
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol. , 10 November 2022
Sec. Modeling of Viral Replication and Pathogenesis
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.968338
This article is part of the Research Topic Women In Modeling of Viral Replication and Pathogenesis:2021 View all 5 articles
 Hyeree Choi1
Hyeree Choi1 Sagar B. Kudchodkar1†
Sagar B. Kudchodkar1† Ziyang Xu1
Ziyang Xu1 Michelle Ho1
Michelle Ho1 Peng Xiao1
Peng Xiao1 Stephanie Ramos2Laurent Humeau2
Stephanie Ramos2Laurent Humeau2 David B. Weiner1
David B. Weiner1 Kar Muthumani1*†
Kar Muthumani1*†Nipah virus (NiV) is a re-emerging pathogen that causes severe disease in animals and humans. Current treatment measures for NiV infection are insufficient, and there is no approved vaccine against NiV for either humans or animals. Nipah virus is listed as a high-priority pathogen for vaccine and therapeutic research by the World Health Organization (WHO). In the present study, we employed synthetic enhanced DNA technologies developed to design and produce novel consensus NiV Fusion (NiV-F) and Glycoprotein (NiV-G) antigen sequences for inclusion in synthetic DNA vaccines for NiV. The expression of each vaccine antigen was confirmed in vitro using immune-binding assays. Electroporation-enhanced intramuscular injection of each NiV-F and NiV-G into mice induced potent cellular immune responses to multiple epitopes of NiV-G and NiV-F that included antigen-specific CD8+ T cells. Both vaccines elicited high antibody titers in mice, with a single immunization sufficient to seroconvert 100% of immunized animals. Additionally, the NiV-F vaccine also induced antibodies to neutralize NiV-F-pseudotyped virus particles. These data support further study of these novel synthetic enhanced NiV nucleic acid-based antigens as potential components of an effective vaccine against the Nipah virus.
Nipah Virus (NiV) is a recently emerged member of the family paramyxoviridae that, together with Hendra virus (HeV), comprises the genus henipavirus (1–3). In contrast to other paramyxoviruses, the henipaviruses cause zoonotic infections exhibiting broad species tropism with the ability to cause fatal disease in humans and in a variety of animal species, including pigs, horses, dogs, and cats (4, 5). These enveloped viruses contain non-segmented, negative-sense RNA genomes (~18.2Kb) comprised of 6 major structural genes (N, P, M, F, G, and L). Members of the Paramyxoviridae has two membrane glycoproteins involved in receptor binding and viral entry, the attachment (G) and fusion (F) proteins, respectively; the G induces viral attachment to two cellular receptors, ephrin-B2 and ephrin-B3, despite a lack of hemagglutination or neuraminidase activity and this subsequently triggers F-mediated membrane fusion between virus and host cells (6). The likely cellular receptors for NiV is Ephrin-B2 and Ephrin-B3 which are predominantly expressed on neurons, smooth muscle, and endothelial cells (7). Both G and F have been implicated in viral pathogenesis and are major targets of neutralizing antibody (nAb) responses in NiV-infected people making them ideal targets for vaccine development (8–10).
Since 1998, outbreaks and/or reported in reservoir species of NiV have been reported in throughout countries in Southeast Asia including Malaysia, Cambodia, Thailand, Singapore, the Philippines, Bangladesh and India, among others, with mortality rates approaching 100% (2, 11–13). The disease has been either associated with naturally occurring outbreaks and/with sporadic outbreaks occurring since the disease was first reported in Malaysia in 1998. NiV isolates from Malaysia and Cambodia clustered together in NiV-MY clade, whereas isolates from Bangladesh and India clustered within NiV-BD clade (12, 14). In the outbreaks in Malaysia and Singapore, the case fatality ratio was around 40% and infected persons were noted to have had close contact with pigs (15). In the outbreaks in Bangladesh and India, case fatality rates have been reported to be from 70-100% and transmission has been seen in people who have ingested food contaminated by bat fecal matter and person to person transmission has also been observed (16).The rate of human-to-human transmission of NiV has increased (17–19), and there is evidence to suggest that the virus is shed in saliva, nasopharyngeal secretions, and urine (20–22). The respiratory epithelium is an early and prominent target for inhaled NiV, resulting in significant histopathological changes including pneumonia, pulmonary edema, and necrotizing alveolitis (21). In addition, NiV infection is associated with acute encephalitis and systemic vasculitis, and direct NiV infection of neurons has been noted in the brain (20). In fatal cases, death occurs between 1-2 weeks following initial symptom onset, and 20% of the survivors suffer from residual neurologic effects. Relapse/late-onset encephalitis occurs in approximately 7.5% of survivors, and 3.4% of individuals who were initially asymptomatic develop symptoms within a span of 4 years following initial exposure (23). Neuronal injury in the brain is extensive and viral inclusions are prominent, suggesting that relapse/late-onset encephalitis results from reactivation of the previous neuronal NiV infection (24). The mortality rate in the case of relapse/late-onset encephalitis is approximately 18%, and 61% of the survivors were found to have neurologic sequelae (23). According to the World Health Organization (WHO), the incubation period ranges from 4-to 14 days; however, incubations up to 45 days have been reported in a few cases.
DNA plasmid immunogens have multiple characteristics that make them an ideal platform for vaccines for emerging and re-emerging infectious disease agents. DNA plasmid vaccines can be designed and produced rapidly and cost-effectively. They are thermostable and do not require a frozen cold chain, making them well suited for field study and deployment in low- and middle-income countries (LMICs). While first-generation DNA vaccines were poorly immunogenic in the clinic, advances in plasmid design and the introduction of novel delivery strategies, including localized in vivo electroporation (EP), have greatly improved immunogenicity of DNA vaccines and have allowed for dose reduction while maintaining the positive safety profile of DNA vaccines seen in the initial clinical trials (25, 26). Several DNA vaccines against EIDs have been advanced into clinical trials, including vaccines against HIV, Zika virus, MERS, SARS-CoV-2 and Ebola virus (25–28). A DNA vaccine has been authorized under an emergency use authorization (EUA) for SARS-CoV2 in India after highly successful efficacy trials (29).
The re-emergence of Nipah in several recent outbreaks is troubling as there are no licensed vaccines or therapeutics currently available to combat NiV infection and/or disease (30). The basis for protective immunity against Nipah is not clear. Vaccines based on henipavirus G protein have been shown to protect small animals and monkeys from lethal henipavirus challenge (31). In another study, vaccines encoding NiV-G or -F genes were both able to protect hamsters from lethal NiV challenge, and passive transfer of antibodies induced by each vaccine was also able to protect animals from disease (32). Both Hendra virus and Nipah virus have continued to repeatedly cause spillover from animals and causes lethal disease across a broad range of vertebrate species including humans. Infection of Hendra virus in horses in Australia has occurred annually and have now been 7 human cases of which 4 have been fatal which was transmitted from infected horses to humans. A fully registered horse anti-Hendra virus subunit vaccine has been in use in Australia since 2012 (33). It has been reported that there is passive antibody protection targeting the F antigen in NiV-infected people (34, 35), making F protein a bona fide vaccine candidate (10, 36). This is also supported by identifying a specific epitope at the ascorbic acid peroxidase (APEX) of the F trimer (37, 38). In this study, we designed two highly optimized, synthetic enhanced DNA vaccines encoding the Nipah virus Glycoprotein (G) or Fusion (F) antigens. Our results show that both immunogens induced NiV-specific cellular and humoral immune responses, including neutralizing antibodies in mice. These data support further testing of these vaccines as a means to prevent NiV infection.
For the vaccine design we used a focused consensus strategy to design Nipah virus Fusion (F) and Glycoprotein (G) antigen cassettes. The consensus sequences were designed through alignment of multiple F and G protein sequences available in the PubMed database including the genetically categorized two genotypes of NiV-Malaysia (NiV-MY) identified in Malaysia and Cambodia (NiV-M clade) and NiV-Bangladesh (NiV-B clade) identified in Bangladesh and India (39). The Nipah Virus proteins were downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/). Protein accession numbers are as follows: AAF73956.1; AAK29087.1; AAK50544.1; AAK50553.1; AAM13405.1; AAY43915.1; ACT32614.1; ADN51995.1; AEZ01388.1; AEZ1396.2; APT69633.1; APT69700.1; AWT50993.1; CAD92350.1; CAD92356.1; CAD92362.1; CAF25496.1; CBM41033.1; NP112026.1; QBQ56704.1; QCY54420.1; QHR78956.1; and QHR79001.1 were selected for the vaccine consensus construction and retrieved from the NCBI database in protein FASTA format. Several modification including plasmid optimizations which include RNA optimization to improve mRNA stability and more efficient translation on the ribosome, the addition of leader sequences to enhance translation efficiency in human cells as well as the creation of consensus immunogens to induce broader more cross-reactive responses to diverse viral isolates. We have focused on this area and reported improved in vivo expression of such designer plasmids, however, additional advances appear to be needed, as alone these approaches do not engender immunity in the range that was induced by other delivery platforms were performed to obtain enhanced expression of each antigen in mouse and human tissues (27, 40–50). Additionally, an enhanced IgE leader sequence was added to the 5’ end of each insert before cloning each into cytomegalovirus (CMV) promoter-containing expression plasmid (pVax1). GenScript Inc., (USA) carried out large-scale DNA amplification of each plasmid, and purified plasmid DNAs were reconstituted in water. The insert sizes were validated by agarose gel electrophoresis.
Six to eight-week-old female C57BL/6 mice (Charles River, USA) were used for all the experiments and were maintained in specific pathogen-free conditions (Wistar IACUC# 112785). Mice were immunized with 25μg of pVax1 empty vector or NiV-F or NiV-G plasmids into the anterior tibialis (TA) muscle, followed by the delivery of two 0.1 Amps of triangulated square-wave pulses per insertion with 3P CELLECTRA® adaptive constant current enhanced electroporation (EP) delivery device (Inovio Pharmaceuticals, USA) as described previously (28). Each group of vaccinated mice received two immunizations of pVax1, NiV-F, or NiV-G at the interval of two weeks. Animals were monitored for signs of distress after immunization once every week. All animals were humanely sacrificed at various times after vaccination as described in the text and shown in the figures. For sacrifice, mice were anesthetized with 2-5% isoflurane before euthanasia by carbon dioxide inhalation followed by confirmation of death by loss of vital signs and absence of corneal reflex. Blood samples were collected from each mouse by submandibular vein bleed using a lancet, and serum was recovered by centrifugation (47, 51).
MaxiSorp high-binding 96-well ELISA plates (Fisher, USA) were coated with recombinant NiV-F or NiV-G protein (1μg/ml in phosphate-buffered saline (PBS) and incubated overnight at 4°C. For the recombinant proteins, the coding sequence of NiV-F and NiV-G proteins, were cloned into the bacterial expression vector including His-Tag and protein cleavage site were fused in frame with target gene for purification. Purified proteins were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis in a 12% acrylamide gel, and the resolved bands were visualized by staining with Coomassie brilliant blue. The recombinant His6-tagged proteins were purified from the soluble fraction of the lysates using a Ni-NTA chromatographic column (GenScript, USA), and the purified protein was eluted. The purified recombinant proteins were analyzed using a UV spectrophotometer and the concentration was determined at 593 nm using a standard curve. Following incubation with specific recombinant proteins, plates were washed with PBS-T (PBS with 0.05% Tween 20) and blocked using PBS containing 10% FBS for 1 hour at room temperature (RT). Subsequently, the plates were washed with PBS-T and incubated with hybridoma, serially diluted in PBS with 1% FBS and 0.1% Tween 20 for 30 minutes on a shaker and 90 minutes at room temperature. After another wash, the plates were treated with goat-anti-mouse IgG H+L conjugated to Horse Radish Peroxidase (Bethyl Laboratories, USA) at a dilution of 1:10000 for 1 hour at room temperature. Post final wash, the plates were developed with SigmaFast OPD substrate (Sigma-Aldrich, USA) for 5-10 min in the dark, and the reaction was stopped using 1N H2SO4. The plates were read using a Synergy2 plate reader (BioTek Instruments, USA) at an optical density of 450nm.
Subsequently, the plates were washed with PBS containing 0.01% Tween-20 (PBST) four times in order to remove any unbound antigen. Then 100μl of immune sera serially diluted (half log fashion) in ELISA dilution buffer (PBS containing 1% fetal bovine serum (FBS)) were added to the plates and then incubated for 2h followed by four washes with PBST. Then, peroxidase-conjugated anti-mouse IgG, IgG1, IgG2a, IgG2b and IgG3 antibodies (Sigma-Aldrich, USA) were added to the plates and incubated for 1h. Following washing with PBST, 100μl of 3,3’5,5’-Tetramethylbenzidine (TMB) substrate (Sigma-Aldrich, USA) solution was added to the plates to detect the amount of bound antibody. The reaction was stopped using 50μl of 2N-H2SO4, and finally the optical density was measured at 450 nm using an ELISA plate reader (Biotek, USA). Titers were determined as the highest dilution to obtain an OD value of 0.2. For calibrating IgG subclass, plates were coated with recombinant target proteins and immune sera; one week post third immunization (day 35), incubated overnight at 4°C, washed with PBST, and then probed with anti-mouse IgG subclass (IgG1, IgG2a, IgG2b and IgG3) horseradish peroxidase-conjugated antibody (Sigma-Aldrich, USA).
We designed libraries of 153 and138 individual synthetic peptides spanning the external domains of NiV-G and NiV-F, respectively, that consisted of 15mer peptides, overlapping by 11 amino acids. All the probable peptide sequences of length 8-11mer were determined in order to map the potential CD8+ T cell epitopes in positive 15mer peptides. SYFPEITHI database was used for the analysis of the binding of theoretical peptides to the mouse major histocompatibility complex (MHC) class I allele H2-b. The peptides of different amino acid sequence lengths were synthesized and tested. All the peptides were synthesized (10 mg scale, 70% purity) by GenScript Inc., (USA) and were dissolved in DMSO for usage.
This assay was carried out using an ELISpot mouse kit (Mabtech, USA) as per manufacturer’s protocol. Briefly, 96-well ELISpot plates were blocked using Roswell Park Memorial Institute Medium (RPMI) with 10% Fetal Calf Serum (FCS) (R10) for 2 h at room temperature. Spleens of the vaccinated mice were dissected and crushed using a Stomacher device (Seward, UK). The splenocyte suspensions obtained after red blood cell lysis treatment were then added to the plate at the concentration of 2 ×105 cells/well, followed by the addition of Nipah-specific F and G peptides pool in R10. For positive control 5 µg/ml Concanavalin A (ConA) in R10 medium and negative control 50µl R10 into indicated wells indicated wells. Following incubation for 24 h at 37°C; 5% CO2, the plates were washed with PBS and then incubated with biotinylated anti-mouse IFN-γ mAb (1:1000) for 2 h at room temperature, followed by washing. Subsequently, streptavidin-conjugated HRP was added to the plates and incubated for 1 h at room temperature. Thorough washing of the wells was done again before developing with the TMB substrate provided in the kit. Spots representing the T cells capable of producing IFN-γ were finally counted with the help of an automated ELISpot reader (CTL Limited, USA), and the results were presented as spot-forming units (SFU) per 106 splenocytes.
Single-cell suspended mouse splenocytes (2x106 cells/well) were added to a U-bottom 96-well plate (ThermoFisher, USA) and were stimulated with or without Nipah-specific peptides (1µg/ml) for 5 hours at 37°C; 5% CO2, along with anti-mouse CD28 (1µg/ml) in the presence of GolgiPlug™ containing Brefeldin A (1µl/ml). Phorbol myristate acetate (PMA, 5 ng/ml; Sigma, USA) and ionomycin (250 ng/ml) were used as positive controls. The cells were fixed using a Cytofix/Cytoperm™ Plus kit (ThermoFisher, USA) as per manufacturer’s protocol, and stained with fluorochrome-conjugated monoclonal antibodies (mAbs) specific for cell surface antigens as well as intracellular cytokines, Extracellular antibodies used for staining included LIVE/DEAD Fixable Violet Dead Cell Stain kit (Invitrogen); anti-CD19 (V450; clone 1D3; BD Biosciences, USA); anti-CD4 (FITC; clone RM4-5; eBioscience, USA); anti-CD8α (APC-Cy7; clone 53–6.7; BD Biosciences, USA); anti-CD44 (A700; clone IM7; Biolegend, USA). For intracellular staining the following antibodies were used: anti-IFN-γ (APC; clone XMG1.2; Biolegend, USA); anti-TNF-α (PE; clone MP6-XT22; eBioscience, USA); anti-CD3ϵ (PerCP/Cy5.5; clone 145-2C11; Biolegend, USA); anti-IL-2 (PeCy7; clone JES6-SH4; eBioscience, USA). Appropriate isotype-matched controls for cytokines were included in case of each staining. The cells, after staining, were analyzed using LSRII flow cytometer (BD Biosciences, USA) and FlowJo software (Tree Star, USA) (51, 52).
NiV-G/F pseudotyped particles were prepared as described previously by co-transfection of HEK293T cells with a 1:1 ratio of DNA plasmids encoding NiV-G/F protein; or the plasmids encoding vesicular stomatitis virus G protein (VSV-G) were co-transfected with pNL4-3.luc.R-E- plasmid (NIH AIDS Reagent) to generate VSV-G pseudovirus as controls (40, 53–55). The supernatant containing pseudovirus was harvested 48hrs post-transfection and enriched with FBS to 12% total volume, sterile filtered, and stored at -80˚C. Pseudotyped viruses were diluted suitably to produce 1500-2000 relative light units (RLUs) in the control wells. Virus titers were determined by measuring RLU.
Pseudovirus inhibition assay was established to detect neutralizing activity of vaccinated mouse sera against infection of NIV-G/F pseudovirus in target cells. Briefly, NiV-G/F pseudovirus-containing supernatants were respectively incubated with serially diluted mouse sera at 37°C for 1h; 5% CO2 for adsorption before adding to confluent monolayers of Vero cells pre-plated in 96-well culture plates (104 cells/well). Twenty-four hours later, the inoculum was removed, cells were refed with fresh medium with 5% FBS and 1% penicillin-streptomycin, which was followed by lysing cells 72hrs later using cell lysis buffer (Promega) and transferring the lysates into 96-well luminometer plates. Luciferase substrate was added to the plates, and relative luciferase activity were performed according to the protocol recommended by the manufacturer with BriteLite reagent (PerkinElmer, USA). The inhibition of NiV-G/F pseudovirus was presented as % inhibition and the percent neutralization were calculated using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA) (40).
The data are presented as mean with standard error (SE) or standard error mean (SEM). Two-tailed student t-test or modified ANOVA was used to determine statistical significance. A p-value less than 0.05 was considered significant. Statistical analyses were performed using Graph Pad Prism software.
We have developed and advanced into the clinic several synthetic DNA vaccines against viral EID targets, including MERS-CoV (26, 40), Zika virus (41, 50), Ebola (52), Mayaro virus (MAYV) (51); Powassan virus (POWV) (44) and recently, SARS-CoV-2 (46). Each of these DNA vaccines induced robust antigen-specific antibodies as well as strong CD8+ and CD4+ T cell responses in clinical trials (25–27, 56). We have been very active over the past decade in improving the tolerability and responses of DNA vaccines. Electroporation-enhanced delivery of DNA vaccines significantly increases in vivo transfection efficiency, which improves antigen production and boosts their immunogenicity (25–27, 40, 51, 52, 57, 58).
The Nipah virus Glycoprotein (NiV-G) and Fusion protein (NiV-F), which mediate virus attachment and fusion, respectively, were selected as antigens for development of DNA vaccines. Both proteins are important targets on the virus envelope for inducing a protective neutralizing antibody response (13, 59, 60). The G protein is responsible for virus attachment to the putative viral receptor while the F protein mediates fusion. We employed a consensus antigen design strategy to identify the most highly conserved regions of F and G for inclusion in antigen cassettes. We performed alignments of multiple NiV-F and NiV-G protein sequences deposited in GenBank that came from divergent strains, including ones from the recent Indian NiV outbreak as described in materials and methods.
Several modifications were included when constructing the vaccine plasmids to improve the production of the antigens when delivered in vivo, including codon and RNA optimization of the consensus antigen sequences and the addition of a highly efficient IgE leader sequence to improve secretion of antigens from cells (Figure 1A). Prior to immunogenicity studies in mice, expression of each antigen was confirmed by Western blot analysis of lysates collected from HEK293T cells transfected with the NiV-G or NiV-F vaccines using specific monoclonal antibodies (Figure 1B). Proteins with estimated molecular masses of approximately 65-70kDa and 55-60kDa were detected in the cell lysates from NiV-G, and NiV-F transfected, respectively, consistent with the reported molecular weights of these antigens. Further, the expression of these constructs was confirmed by a green fluorescence protein (GFP)-based flow cytometry method. Specifically, HEK293T cells were transiently transfected with plasmids in which GFP was fused in-frame with the coding sequences of each antigen. Transfected cells were harvested 48 hours post-transfection and tested for protein expression by flow cytometry with GFP and specific antibodies. As shown in Figure 1C, increased antigen expression was observed in the transfected cells. This data confirms that the synthetic full-length NiV-G and NiV-F protein encoded in each vaccine is expressed by cells and readily detectable.
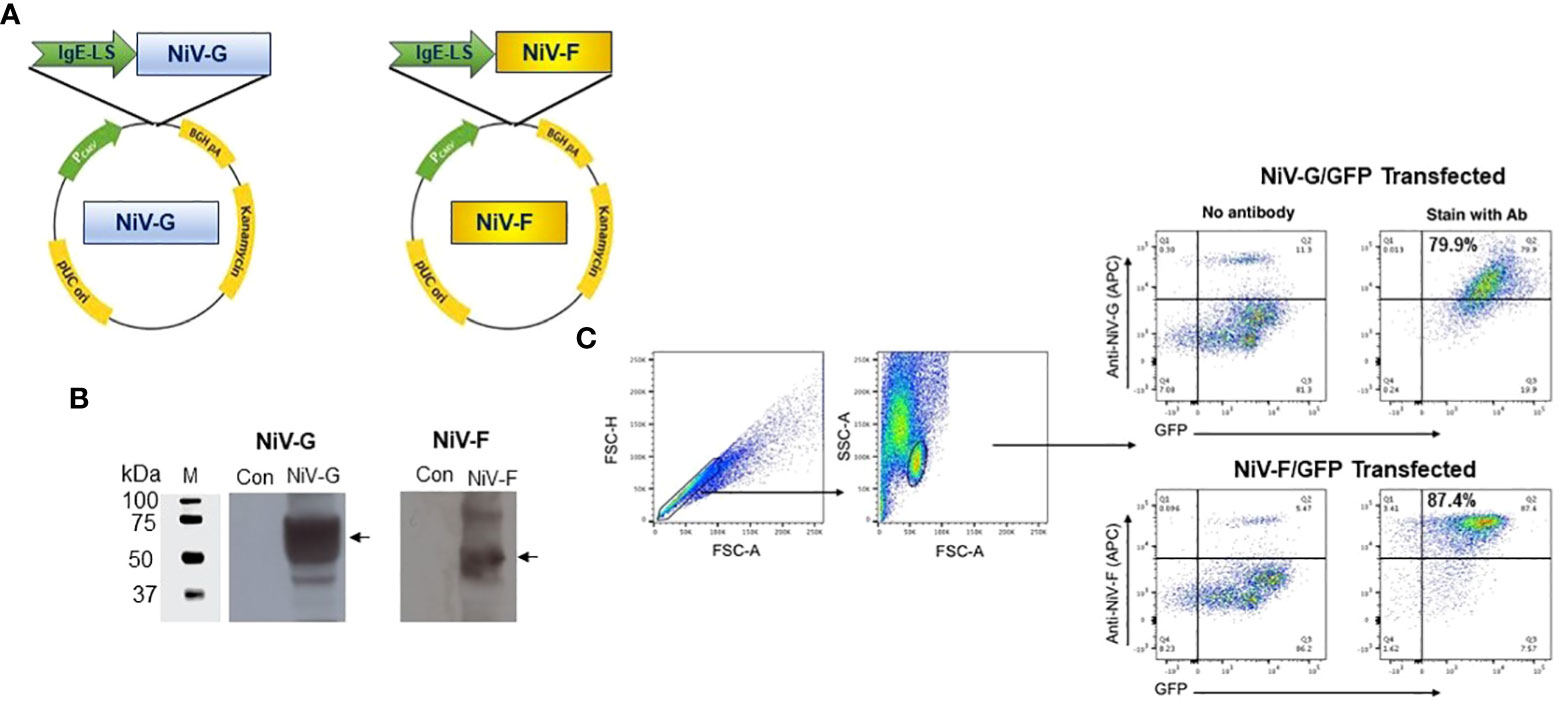
Figure 1 Generation and characterization of NiV-F and NiV-G specific vaccine. (A) Schematic diagram of NiV-vaccine encoding either synthetic consensus NiV-F or NiV-G antigens. (B) Western analyses of cell lysates from cells transfected with pNiV-G and pNiV-F vaccines probed with antibodies specific to each transfected cell lysates. The lanes were loaded with 20μg of total protein from the vaccine-transfected cell lysates. Negative control was established with pVax1 transfected cells (Con). (C) Flow cytometric determination of transgene expression. HEK293T cells were transfected with pNiV-G-GFP or pNiV-F-GFP and then analyzed by flow cytometry two days post-transfection cells to detect the surface expression of NiV-G and NiV-F protein using an antibody specific to each vaccine. Representative plots for flow cytometry gating strategy were shown for forward and side scatter and discrimination of doublets and percentage of unstained cells (negative control/GFP only) and cells without specific antibodies are added. In this plot, Q2 quadrant demonstrates cells that express target proteins (NiV-G-GFP or NiV-F-GFP) and show GFP fluorescence. The percentage of positive cells is indicated in Q2 gate.
Immune responses to NiV-G and NiV-F correlates with protection from Nipah virus infection in several susceptible species (11, 19, 61, 62). The goal of this study was to evaluate comprehensively and empirically the ability of our NiV-G and NiV-F vaccines to induce NiV-specific cellular and humoral responses. Immunogenicity evaluations were carried out in mice (n=4/group) vaccinated twice, 14 days apart, with the individual NiV-F or NiV-G vaccine constructs or an empty pVax1 vector. Sera were collected from mice prior to each immunization (days 0 and 14) as well as one-week post-second immunization (day 28) when spleens were also harvested from each animal. The induction of antibodies to NiV-G or NiV-F by each vaccine was measured using ELISA. The cellular responses were evaluated by performing IFN-γ ELISpot assay and intracellular cytokine staining (ICS) flow cytometric assays on splenocytes collected from vaccinated animals that were ex-vivo stimulated with pools of linear 15-mer peptides spanning the extracellular domain of NiV-G or NiV-F immunogens.
The induction of cellular responses in NiV-G vaccine-immunized mice was evaluated by performing an IFN-γ ELISpot assay on splenocytes collected two weeks after a second injection with the NiV-G vaccine (Figure 2A). Splenocytes were stimulated with linear pools of NiV-G peptides overnight. Figure 2B shows that NiV-G vaccine immunization induced a significant level of INF-γ spot-forming units (SFU) to multiple regions of the NiV-G. We went on to map the murine T-cell epitopes by screening these splenocytes with matrixed NiV-G peptide pools using ELISpot. All of the peptides from the NiV-G pools were assigned to matrix configuration (Table 1). In this ELISpot assay, splenocytes stimulated with matrix pools 8, 23, and 24 had an IFN-γ SFU response approximately 2-fold over the highest subdominant response. Based on the matrix pool composition, this result narrowed the dominant response to overlapping peptides 94 and 95 (aa 394-408 of NiV-G) which led to the sequence “YNDSNCPITKC” being identified as an immunodominant epitope of NiV-G (Figure 2C). Intracellular cytokine staining for IFN-γ, IL-2, and TNF-α on NiV-G peptide-stimulated splenocytes showed that vaccination with NiV-G vaccine increased the percentage of NiV-G-specific cytotoxic (i.e., CD8+) T cells in mice (Figure 2D).
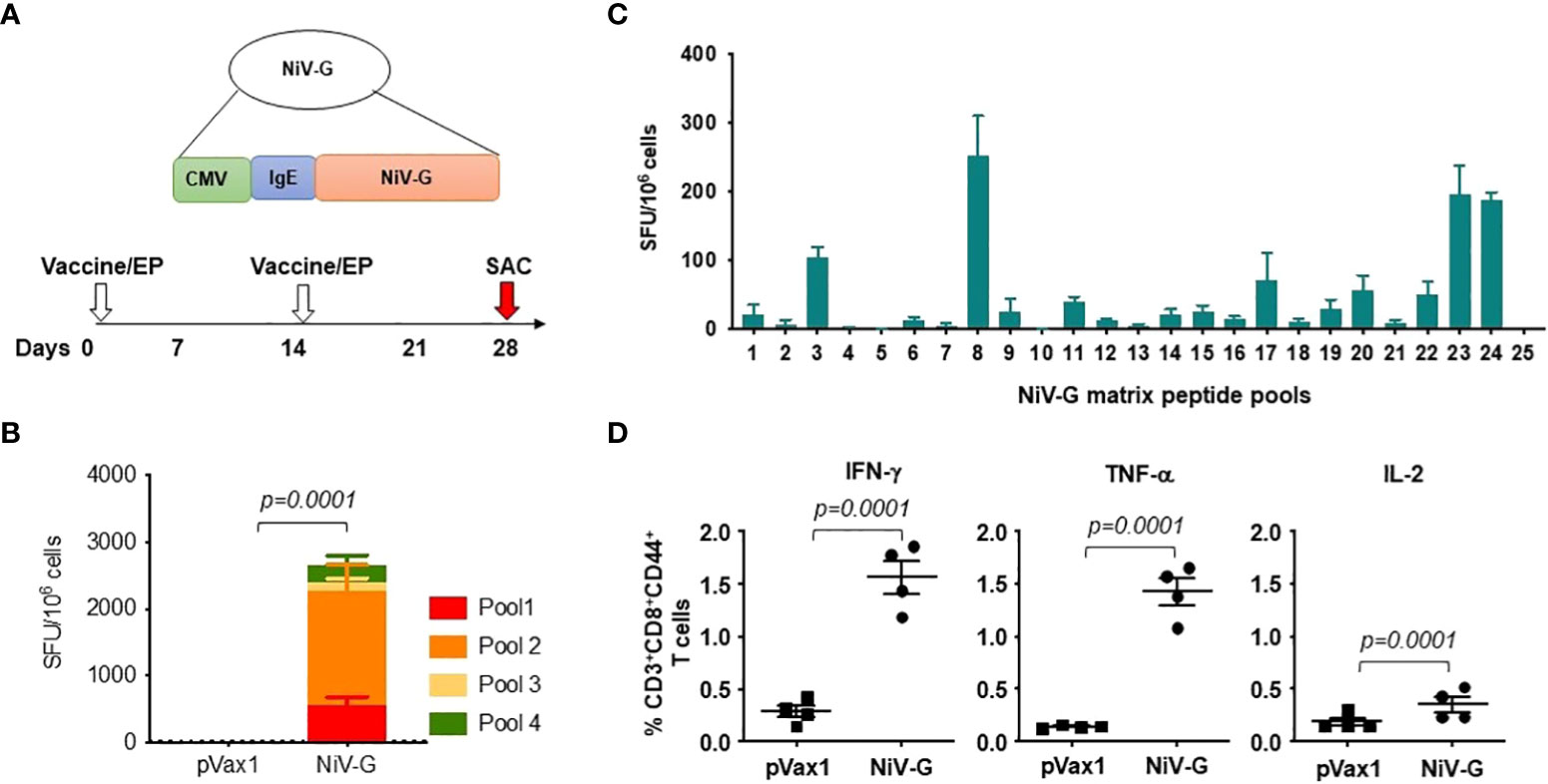
Figure 2 Functional profile of cellular responses elicited by NiV-G vaccine in mice. (A) Schematic representation of EP mediated immunization and immune analysis. C57BL/6 mice (n=4 per group) were immunized with 25μg of pNiV-G vaccine using EP mediated enhanced delivery. (B, C) Immunization of mice with NiV-G vaccine induced robust T cell responses, as assessed by total IFN-γ ELISpot assay on splenocytes stimulated with each of 4 pools of linear overlapping peptides spanning the whole NiV-G (B) and characterization of NiV-G-specific dominant epitopes in C57BL/6 mice. IFN-γ responses were assessed by ELISpot assays with matrix pools of NiV-G peptides where each 15-mer peptide was present in two pools (C). The data represents the total IFN-γ response of mean±SE in each group two weeks after the second immunization (day 28). Values represent mean responses in each group (n=4) ± SEM. (D) Immunization with NiV-G vaccine induced activated CD8+ T cells capable of producing IFN-γ, TNF-α and IL-2 secreting cells when stimulated by NiV-G peptides. Two weeks after the second immunization with the NiV-G vaccine, splenocytes were cultured in the presence of pooled NiV-G peptides or R10 medium only. Flow cytometry measured the frequency of NiV-G -specific IFN-γ, TNF-α and IL-2 producing CD8+ T cells. Single function gates were set based on negative control (unstimulated) and were placed consistently across the samples. The percentage of the total CD8+ T-cell responses are shown. These data are representative of two independent immunization experiments.
Table 1 The corresponding list of NiV-F/G liner and Matrix peptide pools.
Immunization with the NiV-G vaccine induced a significant level of antigen-specific antibodies in mice one week after a second immunization with the construct (Figure 3A). All immunized animals seroconverted after their first immunization, and a little increase in antibody titer was seen after a second immunization (Figure 3B). Mice vaccinated with the control pVax1 vector elicited no anti-G antibody. The IgG induced in vaccinated mice was primarily of the IgG2a and IgG1 subclasses (Figure 3C). Antibodies present in sera from NiV-G vaccine-vaccinated animals bound to native forms of NiV-G in an indirect fluorescence assay (IFA) of HEK293T cells transfected with the NiV-G vaccine plasmid (Figure 3D). To exclusively measure NiV-G specific neutralization responses, we made use of pseudoviral based neutralization to NiV-G-specific responses. As indicated in Figure 3E, antisera from immunized with the NiV-G vaccine mice (n=4) we observed that groups vaccinated with NiV-G, retained some levels of neutralization by the pseudoviral tested. These data show that the consensus NiV-G vaccine is a potent immunogen for generating both cellular and humoral immune responses against NiV.
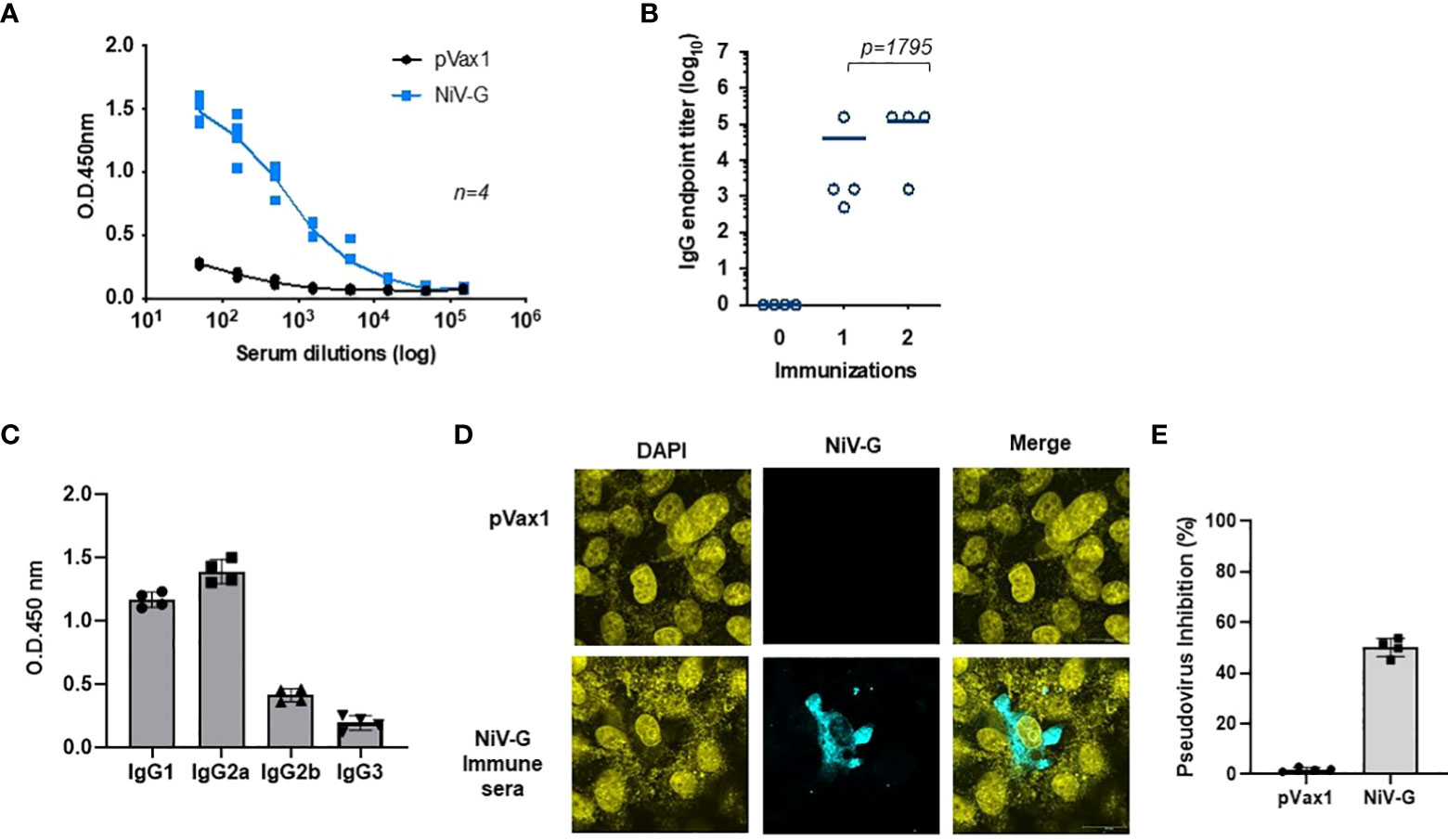
Figure 3 Evaluation of NiV-G antigen-specific antibody responses following immunization of mice with NiV-G vaccine. (A) NiV-G specific-antibody responses were measured in sera collected two weeks after the second immunization (day 28). Recombinant NiV-G protein was used in ELISA to assess the antigen-specific antibody responses. (B) Endpoint titers for the single and two immunization samples were measured in sera collected on day 14 (indicated as immunization 1) and 28 (indicated as immunization 2) post-immunization (n=4 mean ± SEM). (C) IgG1, IgG2a, IgG2b, and IgG3 subclass of mouse sera were determined by ELISA in samples collected two weeks post second immunization (day 28). (D) Immunofluorescence assay demonstrated that IgG generated from NiV-G vaccine-administered mice was capable of binding to NiV-G vaccine-transfected cells. (E) Day 28 immune sera from each mouse were diluted at 1:20 and used to measure inhibition ability for NiV-F/G pseudovirus. Sera samples were measured in duplicates and the data shows one of two representative experiments.
We next evaluated cellular responses induced in mice after vaccination with our NiV-F vaccine (Figure 4A). An IFN-γ ELISpot assay performed on splenocytes from NiV-F vaccine-immunized mice showed a significant increase in IFN-γ-SFUs following culturing of the splenocytes with linear pools of peptides spanning the extracellular domain of NiV-F compared to splenocytes from control vector-immunized mice (Figure 4B). To identify specific epitopes within NiV-F that elicited cellular response, an IFN-γ ELISpot assay was performed on splenocytes using matrixed pools of NiV-F peptides (Table 1). There were six matrix pools that elicited more than 100 spots, indicating cellular responses generated by NiV-F vaccine recognized multiple regions of NiV-F. Based on this result and the composition of the matrix pools, the sequence “IQELLPVSFNNDNSE” was identified as an immunodominant epitope of NiV-F (Figure 4C). It was confirmed to contain one H2-Db-restricted epitope by using the IEDB analysis resource consensus tool (http://tools.immuneepitope.org). Vaccination with the NiV-F vaccine also increased the percentage of CD8+ T cells that could produce IFN-γ, IL-2, and TNF-α upon stimulation with NiV-F peptides (Figure 4D).
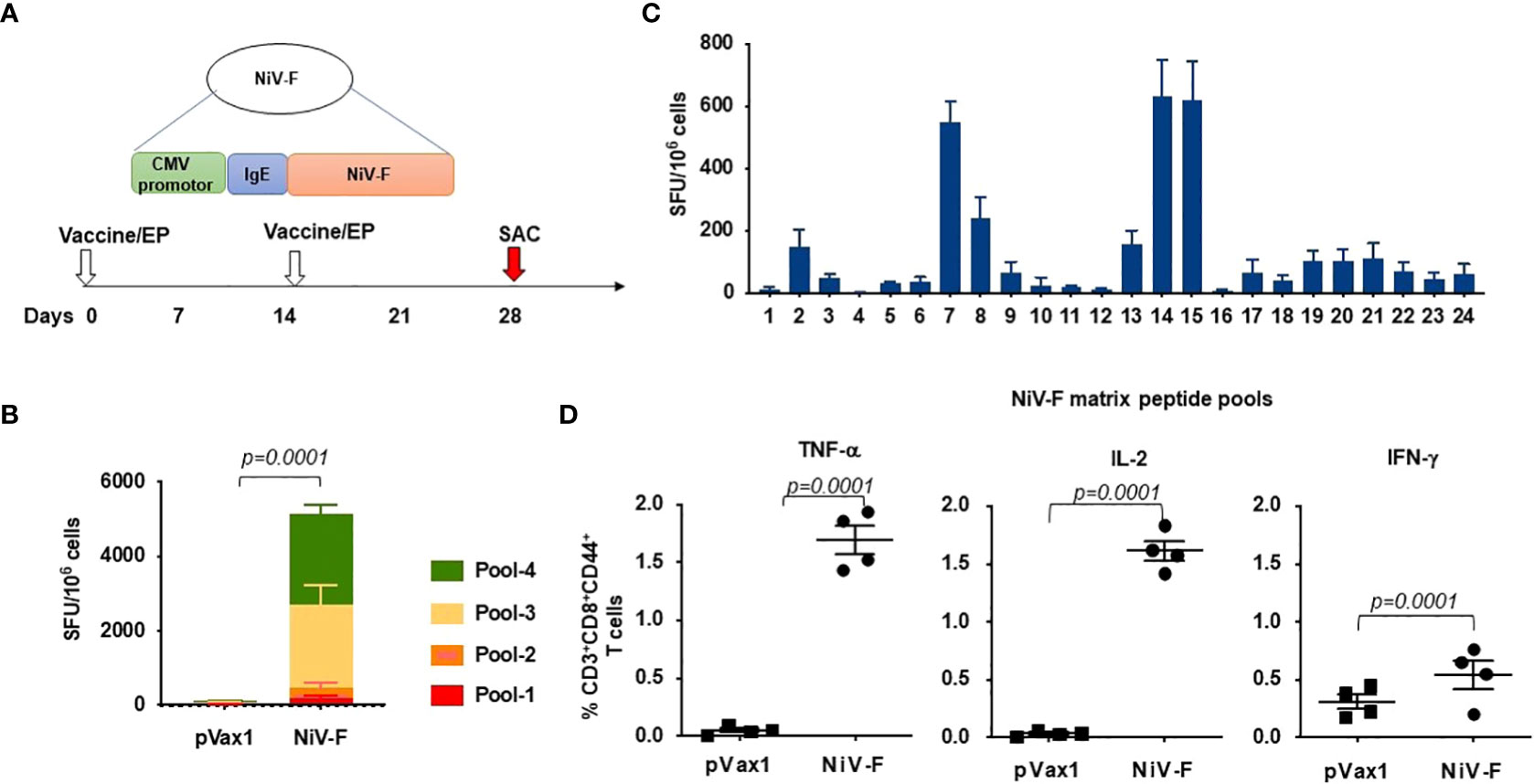
Figure 4 Functional profile of cellular immune responses elicited by NiV-F vaccine in mice. (A) Schematic representation of different time points of EP mediated immunization and immune analysis of this study. C57BL/6 mice (n=4 per group) were immunized with 25μg of NiV-F vaccine using EP mediated enhanced delivery. (B) Immunization of mice with NiV-F vaccine induced robust CD8+ T cell responses. Nipah-specific CD8+ T-lymphocyte responses were assessed by IFN-γ ELISpot assay to each of 4 pools of linear overlapping peptides spanning the whole NiV-F. Data represents the responses Mean±SD in each group one week after the second immunization. (C) Characterization of NiV-specific dominant epitopes in C57BL/6 mice. IFN-γ responses were assessed by ELISpot assays with matrix pools of peptides, indicating the presence of immunodominant epitopes comprising of the NiV-F. Values represent mean responses in each group (n=4) ± SEM. (D) Immunization with NiV-F vaccine induced a higher number of activated CD8+ T cells capable of producing IFN-γ, TNF-α and IL-2 when stimulated by Nipah-F peptides. One week after the last immunization with the NiV-F vaccine, splenocytes were cultured in the presence of pooled NiV-F peptides or RPMI media + 10% FBS (R10) only. Flow cytometry measured the frequency of NiV-F -specific IFN-γ, TNF-α and IL-2 secreting CD8+ T cells. Single function gates were set based on negative control (unstimulated) and were placed consistently across the samples. The percentage of the total CD8+ T-cell responses are shown. These data are representative of two independent immunization experiments.
ELISA evaluation of sera collected from immunized mice showed that the NiV-F-vaccine vaccine-induced significant NiV-F specific antibodies in all mice by two weeks following the first dose, and a second immunization with the vaccine boosted antibody titers (Figures 5A, B). Antibodies generated in NiV-F vaccine-immunized animals were primarily of the IgG2a subclass, suggesting that the NiV-F vaccine induces a dominant Th1-type immune response (Figure 5C). In addition, sera from vaccinated mice could also readily detect F proteins in Western analyses and in IFA of NiV-F vaccine-transfected HEK293T cells (Figures 5D, 5E). To evaluate the neutralization potential of sera collected from NiV-F vaccine-immunized mice, we generated NiV-F-pseudotyped recombinant lentiviral particles that expresses the luciferase reporter gene. Preincubation of these pseudotyped particles with sera from NiV-F vaccine-immunized mice efficiently inhibited infection of Vero cells by the pseudovirus tested (Figure 5F), suggesting that the antibodies induced by this vaccine could neutralize NiV infection.
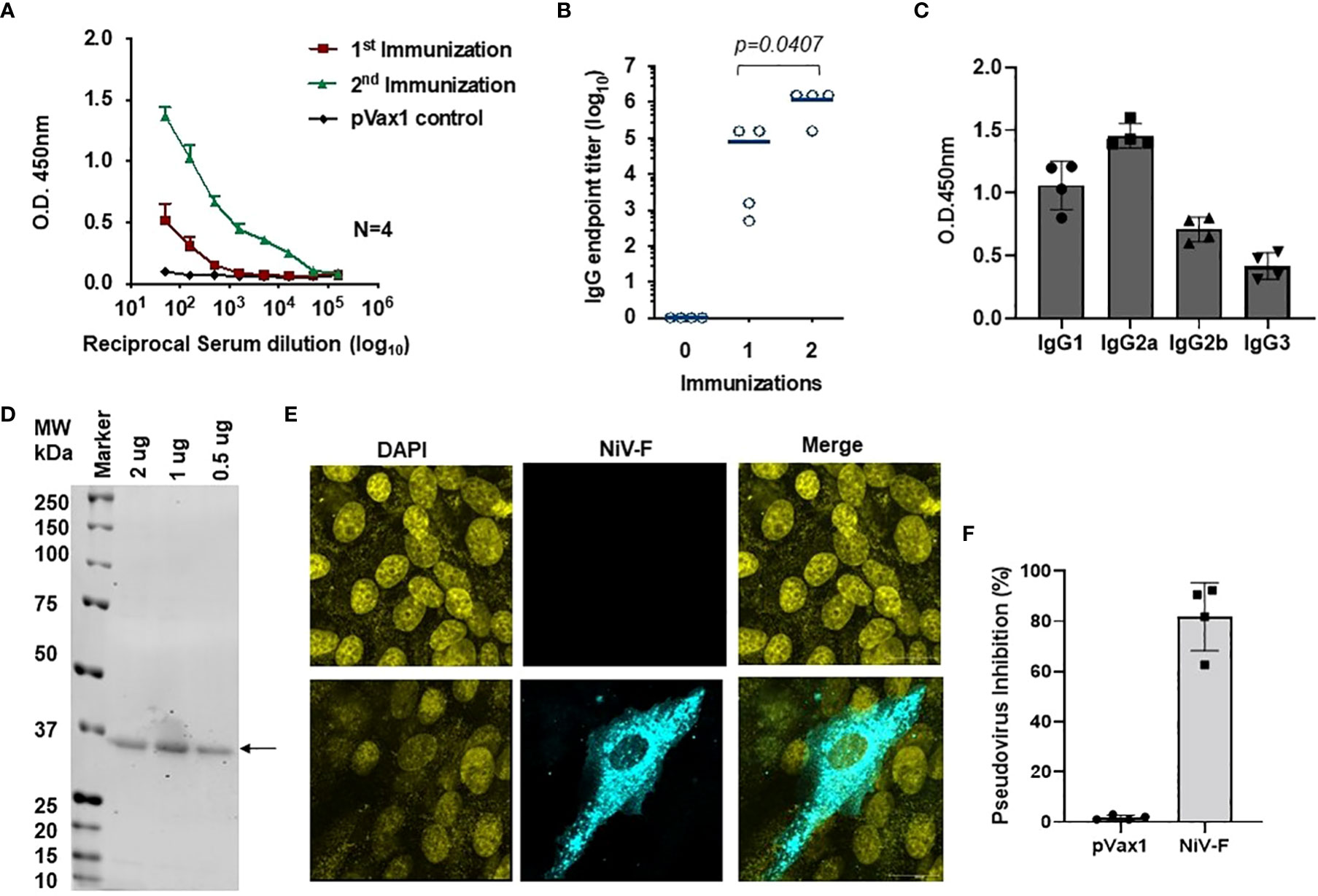
Figure 5 Antigen-specific humoral immunity is induced by synthetic vaccine candidates expressing NiV-F. (A) Sera collected 14 days after the first (prime) immunization and 7 days after the second (boost) immunization (day 28) were analyzed for NiV-F-specific IgG. (B) Endpoint titers for the single and two immunization samples were measured in sera collected on day 14 (indicated as immunization 1) and 28 (indicated as immunization 2) post-immunization (n = 4 mean ± SEM). (C) IgG1, IgG2a, IgG2b, and IgG3 subclass of mouse sera were determined by ELISA in samples collected one week post second immunization (day 28). (D) Western blot of NiV-F protein detected with immune sera from mice immunized with NiV-F vaccine. The arrow indicates molecular mass. Lane M: protein size marker. (E) IFA analysis for NiV-F protein expression in Vero cells. 24h after transfection with NiV-F vaccine, immunofluorescence labeling was performed by incubation with the immune sera (1:100). (F) Day 28 immune sera from NiV-F vaccine-immunized mice were diluted at 1:20 and used to measure inhibition ability for NiV-F/G pseudovirus. Sera samples were measured in duplicates and the data shows one of two representative experiments.
In this study, we applied synthetic DNA vaccine technologies to address the major need for a vaccine against Nipah virus. Both the NiV-G and NiV-F vaccines designed and tested in this study induced robust cellular and humoral immune responses specific to each NiV protein. Both vaccines activated polyfunctional cytotoxic CD8+ T cells specific for NiV as well as full seroconversion with robust antigen-specific antibody titers after a single immunization. Antibodies induced by both vaccines in mice inhibited entry of NiV-F/G-pseudotyped viral particles into cells, supporting the hypothesis that these vaccines are capable of neutralizing NiV infection. The membrane-distal region of NiV-F has been identified as a site of vulnerability on the NiV surface and monoclonal antibodies have been developed against NiV-F that block NiV infection in vitro (19, 36). Additionally, neutralizing antibodies induced by recombinant VSV-based NiV-F vaccines offered protection against infection in ferret and hamster NiV challenge models (63, 64). While the correlations of protection for NiV infection are unknown, vaccines that can induce or engage multiple effector mechanisms like our NiV-G and NiV-F vaccines have a better chance of providing protection from NiV infection and/or severe disease.
Some desirable characteristics of a NiV vaccine to be used during an outbreak include an ability to rapidly induce protective immunity after a single dose with a good safety profile, thermostability (2-8°C), and an ability to confer immunity against both NiV M and B clade viruses (65). The DNA vaccine platform used to create the NiV-G and -F vaccines described here has been evaluated clinically for over 25 years and has maintained a good safety profile over this time. DNA plasmids can be designed rapidly; manufactured in large quantities cheaply; and are thermostable at room temperature for extended periods - all helpful properties for a NiV vaccine considering that several low- and middle-income countries (LMICs) are present in the danger zone for where a NiV outbreak may occur (25, 40, 66–68). The use of synthetic DNA technology to create consensus NiV-G and NiV-F sequences for the vaccines described here should provide broader protection against both NiV-M and B clade viruses, although further testing is needed to confirm this. Furthermore, the consensus sequence antigens should maintain the efficacy of these vaccines against strains of NiV that emerge in the future. In mice experiments, seroconversion to NiV-G and NiV-F were noted after a single immunization, and the electroporation-enhanced delivery protocol allowed for dose-sparing as only 25μg of each vaccine induced significant humoral and cellular responses. Further studies are also needed to evaluate whether these individual vaccine responses are sufficient to protect against NiV infection and disease and if delivering the vaccines as a cocktail enhances their ability to protect against NiV infection.
This report is the third to describe the development of DNA vaccines against NiV. Wang et al. created vaccine plasmids with codon optimized sequences of NiV F and G cloned into the pCAGGS vector and observed that their NiV G vaccine induced higher specific IgG and neutralizing antibodies that their NiV F vaccine in mice (69). The sequences of each protein in their vaccines came from a NiV M clade virus. Nie et al. also generated DNA vaccines using the G and F sequences from a NiV-M clade virus, using the CMV promoter-containing pDRV1.0 vector as a backbone (70). They noted that their vaccines generated neutralizing antibodies in mice and guinea pigs, when administered separately or together, after electroporation-enhanced immunization. They also observed that their NiV G vaccine induced higher nAb than their NiV F vaccine, but in a NiV F/G pseudotyped particle mouse challenge model that they developed, immunization with their NiV-G vaccine induced equivalent protection as immunization with their NiV-F vaccine and immunization with both together had no significant effect on protection. Neither group reported on the generation of cellular immune responses by their vaccines. In addition to these DNA vaccines, at least 38 other NiV vaccines have been reported on (71). More than half of these were made using viral vector platforms with protein subunit, virus-like particles, and mRNA platforms also being employed. Only three of these reported vaccines were made using the G and F proteins from NiV B clade viruses while the remainder encode G and/or F sequences from NiV-M clade viruses or HeV. While several of these vaccines were demonstrated to offer protection against NiV challenge in small animal models, only two have advanced into phase I trials.
Although extensive studies have been carried out over the years to understand the pathogenicity of NiV infection, little progress has been made in the development of specific anti-NiV compounds (8, 72). As seen with the Ebola virus in 2014-15 and SARS-CoV-2 in 2019-present, not having a vaccine or a viable vaccine candidate available when an EID pathogen appears can lead to significant disruption to everyday life throughout the globe as well as innumerable and tragic loss of life. The appearance of EID agents has become more frequent over the last few decades, and their ability to spread around the world within hours and days means that no one is truly safe from them. Preemptively developing vaccines for specific pathogens or pathogen families that are likely to cause widespread epidemics is a global priority to minimize or hopefully prevent the damage that can be caused by an EID. The data presented in this paper support further evaluation of these immunogenic NiV-G and NiV-F vaccines as a viable strategy to prevent NiV infection and disease in expanded animal studies. These tools likely could be important for possible deployment if and when a NiV outbreak occurs.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Wistar Institute IACUC# 112785.
DW and KM were involved in study concept and design. HC, SK, ZX, MH, and PX were involved in the acquisition, the analysis and the interpretation of data. SR and LH contributed crucial reagents. SK and KM wrote the paper. All authors were involved in critically revising and approved of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by WW Smith Charitable Trust 6743101374 awarded to DBW. Support for Shared Resources utilized in this study was provided by Cancer Center Support Grant (CCSG) P30CA010815 to The Wistar Institute. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No writing assistance was utilized in the production of this manuscript. No writing assistance was utilized in the production of this manuscript.
We would like to thank the Animal Facility staff at the Wistar Institute for providing house and care to the animals. We thank the Wistar Flow Core for assistance with the flow/sorting experiments. The following reagent was obtained through the NIH-AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 pNL4-3 ΔEnv-ΔVpr Luciferase Reporter Vector (pNL4-3.Luc.R-E-: Cat #3418).
K.M., and D.B.W. are named inventors of the PCT/US2019/047410 patent application titled “Vaccines against NIPAH virus, and Methods of Using Same”. KM discloses grant funding, industry collaborations, speaking honoraria, and fees for consulting from Inovio Pharmaceuticals related to DNA and DMAb vaccine development. He has a patent application for DNA vaccine development and delivery of DNA-encoded monoclonal antibodies pending to Inovio Pharmaceuticals. Remuneration includes direct payments. DBW discloses grant funding, SAB and Board service, industry collaborations, speaking honoraria, and fees for consulting. His service includes serving on scientific review committees and advisory boards. Remuneration includes direct payments and/or stock or stock options. He notes potential conflicts associated with this work with Pfizer, Bristol Myers Squibb, Inovio Pharmaceuticals, Merck, VGXI, Geneos, AstraZeneca, and potentially others. Licensing of technology from this laboratory has created over 150 jobs in the biotech/pharma industry. S.R. and L.H are employer of Inovio Pharmaceuticals and as such receive salary and benefits, including ownership of stock and stock options. The other authors declare no competing financial interests.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Meulemans G. [Significance of paramyxovirinae protein f in physiopathology and immunity]. Bull Acad Natl Med (2000) 184:1255–1264; discussion 1265.
2. Enserink M. Emerging infectious diseases. Nipah virus (or a cousin) strikes again. Science (2004) 303:1121. doi: 10.1126/science.303.5661.1121b
3. Von Messling V, Cattaneo R. Virology. A henipavirus vaccine in sight. Science (2012) 337:651–2. doi: 10.1126/science.1227810
4. Rockx B. Recent developments in experimental animal models of henipavirus infection. Pathog Dis (2014) 71:199–206. doi: 10.1111/2049-632X.12149
5. Sharma V, Kaushik S, Kumar R, Yadav JP, Kaushik S. Emerging trends of nipah virus: A review. Rev Med Virol (2019) 29:e2010. doi: 10.1002/rmv.2010
6. Wong JJ, Chen Z, Chung JK, Groves JT, Jardetzky TS. EphrinB2 clustering by nipah virus G is required to activate and trap f intermediates at supported lipid bilayer-cell interfaces. Sci Adv (2021) 7:eabe1235. doi: 10.1126/sciadv.abe1235
7. Watkinson RE, Lee B. Nipah virus matrix protein: expert hacker of cellular machines. FEBS Lett (2016) 590:2494–511. doi: 10.1002/1873-3468.12272
8. Geisbert TW, Mire CE, Geisbert JB, Chan YP, Agans KN, Feldmann F, et al. Therapeutic treatment of nipah virus infection in nonhuman primates with a neutralizing human monoclonal antibody. Sci Transl Med (2014) 6:242ra282. doi: 10.1126/scitranslmed.3008929
9. Gomez Roman R, Wang LF, Lee B, Halpin K, De Wit E, Broder CC, et al. Nipah@20: Lessons learned from another virus with pandemic potential. mSphere (2020) 5:e00602-20. doi: 10.1128/mSphere.00602-20
10. Loomis RJ, Stewart-Jones GBE, Tsybovsky Y, Caringal RT, Morabito KM, Mclellan JS, et al. Structure-based design of nipah virus vaccines: A generalizable approach to paramyxovirus immunogen development. Front Immunol (2020) 11:842. doi: 10.3389/fimmu.2020.00842
11. Walpita P, Cong Y, Jahrling PB, Rojas O, Postnikova E, Yu S, et al. A VLP-based vaccine provides complete protection against nipah virus challenge following multiple-dose or single-dose vaccination schedules in a hamster model. NPJ Vaccines (2017) 2:21. doi: 10.1038/s41541-017-0023-7
12. Epstein JH, Anthony SJ, Islam A, Kilpatrick AM, Ali Khan S, Balkey MD, et al. Nipah virus dynamics in bats and implications for spillover to humans. Proc Natl Acad Sci U.S.A. (2020) 117:29190–201. doi: 10.1073/pnas.2000429117
13. Majee P, Kumar Mishra S, Pandya N, Shankar U, Pasadi S, Muniyappa K, et al. Identification and characterization of two conserved G-quadruplex forming motifs in the nipah virus genome and their interaction with G-quadruplex specific ligands. Sci Rep (2020) 10:1477. doi: 10.1038/s41598-020-58406-8
14. Whitmer SLM, Lo MK, Sazzad HMS, Zufan S, Gurley ES, Sultana S, et al. Inference of nipah virus evolution 1999-2015. Virus Evol (2021) 7:veaa062. doi: 10.1093/ve/veaa062
15. Ang BSP, Lim TCC, Wang L. Nipah virus infection. J Clin Microbiol (2018) 56(6):e01875-17. doi: 10.1128/JCM.01875-17
16. Aditi, Shariff M. Nipah virus infection: A review. Epidemiol Infect (2019) 147:e95. doi: 10.1017/S0950268819000086
17. Diederich S, Maisner A. Molecular characteristics of the nipah virus glycoproteins. Ann N Y Acad Sci (2007) 1102:39–50. doi: 10.1196/annals.1408.003
18. Johnson JB, Borisevich V, Rockx B, Parks GD. A novel factor I activity in nipah virus inhibits human complement pathways through cleavage of C3b. J Virol (2015) 89:989–98. doi: 10.1128/JVI.02427-14
19. Dang HV, Chan Y-P, Park Y-J, Snijder J, Da Silva SC, Vu B, et al. An antibody against the f glycoprotein inhibits nipah and hendra virus infections. Nat Struct Mol Biol (2019) 26:980–7. doi: 10.1038/s41594-019-0308-9
20. Gurley ES, Montgomery JM, Hossain MJ, Bell M, Azad AK, Islam MR, et al. Person-to-person transmission of nipah virus in a Bangladeshi community. Emerg Infect Dis (2007) 13:1031–7. doi: 10.3201/eid1307.061128
21. Escaffre O, Borisevich V, Carmical JR, Prusak D, Prescott J, Feldmann H, et al. Henipavirus pathogenesis in human respiratory epithelial cells. J Virol (2013) 87:3284–94. doi: 10.1128/JVI.02576-12
22. Goh GK, Dunker AK, Foster JA, Uversky VN. Nipah shell disorder, modes of infection, and virulence. Microb Pathog (2020) 141:103976. doi: 10.1016/j.micpath.2020.103976
23. Tan CT, Goh KJ, Wong KT, Sarji SA, Chua KB, Chew NK, et al. Relapsed and late-onset nipah encephalitis. Ann Neurol (2002) 51:703–8. doi: 10.1002/ana.10212
24. Wong KT, Shieh WJ, Kumar S, Norain K, Abdullah W, Guarner J, et al. Nipah virus infection: pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am J Pathol (2002) 161:2153–67. doi: 10.1016/S0002-9440(10)64493-8
25. Tebas P, Roberts CC, Muthumani K, Reuschel EL, Kudchodkar SB, Zaidi FI, et al. Safety and immunogenicity of an anti-zika virus DNA vaccine - preliminary report. N Engl J Med (2017). doi: 10.1056/NEJMoa1708120
26. Modjarrad K, Roberts CC, Mills KT, Castellano AR, Paolino K, Muthumani K, et al. Safety and immunogenicity of an anti-middle East respiratory syndrome coronavirus DNA vaccine: a phase 1, open-label, single-arm, dose-escalation trial. Lancet Infect Dis (2019) 19:1013–22. doi: 10.1016/S1473-3099(19)30266-X
27. Bagarazzi ML, Yan J, Morrow MP, Shen X, Parker RL, Lee JC, et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci Transl Med (2012) 4:155ra138. doi: 10.1126/scitranslmed.3004414
28. Broderick KE, Humeau LM. Enhanced delivery of DNA or RNA vaccines by electroporation. Methods Mol Biol (2017) 1499:193–200. doi: 10.1007/978-1-4939-6481-9_12
29. Abbasi J. India’s new COVID-19 DNA vaccine for adolescents and adults is a first. JAMA (2021) 326:1365–5. doi: 10.1001/jama.2021.16625
30. Butler D. Billion-dollar project aims to prep vaccines before epidemics hit. Nature (2017) 541:444–5. doi: 10.1038/nature.2017.21329
31. Bossart KN, Crameri G, Dimitrov AS, Mungall BA, Feng YR, Patch JR, et al. Receptor binding, fusion inhibition, and induction of cross-reactive neutralizing antibodies by a soluble G glycoprotein of hendra virus. J Virol (2005) 79:6690–702. doi: 10.1128/JVI.79.11.6690-6702.2005
32. Guillaume V, Wong KT, Looi RY, Georges-Courbot MC, Barrot L, Buckland R, et al. Acute hendra virus infection: Analysis of the pathogenesis and passive antibody protection in the hamster model. Virology (2009) 387:459–65. doi: 10.1016/j.virol.2009.03.001
33. Balzer M. Hendra vaccine success announced. Aust Vet J (2011) 89:N2–3. doi: 10.1111/j.1751-0813.2011.news_v89_i7.x
34. Bossart KN, Rockx B, Feldmann F, Brining D, Scott D, Lacasse R, et al. A hendra virus G glycoprotein subunit vaccine protects African green monkeys from nipah virus challenge. Sci Transl Med (2012) 4:146ra107. doi: 10.1126/scitranslmed.3004241
35. Geisbert JB, Borisevich V, Prasad AN, Agans KN, Foster SL, Deer DJ, et al. An intranasal exposure model of lethal nipah virus infection in African green monkeys. J Infect Dis (2020) 221:S414–8. doi: 10.1093/infdis/jiz391
36. Avanzato VA, Oguntuyo KY, Escalera-Zamudio M, Gutierrez B, Golden M, Kosakovsky Pond SL, et al. A structural basis for antibody-mediated neutralization of nipah virus reveals a site of vulnerability at the fusion glycoprotein apex. Proc Natl Acad Sci U.S.A. (2019) 116:25057–67. doi: 10.1073/pnas.1912503116
37. Mclellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science (2013) 340:1113–7. doi: 10.1126/science.1234914
38. Gilman MSA, Furmanova-Hollenstein P, Pascual G, van 't Wout AB, Langedijk JPM, Mclellan JS. Transient opening of trimeric prefusion RSV f proteins. Nat Commun (2019) 10(1):2105. doi: 10.1038/s41467-019-09807-5
39. Arunkumar G, Chandni R, Mourya DT, Singh SK, Sadanandan R, Sudan P, et al. Outbreak investigation of nipah virus disease in kerala, indi. J Infect Dis (2019) 219:1867–78. doi: 10.1093/infdis/jiy612
40. Muthumani K, Falzarano D, Reuschel EL, Tingey C, Flingai S, Villarreal DO, et al. A synthetic consensus anti-spike protein DNA vaccine induces protective immunity against middle East respiratory syndrome coronavirus in nonhuman primates. Sci Transl Med (2015) 7:301ra132. doi: 10.1126/scitranslmed.aac7462
41. Muthumani K, Griffin BD, Agarwal S, Kudchodkar SB, Reuschel EL, Choi H, et al. In vivo protection against ZIKV infection and pathogenesis through passive antibody transfer and active immunisation with a prMEnv DNA vaccine. NPJ Vaccines (2016) 1:16021. doi: 10.1038/npjvaccines.2016.21
42. Patel A, Park DH, Davis CW, Smith TRF, Leung A, Tierney K, et al. In vivo delivery of synthetic human DNA-encoded monoclonal antibodies protect against ebolavirus infection in a mouse model. Cell Rep (2018) 25:1982–1993 e1984. doi: 10.1016/j.celrep.2018.10.062
43. Perales-Puchalt A, Duperret EK, Yang X, Hernandez P, Wojtak K, Zhu X, et al. DNA-Encoded bispecific T cell engagers and antibodies present long-term antitumor activity. JCI Insight (2019) 4(8):e126086. doi: 10.1172/jci.insight.126086
44. Choi H, Kudchodkar SB, Ho M, Reuschel EL, Reynolds E, Xu Z, et al. A novel synthetic DNA vaccine elicits protective immune responses against powassan virus. PloS Negl Trop Dis (2020) 14:e0008788. doi: 10.1371/journal.pntd.0008788
45. Schultheis K, Pugh HM, Oh J, Nguyen J, Yung B, Reed C, et al. Active immunoprophylaxis with a synthetic DNA-encoded monoclonal anti-respiratory syncytial virus scFv-fc fusion protein confers protection against infection and durable activity. Hum Vaccin Immunother (2020) 1-11. doi: 10.1080/21645515.2020.1748979
46. Smith TRF, Patel A, Ramos S, Elwood D, Zhu X, Yan J, et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat Commun (2020) 11:2601. doi: 10.1038/s41467-020-16505-0
47. Wise MC, Xu Z, Tello-Ruiz E, Beck C, Trautz A, Patel A, et al. In vivo delivery of synthetic DNA-encoded antibodies induces broad HIV-1-neutralizing activity. J Clin Invest (2020) 130:827–37. doi: 10.1172/JCI132779
48. Xu Z, Wise MC, Chokkalingam N, Walker S, Tello-Ruiz E, Elliott STC, et al. In vivo assembly of nanoparticles achieved through synergy of structure-based protein engineering and synthetic DNA generates enhanced adaptive immunity. Adv Sci (Weinh) (2020) 7:1902802. doi: 10.1002/advs.201902802
49. Patel A, Walters JN, Reuschel EL, Schultheis K, Parzych E, Gary EN, et al. Intradermal-delivered DNA vaccine induces durable immunity mediating a reduction in viral load in a rhesus macaque SARS-CoV-2 challenge model. Cell Rep Med (2021) 2:100420. doi: 10.1016/j.xcrm.2021.100420
50. Tebas P, Roberts CC, Muthumani K, Reuschel EL, Kudchodkar SB, Zaidi FI, et al. Safety and immunogenicity of an anti-zika virus DNA vaccine. N Engl J Med (2021) 385:e35. doi: 10.1056/NEJMoa1708120
51. Choi H, Kudchodkar SB, Reuschel EL, Asija K, Borole P, Ho M, et al. Protective immunity by an engineered DNA vaccine for mayaro virus. PloS Negl Trop Dis (2019) 13:e0007042. doi: 10.1371/journal.pntd.0007042
52. Patel A, Reuschel EL, Kraynyak KA, Racine T, Park DH, Scott VL, et al. Protective efficacy and long-term immunogenicity in cynomolgus macaques by Ebola virus glycoprotein synthetic DNA vaccines. J Infect Dis (2019) 219:544–55. doi: 10.1093/infdis/jiy537
53. Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, Tajyar S, et al. EphrinB2 is the entry receptor for nipah virus, an emergent deadly paramyxovirus. Nature (2005) 436:401–5. doi: 10.1038/nature03838
54. Porotto M, Rockx B, Yokoyama CC, Talekar A, Devito I, Palermo LM, et al. Inhibition of nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entry. PloS Pathog (2010) 6:e1001168. doi: 10.1371/journal.ppat.1001168
55. Wolf MC, Freiberg AN, Zhang T, Akyol-Ataman Z, Grock A, Hong PW, et al. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc Natl Acad Sci U.S.A. (2010) 107:3157–62. doi: 10.1073/pnas.0909587107
56. Kalams SA, Parker SD, Elizaga M, Metch B, Edupuganti S, Hural J, et al. Safety and comparative immunogenicity of an HIV-1 DNA vaccine in combination with plasmid interleukin 12 and impact of intramuscular electroporation for delivery. J Infect Dis (2013) 208:818–29. doi: 10.1093/infdis/jit236
57. Trimble CL, Morrow MP, Kraynyak KA, Shen X, Dallas M, Yan J, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet (2015) 386:2078–88. doi: 10.1016/S0140-6736(15)00239-1
58. Griffin BD, Muthumani K, Warner BM, Majer A, Hagan M, Audet J, et al. DNA Vaccination protects mice against zika virus-induced damage to the testes. Nat Commun (2017) 8:15743. doi: 10.1038/ncomms15743
59. Xu K, Rockx B, Xie Y, Debuysscher BL, Fusco DL, Zhu Z, et al. Crystal structure of the hendra virus attachment G glycoprotein bound to a potent cross-reactive neutralizing human monoclonal antibody. PloS Pathog (2013) 9:e1003684. doi: 10.1371/journal.ppat.1003684
60. Baseler L, Scott DP, Saturday G, Horne E, Rosenke R, Thomas T, et al. Identifying early target cells of nipah virus infection in Syrian hamsters. PloS Negl Trop Dis (2016) 10:e0005120. doi: 10.1371/journal.pntd.0005120
61. Dhondt KP, Horvat B. Henipavirus infections: Lessons from animal models. Pathogens (2013) 2:264–87. doi: 10.3390/pathogens2020264
62. Mclean RK, Graham SP. Vaccine development for nipah virus infection in pigs. Front Vet Sci (2019) 6:16. doi: 10.3389/fvets.2019.00016
63. Mire CE, Versteeg KM, Cross RW, Agans KN, Fenton KA, Whitt MA, et al. Single injection recombinant vesicular stomatitis virus vaccines protect ferrets against lethal nipah virus disease. Virol J (2013) 10:353. doi: 10.1186/1743-422X-10-353
64. Debuysscher BL, Scott D, Marzi A, Prescott J, Feldmann H. Single-dose live-attenuated nipah virus vaccines confer complete protection by eliciting antibodies directed against surface glycoproteins. Vaccine (2014) 32:2637–44. doi: 10.1016/j.vaccine.2014.02.087
65. Gomez Roman R, Tornieporth N, Cherian NG, Shurtleff AC, L’azou Jackson M, Yeskey D, et al. Medical countermeasures against henipaviruses: a review and public health perspective. Lancet Infect Dis (2022) 22:e13–27. doi: 10.1016/S1473-3099(21)00400-X
66. Kudchodkar SB, Choi H, Reuschel EL, Esquivel R, Jin-Ah Kwon J, Jeong M, et al. Rapid response to an emerging infectious disease - lessons learned from development of a synthetic DNA vaccine targeting zika virus. Microbes Infect (2018) 20:676–84. doi: 10.1016/j.micinf.2018.03.001
67. Jeyanathan M, Afkhami S, Smaill F, Miller MS, Lichty BD, Xing Z. Immunological considerations for COVID-19 vaccine strategies. Nat Rev Immunol (2020) 20:615–32. doi: 10.1038/s41577-020-00434-6
68. Krammer F. SARS-CoV-2 vaccines in development. Nature (2020) 586:516–27. doi: 10.1038/s41586-020-2798-3
69. Wang X, Ge J, Hu S, Wang Q, Wen Z, Chen H, et al. Efficacy of DNA immunization with f and G protein genes of nipah virus. Ann N Y Acad Sci (2006) 1081:243–5. doi: 10.1196/annals.1373.029
70. Nie J, Liu L, Wang Q, Chen R, Ning T, Liu Q, et al. Nipah pseudovirus system enables evaluation of vaccines in vitro and in vivo using non-BSL-4 facilities. Emerg Microbes Infect (2019) 8:272–81. doi: 10.1080/22221751.2019.1571871
71. Liew YJM, Ibrahim P, Ong HM, Chong CN, Tan CT, Schee JP, et al. The immunobiology of nipah virus. Microorganisms (2022) 10. doi: 10.3390/microorganisms10061162
Keywords: Nipah virus, synthetic DNA vaccine, immune response, electroporation, neutralization
Citation: Choi H, Kudchodkar SB, Xu Z, Ho M, Xiao P, Ramos S, Humeau L, Weiner DB and Muthumani K (2022) Elicitation of immune responses against Nipah virus by an engineered synthetic DNA vaccine. Front. Virol. 2:968338. doi: 10.3389/fviro.2022.968338
Received: 13 June 2022; Accepted: 13 October 2022;
Published: 10 November 2022.
Edited by:
Fan Zhou, University of Bergen, NorwayReviewed by:
Glenn Marsh, Commonwealth Scientific and Industrial Research Organisation (CSIRO), AustraliaCopyright © 2022 Choi, Kudchodkar, Xu, Ho, Xiao, Ramos, Humeau, Weiner and Muthumani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kar Muthumani, a211dGh1bWFuaUBnbWFpbC5jb20=
†Present addresses: Sagar B. Kudchodkar, GeneOne Life Science, Inc. Seoul, South Korea
Kar Muthumani, GeneOne Life Science, Inc. Seoul, South Korea
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.