
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol., 14 September 2022
Sec. Viral Diversification and Evolution
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.935131
 Peter Mwangi1†
Peter Mwangi1† Javan Okendo2,3†
Javan Okendo2,3† Milton Mogotsi1
Milton Mogotsi1 Ayodeji Ogunbayo1
Ayodeji Ogunbayo1 Olusesan Adelabu1
Olusesan Adelabu1 Hlengiwe Sondlane1Makgotso Maotoana4
Hlengiwe Sondlane1Makgotso Maotoana4 Lutfiyya Mahomed4Molefi Daniel Morobadi4,5Sabeehah Vawda4,6Anne von Gottberg7,8
Lutfiyya Mahomed4Molefi Daniel Morobadi4,5Sabeehah Vawda4,6Anne von Gottberg7,8 Jinal Bhiman7,8Houriiyah Tegally9Eduan Wilkinson9
Jinal Bhiman7,8Houriiyah Tegally9Eduan Wilkinson9 Jennifer Giandhari9Sureshnee Pillay9Yeshnee Naidoo9
Jennifer Giandhari9Sureshnee Pillay9Yeshnee Naidoo9 Upasana Ramphal9Tulio de Oliveira9,10,11
Upasana Ramphal9Tulio de Oliveira9,10,11 Armand Bester4,6
Armand Bester4,6 Dominique Goedhals4,6,12
Dominique Goedhals4,6,12 Martin Nyaga1*
Martin Nyaga1*Since the COVID-19 outbreak emerged, SARS-CoV-2 has continuously evolved into variants with underlying mutations associated with increased transmissibility, potential escape from neutralizing antibodies, and disease severity. The SARS-CoV-2 pandemic in South Africa has been characterized by periods of infections with four major epidemic waves. To determine whether the variants driving the epidemic waves at the national level were also driving the epidemic waves at the local level, we performed analysis of a total of 1287 samples from qPCR confirmed SARS-CoV-2 positive individuals. The samples were subjected to viral RNA extraction, genomic amplification, and sequencing. Variant assignment of the viral sequences and mutation identification were conducted using PANGOLIN and SARS-CoV-2 genome annotator, respectively. Our analysis revealed that during the initial part of the first wave, B.1, B.1.1, B.1.1.53, B.1.1.448 and B.1.237 circulated in the Free State province, followed by Beta variant, B.1.351 later in the wave. Although most of the initially detected variants disappeared during the second wave, the Beta variant, B.1.351, persisted. Early in the third wave, the Beta variant, B.1.351, predominated but was replaced by the Delta sub-lineage, AY.45. The fourth wave was characterized by unique emergence of the Omicron sub-variant, BA.1. The data further indicates that SARS-CoV-2 variants driving the epidemic waves in the Free State at the local level correlated with the ones driving the epidemic waves at the national level. Findings from this study highlight the importance of continued genomic surveillance and monitoring of the circulating SARS-CoV-2 variants to inform public health efforts and ensure adequate control of the ongoing pandemic.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in Wuhan, China, in December 2019 and spread rapidly worldwide (1, 2). The SARS-CoV-2 is an enveloped RNA virus with a positive-sense, single-stranded RNA genome of approximately 30 kb (3). Coronavirus disease 2019 (COVID-19) is the clinical syndrome associated with SARS-CoV-2 and is characterized by respiratory or gastrointestinal symptoms. In severe cases, there is the potential for cardiovascular and neurological complications, thrombosis and renal failure (4, 5). Approximately 544 million COVID-19 cases with over 6.3 million deaths have been reported globally as of 27th June 2022 (https://covid19.who.int/). In Africa, the precise impact of the SARS-CoV-2 pandemic is not fully known (6), although 252,560 deaths have been confirmed so far with five countries, South Africa (100,147), Tunisia (28,509), Egypt (24,613), Morocco (16,063) and Ethiopia (7,509) reporting the largest number of deaths (https://covid19.who.int/) at the time of writing this work. The number of SARS-CoV-2 cases and deaths varies across the nine provinces of South Africa (Western Cape n = 700,090 (22,257), Gauteng n = 1,320,982 (20,973), Eastern Cape n = 363,529 (16, 847), KwaZulu-Natal n = 715, 652 (16,228), North West n = 201,653 (4,984), Limpopo n = 159, 429 (4,671), Northern Cape n = 115,122 (3,249), Mpumalanga n = 201,665 (4758)) with the Free State having 215, 721 (7,826) confirmed cases and deaths, respectively (www.sacoronavirus.co.za).
SARS-CoV-2 has complex anatomy, consisting of nonstructural proteins (nsps), structural proteins, and accessory proteins (7). The structural proteins are numbered from 1 to 16 (nsp1-16) while the structural proteins are also subdivided into spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins (7). Genomic investigations have been integral in SARS-CoV-2 surveillance and the Network for Genomic Surveillance South Africa (NGS-SA) consortium has been at the forefront in real-time tracking of the SARS-CoV-2 epidemiology (8). The inherent mutational ability of SARS-CoV-2 has led to multiple variants classified into four groups: variants of concern (VOC), variants of interest (VOI), variants being monitored (VBM) and variants of high consequence (VOHC) (www.cdc.gov). The SARS-CoV-2 variants are further classified by the use of the letters of the Greek alphabet e.g. Alpha, Beta, Delta Gamma, Iota, Kappa, Lambda, Omicron, etc. for easy-to-say labeling (www.who.int). Currently, three VBM’s (Alpha-B.1.1.7, Beta-B.1.351 and Gamma-P.1) and two VOC’s (Delta - B.617.2 including AY sub-lineages and Omicron - B.1.1.529 including BA lineages) are in circulation worldwide (www.cdc.gov). The Omicron variant has predominated over other variants globally (9). Viral evasion due to mutations in the spike protein region of the SARS-CoV-2 arising in immunocompromised patients with prolonged infection is hypothesized to contribute to the emergence of the global variants of concern (10–12).
The SARS-CoV-2 pandemic in South Africa has been characterized by periods of infections with four major epidemic waves. During the first wave of the epidemic (March 2020 to September 2020), a total of 42 phylogenetic lineages were identified with three lineages, B.1.1.54, B.1.1.56 and C.1, causing half of the infections (13). The second wave of the SARS-CoV-2 epidemic began around October 2020 to March 2021 characterized mainly by the Beta/501Y.V2 lineage (B.1.351) that superseded the B.1.1.54, B.1.1.56 and C.1 lineages that were prevalent during the first wave of infections (13). The third wave that began in May 2021 was associated with the Delta variant (also referred to as lineage B.1.617.2) that was detected first in India in late 2020 (www.cdc.gov). In mid-November 2021, in what appeared to be the emergence of a fourth wave, there was a resurgence of infections in South Africa’s Gauteng province driven by the omicron variant which has since spread globally (9).
The epidemiological profile of SARS-CoV-2 in South Africa has been influenced greatly by government response in the implementation of lockdown measures which helped minimize transmissions (8). Additionally, the government also promoted preventative measures such as frequent washing of hands, wearing of face masks in public, avoidance of physical contact and gathering and quarantine and isolation measures (for individuals with COVID-19 symptoms) (14). More importantly, the government rolled out the administration of three SARS-CoV-2 vaccines (Johnson & Johnson, Pfizer-BioNTech and the Oxford-AstraZeneca). The Oxford-AstraZeneca vaccine was, however, suspended prior to implementation due to its suboptimal effectiveness against the Beta variant (15). As of 4th July 2022, 36, 861, 272 vaccines have been administered across South Africa which have helped ease the burden of SARS-CoV-2 disease (https://sacoronavirus.co.za/latest-vaccine-statistics/). Both the Johnson & Johnson vaccine and Pfizer-BioNTech vaccine were administered in the Free State province. As per the time of writing, the total number of vaccines administered in the Free State province was 2,217,104 with the number of individuals vaccinated as percentage of the population at 60.30% (https://sacoronavirus.co.za/latest-vaccine-statistics/).
We sought to investigate the epidemiological profile of SARS-CoV-2 variants circulating in the Free State province during the 2020-2021 genomic surveillance period with sequenced data (between July 2020 – December 2021) available in the GISAID database. The main aim was to evaluate whether the variants driving the epidemic waves at the national level were also driving the epidemic waves at the local level, in the context of the Free State province.
The Health Sciences Research Ethics Committee (HSREC) of the University of the Free State (UFS), Bloemfontein, South Africa, approved the study under ethics number HSD2020/1860/2710. The patients’ identities and demographics were de-linked from their unique laboratory identities to ensure confidentiality. Participant consent was not required for genomic surveillance. All generated sequences were uploaded to the GISAID database, and their use is subject to the database terms and conditions.
In our analysis, the samples were divided into four different waves of infections. The nasopharyngeal and oropharyngeal swab samples of individuals who tested positive for SARS-CoV-2 in the first-wave infection in the Free State province (between July 2020-October 2020, n = 176) were sequenced and submitted to the GISAID database (https://www.gisaid.org/). The SARS-CoV-2 positive samples collected during the second wave of infection (November 2020 - March 2021, n = 156) were regarded as the second-wave infection samples. The third wave SARS-CoV-2 genomic sequences were submitted to GISAID between April 2021 – October 2021, n = 904 while the fourth wave SARS-CoV-2 sequences entailed samples collected from November to December 2021, n = 51).
The National Health Laboratory Service (NHLS) Universitas Academic Laboratories in conjunction with the University of the Free State-Next Generation Sequencing (UFS-NGS) Unit received randomly selected nasopharyngeal and oropharyngeal swab samples from qPCR confirmed SARS-CoV-2 positive individuals for whole-genome sequencing as part of the Network for Genomic Surveillance South Africa (NGS-SA). The samples were collected from several health centers around the Free State from the following municipalities: Dihlabeng, Fezile Dabi, Kopanong, Lejweleputswa, Letsemeng, Maluti A Phofung, Mangaung Metro, Mantsopa, Masilonyana, Matjhabeng, Metsimaholo, Mohokare, Moqhaka, Ngwathe, Nketoana, Phumelela, Setsoto, Thabo Mofutsanyana, Tokologo, Tswelopele and Xhariep. In total, the number of sequenced samples from Free State available in Global Initiative on Sharing Avian Influenza Data (GISAID) from July 2020 to December 2021 at the time of analysis of the data was 1,461 sequences.
Automated RNA extraction was performed using the NUCLISENS® EASYMAG® instrument (Biomerieux, Marcy I’Etoile, France) as per the manufacturer’s instructions.
The extracted RNA was subjected to cDNA synthesis using Lunascript RT SuperMix kit (New England Biolabs, Ipswich, MA, USA). The cDNA reaction was prepared by adding 4 µL of LunaScript RT Mix (5X) to 10 µL of extracted RNA, and 6 µL of nuclease-free water was added to the mix for a total volume of 20 µL. The mix was then incubated in a thermocycler as follows: 25°C for 2 minutes, 55°C for 20 minutes, 95°C for 1 minute and hold at 4°C. Whole genome amplification by multiplex PCR was carried out using primer pools to generate 400 bp amplicons covering the 30-kb SARS-CoV-2 genome (16). Briefly, 20 µL Mastermix comprising 5 µL of Q5 reaction buffer, 0.5 µl of 10 nM dNTPs, 0.25 µl of Q5 Hotstart DNA polymerase, 3.6 µL of primer pool and 10.65 µL of nuclease free water was added to 5 µL of the synthesized cDNA. The amplification reaction was performed under the following conditions: Heat activation at 98°C for 30 seconds, 35 cycles of denaturation and annealing at 98°C for 15 seconds, 65°C for 5 minutes, respectively, and a hold step at 4°C.
The PCR amplicons were cleaned up using Ampure XP beads (Beckman Coulter, Pasadena, CA, USA) and 70% ethanol (Sigma-Aldrich®, St Louis, MO, USA). Quantification was performed on Qubit™ 3.0, using Qubit dsDNA High Sensitivity Assay kit (Invitrogen, Carlsbad, CA, USA). DNA libraries were then prepared using the Nextera Flex DNA Library Preparation Kit and Nextera CD Index kit (Illumina, San Diego, CA, USA), following manufacturer’s instructions. The fragment sizes were determined on Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA), using dsDNA High Sensitivity Assay Kit. The uniquely indexed DNA libraries were normalized to a concentration of 4 nM, pooled and denatured with 0.2 N sodium hydroxide (NaOH). A final library diluted to 8 pM concentration was spiked with 0.5% PhiX control and sequenced on an Illumina MiSeq platform using V2 Standard sequencing cartridge (Illumina, San Diego, CA, USA) for 500 cycles to generate 250 x 2 paired end reads.
Raw reads coming from Illumina sequencing were assembled using Genome Detective 1.126 and the Coronavirus Typing Tool (17, 18). The initial assembly obtained from Genome Detective was polished by aligning mapped reads to the references and filtering out low-quality mutations using the bcftools 1.7-2 mpileup method (19). The sequence variant assignment was done using Phylogenetic Assignment of Named Global Outbreak LINeages (Pangolin COVID-19 Lineage Assigner) (20). SARS-CoV-2 genome annotator (21) was used to identify the single amino acid mutations using Wuhan_1 (NC_045512.2) as the reference sequence. The ggplot2 (22), Bioconductor library was used in the visualization of the date and all the statistical analyses conducted using R programming language (23).
The SARS-CoV-2 variants circulating in the Free State province during the four waves are summarized (Figure 1). During the initial part of the first wave, B.1, B.1.1, B.1.1.53, B.1.1.448 and B.1.237 circulated in the Free State province, followed by Beta variant, B.1.351 later in the wave. In terms of the number of isolates sequenced, the Beta variant, B.1.351, (52.9%) predominated in the Free State province, followed by B.1.237 (10.3%), B.1.1.448 (9.03%), B.1(8.4%) and B.1.1.53 (6.5%), respectively. Interestingly, in the second wave, most of the variants detected in the first wave became extinct. During this wave, the Beta variant, B.1.351, (95.5%), persisted (Figure 1). The Zeta variant, P.2, first detected in Brazil, was also found in 0.65% proportion of sequenced genomes during the second peak of infections (Figure 1). During the third wave, variant B.1.1 became extinct. Early in the third wave, the Beta variant predominated until it was replaced by the Delta variant, AY.45. In total, Beta variant accounted for 41.7% of isolates sequenced during this period, while 44% were Delta variant (Figure 1). The UK Alpha variant, B.1.1.7 and the Delta variant, AY.11, was also identified in 1.36% and 5.67% of sequenced genomes during the third wave of infections, respectively. The C.1 variant, which was circulating during the first wave, underwent mutation to C.1.2, and was identified in 1.36% proportion of sequenced genomes during the third wave (Figure 1). The fourth wave was predominantly characterized by AY.45 which was identified from 41.28% of the data we analyzed followed by BA.1 which was identified in 37.1% of the samples (Figure 1).
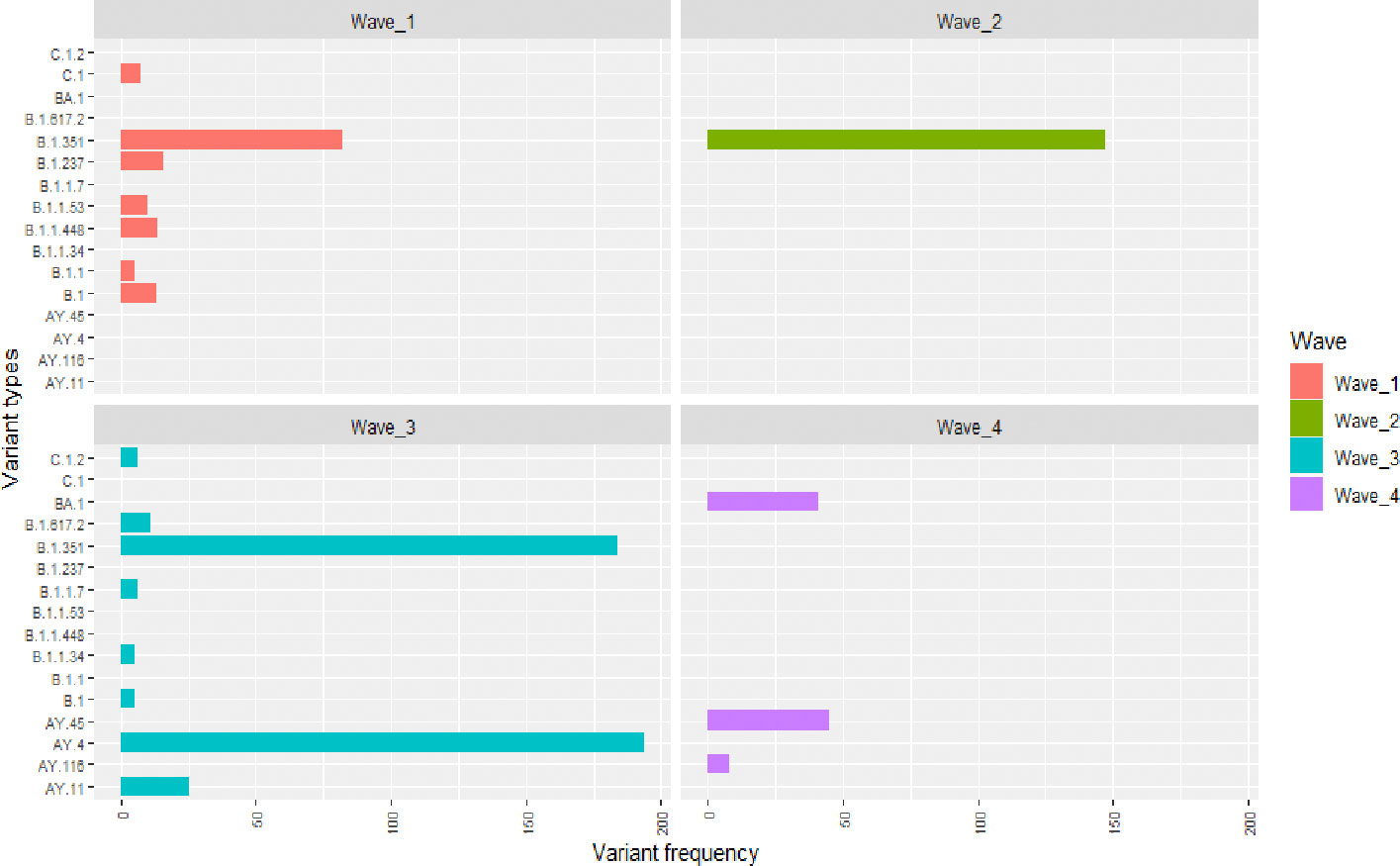
Figure 1 The SARS-CoV-2 variants and their frequency in Free State during the course of four epidemic waves based on genomic surveillance data analysed between July 2020 and December 2021. The y-axis indicates the SARS-CoV-2 variants while the x-axis shows the occurrence frequency. The first, second, third and fourth waves are denoted by the red, green, blue and purple colours, respectively. Unique variants that were identified in each wave are summarized in Table 1.
Our data from the first wave of infection shows that the SARS-CoV-2 in the Free State province in South Africa accumulated amino acid mutations on the spike protein (S) (Figure 2). The Beta variant related mutations: D80A, D215G, K417N, E484K, N501Y, D614G, A701V were the predominant spike protein mutations with the D614G being identified in the majority of the sequenced SARS-CoV-2 viruses in the Free State province (Figure 2). The second wave of infection was characterized by some new amino acid mutations such as L18F and G769V that were not detected in the first wave even though D614G was still present in high frequency during the second wave of infection (Figure 2). Notably, the E484K mutations decreased, and we identified a deletion at position L242 on the spike protein. The SARS-CoV-2 variants circulating in the Free State province in South Africa had accumulated relatively more amino acid mutations than the ones in the first wave (Figure 2). During the third wave, there was an upsurge of mutations across the spike region of the SARS-CoV-2 (Figure 3). Notably, amino acid mutations, L452R, T478K, P681R and D950N were observed in relatively higher frequencies during the third wave and were not present during the first and second waves (Figure 2). The third wave saw an increase in the amino acid mutations which were characterized by the Delta variant of SARS-CoV-2 virus (Figure 1). Each wave of infection was driven by a unique set of SARS-CoV-2 variants (Table 1; Figure 3).
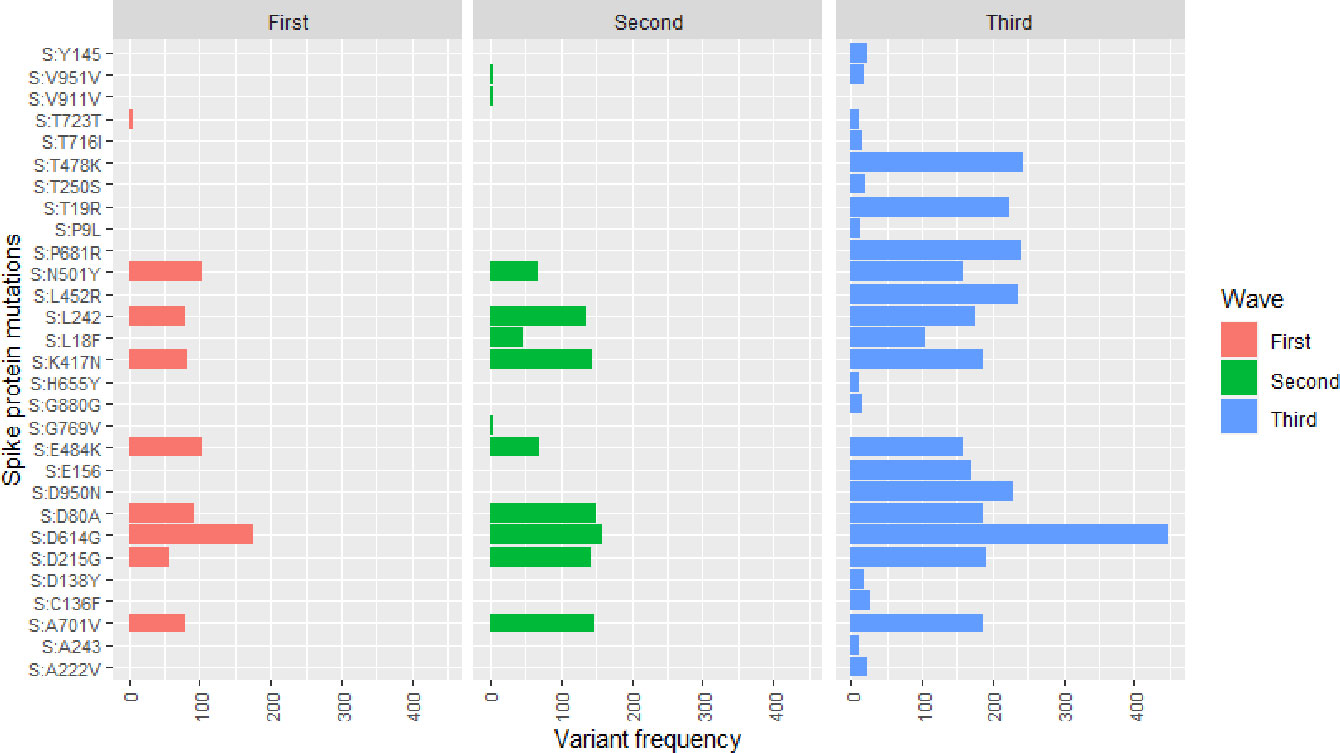
Figure 2 The frequency of spike protein mutations identified in SARS-CoV-2 variants during the epidemic waves. The x-axis indicates the frequency of the SARS-CoV-2 variants while the y-axis indicates the amino acid point mutation and the exact location identified within the SARS-CoV-2 protein structure. The first, second and third waves are denoted by the red, green and blue colours, respectively.
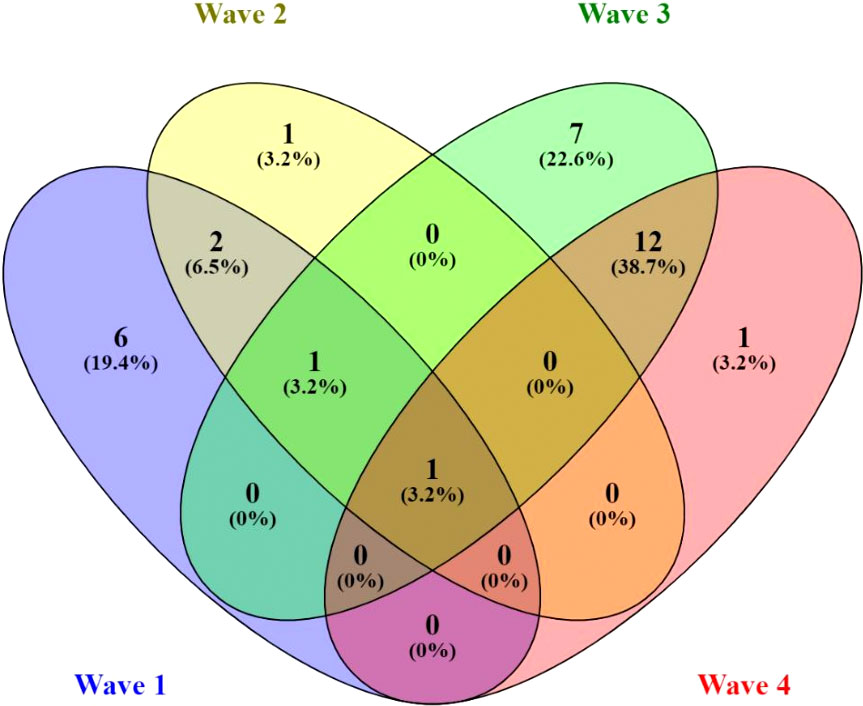
Figure 3 Venn diagram indicating the unique and the overlapping SARS-CoV-2 variants in each epidemic wave. In each wave, the number of unique and overlapping genome variants identified during the analysis of each peak infections are indicated in absolute numbers and percentages. The first, second, third and fourth waves are denoted by blue, yellow, green and red colours, respectively.

Table 1 Unique variants circulating in the Free State province in South Africa during the first, second, third and fourth wave of infections.
Genomic surveillance has become an invaluable approach in combating the spread of SARS-CoV-2. It has also been effective in identifying different variants with multiple mutations at the immunodominant spike protein that facilitates viral cell entry through the angiotensin-converting enzyme-2 (ACE2) receptor (24–26). We describe the epidemiological profile of SARS-CoV-2 variants identified from July 2020 to December 2021 in the Free State province, South Africa. Each epidemic wave had distinct variants. The predominant variants driving the epidemic waves at the local level in the Free State province were similar those driving the epidemic waves at the national level. The Free State data also showed an increase in the number of amino acid mutations in the spike region of the SARS-CoV-2 genome with each infection wave.
While the first wave featured a mixture of SAR-CoV-2 lineages, the Beta variant, B.1.351 predominated in the Free State in the second and third waves and this variant was first reported in Nelson Mandela Bay, the worst-affected metropolitan area in Eastern Cape province, South Africa, in early August 2020 (27). The D614G mutation identified in the main first-wave lineages (B.1.1.54, B.1.1.56 and C.1) in South Africa has been associated with increased transmissibility while the combination of E484K, K417N and N501Y mutations in the B.1.351 lineage has been hypothesized to result in potential escape mutants (28–31). The first wave variants (B.1.1.117, B.1.1.53, B.1.1.54, B.1.237 and B.1.381) containing only D614G mutation, later became extinct in the subsequent epidemic waves due to the mutational fitness effects and the consequent emergence of other more transmissible variants.
The P.2 variant, a descendant of the B.1.1.28 strain, was the unique variant identified only during the second wave in the Free State. This variant emerged in July 2020 in the Brazilian state of Rio de Janeiro and exhibited the E484K mutation in the spike region (with the absence of N501Y mutation) (32). Lineages carrying the E484K mutation, such as B.1351 and P.1, are suggested to escape neutralizing antibodies (33). The P.2 variant has also been associated with a symptomatic case of re-infection underscoring the need for its continuous monitoring (34).
The third wave in the Free State province was driven by the Delta variant and this variant has a higher replication efficacy and fitness advantage than the Alpha and Beta variant in both airway organoid and human airway epithelial systems (35, 36). The Alpha variant arose from the UK and spread globally and was also identified during the third wave in the Free State (37). This variant contains a combination of N501 and 69-70del mutations in the spike protein region (38). While previous investigations have shown that the UK Alpha variant is more transmissible, there is no evidence that the variant is more clinically severe or has the capacity for vaccine escape (39).
The C.1.2 lineage that was first detected in the Mpumalanga and Gauteng provinces of South Africa in May 2021 (9) was also found in the third wave in the Free State province. This lineage evolved from C.1, one of the dominant first-wave lineages in South Africa (40). This variant contains multiple substitutions (R190S, D215G, N484K, N501Y, H655Y, T859 including deletions Y144del and L242-A243del) in the spike region (9) that are associated with increased transmissibility and reduced neutralization sensitivity (33, 41–44). The C.1.2 variant was reportedly on the rise in South Africa in November 2021 (9) and the mutations raised concerns of potential gain of replicative advantage over the Delta variant (40).
BA.1, a sub-lineage of the omicron variant, emerged to drive the fourth wave of infections in the Free State province. The omicron variant was detected in November 2021 by Southern African genomic researchers after being linked to a rapid resurgence of infections in South Africa’s Gauteng province (9). This variant contains over 30 mutations in the spike protein that are predicted to affect antibody neutralization and spike function, resulting in increased transmission (45).
The study was limited by a lack of correlation in the number of sequenced samples and the proportion of infected individuals in the Free State. Regardless, our findings indicate that continuous monitoring of concerning variants such as the omicron is imperative to ensure sustained public health response efforts.
The data presented in the study are deposited in the GISAID repository and accession numbers are provided as Supplementary 1. The data is publicly available via the link: https://doi.org/10.6084/m9.figshare.20231826.v2.
The studies involving human participants were reviewed and approved by Health Sciences Research Ethics Committee (HSREC) of the University of the Free State (UFS), Bloemfontein, South Africa. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
PM, MiM, AO, OA, AB, DG, HS and MN produced SARS-CoV-2 genome data. MaM, LM, MD, SV, AB and DG were involved in sample extraction and meta data curation. AB and MN helped with data interpretation. PM, JO, and MN wrote the initial manuscript and concept development. All authors participated in the review of the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by the South African Medical Research Council (SAMRC) and the Department of Science and Innovation of South Africa. Other funds that supported this work include the Bill and Melinda Gates Foundation (BMGF-OPP1180423_2017), Poliomyelitis Research Fund (PRF Grant 19/16 and PRF Grant 21/77) and National Research Foundation (NRF-120814).
We acknowledge all the participating health centers in the Free State province where the nasopharyngeal swab samples were collected.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.935131/full#supplementary-material
1. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature (2020) 579(7798):265–9. doi: 10.1038/s41586-020-2008-3
2. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med (2020) 382(8):727–33. doi: 10.1056/NEJMoa2001017
3. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol (2020) 5(4):536–44. doi: 10.1038/s41564-020-0695-z
4. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
5. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in wuhan, China. JAMA Neurol (2020) 77(6):683–90. doi: 10.1001/jamaneurol.2020.1127
6. Wilkinson E, Giovanetti M, Tegally H, San JE, Lessells R, Cuadros D, et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Science (2021) 374(6566):423–431. doi: 10.1126/science.abj4336
7. Gorkhali R, Koirala P, Rijal S, Mainali A, Baral A, Bhattarai HK. Structure and function of major SARS-CoV-2 and SARS-CoV proteins. (2021) 15:11779322211025876. doi: 10.1177/11779322211025876
8. Msomi N, Mlisana K, de Oliveira T, Willianson C, Bhiman JN, Goedhals D, et al. A genomics network established to respond rapidly to public health threats in south Africa. Lancet Microbe (2020) 1(6):e229–30. doi: 10.1016/S2666-5247(20)30116-6
9. Viana R, Moyo S, Amoako DG, Tegally H, Scheepers C, Althaus CL, et al. Rapid epidemic expansion of the SARS-CoV-2 omicron variant in southern Africa. Nature (2022) 7:1–0. doi: 10.1038/s41586-022-04411-y
10. Choi B, Choudhary MC, Regan J, Sparks JA, Padera RF, Qiu X, et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. New Engl J Med (2020) 383(23):2291–3. doi: 10.1056/NEJMc2031364
11. Greaney AJ, Loes AN, Crawford KH, Starr TN, Malone KD, Chu HY, et al. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe (2021) 29(3):463–76. doi: 10.1016/j.chom.2021.02.003
12. Cele S, Karim F, Lustig G, San JE, Hermanus T, Tegally H, et al. SARS-CoV-2 prolonged infection during advanced HIV disease evolves extensive immune escape. Cell Host Microbe (2022) 30(2):154–62. doi: 10.1016/j.chom.2022.01.005
13. Tegally H, Wilkinson E, Lessells RJ, Giandhari J, Pillay S, Msomi N, et al. Sixteen novel lineages of SARS-CoV-2 in south Africa. Nat Med (2021) 27(3):440–6. doi: 10.1038/s41591-021-01255-3
14. Giandhari J, Pillay S, Wilkinson E, Tegally H, Sinayskiy I, Schuld M, et al. Early transmission of SARS-CoV-2 in South Africa: An epidemiological and phylogenetic report. Int J Infect Dis (2021) 103:234–41. doi: 10.1016/j.ijid.2020.11.128
15. Madhi SA, Baillie V, Cutland CL, Voysey M, Koen AL, Fairlie L, et al. Efficacy of the ChAdOx1 nCoV-19 covid-19 vaccine against the b. 1.351 variant. New Engl J Med (2021) 384(20):1885–98. doi: 10.1056/NEJMoa2102214
16. Quick J. nCoV-2019 sequencing protocol. protocols. io. (2020). Available at: https://protocols.io/view/ncov-2019-sequencing-protocol-bbmuik6w
17. Vilsker M, Moosa Y, Nooij S, Fonseca V, Ghysens Y, Dumon K, et al. Genome detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics (2019) 35(5):871–3. doi: 10.1093/bioinformatics/bty695
18. Cleemput S, Dumon W, Fonseca V, Karim WA, Giovanetti M, Alcantara LC, et al. Genome detective coronavirus typing tool for rapid identification and characterization of novel coronavirus genomes. Bioinformatics (2020) 36(11):3552–5. doi: 10.1093/bioinformatics/btaa145
19. Danecek P, McCarthy SA. BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics (2017) 33(13):2037–9. doi: 10.1093/bioinformatics/btx100
20. Rambaut A, Holmes EC, Hill V, Toole ÁO, Mccrone JT, Ruis C. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. Nat Microbiol (2020) 5(11):1403–07. doi: 10.1101/2020.04.17.046086
21. Mercatelli D, Triboli L, Giorgi FM, Fornasari E, Ray F. Coronapp: A web application to annotate and monitor SARS-CoV-2 mutations. J Med Virol. (2021) 93(5):3238–45. doi: 10.1002/jmv.26678
22. Wickham H, Ginestet C. ggplot2: Elegant graphics for data analysis. J r stat soc ser a (Statistics soc [Internet] (2011). Available at: http://had.co.nz/ggplot2/book.
23. R Foundation for Statistical Computing. R: a language and environment for statistical computing (2018). Available at: http://www.R-project.org/.
24. Supasa P, Ren J, Stuart DI, Screaton GR. Article evidence of escape of SARS-CoV-2 variant b. 1. 351 from natural and vaccine-induced sera ll ll evidence of escape of SARS-CoV-2 variant b. 1. 351 from natural and vaccine-induced sera. Cell (2021) 184(9):2348–2361.e6. doi: 10.1016/j.cell.2021.02.037
25. Sewell BT, Lourenço J, Carlos L, Alcantara J. Detection of a SARS-CoV-2 variant of concern in south Africa. Nature (2021) 592(7854):438–43. doi: 10.1038/s41586-021-03402-9
26. Ramundo MS, De Jesus JG, Andrade PS, Coletti TM. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in manaus, Brazil. Science (2021) 372(6544):815–21. doi: 10.1126/science.abh2644
27. Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Detection of a SARS-CoV-2 variant of concern in south Africa. Nature (2021) 592(7854):438–43. doi: 10.1038/s41586-021-03402-9
28. Korber B, Fischer WM, Gnanakaran S, Labranche CC, Saphire EO, Montefiori DC, et al. Article tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID- ll tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell (2020) 182(4):812–27.e19. doi: 10.1016/j.cell.2020.06.043
29. Yurkovetskiy L, Wang X, Pascal KE, Dudkina NV, Shen K, Luban J, et al. Article structural and functional analysis of the D614G SARS-CoV-2 spike protein variant ll article structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell (2020) 183(3):739–751.e8. doi: 10.1016/j.cell.2020.09.032
30. Plante JA, Mitchell BM, Plante KS, Debbink K, Weaver SC. Review the variant gambit: COVID-19 ‘ s next move. Cell Host Microbe (2021) 29(4):508–15. doi: 10.1016/j.chom.2021.02.020
31. Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature (2021) 593(7858):266–9. doi: 10.1038/s41586-021-03470-x
32. De Janeiro R, Gerber AL, Guimarães PDC, Mariani D, Ferreira OC, Workgroup C, et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. J Virol (2021) 95(10):e00119-21. doi: 10.1128/JVI.00119-21
33. Wang P, Casner RG, Shapiro L, Ho DD. Brief report increased resistance of SARS- CoV-2 variant p. 1 to antibody neutralization increased resistance of SARS-CoV-2 variant p. 1 to antibody neutralization. Cell Host Microbe (2021) 29(5):747–751.e4. doi: 10.1016/j.chom.2021.04.007
34. Nonaka CKV, Franco MM, Gräf T, de Lorenzo Barcia CA, de Ávila Mendonça RN, de Sousa KAF, et al. Genomic evidence of SARS-CoV-2 reinfection involving E484K spike mutation, Brazil. Emerg Infect Dis (2021) 27(5):1522–4. doi: 10.3201/eid2705.210191
35. Youk J, Kim T, Evans KV, Choi B, Ju YS, Lee J. Article three-dimensional human alveolar stem cell culture models reveal infection response to SARS- ll ll three- dimensional human alveolar stem cell culture models reveal infection response to SARS-CoV-2. Stem Cell (2020) 27(6):905–919.e10. doi: 10.1016/j.stem.2020.10.004
36. Mlcochova P, Kemp SA, Dhar MS, Papa G, Meng B, Pandey R, et al. Delta variant replication and immune evasion. Nature (2021) 599(7883):114–9. doi: 10.1038/s41586-021-03944-y
37. Kirby T. Country in focus new variant of SARS-CoV-2 in UK causes surge of COVID- 19. Lancet Respir (2021) 9(2):e20–1. doi: 10.1016/S2213-2600(21)00005-9
38. Meng B, Kemp SA, Papa G, Robertson DL, Bailey D, Gupta RK. Article recurrent emergence of SARS-CoV-2 spike deletion ll recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role. Cell Rep (2021) 35(13):109292. doi: 10.2139/ssrn.3780277
39. Tang JW, Tambyah PA, Hui DSC. Emergence of a new SARS-CoV-2 variant in the UK. J Infect (2021) 82(4):e27–8. doi: 10.1016/j.jinf.2020.12.024
40. Scheepers C, Everatt J, Amoako DG, Mnguni A, Ismail A. The continuous evolution of SARS-CoV-2 in south Africa: a new lineage with rapid accumulation of mutations of concern and global detection. medRxiv (2021) doi: 10.1101/2021.08.20.21262342
41. Jangra S, Ye C, Rathnasinghe R, Stadlbauer D, Personalized Virology Initiative study group, Krammer F, et al. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. Lancet Microbe (2020) 2(7):e283–4. doi: 10.1016/S2666-5247(21)00068-9
42. Johnson BA, Xie X, Kalveram B, Lokugamage KG, Muruato A, Zou J, et al. Furin cleavage site is key to SARS-CoV-2 pathogenesis. Cell Host Microbe (2020) 29(3):477–88.e4. doi: 10.1016/j.chom.2021.01.014
43. Liu Z, Vanblargan LA, Bloyet L, Fremont DH, Diamond MS, Whelan SPJ, et al. Article identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization ll article identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe (2021) 29(3):477–488.e4. doi: 10.1016/j.chom.2021.01.014
44. Supasa P, Zhou D, Dejnirattisai W, Liu C, Mentzer AJ, Ginn HM, et al. Reduced neutralization of SARS-CoV-2 B.1.1.7 variant by convalescent and vaccine sera. Cell (2021) 184(8):2201–11. doi: 10.1016/j.cell.2021.02.033
45. Martin DP, Lytras S, Lucaci AG, Maier W, Grüning B, Shank SD. Selection analysis identifies significant mutational changes in omicron that are likely to influence both antibody neutralization and spike function (Part 1 of 2). bioRxiv [Preprint]. (2022) 2022.01.14.476382. doi: 10.1101/2022.01.14.476382
Keywords: Beta variant, Delta variant, epidemiological profile, Omicron variant, Free State, SARS-CoV-2 variants, South Africa
Citation: Mwangi P, Okendo J, Mogotsi M, Ogunbayo A, Adelabu O, Sondlane H, Maotoana M, Mahomed L, Morobadi MD, Vawda S, von Gottberg A, Bhiman J, Tegally H, Wilkinson E, Giandhari J, Pillay S, Naidoo Y, Ramphal U, de Oliveira T, Bester A, Goedhals D and Nyaga M (2022) SARS-CoV-2 variants from COVID-19 positive cases in the Free State province, South Africa from July 2020 to December 2021. Front. Virol. 2:935131. doi: 10.3389/fviro.2022.935131
Received: 03 May 2022; Accepted: 24 August 2022;
Published: 14 September 2022.
Edited by:
Chris Illingworth, School of Clinical Medicine, University of Cambridge, United KingdomReviewed by:
Saheed Sabiu, Durban University of Technology, South AfricaCopyright © 2022 Mwangi, Okendo, Mogotsi, Ogunbayo, Adelabu, Sondlane, Maotoana, Mahomed, Morobadi, Vawda, von Gottberg, Bhiman, Tegally, Wilkinson, Giandhari, Pillay, Naidoo, Ramphal, de Oliveira, Bester, Goedhals and Nyaga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Nyaga, bnlhZ2FtbUB1ZnMuYWMuemE=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.