle and goat RVBs. The phylogenetic analyses also showed that the ERVB shared a common ancestor with the bovine and caprine RVB strains, suggesting ERVB originated in ruminants. In the current study, an RVB outbreak was identified in a goat farm, and the genetic and phylogenetic relationship was investigated to understand its evolutionary relatedness to the global RVB strains and the recently identified ERVB.
In March 2021, the University of Kentucky Veterinary Diagnostic Laboratory (UKVDL), Lexington, Kentucky, received a fecal sample from a 1-week-old goat kid with diarrhea from a farm with 5 additional diarrheic kids. All goat kids experienced similar clinical signs with the pattern of disease being consistent with a contagious agent. This farm was in Harrison County, a county that borders five counties around Lexington that constitute the vast majority of the central Kentucky Thoroughbred breeding industry. Harrison county borders Bourbon County where multiple Thoroughbred farms experienced outbreaks of ERVB neonatal diarrhea in the spring of 2021 (19).
The fecal sample was extracted for total nucleic acids and tested for Bovine coronavirus, Bovine rotavirus Group A, E. coli K99+, Cryptosporidium parvum and Salmonella spp by multiplex real-time PCR assays (18). Both remaining fecal swab sample and extracted nucleic acid were archived and stored at -80C for any future studies. Once the ERVB-specific real-time PCR assay became available (19), the total nucleic acid extract from the goat kid was tested for ERVB according to the Assay 2 as described (19).
Shotgun metagenomic sequencing of the fecal sample from the goat kid was performed using Illumina MiSeq. Briefly, the stored fecal sample was thawed, resuspended in PBS solution and, then centrifuged at 14,000 rpm for 10 minutes. 80 µl of clarified fecal sample was then treated with nucleases to eliminate host nucleic acids (19). Following the nuclease pre- treatment, total nucleic acids were extracted using MagMax Microbiome Ultra Nucleic Acid Isolation Kit, with bead tubes (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Reverse transcription was performed using SuperScript III First Strand Synthesis System (ThermoFisher Scientific, Waltham, MA, USA), followed by RNase H digestion. The second strand was synthesized using Sequenase 2.0 DNA Polymerase (Afflymetrix, Santa Clara, CA, USA). Sequencing libraries were constructed from purified 1 ng cDNA using the Nextera XT library preparation kit (Illumina, San Diego, CA, USA) followed by sequencing on a MiSeq instrument for 300 cycles (2 X 150 paired end). The raw reads were adapter-trimmed and mapped against each of the individual viral segments of published caprine RVB (16) by using CLC Genomics (Qiagen, Hilden, Germany) to obtain the number of reads mapping to RVB. Contigs were assembled de novo using CLC Genomics by setting the bubble size, word size graph and detection of paired distances parameters to automatic and, the minimum allowed contig length to 500 bp. Contigs encoding nucleotides with homology to RVB were identified by BLAST2Go plugin analysis. In addition, the identified contigs were individually confirmed by BLASTn in NCBI. The partial or complete genome sequences of VP1-VP4, VP7, NSP1, NSP2, NSP4, NSP5, (Segments 1-5 and 8-11); VP6 (Segment 6) and NSP3 (Segment 7) of RVB/Goat/KY/03/2021 were submitted to GenBank under accession numbers of ON565197-ON565205, OQ832070 and OQ832069, respectively.
Phylogenetic analyses were performed using MEGA11 software for protein coding nucleotide sequences. Evolutionary analyses of open reading frames (ORF) of 11 segments of RVB/Goat-wt/USA/KY/03/2021 were conducted individually using the maximum-likelihood algorithm with best nucleotide substitution models by performing 1000 bootstrap replicates as reported recently (19). Complete or near-complete nucleotide sequences of the genes from RVB strains of swine, human, caprine, bovine, ovine and murine origin were included in the analysis. Alignment of protein coding sequences for each gene was performed by MUSCLE in MEGA11. The trees were constructed as the general time-reversible model with gamma distribution and invariant sites (GTR+G+I) for VP1, VP2, VP3, VP6, and NSP1; the Hasegawa-Kishino-Yano model with gamma distribution and invariant sites (HKY+G+I) for VP4 and NSP4; the Tamura 3-parameter with gamma distribution and invariant sites (T92+G+I) for VP7; and the Tamura-Nei with gamma distribution and invariant sites (TN93+G+I) for NSP2, NSP3, and NSP5 genes. Genotypes were assigned based on the phylogenetic tree, and nucleotide percentage identity analysis for protein coding sequences in each segment. Nucleotide cut-off percentage established previously were used for assigning genotypes for both ERVB and RVB/Goat-wt/USA/KY/03/2021 (19, 20).
The fecal sample from the goat kid was negative for Bovine coronavirus, Bovine rotavirus Group A, E. coli K99+, Cryptosporidium parvum and Salmonella spp by multiplex real-time PCR assays. The total nucleic acid extract from the fecal sample of the goat kid was tested for ERVB and found positive with cycle of threshold (Ct) value 27.5.
Sequencing of the caprine fecal sample generated 2,394,488 reads, mapping 5,460 reads to the previously published goat RVB (16). Distribution of mapped reads for each genome segment was 95 (VP7), 1283 (VP4), 26 (VP6), 1723 (VP1), 379 (VP2), 1025 (VP3), 361 (NSP1), 97 (NSP2), 275 (NSP3), 92 (NSP4) and 104 (NSP5). All the 11 gene segments of RVB/Goat-wt/USA/KY/03/2021 were sequenced either completely or partially following de novo assembly of the reads. While VP7, VP4, VP1, NSP1-NSP5 were sequenced completely, VP6, VP2 and VP3 were partially sequenced (Table 1). VP7, VP4, VP6, VP1, NSP2 and NSP4 had the highest nucleotide identities to ERVB with 96.92%, 97.75%, 98.37%, 98.07%, 98.89% and 98.54%, respectively (Table 1). VP2, VP3, NSP1, NSP3 and NSP5 had the highest nucleotide identity to previously sequenced caprine RVBs with 98.66%, 94.72%, 96.98%, 95.17% and 96.80%, respectively (Table 1).
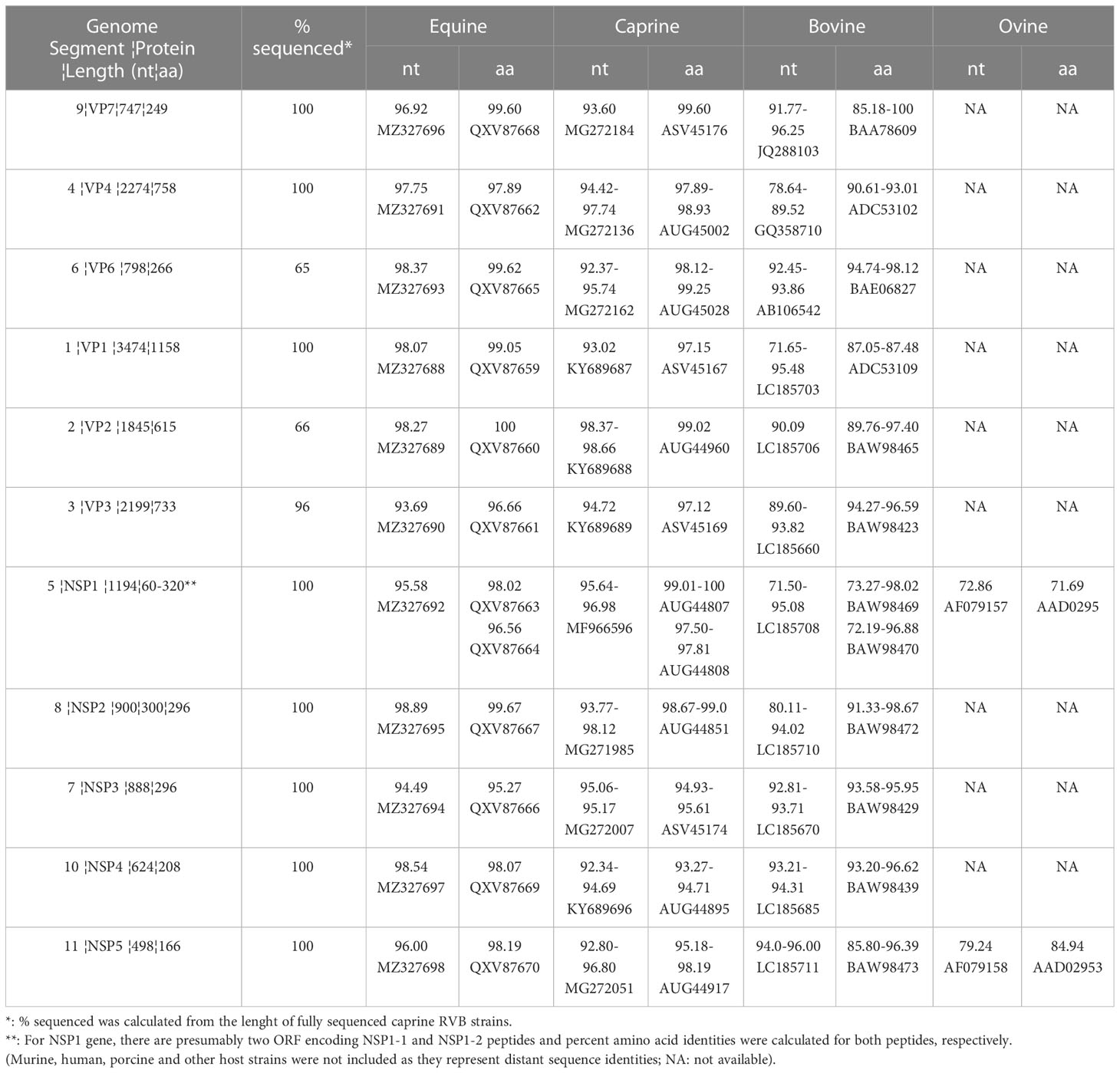
Table 1 Gene segments and ranges of percent nucleotide (nt) and amino acid (aa) identities (with best BLAST hit accession numbers) of the RVB/Goat-wt/USA/KY/03/2021 with those of the equine, caprine, bovine, and ovine host species.
VP6, VP1, VP2, NSP2, and NSP4 of RVB/Goat-wt/KY/03/2021 had highest amino acid identities to ERVBs (Table 1). While VP4, VP3 and NSP1 had the highest amino acid identities to previously sequenced caprine RVBs, VP7 and NSP3 had the highest amino acid identities to bovine RVBs (Table 1). NSP5 had the highest amino acid identities to both equine and caprine RVBs. The amino acid percent identities of the individual viral proteins of RVB/Goat-wt/USA/KY/03/2021 were between 96.66-100% to ERVB, 93.27-99.60% to goat RVBs, and 72.19-100% to cattle RVBs (Table 1).
RVB/Goat-wt/USA/KY/03/2021 had the genome constellations of G3-P[3]-I3-R3-C3-M3-A4-N3-T3-E3-H3, identical to ERVB and previously published two American caprine RVBs (Table 2). RVB strains from the same genotypes clustered in the phylogenetic trees confirming genome constellations. VP7, VP4, VP6, VP1, NSP2 and NSP4 shared a common ancestor with the ERVB strains, albeit with relatively low bootstrap values at the nodes in the tree of VP6. (Supplemental Figures 1, 2). NSP3, VP2 and VP3 shared a common ancestor with the ERVB and previously sequenced caprine strains (Supplemental Figures 1, 2), The caprine, equine, and a Japanese bovine strain shared a common ancestor for NSP1 while the US caprine strain from California CA22, ERVB, and our strain shared a common ancestor for NSP5. RVB/Goat-wt/USA/KY/03/2021 was more closely related to the Japanese bovine RVB strains than the Indian bovine RVB strains.
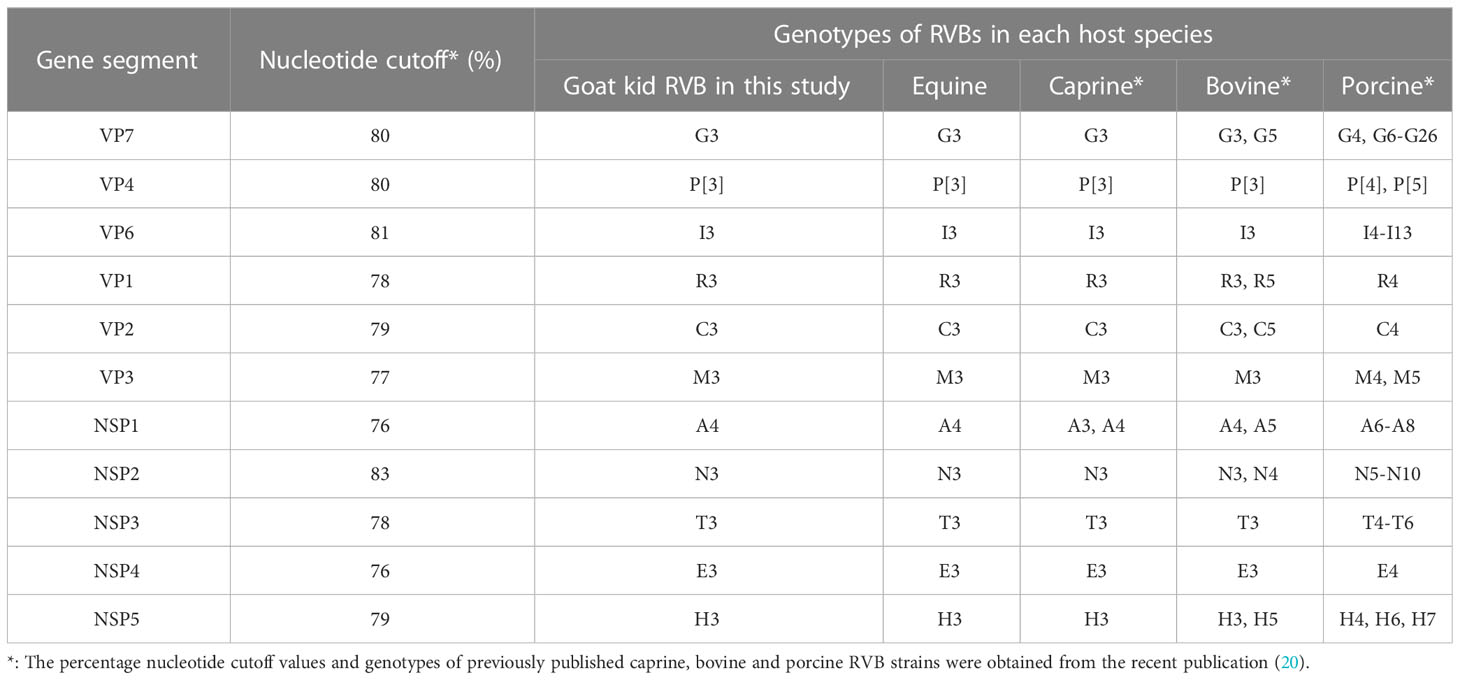
Table 2 Genotypes of RVB/Goat-wt/USA/KY/03/2021 and other RVBs from equine, caprine, bovine, and porcine host species.
Rotavirus B strains cause clinical disease in a variety of animals, including humans. Previous phylogenetic studies indicated bovine and caprine RVB strains formed a distant cluster from rat, human, and porcine RVB strains (14–16). We present the third case of a RVB in goat kids with diarrhea in the United States, suggesting RVB may cause diarrhea in goat kids more commonly than previously thought. Additional surveillance studies would further enhance our understanding of caprine RVB prevalence and its association with clinical disease in goats.
Analysis of phylogenetic trees constructed with the sequenced gene segments indicated that all gene segments of RVB/Goat-wt/USA/KY/03/2021 (except NSP1) shared a common ancestor with equine alone or both equine and caprine RVB strains confirming an earlier study (19). The NSP1 clustered with both equine, caprine, and Japanese bovine strains, again confirming the earlier study (19). It should be noted that relatively low bootstrap value for VP6 was generated, most likely due to the low sequence coverage (65%). Overall, the caprine strains did not cluster together in all the gene segments (most evidently VP7), indicating reassortment and a complex evolutionary relationship with bovine and equine strains.
Central Kentucky, generally comprised of five counties surrounding Lexington, is considered as the capital of Thoroughbred equine breeding industry. For example, in 2021 approximately 16,500 Thoroughbred mares (approximately 75% of registered Thoroughbred broodmares in North America) were bred natural by Kentucky stallions. Of note, the farm with the diarrheic goat kids is around 25 kilometers away from the Thoroughbred farms that had the ERVB associated neonatal foal diarrhea outbreaks in the Spring of 2021 (19). Given the geographical and temporal relationship of RVB/Goat-wt/USA/KY/03/2021 and ERVB, it is possible that a single RVB strain may be the source of the infection in both neonatal foals and goat kids. The outer and inner capsid genes (VP7, VP4, and VP6) of RVB/Goat-wt/USA/KY/03/2021 shared a common ancestor with the ERVB highlighting the potential for interspecies transmission. However, the directionality or host species of the infection is indecipherable due to the lack of temporal and geographical caprine, equine, and bovine RVB strains. There are only three caprine and a single equine RVB strains in the publicly available databases and are only from USA, which limits our ability to unravel the complex evolutionary relationships among caprine, equine and bovine strains.
Emergence of new variants of concern can arise because the segmented nature of RV genome allows for both genomic reassortment and recombination (2, 21, 22). It has been shown that experimental infection of animals with different rotavirus strains results in the development of illness indicating interspecies transmission (23–25). In addition, several groups have reported naturally occurring interspecies transmission and reassortment events involving human and porcine RVA (26), human and bat RVA (9, 27), equine and bat RVA (27), feline and bovine RVA (22), caprine and bovine RVB (16), and turkey RVG and human RVB strains (28). Interspecies transmission between foals and goat kids is highly possible, especially since some farms house both species. Moreover, natural factors like snowmelt, rainwater runoff and floods might disseminate the excreted viruses to the environment. Animal transmission and pathogenesis studies are needed to understand the genetic signatures that drive the interspecies transmission of rotavirus B between equine and caprine species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethical review and approval was not required for the study on animals in accordance with the local legislation and institutional requirements.
LP performed the research, analyzed the data and contributed writing the manuscript. JM, GP performed research. TU performed research and analyzed the data. FL, BH, EA and CC critically reviewed the manuscript. DM performed the research, analyzed the data, contributed writing the manuscript and critically reviewed the manuscript. EE conceptualized the study, performed the research, analyzed the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by internal sources from University of Kentucky Veterinary Diagnostic Laboratory.
We thank practitioners, and faculty and staff of University of Kentucky Veterinary Diagnostic Laboratory for their dedication and meticulous work.
Author DM was employed by the company Indical Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2023.1181017/full#supplementary-material
Supplementary Figure 1 | Phylogenetic trees (Original trees) of the protein-coding nucleotide sequences of RVB/Goat/KY/03/2021 NSP1, NSP2, NSP3, NSP4, NSP5, VP1, VP2, VP3, VP4, VP6 and VP7 (I) genes. Maximum-likelihood analysis in combination with 1000 bootstrap replicates was used to derive trees based on the nucleotide sequences encoding respective proteins. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Evolutionary analyses were conducted in MEGA11. Human and porcine RVBs were condensed for simplicity.
Supplementary Figure 2 | Bootstrap consensus trees of the RVB/Goat/KY/03/2021. In addition to the original trees, this figure is provided to better visualize the collapsed genes and the evolution of each of the gene segment of RVBs.
1. Matthijnssens J, Otto PH, Ciarlet M, Desselberger U, Ranst V, Johne R. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch Virol (2012) 157:1177–82. doi: 10.1007/s00705-012-1273-3
2. Martella V, Banyai K, Matthijnssens J, Buonavoglia C, Ciarlet M. Zoonotic aspects of rotaviruses. Vet Microbiol (2010) 140:246–55. doi: 10.1016/j.vetmic.2009.08.028
3. Mukherjee A, Mullick S, Deb AK, Panda S, Chawla-Sarkar M. First report of human rotavirus G8P(4) gastroenteritis in India: evidence of ruminants-to-human zoonotic transmission. J Med Virol (2013) 85:537–45. doi: 10.1002/jmv.23483
4. Bwogi J, Jere KC, Karamagi C, Byarugaba DK, Namuwulya P, Baliraine FN, et al. Whole genome analysis of selected human and animal rotaviruses identified in Uganda from 2012 to 2014 reveals complex genome reassortment events between human, bovine, caprine and porcine strains. PloS One (2017) 12:e0178855. doi: 10.1371/journal.pone.0178855
5. Thanh HD, Tran VT, Lim I, Kim W. Emergence of human G2P [4] rotaviruses in the post-vaccination era in south Korea: footprints of multiple interspecies re-assortment events. Sci Rep (2018) 8:6011. doi: 10.1038/s41598-018-24511-y
6. Sircar S, Malik YS, Kumar P, Ansari MI, Bhat S, Shanmuganathan S, et al. Genomic analysis of an Indian G8P[1] caprine rotavirus-a strain revealing artiodactyl and DS-1-Like human multispecies reassortment. Front Vet Sci (2021) 7:606661. doi: 10.3389/fvets.2020.606661
7. Matthijnssens J, Ciarlet M, Rahman M, Attoui H, Banyai K, Estes MK, et al. Recommendations for the classification of group a rotaviruses using all 11 genomic RNA segments. Arch Virol (2008) 153(8):1621–9. doi: 10.1007/s00705-008-0155-1
8. Walker PJ, Siddell SG, Lefkowitz EJ, Mushegian AR, Dempsey DM, Dutilh BE, et al. Changes to virus taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses. Arch Virol (2019) 164:2417–29. doi: 10.1007/s00705-019-04306-w
9. Komoto S, Tacharoenmuang R, Guntapong R, Upachai S, Singchai P, Ide T, et al. Genomic characterization of a novel G3P[10] rotavirus strain from a diarrheic child in Thailand: evidence for bat-to-human zoonotic transmission. Infect Genet Evol (2021) 87:104667. doi: 10.1016/j.meegid.2020.104667
10. Hung T, Chen GM, Wang CG, Chou ZY, Chao TX, Ye WW, et al. Rotavirus-like agent in adult non-bacterial diarrhea in China. Lancet (1983) 2:1078–9. doi: 10.1016/S0140-6736(83)91058-9
11. Muñoz M, Alvarez M, Lanza I, Cármenes P. An outbreak of diarrhoea associated with atypical rotaviruses in goat kids. Res Vet Sci (1995) 59:180–2. doi: 10.1016/0034-5288(95)90057-8
12. Chang KO, Parwani AV, Smith D, Saif LJ. Detection of group b rotaviruses in fecal samples from diarrheic calves and adult cows and characterization of their VP7 genes. J Clin Microbiol (1997) 35:2107–10. doi: 10.1128/jcm.35.8.2107-2110.1997
13. Papp H, Malik YS, Farkas SL, Jakab F, Martella V, Banyai K. Rotavirus strains in neglected animal species including lambs, goats, camelids. Virus Dis (2014) 25:215–22. doi: 10.1007/s13337-014-0203-2
14. Barman P, Ghosh S, Das S, Varghese V, Chaudhuri S, Sarkar S, et al. Sequencing and sequence analysis of VP7 and NSP5 genes reveal emergence of a new genotype of bovine group b rotaviruses in India. J Clin Microbiol (2004) 42:2816–8. doi: 10.1128/JCM.42.6.2816-2818.2004
15. Hayashi-Miyamoto M, Murakami T, Minami-Fukuda F, Tsuchiaka S, Kishimoto M, Sano K, et al. Diversity in VP3, NSP3, and NSP4 of rotavirus b detected from Japanese cattle. Infect Genet Evol (2017) 49:97–103. doi: 10.1016/j.meegid.2017.01.003
16. Chen F, Knutson TP, Ciarlet M, Sturos M, Marthaler DG. Complete genome characterization of a rotavirus b (RVB) strain identified in alpine goat kids with enteritis reveals inter-species transmission with RVB bovine strains. J Gen Virol (2018) 99:457–63. doi: 10.1099/jgv.0.001022
17. Otto PH, Rosenhain S, Elschner MC, Hotzel H, Machnowska P, Trojnar E, et al. Detection of rotavirus species a, b and c in domestic mammalian animals with diarrhoea and genotyping of bovine species a rotavirus strains. Vet Microbiol (2015) 179:168–76. doi: 10.1016/j.vetmic.2015.07.021
18. Cho Y, Kim W, Liu S, Kinyon JM, Yoon KJ. Development of a panel of multiplex real-time polymerase chain reaction assays for simultaneous detection of major agents causing calf diarrhea in feces. J Vet Diagn Invest (2010) 22:509–17. doi: 10.1177/104063871002200403
19. Uprety T, Sreenivasan CC, Hause BM, Li G, Odemuyiwa SO, Locke S, et al. Identification of a ruminant origin group b rotavirus associated with diarrhea outbreaks in foals. Viruses (2021) 13(7):1330. doi: 10.3390/v13071330
20. Shepherd FK, Herrera-Ibata DM, Porter E, Homwong N, Richard Hesse N, Jianfa Bai J, et al. Whole genome classification and phylogenetic analyses of rotavirus b strains from the united states. Pathogens (2018) 7:44. 18. doi: 10.3390/pathogens7020044PMCID:
21. Marthaler D, Suzuki T, Rossow K, Culhane M, Collins J, Goyal S, et al. VP6 genetic diversity, reassortment, intragenic recombination and classification of rotavirus b in American and Japanese pigs. Vet Microbiol (2014) 172:359–66. doi: 10.1016/j.vetmic.2014.05.015
22. Jeong S, Than VT, Lim I, Kim W. Whole-genome analysis of a rare human Korean G3P rotavirus strain suggests a complex evolutionary origin potentially involving reassortment events between feline and bovine rotaviruses. PloS One (2014) 9:e97127. doi: 10.1371/journal.pone.0097127
23. Azevedo MS, Yuan L, Jeong KI, Gonzalez A, Nguyen TV, Pouly S, et al. Viremia and nasal and rectal shedding of rotavirus in gnotobiotic pigs inoculated with wa human rotavirus. J Virol (2005) 79:5428–36. doi: 10.1128/JVI.79.9.5428-5436.2005
24. Fujii Y, Hirayama M, Nishiyama S, Takahashi T, Okajima M, Izumi F, et al. Characterization of an avian rotavirus a strain isolated from a velvet scoter (Melanitta fusca): implication for the role of migratory birds in global spread of avian rotaviruses. J Gen Virol (2022) 103. doi: 10.1099/jgv.0.001722
25. Ciarlet M, Estes MK, Conner ME. Simian rhesus rotavirus is a unique heterologous (non-lapine) rotavirus strain capable of productive replication and horizontal transmission in rabbits. J Gen Virol (2000) 81:1237–49. doi: 10.1099/0022-1317-81-5-1237
26. Wandera EA, Hatazawa R, Tsutsui N, Kurokawa N, Kathiiko C, Mumo M, et al. Genomic characterization of an African G4P[6] human rotavirus strain identified in a diarrheic child in Kenya: evidence for porcine-to-human interspecies transmission and reassortment. Infect Genet Evol (2021) 96:105133. doi: 10.1016/j.meegid.2021.105133
27. Simsek C, Corman VM, Everling HU, Lukashev AN, Rasche A, Maganga GD, et al. At Least seven distinct rotavirus genotype constellations in bats with evidence of reassortment and zoonotic transmissions. mBio (2021) 12:e02755–20. doi: 10.1128/mBio.02755-20
Keywords: foal, goat kid, phylogenetic, rotavirus B, sequencing
Citation: Paul L, Morgan J, Pulley G, Uprety T, Hause BM, Adam E, Li F, Carter CN, Marthaler D and Erol E (2023) Genetic and phylogenetic relationship of an American caprine rotavirus B strain with equine rotavirus B. Front. Virol. 3:1181017. doi: 10.3389/fviro.2023.1181017
Received: 06 March 2023; Accepted: 12 May 2023;
Published: 25 May 2023.
Edited by:
Burtram Clinton Fielding, University of the Western Cape, South AfricaReviewed by:
Tsutomu Omatsu, Tokyo University of Agriculture and Technology, JapanCopyright © 2023 Paul, Morgan, Pulley, Uprety, Hause, Adam, Li, Carter, Marthaler and Erol. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erdal Erol, ZXJkYWwuZXJvbEB1a3kuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.