Abstract
Human respiratory syncytial virus (RSV) is the primary cause of acute lower respiratory tract infections in children and the elderly worldwide, for which neither a vaccine nor an effective therapy is approved. The entry of RSV into the host cell is mediated by stepwise structural changes in the surface RSV fusion (RSV-F) glycoprotein. Recent progress in structural and functional studies of RSV-F glycoprotein revealed conformation-dependent neutralizing epitopes which have become attractive targets for vaccine and therapeutic development. As RSV-F is present on viral surface in a trimeric form, a trivalent binding interaction between a candidate fusion inhibitor and the respective epitopes on each of the three monomers is expected to prevent viral infection at higher potency than a monovalent or bivalent inhibitor. Here we demonstrate a novel RSV entry inhibitory approach by implementing a trimeric DNA nanostructure as a template to display up to three linear peptide moieties that simultaneously target an epitope on the surface of the prefusion RSV-F protein. In order to design synthetic binding peptides that can be coupled to the DNA nanostructure, the prefusion RSV-F-specific monoclonal antibody (D25) was selected. Complementarity-determining region 3 (CDR3) derived peptides underwent truncation and alanine-scanning mutagenesis analysis, followed by systematic sequence modifications using non-canonical amino acids. The most effective peptide candidate was used as a binding moiety to functionalize the DNA nanostructure. The designed DNA-peptide construct was able to block RSV infection on cells more efficiently than the monomeric peptides, however a more moderate reduction of viral load was observed in the lungs of infected mice upon intranasal application, likely due to dissociation or absorption of the underlying DNA structure by cells in the lungs. Taken together, our results point towards the inhibitory potential of a novel trimeric DNA-peptide based approach against RSV and open the possibility to apply this platform to target other viral infections.
1 Introduction
Human respiratory syncytial virus (RSV) is a viral pathogen that circulates seasonally in the human population and is transmitted by aerosols, direct contact with an infected person or contaminated surfaces (1). Despite most people receiving multiple exposures, RSV infection induces poor long-term protective immunity, leading to common recurrent infections. While the course of infection with RSV in healthy patients is usually limited to mild upper respiratory tract infections, it can progress to complicated lower respiratory tract infections and pneumonia in high-risk populations, including infants, immunocompromised patients, and the elderly (2–4). Among children under five years old, RSV is the leading respiratory pathogen requiring hospital admission (5). Worldwide, hospitalizations due to RSV infection are estimated at 3.2 million cases, causing up to 118,000 deaths annually (6). However, the accurate assessment of RSV burden is challenging since most of the cases occur in developing countries (7). Since the first isolation and description of RSV in 1955, neither a vaccine, nor an effective antiviral therapy against RSV have been approved (8, 9). All attempts for the development of safe and effective vaccines against RSV failed either at preclinical or clinical stages. Current prophylactic options are limited to the humanized monoclonal antibody palivizumab, which targets the fusion glycoprotein (RSV-F) on viral surface and prevents infection at its early stage. Prophylaxis with palivizumab requires monthly intramuscular administrations of 15 mg per kg body weight (mg/kg) via injection in RSV-infection season. Due to its high cost, its use is limited to high risk infants, although its efficacy is still debatable (10–14).
RSV belongs to the family Pneumoviridae and is a non-segmented negative-strand RNA virus. The trimeric RSV-F protein is the major surface antigen that mediates RSV entry into host cells and is the prime target for antiviral interventions against RSV infection (15–17). Similar to other class I fusion proteins, RSV-F is present on the surface of infectious viral particles in a metastable pre-fusion conformation containing a hydrophobic fusion peptide, which is inserted into the host cell membrane during the fusion process (18). This triggers the formation of the stable six-helix bundle hairpin-like structure, which brings viral and cell membranes into close proximity leading to membrane fusion and viral entry (19). Following the irreversible structural rearrangements, RSV-F adopts a stable inactive postfusion conformation. Progress in understanding the structural functionality and resulting strategies for the stabilization of RSV-F protein in its prefusion state allowed the characterization of this metastable conformation and revealed new, neutralization-sensitive epitopes present exclusively on the prefusion RSV-F that are targets for potent neutralizing antibodies (20, 21). Since then, numerous antibodies and inhibitors targeting prefusion RSV-F, which prevent the conformational changes required for RSV entry have been identified (20, 22, 23). One of them is the human antibody D25, which is directed against the prefusion-specific antigenic site Ø and neutralizes RSV infection with higher potency than palivizumab. The D25 antibody passed the preclinical tests and its optimized variant (MEDI8897) has already shown promising results in phase II clinical trial (24, 25).
Many viral and cellular proteins are often present in a multimeric form with several recognition sites. Thus, viruses including RSV exploit the principle of multivalence to enter the host cell via cooperative binding between multiple target ligands and geometrically complementary viral surface glycoproteins. In a similar multivalent manner, antibodies bind to viral targets with their two variable domains, leading to blockage of their function (26–28). This principle has also been exploited to design synthetic, multivalent nanoscaffolds for an enhancing efficacy in biological and biomedical applications (29), including many approaches specifically geared towards the inhibition of viral infections (30, 31). In the same line of thought, biomolecules have been presented multivalently in rationally-designed arrangements on synthetic scaffolds consisting of DNA-based nanostructures to form functionalized constructs, which improved the potency of single bioactive ligands toward their targets (32–36). For an oncological application, peptide mimics of the natural ephrin1 ligand of the dimeric Ephrin-A2 (EphA2) receptor that is overexpressed in tumors were linked to DNA nanostructures, which activated EphA2-mediated downstream pathways in cancer cells more efficiently than single peptides (37). In the context of proteins on the surface of viruses, conjugation of small ligands targeting the trimeric hemagglutinin protein onto branched DNA nanostructures has enabled templated, multivalent interactions. In several approaches, the multivalent presentation of sialic acid, the natural targeted ligand for hemagglutinin, on DNA nanostructures has led to increased affinity when compared to single ligands (38–41). Recent studies have also extended this approach to short, synthetic peptides, derived from the complementarity determining region (CDR) of a monoclonal antibody against influenza A hemagglutinin protein, leading to a significantly enhanced inhibition of hemagglutination in comparison to the monomeric peptides (42) and can be integrated into analytical systems for testing peptide-based virus-binding ligands (43).
In both a specialized ELISA-based assay and a quantitative method based on oscillating lever arms to assess binding kinetics, it was demonstrated that the geometrically complementary, trivalent presentation of fusion-inhibiting peptides significantly enhances their binding to the trimeric SARS-CoV-2 spike protein, compared to the monovalent presentation of peptides (44).
The multivalent presentation of dengue-inhibiting aptamers on a more complex DNA nanostructure has also been shown to enhance in vitro inhibition, however with a slightly different approach of simultaneously targeting multiple ED3 protein clusters over the virus surface, each with single ligands, rather than exploiting the multimeric structure of ED3 itself (34). Another approach used large, icosahedral shells, ranging from several tens to hundreds of megadaltons in size, based on DNA origami structures functionalized with virus-binding antibodies to encase whole hepatitis and adeno-associated viruses (45). In the presented study we adapted the same approach as described above for influenza to address RSV infection, with small, DNA-peptide conjugates. For this purpose, we targeted the trimeric RSV-F protein by trimeric DNA nanostructures functionalized with three antigen-binding peptide fragments capable of cooperatively blocking all three subunits of RSV-F. For designing peptide binding moieties, we chose the CDR3 sequence of D25 antibody as a starting point. By functional screening of different sequence regions and subsequent optimizations, we identified the most potent peptide candidate with in vitro and in vivo activity against RSV. The designed trimeric peptide-DNA-scaffold was evaluated as a novel inhibitor of RSV entry.
2 Materials and methods
2.1 Computational analysis
The crystal structure of D25 Antibody in complex with RSV-F protein (PDB ID: 4JHW) was loaded from the RCSB Protein Data Bank (http://www.rcsb.org/) and prepared for analysis, using MOE 2016.08_02 (46). The overall structure is depicted in Figure 1A. Subsequently, a contact analysis was performed, focusing on a maximum sequence of consecutive amino acids (linear) and smallest possible gaps. The results were visualized and manually scored. The essential structural region, from which a main motif was derived, that subsequently served as the basis for following mutation, deletion and permutation experiments, is shown in Figures 1B, C.
Figure 1
2.2 Peptide synthesis
The investigated peptides were either synthesized by standard Fmoc-solid phase peptide synthesis or purchased from Peptide Specialty Laboratories (Heidelberg, Germany). Fmoc-SPPS was performed on a Tetras peptide synthesizer (Advanced ChemTech, Louisville, USA) at 60-μmol scale as C-terminal amides on Rink amide resin (Iris Biotech) using standard Fmoc/tBu-protected amino acids (Iris Biotech). Coupling was performed using O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) and N-methylmorpholine (NMM). Fmoc-deprotection was carried out using 20% piperidine in DMF. Final cleavage and deprotection of the peptides was performed using TFA:DOTA (or EDT):H2O:TIS (30:2:2:1 v/v). After precipitation with cold diethylether, the peptides were purified by preparative RP-HPLC (Phenomenex Luna C18 (2) column, eluents: water and acetonitrile containing 0.04% TFA). Peptide purity was confirmed > 95% as evaluated by HPLC. Identity of the peptides was confirmed by ESI-MS. The peptides were dissolved in dimethylsulfoxide (DMSO) and subsequently diluted in phosphate-buffered saline or cell culture medium for the experiments.
2.3 Cells and virus culture
HEp-2 (human epithelial type-2) cells were obtained from ATCC and cultured in Dulbecco’s Modified Eagle’s medium (DMEM, Thermo Fisher Scientific, Germany) supplemented with 10% (v/v) heat inactivated fetal calf serum (FCS) and 100 U/ml Penicillin with 100 µg/ml Streptomycin (Pen/Strep) (Thermo Fisher Scientific, Germany) at 37°C in 5% CO2 incubator.
RSV laboratory strain A long (VR-26, ATCC) and rgRSV (Recombinant RSV strain A2 expressing green fluorescent protein (GFP), kindly provided by M.E. Peeples) were propagated in HEp-2 cells. Cells were infected with a multiplicity of infection (MOI) of 0.1 focus forming unit (FFU)/cell and incubated for 2-3 days at 37°C and 5% CO2 until cytopathic effects were observed. The virus was purified from cell culture supernatant by ultracentrifugation in a SureSpin 630 (Thermo scientific) swing-out rotor at 10,600 rpm (21,000 xg) through a 20% (w/v) sucrose cushion in PBS for 3 h at 4°C. After centrifugation, the supernatant was discarded and the pelleted virus was resuspended in 10% sucrose in PBS and stored at -80°C.
RSV stocks were titered by infecting monolayers of HEp-2 cells with serial dilutions of the virus in a 96-well microtiter plate. Infected cells were incubated for 48 h at 37°C. Titers of rgRSV and RSV-Long were analyzed either by fluorescent microscopy or immunocytochemical staining using anti-RSV antibody (AB1128; Sigma-Aldrich), respectively (47, 48).
2.4 DNA-peptide trimer synthesis and purification
Partially complementary strands (Table 1) were purchased with 5′ end amino-group (Biomers.net, Germany), delivered in dry and HPLC-purified form and were resuspended in water.
Table 1
| Name | ssDNA | DNA sequence 5′→3′ |
|---|---|---|
| 30 bp construct | forward | ATAATCTTGGATACGATATCACCATACGTC |
| reverse | GACGTATGGTGATATCGTATCCAAGATTAT | |
| DNA trimer | ab | ACTATCTTTGGTCTATTATCTTGAGTCATC |
| b*c | GATGACTCAAGATAAACACACACACAACTA | |
| c*a* | TAGTTGTGTGTGTGTTAGACCAAAGATAGT |
Oligonucleotide sequences of DNA constructs.
Complementary segments are denoted with a star, i.e. “a” as part of strand “ab” is complementary to “a*” in strand “c*a*”.
Amino-modified ssDNA strands were incubated with a 100-fold molar excess of DBCO-PEG4-NHS ester (Jena Bioscience, Jena, Germany) in 1x PBS pH 7.4 at room temperature (RT) overnight. The next day, DNA trimers were purified from unconjugated DBCO-NHS esters by ethanol precipitation and resuspended in DMSO. DNA strands were mixed in equimolar amounts (25 µM each) to guarantee for optimal stoichiometry and thus high yields. To assemble DNA structures, the mixture of DNA strands was heated to 95°C for 2 min, hybridized at 48°C for 15 min and cooled down to 4°C. For trimers carrying no peptides, amino-modified DNA strands were replaced by three unmodified strands.
In case of 30 bp dsDNA construct, DNA was heated to 95°C for 2 min and then cooled down to 4°C. Folded DNA trimers or dsDNA constructs were functionalized with peptides by incubating them with a 20-fold molar excess of azide-containing Pep30 [Chg][Cpa]VSTT[Tyr3,5-I2]LPHYFDN-azidolysin (purchased from Peptide Specialty Laboratories, Heidelberg, Germany). The reaction was left to incubate for 73 h at RT. Unconjugated Pep30 was removed via ethanol precipitation of functionalized DNA-structures. To guarantee an efficient removal of free Pep30, the DNA trimer control (consisting of unmodified strands) was also incubated with Pep30 and purified via ethanol precipitation. Native polyacrylamide gel electrophoresis (PAGE) was used to analyze folding and functionalization.
2.5 ELISA
Nunc-Immuno 96 well MicroWell™ MaxiSorp™ plates were coated with 500 ng/well NeutrAvidin™ in coating buffer (0.1 M NaHCO3, pH 8.0) overnight at 4°C. The plates were washed three times with PBS-T (1x PBS, 10 mM MgCl2, 0.1% (v/v) Tween20, pH 7.4) and 46 nM Biotin- and peptide-carrying 30 bp dsDNA structures or appropriate controls (buffer only, 30bp dsDNA) in PBS-T were applied for 1 h at RT. Next, plates were washed three times with PBS-T and then active RSV-Long was applied for 1 h at RT. The wells were washed with blocking buffer (1x PBS, 10 mM MgCl2, 0.1% (v/v) Tween 20, 1% (v/v) BSA, pH 7.4) and polyclonal HRP conjugated RSV antibody (Thermo Fisher Scientific, Germany) was added to the wells at a dilution of 1:500 in blocking buffer for 1 h at RT. This was followed by washing with blocking buffer. For the detection, TMB substrate (Biozol, Germany) was added and the reaction was stopped by the addition of 1M H2SO4 at 50 µl/well. The absorbance was measured at 450 nm and 520 nm reference wavelength using microplate reader Tecan Infinite M1000 (Tecan, Switzerland).
2.6 Antiviral activity assay
15,000 HEp-2 cells were plated into each well of a 96-well microtiter plate in a total volume of 200 μl DMEM supplemented with 10% FCS and Pen/Strep and incubated overnight at 37°C in a 5% CO2 incubator. Cells were washed with PBS and the medium was replaced with 150 μl DMEM supplemented with 1% FCS and Pen/Strep. Peptides or peptide-DNA trimers dissolved in DMSO were diluted in FCS-free medium, mixed with rgRSV at 150 FFU/well and incubated for 10 min at 37°C. After incubation, virus-peptide mixtures were applied on cell monolayers in triplicates and incubated for 48 h at 37°C in a 5% CO2 incubator. RSV infection was quantified by counting green fluorescent infected cells using the AID EliSpot reader (AID Diagnostika, Strassberg, Germany). The percentage of infected cells was normalized to the corresponding vehicle-treated control. The 50% inhibitory concentrations (IC50) and Hill slopes were determined using the nonlinear regression analysis of the dose-response curves (log(inhibitor) vs response-variable slope) in GraphPad Prism 6.0.7.
2.7 Cell viability assay
Effects of peptides and peptide-DNA trimer on cell viability were tested using CellTiter-Glo Luminescent Cell Viability Assay (Promega) following manufacturer’s instructions. Briefly, HEp-2 cells were seeded at 15,000 cells/well in a 96-well microtiter plate. Next day, compounds and vehicle controls were applied to cell monolayers at different concentrations in triplicates. 48 h post treatment, cell culture supernatants were removed, CellTiter-Glo reagent diluted 1:1 with PBS was applied on cells and incubated for 10 min in the dark at RT to allow cell lysis and the release of ATP. Luminescence signals in supernatants were measured for 0.1 s using Berthold Centro XS3 luminometer. Values were normalized to sham-treated cells.
2.8 Antiviral activity analysis in mice
Female BALB/c mice were purchased from Charles River (Germany) at eight weeks of age and maintained in a specific pathogen-free environment in isolated ventilated cages. All animal experiments were carried out in accordance with the EU Directive 2010/63/EU for animal experiments and were approved by local authorities. For the simultaneous treatment, mice received under isoflurane anesthesia a mixture of either Pep30, DNA-Pep30 trimeric construct either mixed with 104 FFU of purified RSV (the Long strain) in 50 µl total volume via intranasal route. The control group was infected intranasally with 104 FFU of RSV mixed with vehicle solution in 50 µl total volume (49).
In another experiments, mice were pretreated intranasally with 50 nmol in 50 µl of Pep30 at 10 min before infection with 104 FFU of RSV and then treated daily for four days post-infection. In both experiments, mice lungs were isolated at day five post-infection and homogenized in gentleMACS™ M Tubes (Miltenyi Biotec, Germany) containing 2 ml of ice-cold PBS using gentleMACS Dissociator (Miltenyi Biotec, Germany). Homogenized tissues were cleared of cell debris by centrifugation at 2,000 xg and 4°C for 5 min and the supernatants were collected for viral RNA isolation.
2.9 Viral RNA extraction and RT-qPCR
Viral RNA was isolated from 140 µl of lung homogenate supernatants using QIAamp-Viral-RNA-Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions. 5 µl of isolated RNA were reverse transcribed and analyzed with the QuantiTect probe RT-PCR kit (Qiagen, Germany) using RSV forward primer (5’-AGATCAACTTCTGTCATCCAGCAA-3’), RSV reverse primer (5’-GCACATCATAATTAGGAGTATCAAT-3’) and SYBR Green as described previously (47). 10-fold dilutions of synthetic RSV-RNA of T7-transcripts were used as standards for the quantification of viral genome copy numbers in mouse samples.
2.10 Statistical analysis
Statistical analysis was performed using GraphPad Prism 6.0.7 (GraphPad Software, Inc., La Jolla, CA, USA). Data were checked for normality using the Shapiro-Wilk test. Data of viral loads in mouse lungs were non-normally distributed and thus analyzed with a Mann-Whitney-U-test. Level of significance is indicated with ** = p < 0.01.
3 Results
3.1 Design of RSV-binding peptide moieties
3.1.1 Structure-activity relationship analysis of CDR-derived peptide inhibitors
By analyzing the published crystal structure of the prefusion RSV-F protein in a complex with the D25 antibody (Figure 1), we selected a motif in the heavy chain of CDR3 of the D25 antibody (ETALVVSTTYLPHYFDN) that binds to a linear epitope on the prefusion conformation of RSV-F as a starting point for further analysis. First, we analyzed truncated CDRH3-derived peptides generated by gradual single amino acid deletions either at the N-terminal or the C-terminal end. Truncations of positions 1-3 at the N-terminus resulted in a gradual increase in the antiviral activity measured in an in vitro infection assay (Figure 2A). However, the removal of the four terminal amino acids at the N-end leads to a complete loss of antiviral activity. These results indicate that the exposure of leucine at the N-terminus end has a positive influence on the overall activity of the peptide. A peptide lacking the terminal amino acid at the C-end still retains its antiviral activity. However, any further deletions of amino acids from the C-terminus lead to activity loss. Therefore, we focused in this study on the 14-mer peptide (LVVSTTYLPHYFDN) comprising three amino acids deletions at the N-terminal. In the next step, we performed an alanine scanning analysis of the selected peptide (Figure 2B). For that, each of the 14 amino acid positions covering the whole peptide sequence was individually exchanged by alanine, and the resulting single amino acid mutated peptides were screened for antiviral activity against RSV in an infection assay. The substitution of the amino acids by alanine at seven positions (1, 2, 3, 7, 8, 9, and 13) led to more than 50% loss in the activity of the parent peptide at the tested concentration of 40 µM, indicating that the residues at these positions are essential for the activity, while alanine mutation at the remaining seven positions did not alter its antiviral effect.
Figure 2
3.1.2 Sequence modifications of the parent peptide
Four amino acid positions in the lead peptide (1, 2, 3 and 7) that are critical for its antiviral activity according to the results of the alanine-scan analysis were selected to introduce modifications into the peptide sequence. The aim of these modifications was the replacement of the original amino acids at the active positions with structurally similar non-canonical amino acid analogs to increase the stability towards proteases while retaining antiviral activity (50–53).
In the first round of modifications, single amino acids at one of the selected positions (1, 2, 3 and 7) were subjected to exchanges with their analogs (Table 2). The non-canonical amino acids share structure similarity with their natural analogs, but differ in some structural properties. The tyrosine at position 7 was modified by increasing alkyl chain length (homo tyrosine), incorporation of one or two iodines into the aromatic ring (iodo-tyrosine), nitration or methylation of the phenolic hydroxyl group.
Table 2
| Parent amino acid | Amino acid substitution | |
|---|---|---|
| Nomenclature and abbreviation | Structure | |
| Tyr | -L-homo-Tyr [hTyr] | 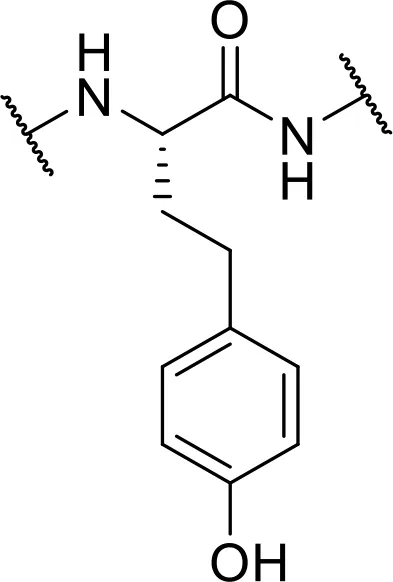 |
| -L-homo-Tyr (Me)-OH [hTyrMe] | 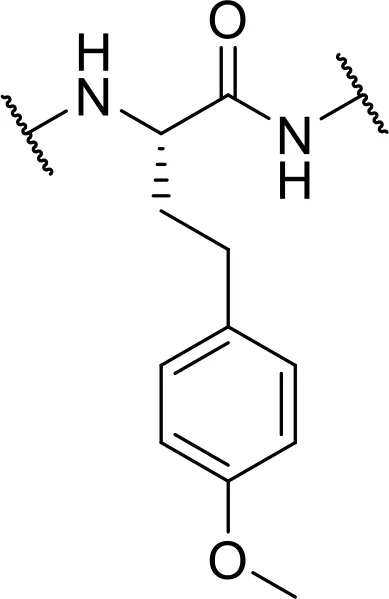 | |
| -3,5-diiodo-Tyr [Tyr3,5-I2] | 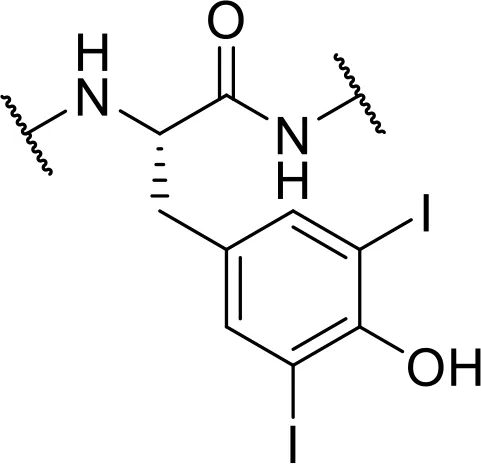 | |
| -3-iod-Tyr [Tyr3-I] | 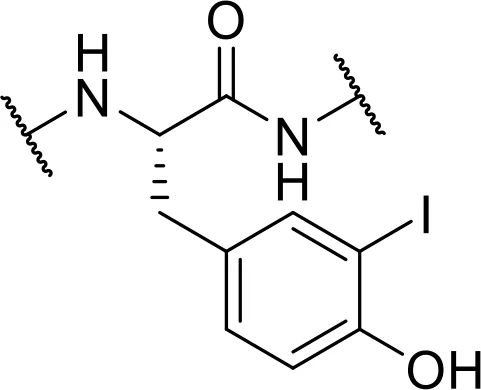 | |
| -3-nitro-Tyr [3-NO2] | 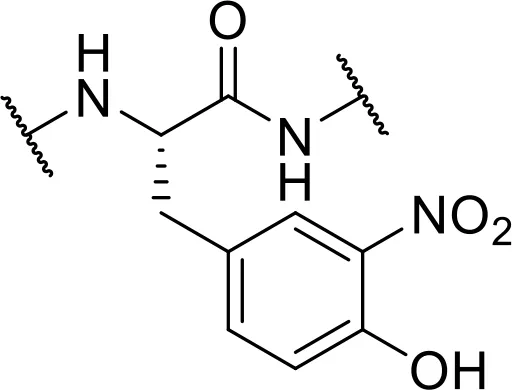 | |
| Val or Leu | Isoleucine [Ile] | 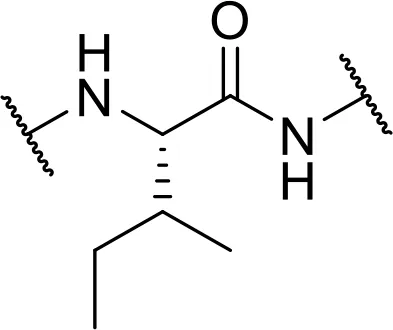 |
| L-Norleucine-OH [Nle] | 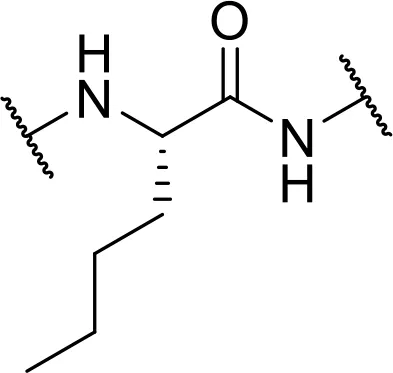 | |
| cyclopropyl-Ala [Cpa] | 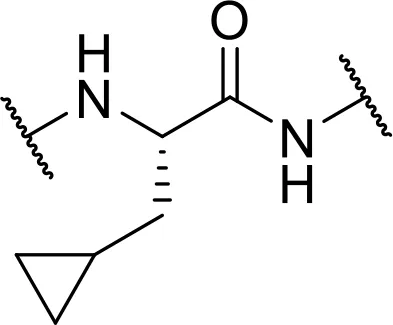 | |
| cyclo-Leucine [AC5C] | 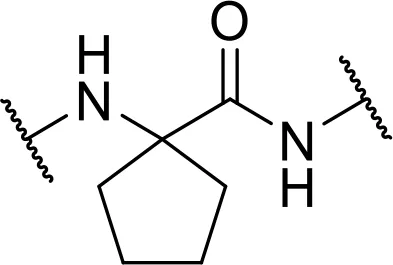 | |
| L-2Abu-OH [2Abu] | 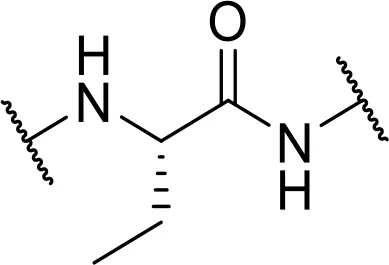 | |
| L-cyclohexyl-Gly-OH [Chg] | 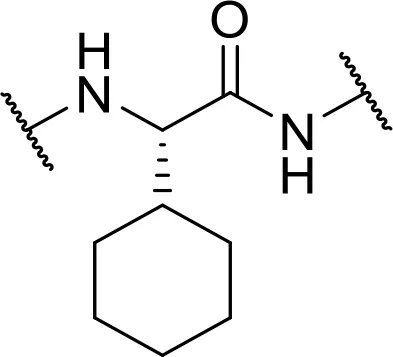 | |
Amino acids and their substitutions examined in this study.
The replacements of the structurally similar valine and leucine located at first three positions of the parent peptide were analogs with modifications in the hydrophobic side chain. This included cyclic amino acid derivates (cyclo-leucin), unbranched carbons in the side chain (Abu and Norleucin) or different alkyl chain branching (Ile).
The antiviral activities of mutant peptides carrying single non-canonical amino acid analogs of valine, leucine or tyrosine were compared in an in vitro assay (Figure 3A). All seven analyzed substitutions of the leucine at the first position led to reduction of the antiviral activity compared to the wild-type peptide. Exchanging the valine at the second and third position with its non-canonical amino acid analogs resulted in varying antiviral activities of the mutant peptides. The most potent peptides showing antiviral activity were those carrying instead of valine either cyclopropylalanine (Cpa) at the second position (Pep10) or cyclohexylglycine (Chg) at the third position (Pep20) with 67.3% and 87.7% inhibition of viral infection at the tested concentration of 10 µM, respectively. Mutations of the tyrosine residue at the seventh position with analogs carrying iodine atom were the most effective in enhancing viral inhibition in comparison to the methylated, nitrated or homo tyrosine, with the diiodotyrosine (Tyr3,5-I2) being superior to the monoiodotyrosine.
Figure 3
Next, peptides with different multiple combinations of non-canonical amino acid mutations at two, three or four positions were synthesized. For that, mutations were chosen that displayed potent activity in the first screening (Figure 3B). Among the peptides with multiple replacement with non-canonical amino acids, Pep30 with the sequence of [Chg][Cpa]VSTT[Tyr3,5-I2]LPHYFDN exerted the highest antiviral activity and was thus selected for further studies. This peptide inhibited RSV infection on HEp-2 cells in a dose-dependent manner with an inhibitory concentration of 50% (IC50) of 5.8 µM (Figure 4A) and showed no detectable cytotoxic effects up to 160 µM (Figure 4D).
Figure 4
To elucidate the prophylactic potential of the peptide candidate against RSV, HEp-2 cells were pretreated with the peptide at different time points up to 24 h before infection, and infection rates were compared. The inhibitory effect remained at similar level at all investigated pretreatment time points (0-24 h before infection) and peptide concentrations (10-80 µM). At 80 µM of Pep30 the inhibition was between 86-92%, while 10 µM of Pep30 resulted in 65-71% inhibition of infection (Figure 4B).
No change in antiviral activity was observed when cells were preincubated with the peptide for 1 hour and then washed before infection to remove the peptide, indicating uptake of the peptide by cells or peptide interaction with the cell membrane (Figure 4C).
3.2 Trivalent peptide-DNA conjugate as RSV inhibitor
Next, we sought to enhance the antiviral activity of the selected peptide candidate (Pep30) by coupling it to DNA nanostructures to form trivalent binding inhibitors. In order to determine whether tethering the peptide to a bulky scaffold eliminated its ability to bind RSV-F, we first constructed a monovalent DNA-peptide structure consisting of linear, double stranded 30 base-pair (30bp) DNA (dsDNA) conjugated to one molecule of Pep30 using NHS ester and copper-free click reaction. To analyze the binding of this initial monovalent construct to active RSV, we performed an ELISA. Avidin-coated ELISA plates were treated with the biotin-labeled 30bp dsDNA carrying Pep30. After applying active RSV and quantifying the amount of captured virus using anti-RSV antibody, we could show that Pep30 coupled to dsDNA is able to bind active RSV, whereas the signals in samples probed with a negative control of non-conjugated dsDNA without Pep30 were at the baseline level (9-fold lower than dsDNA-Pep30) (Figure 5B).
Figure 5
In the next step, we attempted to address the trimeric state of the RSV-F protein by implementing trivalent DNA-nanostructures, formed by the hybridization of three DNA oligonucleotides into a three-armed “Holliday junction” motif, functionalized by coupling Pep30 via NHS ester and copper-free click reactions, to produce a trimeric DNA scaffold with three arms displaying the peptide inhibitors (Figure 5A). The assembly of the DNA construct and the conjugation of Pep30 were analyzed using native PAGE (Supplementary Figure 1). Since the band corresponding to Pep30 incubated with unmodified DNA trimers has the same size as the unmodified DNA trimers, unspecific binding of Pep30 to DNA can be excluded. For a successful conjugation of Pep30, DNA was first modified with DBCO-PEG4, which served as a linker molecule and resulted in an upward shift of the construct size. The conjugation of Pep30 to the modified DNA led to a further increase in the molecular weight (Supplementary Figure 1). The conjugation yields were estimated by analyzing the intensity of the bands, which showed that approximately 60% of the conjugated DNA-Pep30 trimers carried three peptides, and a smaller portion of approximately 40% carried two peptides.
The constructed trimeric DNA-peptide structure was tested for inhibiting RSV infection on cells, and was shown to be able to block RSV infection in a concentration-dependent manner with an IC50 value of 27 nM, demonstrating a 214-fold higher potency than the uncoupled monomeric peptide Pep30 (IC50 5.8 µM) (Figures 5C, D). Since the overall activity is higher than 3-fold, which would correspond to only the additive activities of three monomeric peptides, these results indicate an enhanced inhibitory effect on RSV infection achieved through the trimeric representation of the single peptides on the trivalent DNA nanostructure. Both Pep30 and DNA-Pep30 exhibited dose-response curves with similar Hill slopes, approximately -2.2 for Pep30 and -2.1 for DNA-Pep30, indicating positive cooperativity of inhibitor binding.
All tested controls that include the different intermediate synthesis and modification steps of the DNA nanostructure before obtaining the final trimeric DNA structure coupled to Pep30 showed no effect of neutralizing RSV infection on cells, indicating specific RSV-inhibition attributable to interaction with DNA-Pep30 construct (Figure 5D).
3.3In vivo activity of designed inhibitors
We further evaluated the in vivo antiviral effect of Pep30 in an RSV mouse infection model (Figure 6A). In a proof-of-concept experiment, we tested a simultaneous intranasal treatment with RSV infection. For that, Pep30 was mixed at 50 nmol (equivalent to 100 µg) together with 104 FFU of RSV, and the mixture was immediately applied into the nose of the mice. The analysis of viral titers in lungs at five days post-infection (dpi) showed a significant (P=0.0079) reduction of 200-fold in the lungs of mice treated with Pep30 compared to the sham-treated control group (Figure 6B). In a second experiment, we tested the in vivo efficacy in a pre-and post-exposure prophylaxis treatment schedule. The peptide was administered prophylactically at 10 minutes before viral inoculation, then daily for four days post-infection (50 nmol/application). A statistically significant (P=0.002) 4-fold reduction of viral load in the lungs of treated mice was observed.
Figure 6
Next, we assessed the inhibitory effects of the DNA-Pep30 trimers against RSV upon intranasal delivery in a similar proof-of-concept experiment as for the monovalent Pep30 (Figure 6A). Mice received 6.5 nmol of the DNA-Pep30 trimer mixed with 104 FFU of RSV via the intranasal route. Treatment with DNA-Pep30 led to only an 11-fold significant (p=0.0022) reduction of viral RNA load detected in the lungs of infected mice at day five post-infection in comparison to the infected and sham-treated control group (Figure 6C). The roughly ten-fold lower concentration of the DNA-Pep30 trimers compared to the monovalent peptide Pep30 was due to practical limitations in the synthesis steps. While these trivalent constructs do lead to a moderate degree of inhibition, it is noticeably less, per nmol of inhibitor construct, than the monovalent peptides, in contrast to the results obtained in cellular models.
4 Discussion
The recent advances in structural studies of F protein-mediated RSV entry into host cells emphasize the importance of targeting conformational epitopes present on the active prefusion state of RSV-F protein for the development of effective vaccines and entry inhibitors of RSV infection (54, 55). McLellan et al. were able to stabilize the soluble version of the prefusion RSV-F trimeric protein by co-expressing it with a monoclonal antibody (D25) that exclusively targets this conformation. Solving the crystal structure of the protein-antibody complex revealed a new antigenic site Ø as a target epitope of D25 antibody and one of the major neutralization-sensitive epitopes that impair the conformational changes required for the fusion process, and thereby offering an attractive target site for interventions against RSV (20).
With this insight, our strategy to inhibit RSV entry aimed to capture RSV fusion protein in the prefusion conformation and block by that its fusion capacity through the application of peptide fragments derived from the interacting region of the aforementioned D25 antibody, as well as the trivalent binding of DNA-peptide conjugates to the antigenic sites Ø. In the latter approach, the DNA nanostructure serves as a scaffold to present three antigen-binding peptides in a geometrically defined pattern, complementary to the targeted sites on the RSV-F homotrimer, to simultaneously lock all three protein subunits in their pre-fusion configuration.
It was previously shown that the variable antigen-binding fragment (Fab) of the D25 antibody neutralizes RSV with a similar potency compared to the whole antibody, and the interaction primarily involves capturing the α4 helix of the RSV-F1 subunit between the H-CDR3 and the light chain (20). This encouraged us to analyze short peptides derived from the H-CDR3 of D25 antibody as RSV inhibitors.
Truncation analysis suggested that a 14 amino acid length segment of the H-CDR3 plays a functional role in the neutralization of RSV and the presence of the hydrophobic leucine residue at the N-terminus is essential for this activity. Alanine replacement analysis confirmed the importance of this leucine residue together with further six amino acids at different positions for the peptide activity, including two hydrophobic residues at the N-terminus, three hydrophobic residues in the middle part and one negatively charged aspartic acid residue at the penultimate position. Together, these results indicate the significance of the hydrophobic regions in combination with the negative charge for the antiviral effect. We anticipate that the inhibitor hydrophobicity contributes to the interactions with exposed hydrophobic sites of RSV-F protein during the fusion transition, which in turn promotes the binding of anionic residues to other proximal positively charged regions on RSV (e.g., heparin-binding sites) (56, 57). By that, negatively charged residues enhance the overall antiviral activity, even if not directly contributing to the interaction with the binding site. This pattern of hydrophobicity combined with a negative charge was previously reported to contribute to the antiviral activity of entry inhibitors targeting RSV and other enveloped viruses (58–60). Generally, CDR-derived peptides might differ at least partially in their binding pattern from the parent monoclonal antibodies (61). This is especially applicable to short peptides that are more prone to aggregation and might display different structural features than the paratope of the monoclonal antibody.
To date, only few peptide-based inhibitors of RSV have been studied, and all of them are still in the development or early preclinical evaluation stage. The majority of the investigated peptides interfere with viral entry into the host cell by either binding to cell surface heparan sulfate proteoglycans that act as binding molecules for RSV attachment (62–64) or targeting the RSV-F fusion protein. Peptide inhibitors derived from the heptad repeats of RSV-F sequence were reported, including two candidates stabilized using the stapling technique of introducing non-typical amino acids for cross-linking and fixation of the peptides in an a-helix form (65–67). The double-stapled HR2-derived peptide SAH-RSVBD developed by Bird and colleagues displayed micromolar efficacy against RSV in vitro, similar to our candidate Pep30, but was able to inhibit RSV infection in mice only when delivered in a polysaccharide-based formulation (65). A similarly optimized shorter peptide 4Ca (20 residues) described by Gaillard et al. blocked RSV infection on cells at nanomolar concentrations and in mice after intranasal application, although its in vivo potency was limited to 3-fold inhibition (67).
Only one group described the implementation of CDRs or their parts of a neutralizing monoclonal antibody as inhibitors against RSV infection (68). In this study, peptides derived from the CDR3 of RS-348 monoclonal antibody, which targets the native form of the fusion protein, were effective in inhibiting RSV infection in cell culture and also in mice when administered intranasally prior to infection. However, to our knowledge no further development of peptides targeting RSV-infection was reported.
In our study, we could identify the essential structural properties of peptides derived from the CDR3 of D25 antibody, that targets the prefusion RSV-F. This enabled systematic site-specific modifications of peptide’s amino acid sequence with non-canonical amino acids analogues to improve the in vivo stability towards proteases. In vitro functional screening of different modified peptides enabled the selection of the most potent candidate Pep30 with an IC50 value of 5.8 µM, which was able to reduce the infection in the lungs of mice by 200-fold. After confirming the inhibitory capacity of the monovalent peptide Pep30, we used it as a binding moiety to construct trimeric DNA-peptide nanostructures, to test whether a geometrically complementary trivalent presentation could enhance their activity. DNA scaffolds were synthesized by assembling three partially complementary DNA strands into a branched nanostructure with three dsDNA arms, each approximately 5 nm in length; these were in turn linked to Pep30 to form a three-armed tweezer-like-molecule. The flexibility of the central joint in the structure allows simultaneous, cooperative binding between the three presented peptides binding and the specific conformation of three subunits of the prefusion F protein.
The trimeric presentation of Pep30 on DNA enhanced its in vitro antiviral activity by 214-fold, giving an IC50 value of 27 nM. We could also show that conjugation of the monomeric peptides on the DNA nano-scaffold reduces the formation of peptide aggregates. However, the trimeric Pep30-DNA nanostructures only reduced viral load by 11-fold in an in vivo setting. Even though no improvement in the inhibitory capacity of the DNA-peptide trimer over the monomeric peptide was achieved in our mouse experiments, it needs to be pointed out that the designed construct still requires optimization to achieve better stability of the underlying DNA nanostructure in the respiratory tract. Since the nasal and pulmonary mucosa are rich in nucleases, integration of nuclease-stable nucleotides for this purpose is one possible solution (69–71). This could also be alleviated through the use of non-canonical nucleotides such as so-called locked nucleic acid (LNA) or the general class of xeno-nucleic acids (XNAs) (72). Nevertheless, our study is the first to report the in vivo potential of multimeric DNA-peptide nanostructures as antivirals.
Besides requiring costly and time-consuming production, one concern in conventional antibody-based therapies is the risk of intensifying the severity of the disease course upon several viral infections (73), including RSV (74), measles (75) and SARS-CoV-2 (76). The underlying mechanism of this phenomenon is antibody-dependent enhancement induced by Fc-mediated effector functions of sub-neutralizing or impaired antibodies that promote enhanced viral entry into the cells with increased immunopathology (77, 78). By contrast, synthetic, nucleotide-based, antibody-like nanostructures offer favorable safety properties, rapid production, straightforward scaling-up and generally low immunogenicity from the underlying synthetically produced nucleotide strands as an antiviral platform (69, 79, 80). Even though the peptide-DNA constructs presented in our study were less efficient in neutralizing infection than the parent antibody D25, likely due to the solvable limitation arising from their degradation in the mucosa, the strategy can still offer cost-effective alternative to monoclonal antibodies with wide-ranging potential for optimization worthy of exploration. Most importantly, the modular design and construction strategy, consisting of easily synthesized molecular building blocks can enable a simple, cost-effective, rapid-prototyping approach to generate potent, prophylactic antiviral compounds based on peptides or other small ligands derived from the numerous screening or in silico approaches available for determining effective target-binders Further efforts have to be performed to translate these innovative molecules into the in vivo situation.
Acknowledgments
The authors thank Tanja Henning, Anne-Kathrin Donner, Isabell Schulz and Eric Possardt (Fraunhofer IZI, Leipzig, Germany) for excellent technician assistance, M.E. Peeples and P. Collins (NIH, Maryland, USA) for providing the rgRSV. Molecular graphics of the overall structure (Figure 1A) was performed with UCSF ChimeraX-1.4, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. This work was funded by Fraunhofer internal funding program.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by Landesdirektion Sachsen.
Author contributions
TG, DS, and MB conceived and supervised the study. CJ and DR performed the computational analysis and peptide design. CM and BA synthesized the peptide-DNA constructs and performed the ELISA. LI performed all other experiments. LI and TG analyzed data and wrote the original draft. LI, CM, CJ, BA, MK, DR, MB, DS, and TG revised and approved the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Peptides described in this manuscript are part of an IP application (WO2020212576A1). The DNA-peptide scaffold technology described in this manuscript is part of an IP application (WO2018215660A1).
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.994843/full#supplementary-material
Supplementary10% (v/v) native PAGE of DNA trimer self-assembly. Lane 1) GeneRulerTM Low Range DNA Ladder, lane 2) unmodified DNA trimer in DMSO, lane 3) unmodified DNA trimer incubated with Pep30, lane 4) DNA trimer-DBCO-PEG4, lane 5) DNA-Pep30 trimer in DMSO. Gel was stained with SYBR® Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific, Waltham, MA, USA) and imaged using the ChemiDoc MP Imaging System with UV excitation (302 nm) and 590/110 nm emission. Gel image was cropped and edited (brightness + 40%, contrast + 20%). GeneRulerTM Low Range DNA Ladder served as control, not as ruler.
References
1
HallCB. Respiratory syncytial virus: Its transmission in the hospital environment. Yale J Biol Med (1982) 55:219–23.
2
HallCBLongCESchnabelKC. Respiratory syncytial virus infections in previously healthy working adults. Clin Infect Dis (2001) 33:792–6. doi: 10.1086/322657
3
FalseyARHennesseyPAFormicaMACoxCWalshEE. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med (2005) 352:1749–59. doi: 10.1056/NEJMoa043951
4
CoultasJASmythROpenshawPJ. Respiratory syncytial virus (RSV): A scourge from infancy to old age. Thorax (2019) 74:986–93. doi: 10.1136/thoraxjnl-2018-212212
5
GlezenWPTaberLHFrankALKaselJA. Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child (1986) 140:543–6. doi: 10.1001/archpedi.1986.02140200053026
6
ShiTMcAllisterDAO’BrienKLSimoesEAMadhiSAGessnerBDet al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet (2017) 390:946–58. doi: 10.1016/S0140-6736(17)30938-8
7
NairHNokesDJGessnerBDDheraniMMadhiSASingletonRJet al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet (2010) 375:1545–55. doi: 10.1016/S0140-6736(10)60206-1
8
Blount REJRMorrisJASavageRE. Recovery of cytopathogenic agent from chimpanzees with coryza. Proc Soc Exp Biol Med (1956) 92:544–9. doi: 10.3181/00379727-92-22538
9
ChanokRRoizmanBMyersR. Recovery from infants with respiratory illness of a virus related to chimpanzee coryza agent (CCA). i. isolation, properties and characterization. Am J Hyg (1957) 66:281–90. doi: 10.1093/oxfordjournals.aje.a119901
10
AndersonEJCarosone-LinkPYogevRYiJSimõesEA. Effectiveness of palivizumab in high-risk infants and children: A propensity score weighted regression analysis. Pediatr Infect Dis J (2017) 36:699–704. doi: 10.1097/INF.0000000000001533
11
PollackPGroothuisJR. Development and use of palivizumab (Synagis): A passive immunoprophylactic agent for RSV. J Infect Chemother (2002) 8:201–6. doi: 10.1007/s10156-002-0178-6
12
HomairaNRawlinsonWSnellingTLJaffeA. Effectiveness of palivizumab in preventing RSV hospitalization in high risk children: A real-world perspective. Int J Pediatr (2014) 2014:571609. doi: 10.1155/2014/571609
13
FeltesTFCabalkaAKMeissnerHCPiazzaFMCarlinDATopFHet al. Palivizumab prophylaxis reduces hospitalization due to respiratory syncytial virus in young children with hemodynamically significant congenital heart disease. J Pediatr (2003) 143:532–40. doi: 10.1067/S0022-3476(03)00454-2
14
American Academy of Pediatrics Committee on Infectious DiseasesAmerican Academy of Pediatrics Bronchiolitis Guidelines Committee. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics (2014) 134:e620–38. doi: 10.1542/peds.2014-1666
15
HeminwayBRYuYTanakaYPerrineKGGustafsonEBernsteinJMet al. G, And SH proteins in cell fusion. Virology (1994) 200:801–5. doi: 10.1006/viro.1994.1245
16
MeleroJAMooreML. Influence of respiratory syncytial virus strain differences on pathogenesis and immunity. Curr Top Microbiol Immunol (2013) 372:59–82. doi: 10.1007/978-3-642-38919-1_3
17
MagroMAndreuDGómez-PuertasPMeleroJAPalomoC. Neutralization of human respiratory syncytial virus infectivity by antibodies and low-molecular-weight compounds targeted against the fusion glycoprotein. J Virol (2010) 84:7970–82. doi: 10.1128/JVI.00447-10
18
ZhaoXSinghMMalashkevichVNKimPS. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc Natl Acad Sci USA (2000) 97:14172–7. doi: 10.1073/pnas.260499197
19
Lawless-DelmedicoMKSistaPSenRMooreNCAntczakJBWhiteJMet al. Heptad-repeat regions of respiratory syncytial virus F1 protein form a six-membered coiled-coil complex. Biochemistry (2000) 39:11684–95. doi: 10.1021/bi000471y
20
McLellanJSChenMLeungSGraepelKWDuXYangYet al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science (2013) 340:1113–7. doi: 10.1126/science.1234914
21
McLellanJSChenMJoyceMGSastryMStewart-JonesGBYangYet al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science (2013) 342:592–8. doi: 10.1126/science.1243283
22
MousaJJKoseNMattaPGilchukPCroweJE. A novel pre-fusion conformation-specific neutralizing epitope on the respiratory syncytial virus fusion protein. Nat Microbiol (2017) 2:16271. doi: 10.1038/nmicrobiol.2016.271
23
ZhaoMZhengZ-ZChenMModjarradKZhangWZhanL-Tet al. Discovery of a prefusion respiratory syncytial virus f-specific monoclonal antibody that provides greater In vivo protection than the murine precursor of palivizumab. J Virol (2017) 91(15):e00176-17. doi: 10.1128/JVI.00176-17
24
ZhuQMcLellanJSKallewaardNLUlbrandtNDPalaszynskiSZhangJet al. A highly potent extended half-life antibody as a potential RSV vaccine surrogate for all infants. Sci Transl Med (2017) 9(388):eaaj1928. doi: 10.1126/scitranslmed.aaj1928
25
GriffinMPKhan AnisAEsser MarkTJensenKTakasTKankam MartinKet al. Safety, tolerability, and pharmacokinetics of MEDI8897, the respiratory syncytial virus prefusion f-targeting monoclonal antibody with an extended half-life, in healthy adults. Antimicrob Agents Chemother (2017) 61:e01714–16. doi: 10.1128/AAC.01714-16
26
HewatEABlaasD. Structure of a neutralizing antibody bound bivalently to human rhinovirus 2. EMBO J (1996) 15:1515–23. doi: 10.1002/j.1460-2075.1996.tb00495.x
27
YanRWangRJuBYuJZhangYLiuNet al. Structural basis for bivalent binding and inhibition of SARS-CoV-2 infection by human potent neutralizing antibodies. Cell Res (2021) 31:517–25. doi: 10.1038/s41422-021-00487-9
28
EdelingMAAustinSKShresthaBDowdKAMukherjeeSNelsonCAet al. Potent dengue virus neutralization by a therapeutic antibody with low monovalent affinity requires bivalent engagement. PloS Pathog (2014) 10:e1004072. doi: 10.1371/journal.ppat.1004072
29
FastingCSchalleyCAWeberMSeitzOHechtSKokschBet al. Multivalency as a chemical organization and action principle. Angew Chem Int Ed Engl (2012) 51:10472–98. doi: 10.1002/anie.201201114
30
DeyPBergmannTCuellar-CamachoJLEhrmannSChowdhuryMSZhangMet al. Multivalent flexible nanogels exhibit broad-spectrum antiviral activity by blocking virus entry. ACS Nano (2018) 12:6429–42. doi: 10.1021/acsnano.8b01616
31
LausterDGlanzMBarduaMLudwigKHellmundMHoffmannUet al. Multivalent peptide-nanoparticle conjugates for influenza-virus inhibition. Angew Chem Int Ed Engl (2017) 56:5931–6. doi: 10.1002/anie.201702005
32
SchüllerVJHeideggerSSandholzerNNickelsPCSuharthaNAEndresSet al. Cellular immunostimulation by CpG-sequence-coated DNA origami structures. ACS Nano (2011) 5:9696–702. doi: 10.1021/nn203161y
33
SellnerSKocabeySNekollaKKrombachFLiedlTRehbergM. DNA Nanotubes as intracellular delivery vehicles in vivo. Biomaterials (2015) 53:453–63. doi: 10.1016/j.biomaterials.2015.02.099
34
KwonPSRenSKwonS-JKizerMEKuoLXieMet al. Designer DNA architecture offers precise and multivalent spatial pattern-recognition for viral sensing and inhibition. Nat Chem (2020) 12:26–35. doi: 10.1038/s41557-019-0369-8
35
LiuSJiangQZhaoXZhaoRWangYWangYet al. A DNA nanodevice-based vaccine for cancer immunotherapy. Nat Mater (2021) 20:421–30. doi: 10.1038/s41563-020-0793-6
36
DouglasSMBacheletIChurchGM. A logic-gated nanorobot for targeted transport of molecular payloads. Science (2012) 335:831–4. doi: 10.1126/science.1214081
37
MöserCLorenzJSSajfutdinowMSmithDM. Pinpointed stimulation of EphA2 receptors via DNA-templated oligovalence. Int J Mol Sci (2018) 19:3482. doi: 10.3390/ijms19113482
38
BandlowVLieseSLausterDLudwigKNetzRRHerrmannAet al. Spatial screening of hemagglutinin on influenza a virus particles: Sialyl-LacNAc displays on DNA and PEG scaffolds reveal the requirements for bivalency enhanced interactions with weak monovalent binders. J Am Chem Soc (2017) 139:16389–97. doi: 10.1021/jacs.7b09967
39
YamabeMKaihatsuKEbaraY. Binding inhibition of various influenza viruses by sialyllactose-modified trimer DNAs. Bioorg Med Chem Lett (2019) 29:744–8. doi: 10.1016/j.bmcl.2018.12.064
40
YamabeMKaihatsuKEbaraY. Sialyllactose-modified three-way junction DNA as binding inhibitor of influenza virus hemagglutinin. Bioconjugate Chem (2018) 29:1490–4. doi: 10.1021/acs.bioconjchem.8b00045
41
YamabeMFujitaAKaihatsuKEbaraY. Synthesis of neuraminidase-resistant sialoside-modified three-way junction DNA and its binding ability to various influenza viruses. Carbohydr Res (2019) 474:43–50. doi: 10.1016/j.carres.2019.01.008
42
MemczakHLausterDKarPDi LellaSVolkmerRKnechtVet al. Anti-hemagglutinin antibody derived lead peptides for inhibitors of influenza virus binding. PloS One (2016) 11:e0159074. doi: 10.1371/journal.pone.0159074
43
KruseMMöserCSmithDMMüller-LandauHRantUHölzelRet al. Measuring influenza a virus and peptide interaction using electrically controllable DNA nanolevers. Adv Mater Technol (2022) 7:2101141. doi: 10.1002/admt.202101141
44
KruseMAltattanBLauxE-MGrasseNHeinigLMöserCet al. Characterization of binding interactions of SARS-CoV-2 spike protein and DNA-peptide nanostructures. Sci Rep (2022) 12:12828. doi: 10.1038/s41598-022-16914-9
45
SiglCWillnerEMEngelenWKretzmannJASachenbacherKLiedlAet al. Programmable icosahedral shell system for virus trapping. Nat Mater (2021) 20:1281–9. doi: 10.1038/s41563-021-01020-4
46
Molecular operating environment (MOE) Vol. 2022. 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 08_02; Chemical Computing Group ULC (2016).
47
KohlmannRSchwanneckeSTipplerBTernetteNTemchuraVVTenbuschMet al. Protective efficacy and immunogenicity of an adenoviral vector vaccine encoding the codon-optimized f protein of respiratory syncytial virus. J Virol (2009) 83:12601–10. doi: 10.1128/JVI.01036-09
48
TernetteNTipplerBUberlaKGrunwaldT. Immunogenicity and efficacy of codon optimized DNA vaccines encoding the f-protein of respiratory syncytial virus. Vaccine (2007) 25:7271–9. doi: 10.1016/j.vaccine.2007.07.025
49
BayerLFerteyJUlbertSGrunwaldT. Immunization with an adjuvanted low-energy electron irradiation inactivated respiratory syncytial virus vaccine shows immunoprotective activity in mice. Vaccine (2018) 36:1561–9. doi: 10.1016/j.vaccine.2018.02.014
50
GentilucciLdeMRCerisoliL. Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr Pharm Des (2010) 16:3185–203. doi: 10.2174/138161210793292555
51
CavacoMCastanho, MiguelARBNevesV. Peptibodies: An elegant solution for a long-standing problem. Biopolymers (2017). doi: 10.1002/bip.23095
52
StoneTADeberCM. Therapeutic design of peptide modulators of protein-protein interactions in membranes. Biochim Biophys Acta Biomembr (2017) 1859:577–85. doi: 10.1016/j.bbamem.2016.08.013
53
DingYTingJPLiuJAl-AzzamSPandyaPAfsharS. Impact of non-proteinogenic amino acids in the discovery and development of peptide therapeutics. Amino Acids (2020) 52:1207–26. doi: 10.1007/s00726-020-02890-9
54
LiljeroosLKrzyzaniakMAHeleniusAButcherSJ. Architecture of respiratory syncytial virus revealed by electron cryotomography. Proc Natl Acad Sci USA (2013) 110:11133–8. doi: 10.1073/pnas.1309070110
55
NgwutaJOChenMModjarradKJoyceMGKanekiyoMKumarAet al. Prefusion f-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci Transl Med (2015) 7:309ra162. doi: 10.1126/scitranslmed.aac4241
56
Money VictoriaAMcPhee HelenKMosely JackieASanderson JohnMYeo RobertP. Surface features of a mononegavirales matrix protein indicate sites of membrane interaction. Proc Natl Acad Sci (2009) 106:4441–6. doi: 10.1073/pnas.0805740106
57
MartínezIMeleroJA. Binding of human respiratory syncytial virus to cells: implication of sulfated cell surface proteoglycans. J Gen Virol (2000) 81:2715–22. doi: 10.1099/0022-1317-81-11-2715
58
Lüscher-MattiiM. Polyanions — a lost chance in the fight against HIV and other virus diseases? Antiviral Chem Chemother (2000) 11:249–59. doi: 10.1177/095632020001100401
59
BadaniHGarryRFWimleyWC. Peptide entry inhibitors of enveloped viruses: the importance of interfacial hydrophobicity. Biochim Biophys Acta (2014) 1838:2180–97. doi: 10.1016/j.bbamem.2014.04.015
60
BudgePJGrahamBS. Inhibition of respiratory syncytial virus by RhoA-derived peptides: implications for the development of improved antiviral agents targeting heparin-binding viruses. J Antimicrob Chemother (2004) 54:299–302. doi: 10.1093/jac/dkh355
61
TimmermanPShochatSGDesmetJBarderasRCasalJIMeloenRHet al. Binding of CDR-derived peptides is mechanistically different from that of high-affinity parental antibodies. J Mol Recognit (2010) 23:559–68. doi: 10.1002/jmr.1017
62
FeldmanSAAudetSBeelerJA. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J Virol (2000) 74:6442–7. doi: 10.1128/jvi.74.14.6442-6447.2000
63
CrimRLAudetSAFeldmanSAMostowskiHSBeelerJA. Identification of linear heparin-binding peptides derived from human respiratory syncytial virus fusion glycoprotein that inhibit infectivity. J Virol (2007) 81:261–71. doi: 10.1128/JVI.01226-06
64
ManuelaDMarcoRValeriaCAndreaCAntonellaBAndreaGet al. Inhibition of human respiratory syncytial virus infectivity by a dendrimeric heparan sulfate-binding peptide. Antimicrob Agents Chemother (2012) 56:5278–88. doi: 10.1128/AAC.00771-12
65
BirdGHBoyapalleSWongTOpoku-NsiahKBediRCrannellWCet al. Mucosal delivery of a double-stapled RSV peptide prevents nasopulmonary infection. J Clin Invest (2014) 124:2113–24. doi: 10.1172/JCI71856
66
WangEX’oSQianYZhaoLTienPGaoGF. Both heptad repeats of human respiratory syncytial virus fusion protein are potent inhibitors of viral fusion. Biochem Biophys Res Commun (2003) 302:469–75. doi: 10.1016/s0006-291x(03)00197-9
67
GaillardVGallouxMGarcinDEléouëtJ-FLe GofficRLarcherTet al. A short double-stapled peptide inhibits respiratory syncytial virus entry and spreading. Antimicrob Agents Chemother (2017) 61:e02241-16. doi: 10.1128/AAC.02241-16
68
BourgeoisCBourJBAhoLSPothierP. Prophylactic administration of a complementarity-determining region derived from a neutralizing monoclonal antibody is effective against respiratory syncytial virus infection in BALB/c mice. J Virol (1998) 72:807–10. doi: 10.1128/JVI.72.1.807-810.1998
69
ChandrasekaranAR. Nuclease resistance of DNA nanostructures. Nat Rev Chem (2021) 5:225–39. doi: 10.1038/s41570-021-00251-y
70
ShawJPKentKBirdJFishbackJFroehlerB. Modified deoxyoligonucleotides stable to exonuclease degradation in serum. Nucleic Acids Res (1991) 19:747–50. doi: 10.1093/nar/19.4.747
71
KimYYinP. Enhancing biocompatible stability of DNA nanostructures using dendritic oligonucleotides and brick motifs. Angew Chem Int Ed Engl (2020) 59:700–3. doi: 10.1002/anie.201911664
72
PinheiroVBTaylorAICozensCAbramovMRendersMZhangSet al. Synthetic genetic polymers capable of heredity and evolution. Science (2012) 336:341–4. doi: 10.1126/science.1217622
73
TiradoSMYoonK-J. Antibody-dependent enhancement of virus infection and disease. Viral Immunol (2003) 16:69–86. doi: 10.1089/088282403763635465
74
KimHWCancholaJGBrandtCDPylesGChanockRMJensenKet al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol (1969) 89:422–34. doi: 10.1093/oxfordjournals.aje.a120955
75
RødTHaugKWUlstrupJC. Atypical measles after vaccination with killed vaccine. Scand J Infect Dis (1970) 2:161–5. doi: 10.3109/inf.1970.2.issue-3.02
76
LeeWSWheatleyAKKentSJDeKoskyBJ. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat Microbiol (2020) 5:1185–91. doi: 10.1038/s41564-020-00789-5
77
PolackFPTengMNCollinsPLPrinceGAExnerMRegeleHet al. A role for immune complexes in enhanced respiratory syncytial virus disease. J Exp Med (2002) 196:859–65. doi: 10.1084/jem.20020781
78
HalsteadSB. Neutralization and antibody-dependent enhancement of dengue viruses. Adv Virus Res (2003) 60:421–67. doi: 10.1016/s0065-3527(03)60011-4
79
PraetoriusFKickBBehlerKLHonemannMNWeuster-BotzDDietzH. Biotechnological mass production of DNA origami. Nature (2017) 552:84–7. doi: 10.1038/nature24650
80
RenSFraserKKuoLChauhanNAdrianATZhangFet al. Designer DNA nanostructures for viral inhibition. Nat Protoc (2022) 17:282–326. doi: 10.1038/s41596-021-00641-y
Summary
Keywords
respiratory syncial virus, antiviral, peptide, DNA nanostructure, multivalence
Citation
Issmail L, Möser C, Jäger C, Altattan B, Ramsbeck D, Kleinschmidt M, Buchholz M, Smith D and Grunwald T (2022) Prefusion-specific antibody-derived peptides trivalently presented on DNA-nanoscaffolds as an innovative strategy against RSV entry. Front. Virol. 2:994843. doi: 10.3389/fviro.2022.994843
Received
15 July 2022
Accepted
15 September 2022
Published
06 October 2022
Volume
2 - 2022
Edited by
Nancy Sullivan, National Institute of Allergy and Infectious Diseases (NIH), United States
Reviewed by
Mauricio Comas-Garcia, Universidad Autónoma de San Luis Potosí, Mexico; Roland G. Huber, Bioinformatics Institute (A*STAR), Singapore
Updates
Copyright
© 2022 Issmail, Möser, Jäger, Altattan, Ramsbeck, Kleinschmidt, Buchholz, Smith and Grunwald.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Grunwald, thomas.grunwald@izi.fraunhofer.de
This article was submitted to Antivirals and Vaccines, a section of the journal Frontiers in Virology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.