 Andrew Purssell1
Andrew Purssell1 Jonathan B. Angel
Jonathan B. Angel- 1Division of Infectious Diseases, Department of Medicine, University of Ottawa, Ottawa, ON, Canada
- 2The Ottawa Hospital Research Institute and Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, ON, Canada
Human immunodeficiency virus (HIV) infection remains a global epidemic. While antiretroviral therapy (ART) suppresses viral replication, cessation of ART results in viral rebound necessitating lifelong treatment. This is a result of a reservoir of latently infected cells, resistant to clearance by ART and the major obstacle in curing HIV. HIV cure strategies have focused on reactivating this latent reservoir with latency reversal agents (LRAs) along with enhancement of anti-HIV immunity to eliminate reactivated HIV. Retinoic acid (RA) derivatives are promising therapeutics that may promote clearance HIV latent reservoir allowing for definitive cure. In addition to plausible mechanisms for depleting the latent reservoir with LRA activity via the p300 acetyl transferase pathway, countering HIV-mediated suppression of RIG-I and IRF-3, and proposed induction of selective apoptosis of HIV-infected cells via RIG-I, RA may also limit HIV spread by augmenting cellular trafficking via CCR7 and CCR9 and induce accumulation of high-affinity effector CD8+ T cells that aid immune clearance of HIV-infected cells. Furthermore, due to their specificity for HIV-infected cells, retinoids are attractive agents to form the basis of multidrug regimens. Altogether, retinoids have many compelling properties as potential novel therapeutics in the cure of HIV.
Human immunodeficiency virus and antiretroviral therapy
Since the early 1980s, more than 35 million people have died of HIV or acquired immune deficiency syndrome. The World Health Organization estimates 37.7 million people currently living with HIV, with 1.5 million more infected last year (1). After the advent of effective antiretroviral therapy (ART) in the mid-1990s, HIV was transformed from a near universally fatal disease to a chronic one requiring life-long therapy to maintain health and prevent transmission. Despite its effectiveness in suppressing viral replication, ART does not eradicate HIV infection. Additionally, initiation and maintenance of lifelong ART remains a global challenge due to cost, stigma, and other important barriers to access. These limitations have spurred considerable effort in finding a cure.
Curing HIV requires eradication of the reservoir of HIV-infected cells that persist during ART (2). One cure strategy is the “Shock and Kill” approach, where active replication of HIV is induced in latently infected cells (i.e. reversing latency), making them more susceptible to immune-mediated clearance, ART-mediated clearance, or apoptotic pathways. Other cure strategies include: 1) “Block and Lock”, using latency promoting agents (LPAs) to drive HIV-infected cells into deeper latency to permanently suppresses viral transcription (3), 2) Introduction of an HIV-resistant T cell population via hematopoietic stem cell transplant (4), and 3) Immunotherapy including immune checkpoint inhibitors (5), Toll-like receptor agonists (6), and broadly neutralizing antibodies (7).
The “Shock and Kill” approach requires two steps: 1) “Shock” or latency reversal via aptly named Latency Reversal Agents (LRAs), and 2) “Kill” or clearance of the reactivated HIV-infected cells. Many potentially useful drug classes have been identified as LRAs (3, 8–10), but there has been limited success in delivering the subsequent kill. One novel class that may possess both “Shock” and “Kill” activities are retinoids, derivatives of Vitamin A which perform important roles in immune homeostasis and modulation affecting T cell signaling, homing, and differentiation. In this review we will explore how the pleiotropic immune effects of retinoids intersects with HIV pathogenesis, and how retinoids can enhance intrinsic antiviral mechanisms to inform potential therapeutic applications of retinoids in HIV cure.
Natural functions of retinoids
Retinoids are defined as Vitamin A and its natural or synthetic analogues that have 4 isoprenoid units linked sequentially head to tail (11, 12). Humans ingest retinoids as retinyl esters or carotenoids (most importantly β-carotene) via intestinal epithelial cells. Vitamin A is then esterified and transported to the liver or target tissues packaged in chylomicrons. At these sites hydrolysis releases retinol which is taken up by the cell. There retinol is hydrolyzed to form retinaldehyde by alcohol dehydrogenase. Retinoic acid (RA), the active metabolite, is then formed by retinaldehyde dehydrogenase which is only present in certain cell types, allowing for restriction of RA activity. RA can then be transported to the nucleus and promote expression of RA-target genes via nuclear Retinoic Acid Receptors (RARs) and retinoid X receptors acting on specific RA response elements (RARE) (13). Vitamin A deficiency has detrimental effects on: stem cell differentiation, embryogenesis, epithelial cell development and barrier function, and innate and acquired immune cell function (12), while excessive Vitamin A can have toxic effects (14, 15). The impact of RA on immune function is complex, often paradoxical, and determined by local concentration, cell types involved, and expression of receptors, cofactors, and other cytokines. RA can therefore have both anti- and pro-inflammatory functions facilitating nuanced control of immunity (13).
Retinoids as therapeutics
Retinoids have long been used in treatment of malignancies (15–18) and dermatological diseases (19–26). All trans retinoic acid (ATRA) (t1/2 0.5-2h) is used in high-dose pulsed regimens to treat acute promyelocytic leukemia (16). Acitretin (t1/2 49h) is effectively used to treat chronic conditions such as psoriasis (20, 23, 27). Moreover, acitretin has been used safely to treat psoriasis in HIV-infected individuals (19, 22, 26). Retinoids have a long record of use, well-documented safety data, and pharmacodynamic flexibility, making them attractive targets for novel therapeutic applications.
Pleiotropic interplay between retinoids and HIV-1 infection
Modulation of CD4+ T cell activity
HIV infection of CD4+ T cell occurs when virions bind to CD4 on the cell surface followed by an interaction with chemokine receptors CCR5 or CXCR4 (28) which act as required co-receptors for viral entry. In CCR6+ T cells, which contribute to mucosal immunity in the gut, exposure to ATRA increases expression of CCR5 and CXCR4 and may lead to increased local permissiveness of HIV infection (29). Another co-receptors that acts as a direct or indirect target for HIV cellular infection is integrin α4β7, whose expression is also upregulated in response to RA (30).
RA has been shown to affect the balance between Th1 and Th2 CD4+ T cells, in a manner dependent on their stage of development. In naïve T cells, RA favors Th2 over Th1 polarization (31), yet, in already polarized Th1 effector cells RA prevents conversion into other T helper subtypes. The effect of RA can be augmented by local cytokines including TGF-β, IL-12, and IL-27 produced by antigen presenting cells, converting CD4+ T helper cells into unique MHC class-II restricted cytotoxic T lymphocytes (CTL) thought to be important for maintaining intestinal mucosal barrier function (32).
RA induces accumulation of high-affinity CD8+ T cells at the mucosal barrier
In a similar paradigm to RA-induced CD4+ MHC class-II restricted CTLs, RA fosters selective accumulation of the most effective CD8+ T cells at the mucosal barrier. In mice, exposure to ATRA during vaccination with a replication-defective recombinant adenovirus vector expressing LCMV glycoprotein (LCMVgp) led to increased effector and memory CD8+ T cells and conferred resistance against further challenge with recombinant vaccinia virus expressing LCMVgp (33). This suggests RA can act as an adjuvant enhancing CTL responses to previously encountered viral epitopes. Exogenous RA may promote a more robust CD8+ T cell response against HIV at mucosal barriers in particular – a critical site of viral reactivation and replication.
RA promotes migration and sequestration of effector CD4+ T cells to the mucosal barrier
Central memory (CM) CD4+ T cells express CCR7 as a homodimer and a heterodimer with CXCR4. CCR7 facilitates migration of T cells to lymph nodes. Viral gp120 expression in HIV-infected cells enhances this migration, promoting escape from the site of initial infection and systemic propagation of HIV (34, 35). In addition, sequestering T cells inside lymph nodes prevents them from acting as effector cells at the mucosal barrier, impairing local mucosal immune response (36). In an in vitro model, dendritic cells expressing CXCR4 and CCR7 exposed to retinoids were prevented from migrating towards CCL19, the chemokine responsible for lymph node homing (37). The mechanism appears to be dual inhibition of CXCR4 and CCR7, suggesting retinoids may prevent migration of HIV-infected T cells to central immune tissue. Similarly, mucosal T cell gut-homing receptor CCR9 is induced in the presence of RA (38). Notably in central lymphoid tissue such as the spleen, the concentration of RA is low, preventing re-trafficking of T cells back to mucosal surfaces (39). Together, these data suggest that supplementation with exogenous retinoids could both induce migration of T cells to mucosal tissue via CCR9, and sequester them at the mucosal barrier by suppressing CXCR4 and CCR7-induced migration, limiting HIV dissemination and improving barrier competency.
In chronic HIV infection, HIV disrupts normal mucosal barrier function by depleting Th17 CCR6+ T cells, inducing anergy against HIV infected cells, and interfering with anti-inflammatory mechanisms leading a pro-inflammatory state favorable to HIV replication (29). Clearly, local physiologic production of RA is insufficient to control HIV infection. However, treatment with exogenous retinoids achieving supraphysiologic concentrations could help restore mucosal immunity via accumulation of high-affinity CD8+ T cells at the mucosal barrier, induction of unique MHC class-II restricted cytotoxic T lymphocytes (CTL), and trafficking of T cells to mucosal tissue that altogether may enhance clearance of reactivated HIV.
RIG-I intracellular immunity is abrogated by HIV
Innate immunity is the first line of defense against infection. It is activated after detection of conserved pathogen-associated molecular patterns by pattern-recognition receptors (PRRs). Multiple PRRs are expressed to effectively survey the interstitial environment and the various intracellular compartments. One family are the retinoic acid-inducible gene-I (RIG-I)-like receptors, named for the prototypical member RIG-I, whose expression is induced by retinoic acid (40). RIG-I serves as a cytoplasmic sensor, detecting a variety of RNA viruses, and initiates a signaling cascade resulting in Type 1 interferon production and antiviral gene expression.
Activated RIG-I functions through two signaling pathways: via TRAF6 resulting in NF-κB activation through the canonical pathway and via TRAF3 leading to activation of IRF3/7. These effectors, along with ATF2, c-jun, and p300/CBP coordinate interferon and pro-inflammatory gene expression and increase expression of RIG-1, allowing for positive feedback (41). Separate from its function as a transcriptional activator, IRF3 can trigger apoptosis via the RLR-induced IRF3-mediated pathway of apoptosis (RIPA) (42).
HIV disrupts RIG-I signaling by decreasing stability of IRF3 through a Vpr-induced ubiquination leading to proteosome degradation (43), yet NF-κB signaling remains intact and capable of driving HIV transcription. RIG-I signaling is also inhibited by HIV via sequestration dependent on the HIV protease, specifically by shuttling RIG-I into the lysosomal compartment for degradation (44). However, HIV viral production is significantly decreased if IRF3 production is restored, suggesting that RIG-I mediated intracellular immunity is capable of controlling HIV (45). Overcoming HIV-mediated attenuation of IRF3 signaling may be critical to controlling HIV infection.
Acitretin induces reactivation of HIV
Activated NF-κB drives HIV-1 transcription through multiple mechanisms. NF-κB can directly bind to regions of the HIV 5’LTR and induce HIV transcription through a cooperative interaction with transcription factor Sp1 (46, 47). In latency, p50/p50 homodimers are bound to the HIV-1 5’ LTR and recruit HDAC-1 resulting in histone deacetylation and chromatin condensation which silences HIV gene expression (48). Activated NF-κB p50/RelA heterodimers displace the p50/p50 homodimer and instead recruit histone acetyl-transferases including p300/CBP allowing for transcription of HIV via histone acetylation (49). This leads to increased expression of HIV-encoded Tat which aids in elongation of the HIV transcript by binding to the TAR sequence of HIV transcripts and recruiting the P-TEFb complex (50); this complex phosphorylates the C-terminal domain of RNA Pol II which increases processivity (51) and phosphorylates DSIF and NELF which removes the block to elongation (52). Tat can also be acetylated at two sites including lysine 50 by CBP/p300, which allows for a switch from early to late phase in HIV transcription elongation (53). As such, NF-κB stimulators are considered a potent class of LRAs. Their clinical utility is limited by a lack of specificity to HIV-infected cells and the potential for severe adverse reactions including life-threatening systemic inflammatory responses from global NF-κB activation (3).
In Li et al., latently HIV infected cell line ACH-2 treated with acitretin or vorinostat, a histone deacetylase inhibitor (HDACi), for 72 hours caused significant HIV reactivation (54). The acitretin-mediated reactivation was blocked by addition of curcumin, an inhibitor of p300. Increased HIV RNA expression was also observed after TZM-bl cells actively infected with GM-HIV (a gag-mutated NL4-3 HIV-1 clone) were treated with acitretin. Western blot analysis revealed increased p300 expression. These data indicate that reactivation of HIV occurs through stimulation of the RIG-I pathway. However, increased HIV expression in acitretin-treated cells was partially maintained in a RIG-I shRNA knockdown, so acitretin may drive expression of HIV through a pathway independent of RIG-I. Garcia-Vidal et al. did not observe significant HIV reactivation in acitretin-treated J-Lat cells [54], but a lack of reactivation after treatment with the potent LRA vorinostat [55, 56] in this study suggests that those results may be due to aspects of their experimental system.
Planas et al. examined the effect of ATRA on Th17-polarized CD4+ T cells, which are important in maintaining the gut barrier (55), and constitute a key reservoir of latent HIV infection (56). The CCR6+ subset of these cells demonstrated increased permissiveness to HIV infection. Stimulation of CCR6+ CD4+ T cells taken from HIV-infected individuals on ART with ATRA led to HIV reactivation (57). The effect was significantly decreased when the cells were treated with INK128, an inhibitor of mTOR1/2, indicating the effect is at least partly mTOR dependent.
Retinoids enhance RIG-I mediated control of HIV infection
Li et al. (54) demonstrated that treatment with acitretin decreased HIV DNA levels in infected cells, reflecting clearance of the reservoir. Specifically, treatment of GM-HIV-infected TZM-bl cells with acitretin resulted in undetectable GM-HIV DNA levels. In an ex vivo model of CD4+ T cells derived from 12 HIV-infected ART-suppressed subjects, acitretin resulted in a significant decrease in HIV DNA levels, which was enhanced by addition of vorinostat suggesting synergy between the two pathways. Analysis of acitretin-treated HIV-infected cells revealed a relative increase of apoptotic cells and caspase-3 activity, indicating that depletion of HIV DNA was at least partly due to acitretin-induced apoptosis of infected cells. Further testing on the TZN-bl cells revealed that acitretin treatment led to increased expression of RIG-I, phosphorylated and non-phosphorylated IRF3, and increased Bax suggesting that depletion of detectable HIV DNA occurred through stimulation of the RIG-I pathway and RIPA-mediated apoptosis. In HIV-uninfected cells, acitretin induced significantly less upregulation of these genes, and cell apoptosis rates were unchanged, suggesting the presence of viral RNA is critical to activation of the enhanced RIG-I pathway and that off-target effects could be minimal in uninfected cells.
Garcia-Vidal et al. (58) examined the effect of acitretin on multiple T cell lines and primary T cells infected with the HIV clone NL4-3 HIG, demonstrating that acitretin could induce increased RIG-I and phosphorylated IRF3, but that this effect was minimal. In J-Hig cells (J-Lat cells latently infected with NL4-3 HIG) the percentage of apoptotic cells was determined after treatment with increasing acitretin concentrations. In this model, the percentage of apoptosis in the cells where the HIV persisted in latency remained unchanged, and, in cells where HIV reactivated, only a small proportion underwent apoptosis. This called into question if acitretin was an effective enhancer of RIG-I mediated HIV clearance.
There are some important differences between the experiments in these studies that may explain the discrepancy. First, the HIV clone used in Garcia-Vidal contained a nef-deletion. HIV-1 Nef interacts with several intracellular proteins involved in kinase signaling pathways and endosomal trafficking. Through interaction with the Itk kinase it activates phospholipase C, eventually resulting in activation of NFAT and NF-κB, both of which drive HIV-1 transcription at the 5’ LTR (59). Loss of Nef could lead to reduction in HIV-1 proviral transcription and failure to activate the primed RIG-I system, avoiding further RIG-I mediated NF-κB activation and RIPA-mediated apoptosis. Furthermore, the apoptosis experiments in Garcia-Vidal examined HIV reactivation and apoptosis together. If acitretin’s latency reversal activity is more modest, as their study suggests, then it is difficult to infer the contribution of acitretin to RIG-I mediated induction of apoptosis. In Li et al., the most dramatic effect of acitretin occurred in ex vivo T cells derived from (nef intact) HIV-infected individuals fully suppressed on ART (54). Subsequent work by Planas et al. showed that RIG-I expression was induced in CCR6 + and CRR6 - T cells in the presence of ATRA, but impact on clearance of HIV-infected cells was not explored (57). Further study is required to illuminate how acitretin’s enhancement of RIG-I functions in vivo and in the presence of established HIV infection.
Potential HIV cure shocktails
To date no approach, let alone a single agent, has been shown to effectively purge the latent HIV reservoir in established infection. Since HIV latency is a multifactorial process, it may be that only multi-drug regimens, which we have termed shocktails, will be able to sufficiently reverse latency (60). The challenge is identifying agents that act synergistically to promote HIV latency reversal and immune clearance with minimal toxicity. Several reviews have been written about the numerous compounds that are being studied as potential cure agents (3), all of which are limited by their lack of specificity; for example, the NF-κB stimulators like prostratin cause global transcriptional activation leading to significant toxicity. Retinoids, in contrast, enhance RIG-I mediated intracellular immunity partly through NF-κB activation which is triggered only in the presence of viral products, for exquisite specificity. This specificity for infected cells makes retinoids attractive agents as the backbone of cure shocktails. By understanding the molecular biology of retinoids, we can speculate about which other candidate compounds could be added to a retinoid-based shocktail as summarized in Figure 1.
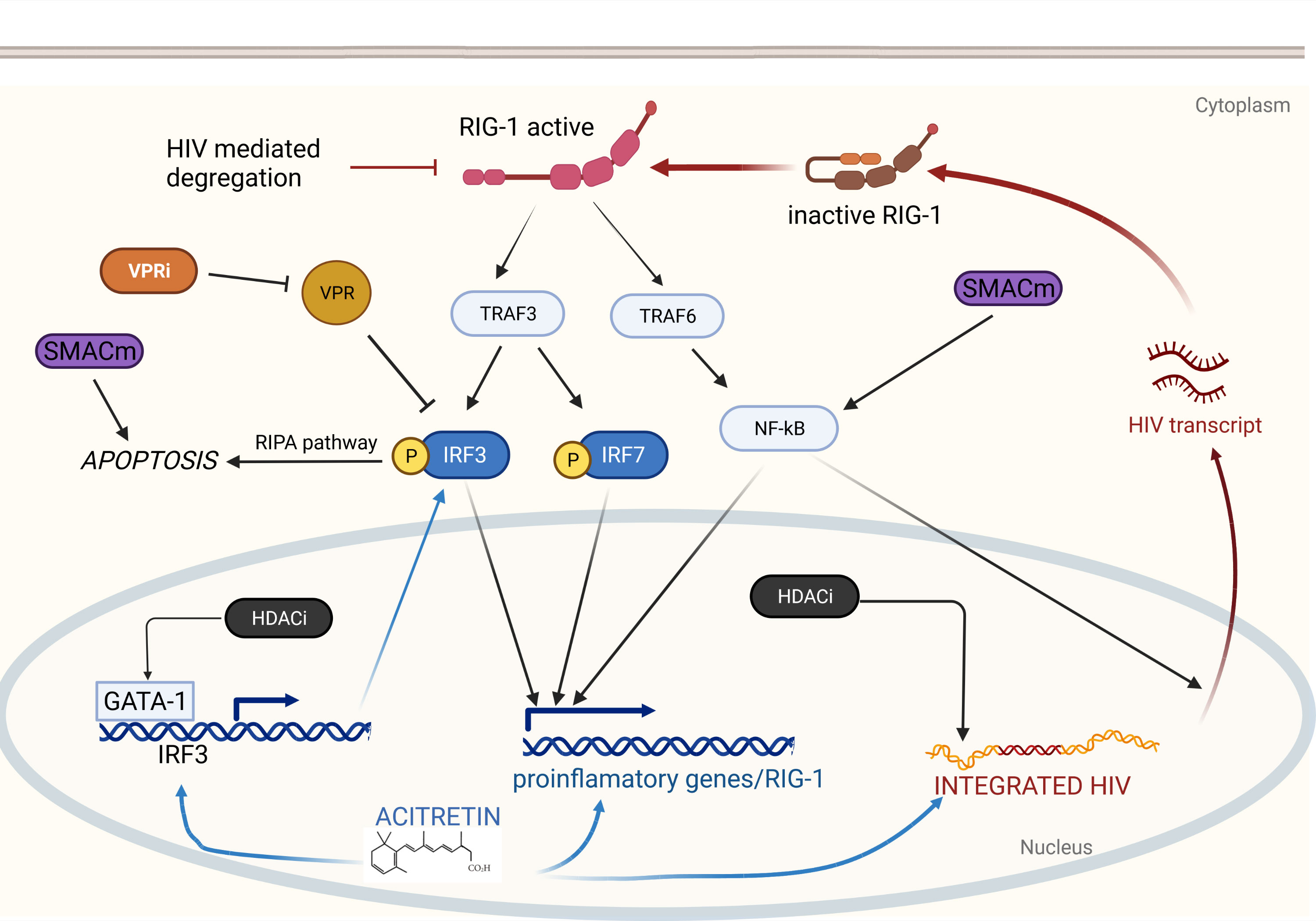
Figure 1 Retinoids induce HIV reactivation and enhance immunity via RIG-I. RIG-I detects intracellular HIV RNA resulting in TRAF3 and TRAF6 activation. TRAF6 elicits a signaling cascade releasing NF-κB which translocates to the nucleus and drives proinflammatory gene expression and HIV transcription. TRAF3 induces phosphorylation of IRF3 and IRF7 which also drive proinflammatory gene expression. Phosphorylated IRF3 can also induce apoptosis via the RIPA pathway. Acitretin enhances expression of IRF3, RIG-I, and proinflammatory genes as well as HIV transcription leading to reactivation of latent HIV infection and immune mediated clearance. Combination with additional therapeutics may enhance acitretin activity allowing for HIV eradication. HDACi increase HIV expression leading to further stimulation of the RIG-I pathway and increase expression IRF3 potentially overcoming HIV-mediated inhibition. VPR inhibitors may directly prevent VPR-mediated suppression of IRF3. SMACm increase NF-κB expression stimulating HIV transcription which can then activate RIG-I. SMACm are also pro-apoptotic and may function synergistically with the RIPA pathway.
HDACi, including FDA-approved agents vorinostat, romidepsin, panobinostat, and valproic acid have all been shown to induce HIV reactivation in vivo (61–64), but have dose-dependent side effects that limit their application. Enhanced clearance of latently-infected cells treated with both vorinostat and acitretin has been observed in vitro and could be explained by stimulation of the RIG-I pathway by HDACi-induced HIV transcription (54). Another HDACi, Trichostatin A (65) was shown to induce expression of IRF-3 when combined with valproic acid (66), which could counter HIV Vpr-mediated IRF-3 depletion and restore RIG-I activity. It remains unclear if increasing IRF-3 expression is a property shared by other HDACi.
Vpr inhibitors are attractive potential agents to combine with retinoids. In addition to IRF-3 suppression, Vpr impacts host cell functions including energy metabolism, oxidative status, and proteasome function that are involved in HIV pathogenesis (67). Vpr inhibition is an attractive target for future therapies, and several potential molecules have been isolated that inhibit Vpr in vitro (68).
Inhibitors of apoptosis (IAPs) prevent apoptosis directly, where XIAP binds and inhibits caspases, or indirectly, where cIAP1/2 prevents the assembly of pro-apoptotic signaling complexes. cIAPs also act as negative regulators of canonical and non-canonical IKKα-dependent NF-κB pathways. SMAC mimetics (SMACm) (69) suppress cIAPs and XIAP leading to NF-κB activation and disinhibition of cell death. SMACm have been shown in vitro and in vivo to induce robust HIV reactivation and exhibit synergy with the HDACi panobinostat (70, 71). Furthermore, compared to prior NF-κB stimulators, SMACm appear to be well tolerated in animal models; the SMACm AZD5582 was shown to reverse latency in SIV infected rhesus macques without inducing global CD4+ T cell activation or significant toxicity (71). By activating the non-canonical IKKα-dependent NF-κB pathway, SMACm could potentially work synergistically with retinoid-induced RIG-I activation via the NF-κB canonical pathway. Since, the synergistic activation would likely be restricted to HIV infected cells, it is expected that the combination would be tolerated. However, there remains theoretical potential for toxicity.
Retinoids: future directions in HIV cure research
Eradicating the latently infected reservoir remains the major hurdle in curing HIV. Retinoids have potent latency reversal activity and are capable of purging HIV-infected cells through enhanced RIG-I-mediated immunity and RIPA-induced apoptosis. Retinoids also influence many aspects of the immune system; including enriching high-affinity CD8+ CTLs with potential enhanced anti-HIV properties, re-trafficking of CD4+ and CD8+ T cells toward depleted mucosal barriers, and halting central spread of HIV. While there is concern that retinoids may increase local permissiveness of HIV infection, enhancement of immune clearance or combination with ongoing ART may render this irrelevant. RA should be further explored as a potential therapy in the pursuit of a cure for HIV. The unique mechanism of retinoids in countering HIV infection and its apparent restriction to infected cells, suggest retinoids could form the backbone of potential shocktails. Given its robust safety data, further research assessing retinoids alone and in combination with other “Shock and Kill” candidates in human trials is the next step in determining the place of retinoids in the arsenal for HIV cure.
Author contributions
AP - first author, conceptualize the review, manuscript preparation, literature search. MM - manuscript preparation. BV - figure preparation, manuscript revisions. JA - corresponding author, manuscript revisions. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. HIV/Aids: World Health Organization. (2020). Available at: https://www.who.int/HIV/data/en/.
2. Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science (1997) 278(5341):1295–300. doi: 10.1126/science.278.5341.1295
3. Abner E, Jordan A. HIV "Shock and kill" therapy: In need of revision. Antiviral Res (2019) 166:19–34. doi: 10.1016/j.antiviral.2019.03.008
4. Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med (2009) 360(7):692–8. doi: 10.1056/NEJMoa0802905
5. Porichis F, Kaufmann DE. Role of pd-1 in HIV pathogenesis and as target for therapy. Curr HIV/AIDS Rep (2012) 9(1):81–90. doi: 10.1007/s11904-011-0106-4
6. Vibholm L, Schleimann MH, Hojen JF, Benfield T, Offersen R, Rasmussen K, et al. Short-course toll-like receptor 9 agonist treatment impacts innate immunity and plasma viremia in individuals with human immunodeficiency virus infection. Clin Infect Dis (2017) 64(12):1686–95. doi: 10.1093/cid/cix201
7. Lu CL, Murakowski DK, Bournazos S, Schoofs T, Sarkar D, Halper-Stromberg A, et al. Enhanced clearance of HIV-1-Infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science (2016) 352(6288):1001–4. doi: 10.1126/science.aaf1279
8. Kim Y, Anderson JL, Lewin SR. Getting the "Kill" into "Shock and kill": Strategies to eliminate latent HIV. Cell Host Microbe (2018) 23(1):14–26. doi: 10.1016/j.chom.2017.12.004
9. Rasmussen TA, Sogaard OS. Clinical interventions in HIV cure research. Adv Exp Med Biol (2018) 1075:285–318. doi: 10.1007/978-981-13-0484-2_12
10. Shang HT, Ding JW, Yu SY, Wu T, Zhang QL, Liang FJ. Progress and challenges in the use of latent HIV-1 reactivating agents. Acta Pharmacol Sin (2015) 36(8):908–16. doi: 10.1038/aps.2015.22
11. Sporn MB, Dunlop NM, Newton DL, Henderson WR. Relationships between structure and activity of retinoids. Nature (1976) 263(5573):110–3. doi: 10.1038/263110a0
12. Conaway HH, Henning P, Lerner UH. Vitamin a metabolism, action, and role in skeletal homeostasis. Endocr Rev (2013) 34(6):766–97. doi: 10.1210/er.2012-1071
13. Larange A, Cheroutre H. Retinoic acid and retinoic acid receptors as pleiotropic modulators of the immune system. Annu Rev Immunol (2016) 34:369–94. doi: 10.1146/annurev-immunol-041015-055427
14. de Die-Smulders CE, Sturkenboom MC, Veraart J, van Katwijk C, Sastrowijoto P, van der Linden E. Severe limb defects and craniofacial anomalies in a fetus conceived during acitretin therapy. Teratology (1995) 52(4):215–9. doi: 10.1002/tera.1420520407
15. Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer (2001) 1(3):181–93. doi: 10.1038/35106036
16. Conley BA, Egorin MJ, Sridhara R, Finley R, Hemady R, Wu S, et al. Phase I clinical trial of all-trans-retinoic acid with correlation of its pharmacokinetics and pharmacodynamics. Cancer Chemother Pharmacol (1997) 39(4):291–9. doi: 10.1007/s002800050575
17. Muindi JR, Frankel SR, Huselton C, DeGrazia F, Garland WA, Young CW, et al. Clinical pharmacology of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Cancer Res (1992) 52(8):2138–42.
18. Sporn MB, Roberts AB. Role of retinoids in differentiation and carcinogenesis. J Natl Cancer Inst (1984) 73(6):1381–7.
19. Buccheri L, Katchen BR, Karter AJ, Cohen SR. Acitretin therapy is effective for psoriasis associated with human immunodeficiency virus infection. Arch Dermatol (1997) 133(6):711–5. doi: 10.1001/archderm.1997.03890420043005
20. Dogra S, Jain A, Kanwar AJ. Efficacy and safety of acitretin in three fixed doses of 25, 35 and 50 mg in adult patients with severe plaque type psoriasis: A randomized, double blind, parallel group, dose ranging study. J Eur Acad Dermatol Venereol (2013) 27(3):e305–11. doi: 10.1111/j.1468-3083.2012.04644.x
21. Goldfarb MT, Ellis CN, Gupta AK, Tincoff T, Hamilton TA, Voorhees JJ. Acitretin improves psoriasis in a dose-dependent fashion. J Am Acad Dermatol (1988) 18(4 Pt 1):655–62. doi: 10.1016/S0190-9622(88)70086-9
22. Jeong YS, Kim MS, Shin JH, Cho JK, Lee HI, Kim HJ, et al. A case of severe HIV-associated psoriasis successfully treated with acitretin therapy. Infect Chemother (2014) 46(2):115–9. doi: 10.3947/ic.2014.46.2.115
23. Kingston TP, Matt LH, Lowe NJ. Etretin therapy for severe psoriasis. evaluation of initial clinical responses. Arch Dermatol (1987) 123(1):55–8. doi: 10.1001/archderm.1987.01660250061018
24. Leal L, Ribera M, Dauden E. [Psoriasis and HIV infection]. Actas Dermosifiliogr (2008) 99(10):753–63. doi: 10.1016/S0001-7310(08)74955-X
25. Nakamura M, Abrouk M, Farahnik B, Zhu TH, Bhutani T. Psoriasis treatment in HIV-positive patients: A systematic review of systemic immunosuppressive therapies. Cutis (2018) 101(1):38;42;56.
26. Valenzuela F, Fernandez J, Sanchez M, Zamudio A. Erythrodermic psoriasis and human immunodeficiency virus: Association and therapeutic challenges. Bras Dermatol (2018) 93(3):438–40. doi: 10.1590/abd1806-4841.20187387
27. Wiegand UW, Chou RC. Pharmacokinetics of acitretin and etretinate. J Am Acad Dermatol (1998) 39(2 Pt 3):S25–33. doi: 10.1016/S0190-9622(98)70441-4
28. Wilen CB, Tilton JC, Doms RW. HIV: Cell binding and entry. Cold Spring Harb Perspect Med (2012) 2(8):1–13. doi: 10.1101/cshperspect.a006866
29. Monteiro P, Gosselin A, Wacleche VS, El-Far M, Said EA, Kared H, et al. Memory CCR6+CD4+ T cells are preferential targets for productive HIV type 1 infection regardless of their expression of integrin Beta7. J Immunol (2011) 186(8):4618–30. doi: 10.4049/jimmunol.1004151
30. Cicala C, Martinelli E, McNally JP, Goode DJ, Gopaul R, Hiatt J, et al. The integrin Alpha4beta7 forms a complex with cell-surface CD4 and defines a T-cell subset that is highly susceptible to infection by HIV-1. Proc Natl Acad Sci USA. (2009) 106(49):20877–82. doi: 10.1073/pnas.0911796106
31. Cantorna MT, Nashold FE, Hayes CE. In vitamin a deficiency multiple mechanisms establish a regulatory T helper cell imbalance with excess Th1 and insufficient Th2 function. J Immunol (1994) 152(4):1515–22.
32. Mucida D, Husain MM, Muroi S, van Wijk F, Shinnakasu R, Naoe Y, et al. Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat Immunol (2013) 14(3):281–9. doi: 10.1038/ni.2523
33. Tan X, Sande JL, Pufnock JS, Blattman JN, Greenberg PD. Retinoic acid as a vaccine adjuvant enhances CD8+ T cell response and mucosal protection from viral challenge. J Virol (2011) 85(16):8316–27. doi: 10.1128/JVI.00781-11
34. Riou C, Yassine-Diab B, Van grevenynghe J, Somogyi R, Greller LD, Gagnon D, et al. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J Exp Med (2007) 204(1):79–91. doi: 10.1084/jem.20061681
35. Hayasaka H, Kobayashi D, Yoshimura H, Nakayama EE, Shioda T, Miyasaka M. The hiv-1 Gp120/CXCR4 axis promotes CCR7 ligand-dependent CD4 T cell migration: CCR7 homo- and CCR7/CXCR4 hetero-oligomer formation as a possible mechanism for up-regulation of functional CCR7. PloS One (2015) 10(2):e0117454. doi: 10.1371/journal.pone.0117454
36. Bucy RP, Hockett RD, Derdeyn CA, Saag MS, Squires K, Sillers M, et al. Initial increase in blood CD4(+) lymphocytes after HIV antiretroviral therapy reflects redistribution from lymphoid tissues. J Clin Invest (1999) 103(10):1391–8. doi: 10.1172/JCI5863
37. Villablanca EJ, Zhou D, Valentinis B, Negro A, Raccosta L, Mauri L, et al. Selected natural and synthetic retinoids impair CCR7- and CXCR4-dependent cell migration in vitro and in vivo. J Leukoc Biol (2008) 84(3):871–9. doi: 10.1189/jlb.0108047
38. Mora JR, von Andrian UH. Retinoic acid: An educational "Vitamin elixir" for gut-seeking T cells. Immunity (2004) 21(4):458–60. doi: 10.1016/j.immuni.2004.10.002
39. Takeuchi H, Yokota A, Ohoka Y, Iwata M. Cyp26b1 regulates retinoic acid-dependent signals in T cells and its expression is inhibited by transforming growth factor-beta. PloS One (2011) 6(1):e16089. doi: 10.1371/journal.pone.0016089
40. Huang SL, Shyu RY, Yeh MY, Jiang SY. Cloning and characterization of a novel retinoid-inducible gene 1(RIG1) deriving from human gastric cancer cells. Mol Cell Endocrinol (2000) 159(1-2):15–24. doi: 10.1016/s0303-7207(99)00207-5
41. Liu Y, Olagnier D, Lin R. Host and viral modulation of RIG-I-mediated antiviral immunity. Front Immunol (2016) 7:662. doi: 10.3389/fimmu.2016.00662
42. Chattopadhyay S, Sen GC. Rig-I-Like receptor-induced IRF3 mediated pathway of apoptosis (Ripa): A new antiviral pathway. Protein Cell (2017) 8(3):165–8. doi: 10.1007/s13238-016-0334-x
43. Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM. Hiv-1 accessory proteins Vpr and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology (2008) 373(1):85–97. doi: 10.1016/j.virol.2007.10.042
44. Solis M, Nakhaei P, Jalalirad M, Lacoste J, Douville R, Arguello M, et al. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of rig-I. J Virol (2011) 85(3):1224–36. doi: 10.1128/JVI.01635-10
45. Doehle BP, Hladik F, McNevin JP, McElrath MJ, Gale M Jr. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J Virol (2009) 83(20):10395–405. doi: 10.1128/JVI.00849-09
46. Kinoshita S, Chen BK, Kaneshima H, Nolan GP. Host control of HIV-1 parasitism in T cells by the nuclear factor of activated T cells. Cell (1998) 95(5):595–604. doi: 10.1016/s0092-8674(00)81630-x
47. Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J (1993) 12(9):3551–8. doi: 10.1002/j.1460-2075.1993.tb06029.x
48. Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB P50 promotes hiv latency through HDAC recruitment and repression of ttranscriptional initiation. EMBO J (2006) 25(1):139–49. doi: 10.1038/sj.emboj.7600900
49. Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/P300 or HDAC-1. Mol Cell (2002) 9(3):625–36. doi: 10.1016/s1097-2765(02)00477-x
50. Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated c-type cyclin interacts directly with hiv-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell (1998) 92(4):451–62. doi: 10.1016/s0092-8674(00)80939-3
51. Kim YK, Bourgeois CF, Isel C, Churcher MJ, Karn J. Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol Cell Biol (2002) 22(13):4622–37. doi: 10.1128/mcb.22.13.4622-4637.2002
52. Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol (2004) 24(2):787–95. doi: 10.1128/mcb.24.2.787-795.2004
53. Ott M, Dorr A, Hetzer-Egger C, Kaehlcke K, Schnolzer M, Henklein P, et al. Tat acetylation: A regulatory switch between early and late phases in HIV transcription elongation. Novartis Found Symp (2004) 259:182–93. doi: 10.1002/0470862637.ch13
54. Li P, Kaiser P, Lampiris HW, Kim P, Yukl SA, Havlir DV, et al. Stimulating the RIG-I pathway to kill cells in the latent HIV reservoir following viral reactivation. Nat Med (2016) 22(7):807–11. doi: 10.1038/nm.4124
55. Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med (2008) 14(4):421–8. doi: 10.1038/nm1743
56. Wong JK, Yukl SA. Tissue reservoirs of HIV. Curr Opin HIV AIDS (2016) 11(4):362–70. doi: 10.1097/COH.0000000000000293
57. Planas D, Zhang Y, Monteiro P, Goulet JP, Gosselin A, Grandvaux N, et al. Hiv-1 selectively targets gut-homing CCR6+CD4+ T cells via mTOR-dependent mechanisms. JCI Insight (2017) 2(15):1–21. doi: 10.1172/jci.insight.93230
58. Garcia-Vidal E, Castellvi M, Pujantell M, Badia R, Jou A, Gomez L, et al. Evaluation of the innate immune modulator acitretin as a strategy to clear the HIV reservoir. Antimicrob Agents Chemother (2017) 61(11):1–11. doi: 10.1128/AAC.01368-17
59. Staudt RP, Alvarado JJ, Emert-Sedlak LA, Shi H, Shu ST, Wales TE, et al. Structure, function, and inhibitor targeting of HIV-1 Nef-effector kinase complexes. J Biol Chem (2020) 295(44):15158–71. doi: 10.1074/jbc.REV120.012317
60. Ruelas DS, Greene WC. An integrated overview of HIV-1 latency. Cell (2013) 155(3):519–29. doi: 10.1016/j.cell.2013.09.044
61. Archin NM, Kirchherr JL, Sung JA, Clutton G, Sholtis K, Xu Y, et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J Clin Invest (2017) 127(8):3126–35. doi: 10.1172/JCI92684
62. Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV (2014) 1(1):e13–21. doi: 10.1016/S2352-3018(14)70014-1
63. Sogaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PloS Pathog (2015) 11(9):e1005142. doi: 10.1371/journal.ppat.1005142
64. Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, et al. Depletion of latent HIV-1 infection in vivo: A proof-of-Concept study. Lancet (2005) 366(9485):549–55. doi: 10.1016/S0140-6736(05)67098-5
65. Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J (1996) 15(5):1112–20. doi: 10.1002/j.1460-2075.1996.tb00449.x
66. Wang LL, Zhou LB, Shu J, Li NN, Zhang HW, Jin R, et al. Up-regulation of IRF-3 expression through GATA-1 acetylation by histone deacetylase inhibitor in lung adenocarcinoma A549 cells. Oncotarget (2017) 8(44):75943–51. doi: 10.18632/oncotarget.18371
67. Gonzalez ME. The HIV-1 Vpr protein: A multifaceted target for therapeutic intervention. Int J Mol Sci (2017) 18(1):1–21. doi: 10.3390/ijms18010126
68. Woo SY, Win NN, Noe Oo WM, Ngwe H, Ito T, Abe I, et al. Viral protein r inhibitors from Swertia chirata of Myanmar. J Biosci Bioeng (2019) 128(4):445–9. doi: 10.1016/j.jbiosc.2019.04.006
69. Molyer B, Kumar A, Angel JB. SMAC mimetics as therapeutic agents in HIV infection. Front Immunol (2021) 12:780400. doi: 10.3389/fimmu.2021.780400
70. Pache L, Dutra MS, Spivak AM, Marlett JM, Murry JP, Hwang Y, et al. BIRC2/cIAP1 is a negative regulator of hiv-1 transcription and can be targeted by Smac mimetics to promote reversal of viral latency. Cell Host Microbe (2015) 18(3):345–53. doi: 10.1016/j.chom.2015.08.009
Keywords: HIV - human immunodeficiency virus, retinoid acid, CD4, antiviral, apoptosis
Citation: Purssell A, McGuinty M, Vulesevic B and Angel JB (2022) Retinoids: novel potential therapeutics in the pursuit of HIV-1 cure. Front. Virol. 2:957124. doi: 10.3389/fviro.2022.957124
Received: 30 May 2022; Accepted: 18 July 2022;
Published: 09 August 2022.
Edited by:
Mayte Coiras, Instituto de Salud Carlos III (ISCIII), SpainReviewed by:
Nancie M. Archin, University of North Carolina Hospitals, United StatesTakeo Kuwata, Kumamoto University, Japan
Copyright © 2022 Purssell, McGuinty, Vulesevic and Angel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan B. Angel, amFuZ2VsQG9ocmkuY2E=