 Gloria Magro
Gloria Magro Arianna Calistri
Arianna Calistri Cristina Parolin
Cristina Parolin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Virol. , 29 July 2022
Sec. Antivirals and Vaccines
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.933418
This article is part of the Research Topic The perpetual challenge of Antiviral drug-resistance View all 4 articles
With nearly 38 million of people worldwide living with HIV-1 and no definitive cure available after almost 40 years of research, AIDS is still a major global public health issue. Modern antiretroviral therapies can achieve viral replication suppression to undetectable levels, thus allowing an almost normal life to HIV-1–infected individuals. However, the virus cannot be fully eradicated. This may lead over time to the accumulation of mutations in the viral genome and, eventually, to the emergence of drug-resistant viruses, which may affect the efficacy of the therapy and the patient’s quality of life. To overcome some of the limitations of the standard antiretroviral therapy, innovative therapeutic approaches such as “shock and kill” and immunotherapies, as well as technologies based on RNA interference and CRISPR-Cas9 genome editing are under investigation. Nevertheless, the virus may find a way to break free even from these novel strategies. In this review, we focus on the mechanisms that enable HIV-1 escape from the most advanced therapies and discuss some of the challenges to prevent this issue.
Few viruses are as elusive and persistent as HIV-1, the etiological agent of AIDS, for which no definitive cure is still available after almost 40 years since its discovery (1). The development of modern combined antiretroviral therapies (ARTs) enables the patients to live an almost normal life, by keeping the viral replication under control and avoiding the onset of severe immunodeficiency. However, this drug regimen does not eliminate the virus from latently infected cells, patient adherence must be lifelong, and ART interruption, even for a short time, can lead to viral rebound (2). Moreover, the emergence of resistance to the administered drugs may render the therapy ineffective, favoring at the same time the spread of the resistant virus to other individuals, limiting their treatment options (3). By the end of 2021, the World Health Organization released the results of a drug resistance survey based on data from 38 countries worldwide. This report not only highlighted the high frequency of resistance toward specific classes of antiretroviral drugs, but also indicated a worrying prevalence of pre-treatment drug resistance. This latter phenomenon is mainly due to the transmission of already resistant viruses or to previous prophylaxis treatment (4).
However, why is HIV-1 able to resist so well to current therapeutic approaches? Much of HIV-1 success in persisting in the host and in escaping from available therapeutic options is due to its distinctive replication cycle and to its tropism for CD4+ T cells. Being a lentivirus, the HIV-1 genome is composed by a single-stranded RNA (ssRNA), enclosed as two identical copies within each viral particle. Viral genome encompasses genes encoding for structural (e.g., gag and env), functional (e.g., pol), and regulatory (e.g., tat and rev) proteins and is characterized by long-terminal repeats (LTRs) at both ends (5). LTRs are essential for the virus to integrate within the host cell genome as a provirus, following reverse transcription of the genomic ssRNA in double-stranded DNA (6). In most of the infected cells, infection is productive, but there is a subset of cells, especially quiescent CD4+ memory T cells, in which the virus is latent. This latently infected cell compartment is established very early during the infection (7) and, as antiretroviral therapies target only the actively replicating virus, it represents one of the main barriers to accomplish a definitive cure. Moreover, as latently infected cells do not express viral antigens and mainly belong to the host immune system, it is not surprising that these HIV-1 reservoirs are extremely difficult to reach and clear.
Despite the current inability to achieve a definitive cure, antiretroviral therapies are very efficient in keeping the viral replication under control. During its rapid replication, HIV-1 genome mutates at a high rate mainly due to transcription errors made by the viral reverse transcriptase (RT), as well as by the host cell RNA polymerase II. The HIV-1 constant evolution leads to a frequent onset of drug resistant mutants, even in the absence of a selective pressure (8). The resulting genomic diversity complicates the design of targeted therapeutic approaches and vaccines, contributing to constant virus evolution with the emergence of variants, as it occurred recently in the Netherlands, where a new and exceptionally virulent HIV-1 variant, characterized by two times faster than the expected decline in CD4+ cell count, was isolated (9). Notably, variants can develop not only in different patients (inter-patient), but also within the same patient (intra-patient).
Single anti–HIV-1 drug regimens can more easily lead to the generation of viral resistant mutants. As there are several classes of antiretrovirals, affecting different steps in the viral life cycle, modern ART includes combinations of two/three compounds. This combinatorial approach reduces the likelihood of emergence of resistance, as this would require the concomitant presence of different mutations within the same viral genome (10). Nevertheless, multi-drug resistant viruses can still appear during treatment and can potentially be transmitted in newly acquired infections. Of note, the limited penetration of the drugs in reservoirs could contribute to the establishment and maintenance of viral sanctuary in tissues in which cells are not exposed to drugs, leading to the development of resistances even after years of successful therapy (10). The most frequently transmitted resistances are those to nucleotides able to interfere with the elongation process of the nascent DNA chain during reverse transcription, and to non-nucleoside reverse transcriptase inhibitors (NNTRIs), which directly interacting with the RT block its activity. By contrast, the most rare transmitted resistances are those toward integrase strand transfer inhibitors (InSTIs) that inhibit provirus integration in the host genome (11). HIV-1 can develop resistances to all the antiretroviral classes, as reviewed in (12), although the evolution of drug-resistant mutations has been shown to come with a fitness cost for the virus (13).
An additional aspect to be taken into consideration is the adherence to the ART. While the optimal level is set to at least 90%, lower percentages can be tolerated for certain drugs (14). However, therapy discontinuation favors the emergence of viral resistance. This issue has been partially overcome by the introduction of single-tablet regimens, easier for the patient to manage (15). A further improvement could be represented by handling long-acting formulations (16), which allow to increase the time between administrations, thus reducing complications related to a daily dosage (17).
In this tug of war between the need to find the best antiretroviral therapy regimen and the emergence of HIV-1 resistances, the development of innovative therapeutic formulations or approaches able to reach viral reservoirs in tissues normally spared by the conventional therapies, as well as to eliminate the integrated viral genome (18), are very promising. Indeed, encouraging results in past and ongoing studies have been reported regarding therapies developed to leverage the immune system against the virus (vaccines and neutralizing antibodies), to reactivate the latent reservoir compartment to clear it (shock and kill), and making use of gene therapy and gene editing approaches to target the host or HIV-1 specific genes (Figure 1).
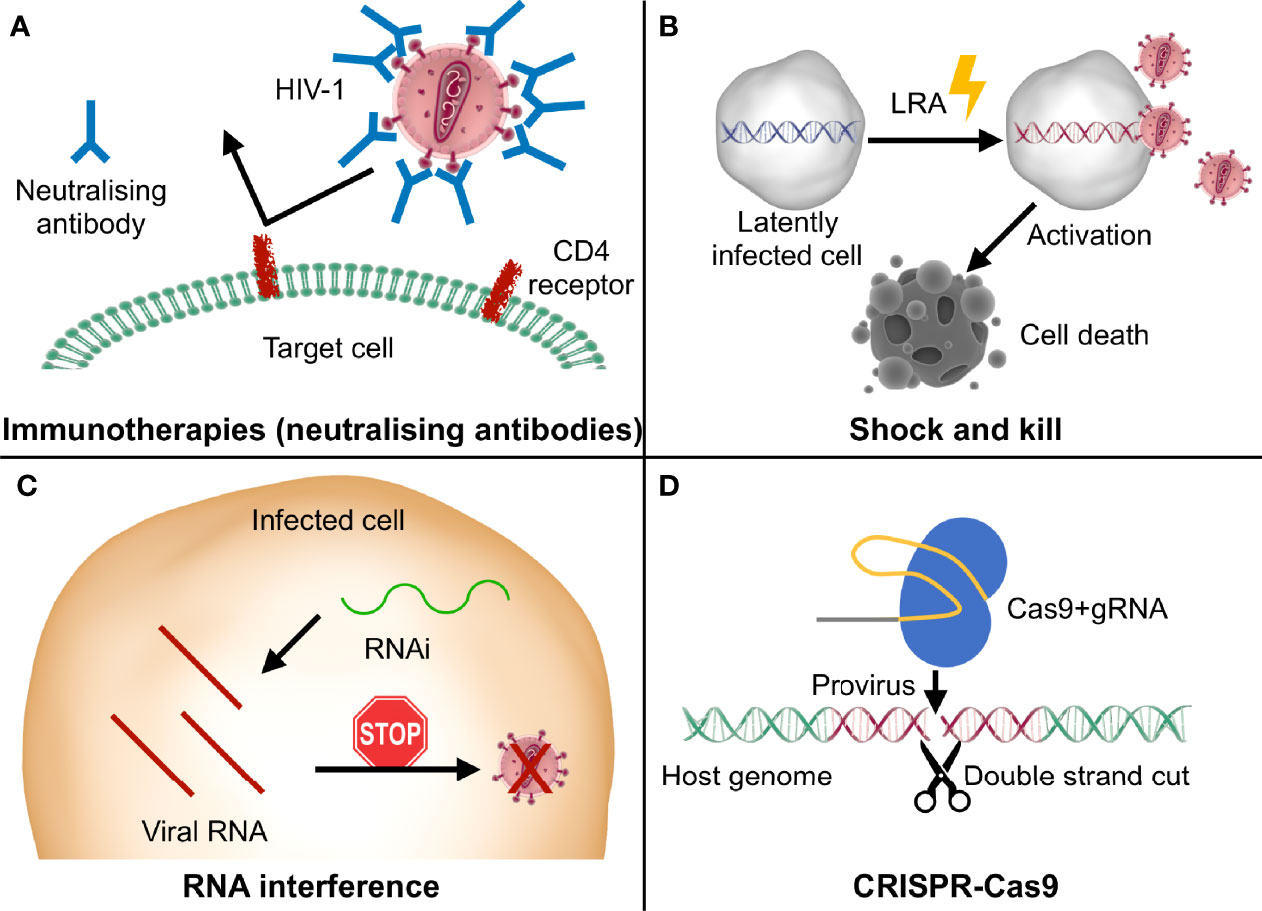
Figure 1 Graphic representation of the main innovative therapeutic strategies discussed in this review. (A) Some immunotherapies exploit neutralizing antibodies to specifically bind viral epitopes and block the infection; (B) shock and kill approaches use LRA (latency reversal agents) to stimulate the reactivation of the latent provirus (“shock”), resulting in virus- or cell-mediated death of the infected cells (“kill”); (C) in RNA interference (RNAi) strategies, small RNAs are designed to bind to the viral RNA and stop viral replication at a post-transcriptional level; (D) the Cas9 nuclease complexed with the gRNA recognizes the specific target sequence in the proviral genome and cuts it, leading to its disruption or elimination and consequently impairing viral replication.
Nevertheless, for each one of these novel therapies, HIV-1 can find different ways to escape or resist, impairing their efficiency. In the following sections, we will provide an overview of the mechanisms enabling HIV-1 to overcome some of the most promising therapeutic approaches that are under development to face the limitations of the currently adopted antiretroviral therapy. We will also outline some possible solutions to hamper HIV-1 evasion from these innovative strategies.
As mentioned in the introduction, HIV-1 targets the cells of the immune system, sentencing them to a slow decrease over time and causing a generalised dysfunction of the immune system. HIV-1, thus, creates the perfect conditions to thrive in the host, while escaping from its immune defences. Eliciting a robust immune response against the virus could be efficient in facing the infection. This conclusion is supported by the observation of a subset of patients, called elite controllers, who are able to keep viral replication under control even in the absence of ARTs (19, 20). These patients are in fact characterized by specific CD8+ T cell responses, typical variations in the class I HLA (human leukocyte antigen), and a particular provirus integration pattern. Indeed, in these individuals proviruses are often located away from transcriptional start sites, into centromeric regions or other regions enriched in repressive chromatin marks, as reported in a recent study (21).
Many approaches under investigation have been directed to elicit HIV-1 specific immune responses, such as the use of vaccines and of neutralizing antibodies, although the viral ability to mutate complicates the establishment of an efficient strategy.
Thanks to vaccines, smallpox has been eradicated and the population has been protected from widespread infections e. g. chicken pox, measles, and rubella; from the potentially cancer-causing HPV and, more recently, from the pandemic SARS-CoV-2, the aetiological agent of COVID-19. However, despite many years of research, no effective vaccine has been developed yet against HIV-1, nor prophylactic nor therapeutic. In fact, even if some of the vaccine candidates were able to generate anti–HIV-1 antibodies, they were not efficient enough to protect from viral infection/replication.
HIV-1 can escape from vaccines by adopting different clever strategies. First, inter- and intra-patient viral genetic variability implies also antigenic variability, with difficulties in the development of a vaccine able to tackle all the variants. Thus, a rational design of optimal antigens is crucial to generate an immune response potent and highly conserved across the variants (22).
Second, HIV-1 has a high mutational rate and can gain mutations in specific epitopes, which may favor its escape from the immune response (in particular from cytotoxic T-lymphocytes, CTL). For example, studies in non-human primates immunized with a DNA vaccine have shown that even those animals in which the viral replication was initially under control, after some time from the treatment, showed viral escape from CTLs. Interestingly, the selective pressure imposed by the CTLs themselves contributed to this escape (23). Vaccines should thus be designed to limit those pathways adopted by the virus to mutate. The implementation of population modeling studies could give further insights in the impact of resistance to different vaccination strategies at a population level (24, 25).
Third, the viral envelope glycoprotein (Env), responsible for viral entry and the only target for neutralizing antibodies, is heavily glycosylated (it is known as “glycan shield”) and is consequently protected from antigen recognition and neutralization (26). Ferreira and colleagues have modeled structural rearrangements in this shield, showing what happens when a glycan important for the shielding of neutralizing antibodies epitopes (N301) is removed. These authors showed that the remaining glycans are able to change their conformation and compensate for the glycan loss, conferring an even increased resistance to neutralizing antibodies (27). Nevertheless, glycosylated Env remains an important target for vaccination, and many studies are investigating the glycan profiles across different viral subtypes (28) and how to mimic, in recombinant Env-based vaccines, the correct glycosylation sites (29), in order to elicit better immune responses.
Among the many developed approaches only one vaccine (RV144) has shown a modest efficacy in HIV-1 prevention in a phase III trial, lowering the infection rate of roughly 30% (30). This vaccine is based on an immune prime and boost strategy with 97% of the treated patients developing an antibody response against the Env antigens used for vaccination (31). The reported efficacy was still too low for a vaccine candidate. However, the data collected from this study are an important starting point to develop more efficient strategies.
Despite the lack of successful vaccine candidates, currently, new hope comes from the mRNA-based vaccines, following the results recently achieved with the ones developed against SARS-CoV-2. The mRNA-based vaccines display some advantages, as (i) the easiness in their design and production, which enables fast adaptation to new emerging variants and (ii) the ability to elicit a robust immune response. These vaccines do not represent a novelty in the HIV-1 field. Indeed, some pre–COVID-19 studies on mRNA-based vaccines against HIV-1 were reported (32–34). However, these early works highlighted important limitations to the adoption of this vaccination strategy to tackle HIV-1 infection, e. g. (i) a complex delivery system, (ii) no significant clinical benefit (32) and (iii) the presence of errors in the RNA sequence encoding for the immunogen (33) and (iv) the instability of the therapeutic nucleic acid (34). In the past few years, modifications of the mRNA structure to ameliorate its stability, the use of lipid nanoparticles (LNPs) for its delivery, and an overall improvement of the vaccine formulation has led to a greater efficacy in eliciting an immune response (34) and, thus, to a rapid translation into the clinical setting, as we observed with the vaccines against SARS-CoV-2. The first phase I clinical trial for an HIV-1 mRNA vaccine started in 2021. This trial conducted by Moderna exploits a combination of priming and boosting immunogens and takes advantage of the most promising LNP-based delivery system (ClinicalTrials.gov Identifier: NCT05001373). Currently, other three anti–HIV-1 mRNA vaccines, designed to evoke an immune response against the envelope trimer (35), are in a phase 1 clinical trial (ClinicalTrials.gov Identifier: NCT05217641). These trials altogether are estimated to be completed by the second half of 2023. Their results on one hand will give further insights in the efficiency of the latest improved mRNA vaccines against HIV-1. On the other hand, the well-known establishment of latently infected cells able to escape from the immune system response may still constitute a barrier to this approach, at least in the case of therapeutic vaccines. To solve this limitation, a novel strategy might be to combine this type of vaccination with latency reversal agents (further described in the "Shock and kill" section), which function by reactivating the virus, thus making it visible to the immune system, along with antibodies designed to block the reactivated virus entry in the cells (ClinicalTrials.gov Identifier: NCT03619278).
As reported in the "Vaccines" section, HIV-1 vaccines aim to elicit broadly neutralizing antibodies (bNAb). These antibodies are defined “neutralizing,” because they are able to neutralize HIV-1 in the early steps of the life cycle, protecting from infection, and “broad” as they are directed to conserved HIV-1 epitopes, to overcome viral variability. The first studies using the administration of single- or first-generation combination of bNAb to block HIV-1 showed a weak and only transient reduction of the viral load, with a fast emergence of antibody-resistant mutants (36–38). Starting from these findings, a study conducted in 2017 reported that the administration of a novel bNAb combination in a non-human primate model of HIV-1 infection led to undetectable level of viral load up to 177 days post-administration. Moreover, a long-term T-cell–mediated immunity was established with only a low level of detectable virus for up to 2 years. By contrast, control primates treated with ART underwent viral rebound upon treatment interruption. Viral suppression duration was related to the half-life of the antibodies used (39). Following this study, the same group conducted a phase I clinical trial, administering a combination of two bNAb directed against Env antigens in patients during ART interruption. Nine patients out of 11 maintained viral suppression for up to 4 months, whereas two of them experienced viral rebound at around 1–2 months. Analysis of the pre-administration viral reservoir in the two resistant individuals highlighted that they harbored mutations, which made them resistant to one of the bNAb, exposing them in fact to a monotherapy rather than to a combinatorial one. Actually, due to HIV-1 variability, this antibody combination has been predicted to function in around 50% of HIV-1 clade B-infected patients, requiring to add or substitute one of the bNAbs to widen its target range (40). The same antibody combination was administered to seven non-virologically suppressed patients. Among these treated individuals, four responded with a decrease of the viral load in the blood and only two of them, which presented the lowest viremia at the beginning of the study, could maintain the suppression for around 3–4 months (41).
Further studies are focusing on improving the bNAb design to prevent the onset of resistance. In this context, Schommers and colleagues were able to design a bNAb targeting the CD4-binding site with one of the most broad activities reported up to date (42). In silico modeling could also help in predicting the resistance of bNAb to HIV-1 variants and guide their rational design (43, 44).
In clinics, Ibalizumab has recently become the first approved monoclonal antibody (mAb) for the treatment of HIV-1 infection (45), used in combination with antiretroviral therapy of multi-drug–resistant patients. Ibalizumab binds to CD4, the primary HIV-1 receptor, and blocks the post-attachment steps involved in viral entry into the cell. The virus can develop resistance to this mAb, leading to a decreased activity. However, it has been observed that the mutants are still dependent on CD4 for the entry process and, therefore, they can be blocked by alternative entry inhibitors. Thus, Ibalizumab does not exert selective pressure toward CD4 resistance (46). Monoclonal antibody approaches could be further improved by the development of antibody conjugates with an increased anti–HIV-1 activity, as reviewed in (47).
“Shock and kill” is one of the most studied functional cure approaches and is designed to work alongside the standard ART to eradicate the latently infected cell compartment. It requires the use of latency reversal agents (LRAs) to induce the provirus expression (“shock”), thus leading to the death of the infected cells due to viral replication or immune clearance (“kill”). LRAs with different mechanisms of action can be employed alone or in combination, as reviewed in (48). These compounds can be epigenetic modifiers (e.g., histone deacetylase [HDAC] inhibitors), agonists of inducible host factors involved in the maintenance of latency (including protein kinase C [PKC], NF-κB, cyclin T1/CDK9 [P-TEFb] complex, Toll-like receptor), activators of the T-cell receptor (TCR), of the PI3K (phosphatidylinositol-3-kinase) pathway, or of AKT/mTOR (mammalian target of rapamycin) (49), Second Mitochondrial-derived Activator of Caspases (SMAC) mimetics, which target the non-canonical NF-κB activation pathway and induce apoptosis (50), or even immunomodulatory agents [including immune checkpoint blockers, cytokines, Toll-like receptor agonists and CD8+ T-cell depletion (51)]. The reactivation of the provirus alone, in most cases, is not sufficient to achieve an effective clearance of the infected cells. Hence, why LRAs are often combined with complementary approaches aimed at inducing apoptosis, as reviewed in (52), or at boosting the HIV-1–specific immune response (53, 54). Recent clinical trials have highlighted that this approach, while effective at reactivating the provirus in vitro, has only a minimal effect on HIV-1 reservoirs in vivo, comparable to the one achieved by using ART alone (55). Even some of the most recent strategies tested in simian models of HIV-1 infection, e.g. the use of a novel drug able to induce HIV-1 replication through the non-canonical NFκB signalling pathway (56) or the one of an interleukin IL-15 superagonist in combination with CD8+ T-cell depletion (57), although showing a robust increase of HIV-1 viral RNA expression, were not able to achieve viral clearance.
Different factors should be taken into consideration to explain why this strategy is not as effective in vivo as it is in vitro and, thus, why HIV-1 is able to resist to shock and kill strategies. One reason is probably to be found in the characteristics of the latent reservoir itself, as HIV-1 latency is, on its own, a mechanism of viral resistance and escape from therapeutic approaches. Viral reservoirs are heterogeneous and contain different cell subsets, so LRAs can have a higher or lower efficiency in reactivating HIV-1 transcription in different subsets. Grau-Expósito and colleagues (58), for example, used a novel fluorescence in situ hybridisation and flow cytometry combined approach (FISH-Flow) to observe HIV-1 RNA and p24 expression in patient-derived cells upon administration of different LRAs, alone or in combination. The advantage of this technique is that it can discern better the differences in expression pattern behavior between cell subsets, on the opposite to what happens, for example, with real-time PCR where the presence of few cells with a higher viral expression could mask differences in the total population. First, they found that the percentage of reactivated cells varied greatly among the different patients. In addition, they observed that different LRAs, even belonging to the same family, impacted in a different way in cell subsets, having a higher efficiency when LRAs were used in combination. Finally, they noticed that T memory stem cells, a long-lived cell subset, were the most difficult to be reactivated (58). Overall these findings prove that shock and kill approaches should take into consideration the heterogeneity of the latent reservoir to achieve a better efficiency, as discussed more extensively in the study by Ait-Ammar and colleagues (59). Furthermore, this strategy may be difficult to be applied to some latently infected cell subsets, such as microglia, as their reactivation could lead to neuroinflammation, a dangerous and unwanted effect (60).
HIV-1 resistance to shock and kill can also be attributed to the fact that HIV-1 latently infected cells are not recognized by the immune system, as they do not express viral antigens, unless they are reactivated and the virus starts replicating. Therefore, if the “shock” is not sufficient to reactivate the majority of the infected cells, these cells can elude immune surveillance, being spared from the “killing” (Figure 2A). Moreover, in vitro studies evidenced that treatment with some LRAs, such as HDAC inhibitors, has immunosuppressive effects by impairing the function of CD8+ T cells, which are the CTL responsible for the recognition and clearance of reactivated latently infected cells (61, 62) (Figure 2B), even though in vivo studies showed no negative effects on T-cell function (63). In addition, some works have particularly focused on natural killer (NK) cells as promising effectors in shock and kill approaches and on the effect that different LRAs have on their function. Studies performed in vitro on patient-derived cells have reported that the impact of different LRAs on these cells is diverse. For example, among HDAC inhibitors, Panobinostat and Romidepsin showed concentration-dependent toxicity and function impairment of NK cells, whereas Vorinostat showed no impact on NK cell function (64). However, in vivo investigations showed that the administration of Vorinostat and Panobinostat did not present negative effects on NK cell function (65). These differences between in vitro and in vivo studies could be related to the drug mechanism itself, to the drug concentration, as sometimes the concentrations used in in vitro studies are not achievable in a clinical setting, as well as to the duration of the treatment (65).
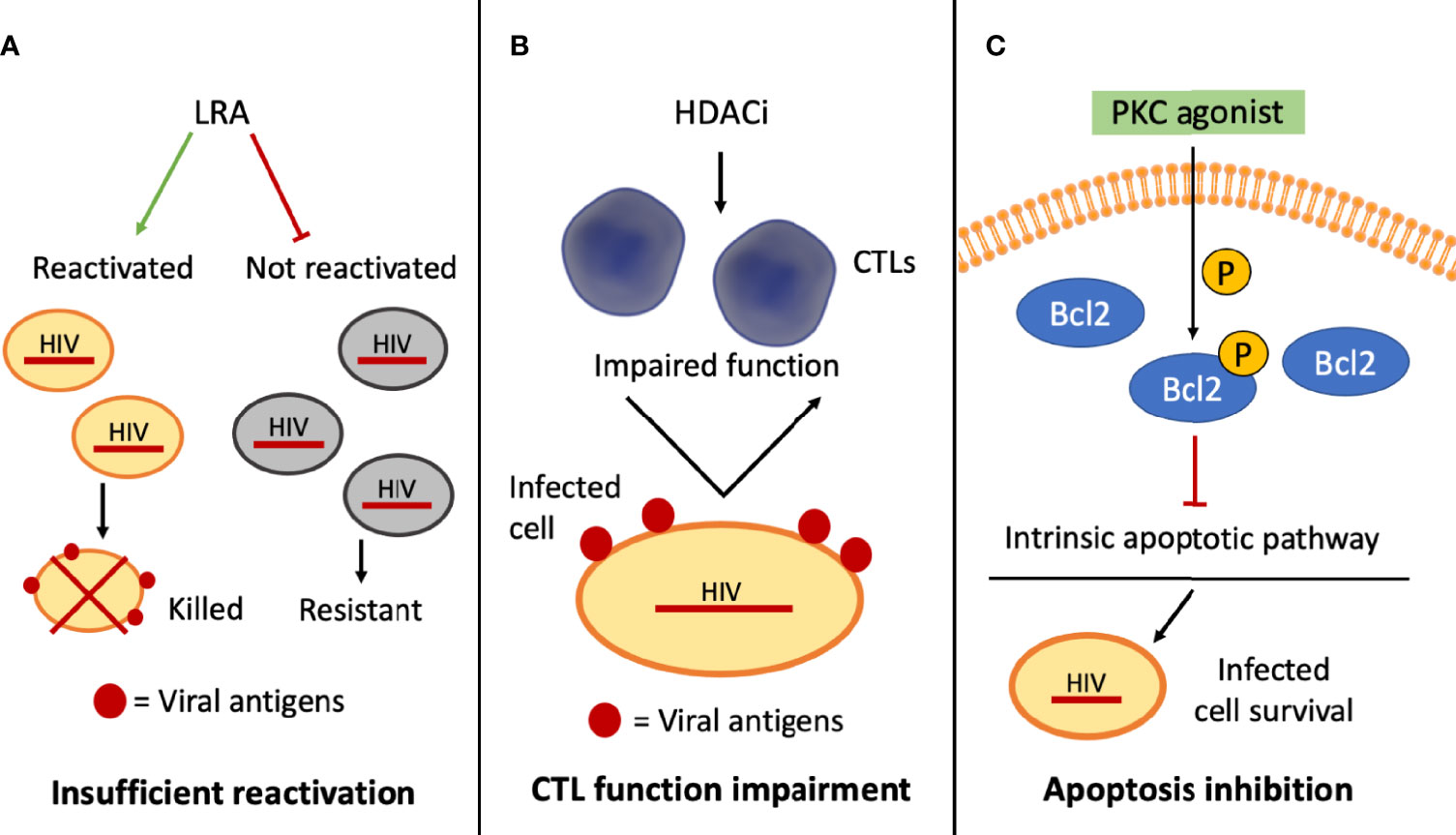
Figure 2 Schematic representation of selected mechanisms of HIV-1 resistance to shock and kill approaches. (A) The inability of the latency reversal agents (LRAs) to activate the provirus in all the infected cells interferes with the exposure of viral antigens on this subset of cells (in gray), which cannot be targeted by cytotoxic T lymphocytes (CTLs)–mediated clearance; (B) histone deacetylase inhibitors (HDACi) may hinder CTL function, affecting CTL-mediated clearance of the infected cells; (C) some PKC agonists can phosphorylate BCL2, blocking the intrinsic apoptotic pathway and promoting the survival of the infected cells.
Another reason why the “kill” might be ineficient could be due to the cellular response to specific LRAs, which enhances resistance to apoptosis and, thus, to clearance. French and collaborators, by adopting patient-derived CD4+ T cells, reported that some PKC agonists, such as bryostatin-1 and prostratin, are able to inhibit the intrinsic apoptotic pathway. These Authors showed that the anti-apoptotic effect was linked to an enhanced phosphorylation of BCL2, responsible for the binding and inhibition of pro-apoptotic proteins (66) (Figure 2C). Some viral proteins themselves, such as Tat, Nef, and Vpr, may act as mediators both of pro- and anti-apoptotic signals (67) and could therefore contribute to the resistance of the infected cells to the “kill”.
The resistance mechanisms presented above are more related to the characteristics of the infected cell compartments rather than to escape mutations of the virus. However, HIV-1 itself can acquire resistant mutations, such as those involving CD8+ T-cell reactive epitopes, which allow the virus to evade CTL recognition and immune clearance (68). It has been shown that not only these mutations are present in the latently infected cell compartment but also that they may represent the dominant population in chronically infected patients (69). A more recent study, by contrast, reported that the prevalence of CTLs resistant mutants may have been overestimated, as the unbiased proteome-wide analysis of the persistent HIV-1 reservoir showed an escape frequency of around 30% (70). Despite this discrepancy, both these studies agree in the conclusion that a strategy to counteract resistant mutants might be the stimulation of the T-cell immunity and the boost of a broad-acting CTL response, for example, by administering viral peptides.
Given the still low efficiency of shock and kill strategies, to overcome HIV-1 current resistance to this therapeutic approach, different studies have tackled the problem by dealing with various aspects.
Some studies have investigated the inefficient reactivation of the provirus by employing alternative strategies to induce latency reversal. Mann and collaborators (71), for example, used an HIV-1–based viral-like particle (VLP) formulation, namely, ACT-VEC, in patient-derived cells, to induce T-cell receptor response in latently infected HIV-1–specific CD4+ T cells and, thus, stimulation of viral transcription. They obtained a significant increase of the viral RNA expression, even greater compared to the one achieved adopting standard LRAs such as HDAC inhibitors and PKC agonists. At the same time, a boost of the immune response was achieved (71). In another investigation, the use of antigen-presenting type 1–polarized monocyte-derived dendritic cells was successful at inducing latency reversal and at activating a cytotoxic T-lymphocyte–mediated response for the clearance of cells harbouring replication-competent virus (72). A further alternative system to the classical LRAs is the use of a “dead” Cas9 (dCas9) nuclease, whose cleavage sites have been inactivated, but which is still able to specifically bind to the DNA, coupled with activators of transcription. This strategy allows to specifically target proviral promoter regions and induce viral expression, with important advantages over classical LRAs as it is potentially less toxic for the cells. Furthermore, a rapid and potent reactivation of the provirus can be obtained, with no need for additional immunotherapy to eliminate the infected cells and only rare off-target effects (73–75).
In addition to these improved approaches, investigations focused on viral reactivation at a single-cell level to understand the heterogenous characteristics of latency could be important to find new LRAs with enhanced efficacy. For example, Golumbeanu and collaborators used single-cell sequencing to identify more than 130 differentially expressed genes in latently infected and reactivated cells (76). In another study, Lu and co-workers adopted fluorescence-activated cell sorting and mathematical modeling to understand the fluctuations and reactivation dynamics at a single-cell level within the same clonal population. The results obtained by Lu’s group confirmed that the use of multiple LRAs with a periodic treatment could be more efficient than a single administration (77), although further experiments using heterogeneous non-clonal populations are needed for a better understanding. Other than the periodic administration of a single LRA, the efficiency of the system could also be boosted by the sequential administration of different LRAs, in a combinatorial approach. In this context, Bouchat and collaborators demonstrated that the sequential administration of the DNA methylation inhibitor 5-AzadC and of histone deacetylase inhibitors (HDACi), compared to their simultaneous administration, displayed a synergistic effect in the reactivation of viral gene expression both in latently infected cell models (in vitro) and in patient-derived cells (ex vivo) (78).
Of note, an alternative and more recently developed method, namely, “block and lock” strategy, could overcome some of the limitations discussed in this section. This approach, in fact, instead of aiming at the latent provirus reactivation, is designed to permanently suppress viral transcription. This might lead to a state of “deep latency,” which possibly helps in avoiding the issue of HIV-1 variability and resistance. To accomplish this goal, latency promoting agents (LPAs) are employed, targeting both viral- and host-specific mechanisms. These compounds are generally more well-tolerated by the patients than LRAs, even though the use of LPAs targeting host-specific mechanisms could, by their function, impair pro-inflammatory signals, thus further weakening the immunocompromised patient, as reviewed in (79). It has been recently proposed that the “shock and kill” and “block and lock” approaches could complement each other. In particular, these strategies might be combined to, on one hand, target and kill those cells, which are easily reached by the shock and kill strategy and, on the other hand, silence and block the residual latently infected cells (80).
Among the innovative therapeutic approaches aimed at controlling viral replication with a single administration, is gene therapy, that is, the introduction of genetic sequences in target cells to produce therapeutic nucleic acids or proteins. RNA interference (RNAi) has been used as a gene therapy strategy against HIV-1 and has shown promising results in different studies and clinical trials (81), as reviewed in (82). Particularly, RNAi works as a post-transcriptional gene regulatory system by using small RNA sequences (short hairpin RNA [shRNA], small interfering RNA [siRNA], micro RNA [miRNA]), which pair specifically with the target messenger RNA (mRNA). This, in turn, affects mRNA translation or, eventually, it may lead to its degradation, thus inhibiting gene expression, reviewed more extensively in (83).
RNAi has been exploited to target and silence the expression of almost all HIV-1 specific genes, among which tat and rev, which encode for regulatory proteins (Tat and Rev, respectively) essential for viral gene expression and for the transcription of full-length viral genomic RNA, structural genes such as gag, encoding for the nucleocapsid proteins, and the LTR. Despite proof of efficiency in the first short-term studies, longer term studies soon evidenced the emergence of resistant mutants due to mutations at the shRNA or siRNA target, depending on the approach used. In one of the first long-term expression investigations, the Authors showed that, in tat-targeting shRNA stably expressing cell lines, viral inhibition lasted only up to 25 days, and then the virus started replicating again, thanks to a point mutation acquired within the shRNA target region (84). The same finding was reported in a long-term study employing nef-targeting siRNA, where the Authors observed escape mutants at around the 23rd–27th day. This result was due, in some of the escape mutants, to a single base mutation at the siRNA target site, and in some others to more extensive deletions (more than 100 bp) of the nef region, which can accommodate bigger nucleotide removals, as even in the absence of Nef, viral replication can occur in vitro (85). These mutations had the signature of mutation introduced by the RT, as this viral enzyme able to transcribe RNA in DNA does not have a proofreading activity. HIV-1 resistance mechanisms are not only limited to the insertion of mutations at the target site, but they can also be related to mutations nearby, which induce an alternative three-dimensional structure in the viral RNA, making it inaccessible to RNAi (86).
As it happens with ART, the use of a combinatorial approach could limit viral escape. In fact, targeting more than one site in the viral genome (or also in the host genome as it will be described below) has been proven to be more effective at inhibiting viral replication than single-gene targeting, while, at the same time, significantly delaying the emergence of resistant mutants (87). However, targeting more than one site does not guarantee that escape will not occur. Indeed, indirect resistance mechanisms are also possible, when mutations at the HIV-1 promoter modulate and enhance transcriptional activity, conferring a generalised resistance to RNAi (88). In addition to using combinatorial strategies, a careful selection of the targets is paramount for the system’s efficiency, as the more HIV-1 target is conserved across different viral strains and subtypes, the less is the chance that they could harbour variants with RNAi resistant mutations (89, 90).
RNAi-based approaches have also been used to target CCR5, a cellular co-receptor used by HIV-1 to enter into host cells, for example, by shRNA-mediated CCR5 knockdown (91). In fact, up to date, only two people resulted functionally cured by HIV-1, the Berlin (92) and London (93) patients, who underwent a bone marrow transplantation from a donor with CCR5 homozygous 32 base pairs deletion, hence why CCR5 represents a very attractive target. Even if this approach has shown encouraging results, HIV-1 can switch its co-receptor usage from CCR5 to CXCR4, with a consequent viral escape from CCR5 knockdown approaches (94).
To solve this problem, a recent investigation reported the combined administration through lentiviral vectors of a shRNA-targeting CCR5 and of maC46, a fusion inhibitor which impedes the viral fusion to the cell membrane, thus possibly blocking CXCR4-tropic virus entry. Infection in Peripheral Blood Mononuclear Cells (PBMCs) was inhibited by 60% compared to the controls, without emergence of resistant mutants for up to 12 days (95). This study has been advanced to a phase 1/2 clinical trial designed as an ex vivo approach, reinfusing patients with hematopoietic stem cells (HSPCs) and CD4+ T lymphocytes modified with the combined strategy (ClinicalTrials.gov identifier: NCT01734850).
The most promising route to avoid the limitations presented in this section is probably the use of combinatorial therapies, which include diverse shRNA/siRNA targeting both host and viral genome as well as additional viral suppressive factors (96–99). Some studies have explored combinations of up to seven shRNAs (100).
Other than its efficiency, the main advantage of RNAi is that it only requires the delivery of small nucleotide sequences, potentially in a single administration, to achieve target-specific expression silencing. A step forward moving from this technique is the development of the CRISPR-Cas gene editing technologies, which can directly edit viral or host DNA and bring forward an alternative to the post-transcriptional regulation provided by RNAi approaches, as explored in the next section (101).
The CRISPR-Cas system is a bacteria-derived gene editing system which uses a nuclease (e.g., Cas9) complexed to a short guide RNA sequence (gRNA) to target specific DNA sequences that will be cut. This approach leads to the knockout of the targeted gene by disruption of the sequence/reading frame, or to the knock-in of the desired sequence when a homology template is provided (102, 103). As already mentioned, one of the main limitations of the current antiretroviral therapies is that they are not able to eliminate the integrated virus, especially in latently infected cells. Thus, the adoption of CRISPR-Cas9 to directly target the provirus is a very promising therapeutic approach. Indeed, many studies have reported the feasibility to disrupt, when targeting HIV-1 functional genes (104), or to excise, when targeting the LTRs (105), the provirus from latently or productively infected cells and from animal models of infection (18, 106, 107). This system has also been used to target the CCR5 host co-receptor, to block viral entry (108), as it had been proposed with RNAi-based strategies. Despite the efficiency of the system, HIV-1 may be able to escape even from this approach, by the introduction of mutations at the CRISPR-Cas9 target site.
In a seminal work from 2016 (109), an infected T-cell line was established stably expressing the CRISPR-Cas9 system. By adopting this tool, breakthrough of viral replication was observed over a time ranging from 10 to more than 40 days after infection, showing that the virus had successfully escaped. The fact that the breakthrough happened faster when less conserved HIV-1 sequences were targeted did not come as a surprise, as it had been already reported when using RNAi (101). What was surprising was that the escape mutants showed a peculiar mutation pattern. While in RNAi approaches escape mutants are mainly due to base substitutions introduced by the RT activity during viral replication, in this case, most of the mutations were concentrated as small indels (insertions/deletions) at the CRISPR-Cas9 cleavage site. Data suggested that the indels introduced by the cell non-homologous end joining (NHEJ) repair mechanisms, occurring at the DNA double-strand break (DSB), led to the formation of a viral sequence, which still retained its function and ability to replicate but was not recognized by gRNAs (Figure 3). In this way, the CRISPR-Cas9 system activity by itself accelerates the generation of escape mutants resistant to a new cut by the nuclease (109).
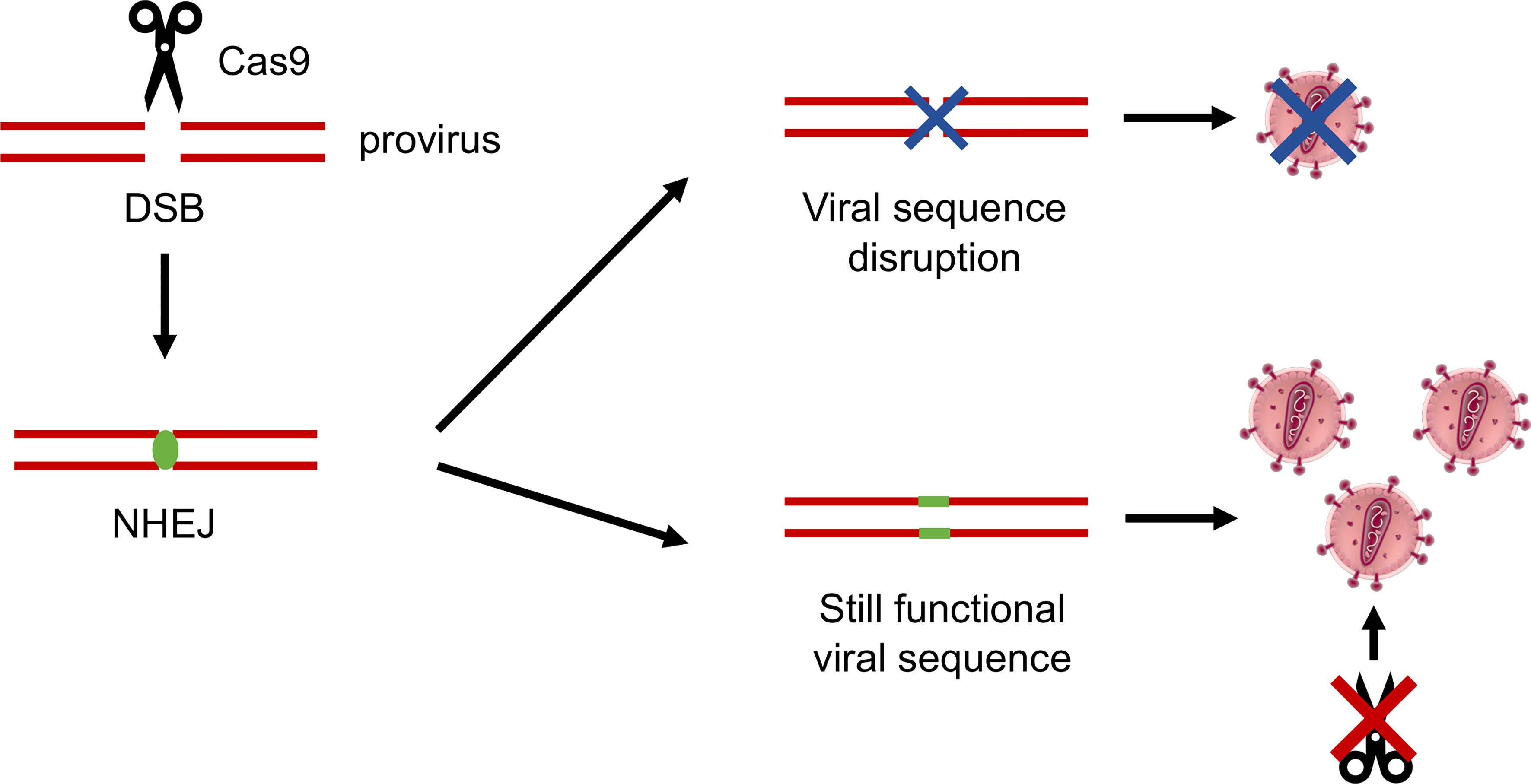
Figure 3 Schematic representation of the HIV-1 mechanisms of resistance to CRISPR-Cas9. The Cas9 nuclease operates a double-strand break (DSB) in the viral DNA sequence, which is repaired by the cellular non-homologous end joining (NHEJ) mechanisms of DNA repair. Indels introduced at the NHEJ site can effectively disrupt the virus, or can generate a sequence which is still functional, but in which the Cas9 recognition site is abolished, successfully generating CRISPR-Cas9–resistant mutant viruses.
The same year, a different research group (110) performed an in-depth analysis of the mutation pattern at the CRISPR-Cas9 target sites of resistant mutants. Different from Wang and co-workers, which hypothesized that most, if not all, the mutations at the target sites were due to the involvement of nuclease-activity–related NHEJ repair mechanisms, the Authors of this study also observed RT activity-derived base substitution escape mutants. In fact, they recognized that transitions were highly favoured upon transversions, signature of mutations introduced by the reverse transcription process. However, in concordance with the previous study, they also observed that the resistant mutants exhibited NHEJ-typical indels at the DSB, with the 5%–10% of them due to ±3 base pairs indels. This mostly happened when HIV-1 coding regions were targeted and lead to the preservation of viral gene reading frame (110).
A second mechanism through which the application of the CRISPR-Cas9 system itself could favor HIV-1 escape, is the formation of circular proviral DNA (111). Even though during the natural infection circular unintegrated forms of the proviral genome are normally present, it has been shown that, by targeting LTRs with a single gRNA, circular forms of the provirus increase both in latently and acutely infected cell models. In rare cases, the Authors observed in latently infected cells the re-integration of the circular DNA in a different region from the original one. This finding might be due to the integration of the excised proviral DNA in off-target DSB sites sharing partial homology with the excised segments, through a homology-directed repair mechanism. However, this phenomenon could not be observed in acutely infected cells, and the Authors did not further investigate its molecular features (111).
In all these studies, HIV-1 escape was faster when a single-guide RNA was used to target the provirus and when the DSB occurred in less conserved regions. Thus, targeting multiple and highly conserved sites in the viral genome seems to be the key to limit the onset of resistances, as it was reported by Wang and collaborators. They compared single and dual gRNAs targeting different HIV-1 regions and found that the latter approach showed no virus replication and no escape for up to 60 days, extended to an even longer period for gRNA combinations targeting highly conserved sites (112). To aid the design of more broad-acting and effective gRNA sequences, in silico predictive pipelines based on sequences shared by genetic variants and viral subtypes have been developed (113, 114). This further optimization may help in reducing viral escape from the CRISPR-Cas9 system. Interestingly, the most employed nuclease in HIV-1 gene targeting experiments has been Cas9 in its different orthologs (e.g., SpCas9 and SaCas9), as it was the first to be characterized and the most widely used. Some studies adopted alternative nucleases, such as Cas12a (115) or the RNA-targeting Cas13 (116), which have distinct sequence recognition and cleavage mechanisms, allowing to target regions different than those adjacent to a Cas9-specific PAM sequence and limiting resistance. Cas12a, in particular, was found to be very effective to inactivate HIV-1 provirus in long-term studies even when using only one gRNA, the opposite of what it had been previously observed with Cas9. This efficiency was attributed to a different mutation pattern introduced by the nuclease (115). While the use of different and optimized Cas nucleases could limit viral escape, it is important to note that older generation gene editing system such as zinc finger nucleases (ZFN) and transcription activator-like effector nucleases (TALEN) could lead to equally promising results, as both the systems were shown to be able to effectively excise HIV-1 provirus in infected cells (117, 118). Nevertheless, escape mechanisms are possible also in this case as reported in a 2016 study where, after targeting the HIV-1 pol sequence with ZFN, a ZFN-resistant virus with mutations in the RT region of pol was identified (119).
Overall, gene editing technologies are a rapidly developing field, and their importance as an innovative HIV-1 therapeutic approach is growing. As the issue of viral resistance could be overcome by exploiting different strategies, and taking into account the promising results obtained in vitro and in vivo, application of gene editing in vivo should be possible in a near future. In fact, clinical trials employing this technology in HIV-1–infected patients are already ongoing (e.g., EBT-101, ClinicalTrials.gov Identifier: NCT05144386). However, while envisioning its broad implementation in vivo, taking into account the potential limitations of this approach is paramount. First, gene editing could lead to unwanted off-target effects, which may be difficult to determine in vivo, although new strategies are under development to tackle this problem (120). In particular, efforts are directed at improving both the nuclease fidelity as well as the design of the gRNAs (121). Second, in vivo applications of this technique, especially in the context of HIV-1 latent infection, are limited by the delivery system. Different methods (both viral and non-viral) have been tested to deliver gene editing components in vivo. Up to now, the most promising results in murine and simian models of HIV-1 infection were accomplished by means of an adeno-associated viral vector (AAV9), able to allow transgene expression in different tissues (18, 107). This vector is currently being tested in the aforementioned EBT-101 clinical trial. Furthermore, there is no known marker of latency which could help to target latently infected cells, especially the even rarer subset harbouring replication-competent viruses (122). In addition, the fact that these cells are possibly hosted in difficult-to-reach tissues and body districts make the delivery of gene editing components where needed very challenging.
The current unfeasibility to achieve a definitive cure against HIV-1 infection has pushed forward the development of therapeutic approaches alternative or complementary to the standard of care. However, high genomic variability and mutational rate provide HIV-1 with the ability to escape from the most varied applications, making the quest to the cure a difficult goal to accomplish.
As discussed in this review, innovative therapeutic approaches do exist that, although promising, still suffer of the viral capability to resist and escape. Different strategies are under evaluation to overcome these issues. HIV-1 replication cycle is complex and deeply connected with the infected cell status and the antiviral immune response. What we have learned from the application of immunotherapies and shock and kill approaches is that the virus is able to hide from the immune response. For example, different studies here presented showed how (i) HIV-1 can mask its epitopes (glycan shield), (ii) the non-exposure of viral antigens in latently infected cells protects them from immune clearance, (iii) escape mutations can be introduced at the target sites of antibodies or CTLs, and (iv) the overall extreme viral heterogeneity requires the use of approaches which encompass antigen variability and their rational design and modelling. Strategies based on RNAi and CRISPR-Cas9 are additional promising technologies, which work at a genetic level by recognizing and targeting viral transcripts or the provirus itself. However, also in this case, experimental evidence demonstrated that the virus can readily escape by introducing mutations at the RNAi/nuclease recognition sites. Targeting highly conserved and multiple sequences may limit the onset of resistant mutants.
Indeed, those strategies which led to a control of viral replication for the longer periods of time suggest that combination is the key to success. Given the high-viral variability, it is difficult to think of one single drug, antibody, gene editing target, or other system that could cover all the possible variants. On the other side, it is challenging to envisage one single approach able to bypass all the strategies that HIV-1 has evolved to persist in different host compartments. Thus, not only combining the targeting of different conserved viral regions in the context of the same therapeutic strategy (e.g., using combinations of antibodies, LRAs, siRNAs, and gRNAs) but also applying different approaches together, with their various mechanisms of action, half-life, and penetration, could reduce, if not overcome, the problem of the viral escape. One clear example of the power of combination comes from a proof of principle study, published in 2019, which has shown, in a humanized mouse model of infection, that the use of a CRISPR-Cas9–based strategy targeting multiple viral sequences together with a novel, highly penetrating ART formulation, could lead to the eradication of the provirus from cells in one third of the animals, with no detectable viral rebound for the time of the experiment (up to 5 weeks) (18).
Altogether, the studies reported here give a bright outlook on the future development of innovative therapeutic approaches against HIV-1, showing how, even if the virus is apt to find ways to escape, numerous strategies may be devised and implemented to target different variants and to limit the emergence of resistances.
Conceptualization, GM, CP; writing—original draft preparation, GM; writing—review and editing, GM, AC, CP. All Authors have read and agreed to the published version of the manuscript.
GM is a recipient of a fellowship funded by PARO_FINA20_01 to CP. The studies of the authors cited in the review were funded by grants from ANRS (Agence Nationale de Recherche sur le Sida et les hépatites virales, Agreement n. 13018), Istituto Superiore di Sanità (Rome-AIDS Project 40H98) and Regione Veneto (Ricerca Sanitaria Finalizzata n.312/10) to CP.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science. (1983) 220(4599):868–71. doi: 10.1126/science.6189183
2. Haberer JE, Musinguzi N, Boum Y, Siedner MJ, Mocello AR, Hunt PW, et al. Duration of antiretroviral therapy adherence interruption is associated with risk of virologic rebound as determined by real-time adherence monitoring in rural Uganda. J Acquir Immune Defic Syndr (2015) 70(4):386–92. doi: 10.1097/QAI.0000000000000737
3. Raymond S, Piffaut M, Bigot J, Cazabat M, Montes B, Bertrand K, et al. Sexual transmission of an extensively drug-resistant HIV-1 strain. Lancet HIV. (2020) 7(8):e529–30. doi: 10.1016/S2352-3018(20)30205-8
4. World Health Organization. HIV Drug resistance report 2021 (2021). Geneva: World Health Organization. Available at: https://apps.who.int/iris/handle/10665/349340 (Accessed 2022 Feb 21).
5. van der Kuyl AC, Berkhout B. The biased nucleotide composition of the HIV genome: a constant factor in a highly variable virus. Retrovirology. (2012) 9(1):92. doi: 10.1186/1742-4690-9-92
6. Craigie R, Bushman FD. HIV DNA Integration. Cold Spring Harbor Perspect Med (2012) 2(7):a006890–a006890. doi: 10.1101/cshperspect.a006890
7. Chavez L, Calvanese V, Verdin E. HIV Latency is established directly and early in both resting and activated primary CD4 T cells. PLoS Pathog (2015) 11(6):e1004955. doi: 10.1371/journal.ppat.1004955
8. Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J Virol (2010) 84(19):9864–78. doi: 10.1128/JVI.00915-10
9. Wymant C, Bezemer D, Blanquart F, Ferretti L, Gall A, Hall M, et al. A highly virulent variant of HIV-1 circulating in the Netherlands. Science (2022) 375(6580):540–5. doi: 10.1126/science.abk1688
10. Feder AF, Harper KN, Brumme CJ, Pennings PS. Understanding patterns of HIV multi-drug resistance through models of temporal and spatial drug heterogeneity. Elife (2021) 10:e69032. doi: 10.7554/eLife.69032.sa2
11. Günthard HF, Calvez V, Paredes R, Pillay D, Shafer RW, Wensing AM, et al. Human immunodeficiency virus drug resistance: 2018 recommendations of the international antiviral society–USA panel. Clin Infect Diseases (2019) 68(2):177–87. doi: 10.1093/cid/ciy463
12. Clavel F, Hance AJ. HIV Drug resistance. N Engl J Med (2004) 350(10):1023–35. doi: 10.1056/NEJMra025195
13. Zanini F, Puller V, Brodin J, Albert J, Neher RA. In vivo mutation rates and the landscape of fitness costs of HIV-1. Virus Evol (2017) 3(1):vex003. doi: 10.1093/ve/vex003
14. Byrd KK, Hou JG, Hazen R, Kirkham H, Suzuki S, Clay PG, et al. Antiretroviral adherence level necessary for HIV viral suppression using real-world data. J Acquir Immune Defic Syndr (2019) 82(3):245–51. doi: 10.1097/QAI.0000000000002142
15. McComsey GA, Lingohr-Smith M, Rogers R, Lin J, Donga P. Real-world adherence to antiretroviral therapy among HIV-1 patients across the United States. Adv Ther (2021) 38(9):4961–74. doi: 10.1007/s12325-021-01883-8
16. Thoueille P, Choong E, Cavassini M, Buclin T, Decosterd LA. Long-acting antiretrovirals: a new era for the management and prevention of HIV infection. J Antimicrobial Chemother (2022) 77(2):290–302. doi: 10.1093/jac/dkab324
17. Swindells S, Andrade-Villanueva JF, Richmond GJ, Rizzardini G, Baumgarten A, Masiá M, et al. Long-acting cabotegravir and rilpivirine for maintenance of HIV-1 suppression. N Engl J Med (2020) 382(12):1112–23. doi: 10.1056/NEJMoa1904398
18. Dash PK, Kaminski R, Bella R, Su H, Mathews S, Ahooyi TM, et al. Sequential LASER ART and CRISPR treatments eliminate HIV-1 in a subset of infected humanized mice. Nat Commun (2019) 10(1):2753. doi: 10.1038/s41467-019-10366-y
19. Yan J, Sabbaj S, Bansal A, Amatya N, Shacka JJ, Goepfert PA, et al. HIV-Specific CD8 + T cells from elite controllers are primed for survival. J Virol (2013) 87(9):5170–81. doi: 10.1128/JVI.02379-12
20. Adams P, Iserentant G, Servais JY, Vandekerckhove L, Vanham G, Seguin-Devaux C, et al. Cytotoxic CD8+ T cells expressing CXCR5 are detectable in HIV-1 elite controllers after prolonged In vitro peptide stimulation. Front Immunol (2021) 11:622343. doi: 10.3389/fimmu.2020.622343
21. Jiang C, Lian X, Gao C, Sun X, Einkauf KB, Chevalier JM, et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature. (2020) 585(7824):261–7. doi: 10.1038/s41586-020-2651-8
22. Nickle DC, Rolland M, Jensen MA, Pond SLK, Deng W, Seligman M, et al. Coping with viral diversity in HIV vaccine design. PLoS Comput Biol (2007) 3(4):e75. doi: 10.1371/journal.pcbi.0030075
23. Barouch DH, Kunstman J, Glowczwskie J, Kunstman KJ, Egan MA, Peyerl FW, et al. Viral escape from dominant simian immunodeficiency virus epitope-specific cytotoxic T lymphocytes in DNA-vaccinated rhesus monkeys. J Virol (2003) 77(13):7367–75. doi: 10.1128/JVI.77.13.7367-7375.2003
24. Herbeck JT, Peebles K, Edlefsen PT, Rolland M, Murphy JT, Gottlieb GS, et al. HIV Population-level adaptation can rapidly diminish the impact of a partially effective vaccine. Vaccine (2018) 36(4):514–20. doi: 10.1016/j.vaccine.2017.12.004
25. Peebles K, Mittler JE, Goodreau SM, Murphy JT, Reid MC, Abernethy N, et al. Risk compensation after HIV-1 vaccination may accelerate viral adaptation and reduce cost-effectiveness: a modeling study. Sci Rep (2021) 11(1):6798. doi: 10.1038/s41598-021-85487-w
26. Rathore U, Saha P, Kesavardhana S, Kumar AA, Datta R, Devanarayanan S, et al. Glycosylation of the core of the HIV-1 envelope subunit protein gp120 is not required for native trimer formation or viral infectivity. J Biol Chem (2017) 292(24):10197–219. doi: 10.1074/jbc.M117.788919
27. Ferreira RC, Grant OC, Moyo T, Dorfman JR, Woods RJ, Travers SA, et al. Structural rearrangements maintain the glycan shield of an HIV-1 envelope trimer after the loss of a glycan. Sci Rep (2018) 8(1):15031. doi: 10.1038/s41598-018-33390-2
28. Wang S, Voronin Y, Zhao P, Ishihara M, Mehta N, Porterfield M, et al. Glycan profiles of gp120 protein vaccines from four major HIV-1 subtypes produced from different host cell lines under non-GMP or GMP conditions. Silvestri G editor. J Virol (2020) 94(7):e01968–19. doi: 10.1128/JVI.01968-19
29. Derking R, Allen JD, Cottrell CA, Sliepen K, Seabright GE, Lee WH, et al. Enhancing glycan occupancy of soluble HIV-1 envelope trimers to mimic the native viral spike. Cell Rep (2021) 35(1):108933. doi: 10.1016/j.celrep.2021.108933
30. Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med (2009) 361(23):2209–20. doi: 10.1056/NEJMoa0908492
31. Karasavvas N, Billings E, Rao M, Williams C, Zolla-Pazner S, Bailer RT, et al. The Thai phase III HIV type 1 vaccine trial (RV144) regimen induces antibodies that target conserved regions within the V2 loop of gp120. AIDS Res Hum Retroviruses (2012) 28(11):1444–57. doi: 10.1089/aid.2012.0103
32. Gay CL, DeBenedette MA, Tcherepanova IY, Gamble A, Lewis WE, Cope AB, et al. Immunogenicity of AGS-004 dendritic cell therapy in patients treated during acute HIV infection. AIDS Res Hum Retroviruses (2018) 34(1):111–22. doi: 10.1089/aid.2017.0071
33. de JW, Leal L, Buyze J, Pannus P, Guardo A, Salgado M, et al. Therapeutic vaccine in chronically HIV-1-Infected patients: A randomized, double-blind, placebo-controlled phase IIa trial with HTI-TriMix. Vaccines (Basel). (2019) 7(4):E209. doi: 10.3390/vaccines7040209
34. Khalid K, Padda J, Khedr A, Ismail D, Zubair U, Al-Ewaidat OA, et al. HIV and Messenger RNA (mRNA) Vaccine. Cureus. (2021) 13(7):e16197. doi: 10.7759/cureus.16197
35. Steichen JM, Kulp DW, Tokatlian T, Escolano A, Dosenovic P, Stanfield RL, et al. HIV Vaccine design to target germline precursors of glycan-dependent broadly neutralizing antibodies. Immunity. (2016) 45(3):483–96. doi: 10.1016/j.immuni.2016.08.016
36. Trkola A, Kuster H, Rusert P, Joos B, Fischer M, Leemann C, et al. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat Med (2005) 11(6):615–22. doi: 10.1038/nm1244
37. Caskey M, Klein F, Lorenzi JCC, Seaman MS, West AP, Buckley N, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature. (2015) 522(7557):487–91. doi: 10.1038/nature14411
38. Lynch RM, Boritz E, Coates EE, DeZure A, Madden P, Costner P, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci Transl Med (2015) 7(319):319ra206. doi: 10.1126/scitranslmed.aad5752
39. Nishimura Y, Gautam R, Chun TW, Sadjadpour R, Foulds KE, Shingai M, et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature. (2017) 543(7646):559–63. doi: 10.1038/nature21435
40. Mendoza P, Gruell H, Nogueira L, Pai JA, Butler AL, Millard K, et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature. (2018) 561(7724):479–84. doi: 10.1038/s41586-018-0531-2
41. Bar-On Y, Gruell H, Schoofs T, Pai JA, Nogueira L, Butler AL, et al. Safety and antiviral activity of combination HIV-1 broadly neutralizing antibodies in viremic individuals. Nat Med (2018) 24(11):1701–7. doi: 10.1038/s41591-018-0186-4
42. Schommers P, Gruell H, Abernathy ME, Tran MK, Dingens AS, Gristick HB, et al. Restriction of HIV-1 escape by a highly broad and potent neutralizing antibody. Cell. (2020) 180(3):471–489.e22. doi: 10.1016/j.cell.2020.01.010
43. Rawi R, Mall R, Shen CH, Farney SK, Shiakolas A, Zhou J, et al. Accurate prediction for antibody resistance of clinical HIV-1 isolates. Sci Rep (2019) 9(1):14696. doi: 10.1038/s41598-019-50635-w
44. Meijers M, Vanshylla K, Gruell H, Klein F, Lässig M. Predicting in vivo escape dynamics of HIV-1 from a broadly neutralizing antibody. Proc Natl Acad Sci USA (2021) 118(30):e2104651118. doi: 10.1073/pnas.2104651118
45. Emu B, Fessel J, Schrader S, Kumar P, Richmond G, Win S, et al. Phase 3 study of ibalizumab for multidrug-resistant HIV-1. N Engl J Med (2018) 379(7):645–54. doi: 10.1056/NEJMoa1711460
46. Jacobson JM, Kuritzkes DR, Godofsky E, DeJesus E, Larson JA, Weinheimer SP, et al. Safety, pharmacokinetics, and antiretroviral activity of multiple doses of ibalizumab (formerly TNX-355), an anti-CD4 monoclonal antibody, in human immunodeficiency virus type 1-infected adults. Antimicrob Agents Chemother (2009) 53(2):450–7. doi: 10.1128/AAC.00942-08
47. Umotoy JC, de Taeye SW. Antibody conjugates for targeted therapy against HIV-1 as an emerging tool for HIV-1 cure. Front Immunol (2021) 12:708806. doi: 10.3389/fimmu.2021.708806
48. Rodari A, Darcis G, Van Lint CM. The current status of latency reversing agents for HIV-1 remission. Annu Rev Virol (2021) 8(1):491–514. doi: 10.1146/annurev-virology-091919-103029
49. Gramatica A, Schwarzer R, Brantley W, Varco-Merth B, Sperber HS, Hull PA, et al. Evaluating a new class of AKT/mTOR activators for HIV latency reversing activity ex vivo and in vivo. J Virol (2021) 3:JVI.02393–20. doi: 10.1128/JVI.02393-20
50. Shin-ichiro H, Matsuda K, Tsuchiya K, Gatanaga H, Oka S, Yoshimura K, et al. Combination of a latency-reversing agent with a Smac mimetic minimizes secondary HIV-1 infection in vitro. Front Microbiol (2018) 9:2022. doi: 10.3389/fmicb.2018.02022
51. Kula-Pacurar A, Rodari A, Darcis G, Van Lint C. Shocking HIV-1 with immunomodulatory latency reversing agents. Semin Immunol (2021) 51:101478. doi: 10.1016/j.smim.2021.101478
52. Kim Y, Anderson JL, Lewin SR. Getting the “Kill” into “Shock and kill”: Strategies to eliminate latent HIV. Cell Host Microbe (2018) 23(1):14–26. doi: 10.1016/j.chom.2017.12.004
53. Cummins NW, Sainski AM, Dai H, Natesampillai S, Pang YP, Bren GD, et al. Prime, shock, and kill: Priming CD4 T cells from HIV patients with a BCL-2 antagonist before HIV reactivation reduces HIV reservoir size. J Virol (2016) 90(8):4032–48. doi: 10.1128/JVI.03179-15
54. Borducchi EN, Liu J, Nkolola JP, Cadena AM, Yu WH, Fischinger S, et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature. (2018) 563(7731):360–4. doi: 10.1038/s41586-018-0600-6
55. Fidler S, Stöhr W, Pace M, Dorrell L, Lever A, Pett S, et al. Antiretroviral therapy alone versus antiretroviral therapy with a kick and kill approach, on measures of the HIV reservoir in participants with recent HIV infection (the RIVER trial): a phase 2, randomised trial. Lancet (2020) 395(10227):888–98. doi: 10.1016/S0140-6736(19)32990-3
56. Nixon CC, Mavigner M, Sampey GC, Brooks AD, Spagnuolo RA, Irlbeck DM, et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature (2020) 578(7793):160–5. doi: 10.1038/s41586-020-1951-3
57. McBrien JB, Mavigner M, Franchitti L, Smith SA, White E, Tharp GK, et al. Robust and persistent reactivation of SIV and HIV by n-803 and depletion of CD8+ cells. Nature. (2020) 578(7793):154–9. doi: 10.1038/s41586-020-1946-0
58. Grau-Expósito J, Luque-Ballesteros L, Navarro J, Curran A, Burgos J, Ribera E, et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. Swanstrom R editor. PLoS Pathog (2019) 15(8):e1007991. doi: 10.1371/journal.ppat.1007991
59. Ait-Ammar A, Kula A, Darcis G, Verdikt R, De Wit S, Gautier V, et al. Current status of latency reversing agents facing the heterogeneity of HIV-1 cellular and tissue reservoirs. Front Microbiol (2019) 10:3060. doi: 10.3389/fmicb.2019.03060
60. Wallet C, De Rovere M, Van Assche J, Daouad F, De Wit S, Gautier V, et al. Microglial cells: The main HIV-1 reservoir in the brain. Front Cell Infect Microbiol (2019) 9:362. doi: 10.3389/fcimb.2019.00362
61. Ruiz A, Blanch-Lombarte O, Jimenez-Moyano E, Ouchi D, Mothe B, Peña R, et al. Antigen production after latency reversal and expression of inhibitory receptors in CD8+ T cells limit the killing of HIV-1 reactivated cells. Front Immunol (2019) 9:3162. doi: 10.3389/fimmu.2018.03162
62. Walker-Sperling VE, Pohlmeyer CW, Tarwater PM, Blankson JN. The effect of latency reversal agents on primary CD8 + T cells: Implications for shock and kill strategies for human immunodeficiency virus eradication. EBioMedicine. (2016) 8:217–29. doi: 10.1016/j.ebiom.2016.04.019
63. Gruell H, Gunst JD, Cohen YZ, Pahus MH, Malin JJ, Platten M, et al. Effect of 3BNC117 and romidepsin on the HIV-1 reservoir in people taking suppressive antiretroviral therapy (ROADMAP): a randomised, open-label, phase 2A trial. Lancet Microbe (2022) 3(3):e203–14. doi: 10.1016/S2666-5247(21)00239-1
64. Garrido C, Spivak AM, Soriano-Sarabia N, Checkley MA, Barker E, Karn J, et al. HIV Latency-reversing agents have diverse effects on natural killer cell function. Front Immunol (2016) 7:356. doi: 10.3389/fimmu.2016.00356/abstract
65. Garrido C, Tolstrup M, Søgaard OS, Rasmussen TA, Allard B, Soriano-Sarabia N, et al. In-vivo administration of histone deacetylase inhibitors does not impair natural killer cell function in HIV+ individuals. AIDS. (2019) 33(4):605–13. doi: 10.1097/QAD.0000000000002112
66. French AJ, Natesampillai S, Krogman A, Correia C, Peterson KL, Alto A, et al. Reactivating latent HIV with PKC agonists induces resistance to apoptosis and is associated with phosphorylation and activation of BCL2. PLoS Pathog (2020) 16(10):e1008906. doi: 10.1371/journal.ppat.1008906
67. Timilsina U, Gaur R. Modulation of apoptosis and viral latency – an axis to be well understood for successful cure of human immunodeficiency virus. J Gen Virol (2016) 97(4):813–24. doi: 10.1099/jgv.0.000402
68. Liu MKP, Hawkins N, Ritchie AJ, Ganusov VV, Whale V, Brackenridge S, et al. Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J Clin Invest (2012) 123(1):380–93. doi: 10.1172/JCI65330
69. Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. (2015) 517(7534):381–5. doi: 10.1038/nature14053
70. Warren JA, Zhou S, Xu Y, Moeser MJ, MacMillan DR, Council O, et al. The HIV-1 latent reservoir is largely sensitive to circulating T cells. Elife. (2020) 9:e57246. doi: 10.7554/eLife.57246
71. Mann JFS, Pankrac J, Klein K, McKay PF, King DFL, Gibson R, et al. A targeted reactivation of latent HIV-1 using an activator vector in patient samples from acute infection. eBioMedicine. (2020) 59:102853. doi: 10.1016/j.ebiom.2020.102853
72. Kristoff J, Palma ML, Garcia-Bates TM, Shen C, Sluis-Cremer N, Gupta P, et al. Type 1-programmed dendritic cells drive antigen-specific latency reversal and immune elimination of persistent HIV-1. EBioMedicine. (2019) 43:295–306. doi: 10.1016/j.ebiom.2019.03.077
73. Ji H, Jiang Z, Lu P, Ma L, Li C, Pan H, et al. Specific reactivation of latent HIV-1 by dCas9-SunTag-VP64-mediated guide RNA targeting the HIV-1 promoter. Mol Ther (2016) 24(3):508–21. doi: 10.1038/mt.2016.7
74. Zhang Y, Arango G, Li F, Xiao X, Putatunda R, Yu J, et al. Comprehensive off-target analysis of dCas9-SAM-mediated HIV reactivation via long noncoding RNA and mRNA profiling. BMC Med Genomics (2018) 11(1):78. doi: 10.1186/s12920-018-0394-2
75. Zhang Y, Yin C, Zhang T, Li F, Yang W, Kaminski R, et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci Rep (2015) 5:16277. doi: 10.1038/srep16277
76. Golumbeanu M, Cristinelli S, Rato S, Munoz M, Cavassini M, Beerenwinkel N, et al. Single-cell RNA-seq reveals transcriptional heterogeneity in latent and reactivated HIV-infected cells. Cell Rep (2018) 23(4):942–50. doi: 10.1016/j.celrep.2018.03.102
77. Lu Y, Singh H, Singh A, Dar RD. A transient heritable memory regulates HIV reactivation from latency. iScience. (2021) 24(4):102291. doi: 10.1016/j.isci.2021.102291
78. Bouchat S, Delacourt N, Kula A, Darcis G, Van Driessche B, Corazza F, et al. Sequential treatment with 5-aza-2’-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol Med (2016) 8(2):117–38. doi: 10.15252/emmm.201505557
79. Abner E, Jordan A. HIV “shock and kill” therapy: In need of revision. Antiviral Res (2019) 166:19–34. doi: 10.1016/j.antiviral.2019.03.008
80. Acchioni C, Palermo E, Sandini S, Acchioni M, Hiscott J, Sgarbanti M. Fighting HIV-1 persistence: At the crossroads of “Shoc-K and b-lock”. Pathogens (2021) 10(11):1517. doi: 10.3390/pathogens10111517
81. DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A, et al. RNA-Based gene therapy for HIV with lentiviral vector–modified CD34 + cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med (2010) 2(36):36ra43. doi: 10.1126/scitranslmed.3000931
82. Scarborough R, Gatignol A. RNA Interference therapies for an HIV-1 functional cure. Viruses. (2017) 10(1):8. doi: 10.3390/v10010008
83. Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discovery (2019) 18(6):421–46. doi: 10.1038/s41573-019-0017-4
84. Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol (2003) 77(21):11531–5. doi: 10.1128/JVI.77.21.11531-11535.2003
85. Das AT, Brummelkamp TR, Westerhout EM, Vink M, Madiredjo M, Bernards R, et al. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol (2004) 78(5):2601–5. doi: 10.1128/JVI.78.5.2601-2605.2004
86. Westerhout EM. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res (2005) 33(2):796–804. doi: 10.1093/nar/gki220
87. ter Brake O, Konstantinova P, Ceylan M, Berkhout B. Silencing of HIV-1 with RNA interference: a multiple shRNA approach. Mol Ther (2006) 14(6):883–92. doi: 10.1016/j.ymthe.2006.07.007
88. Shah PS, Pham NP, Schaffer DV. HIV Develops indirect cross-resistance to combinatorial RNAi targeting two distinct and spatially distant sites. Mol Ther (2012) 20(4):840–8. doi: 10.1038/mt.2012.3
89. Herrera-Carrillo E, Berkhout B. The impact of HIV-1 genetic diversity on the efficacy of a combinatorial RNAi-based gene therapy. Gene Ther (2015) 22(6):485–95. doi: 10.1038/gt.2015.11
90. Kretova OV, Fedoseeva DM, Gorbacheva MA, Gashnikova NM, Gashnikova MP, Melnikova NV, et al. Six highly conserved targets of RNAi revealed in HIV-1-Infected patients from Russia are also present in many HIV-1 strains worldwide. Mol Ther Nucleic Acids (2017) 8:330–44. doi: 10.1016/j.omtn.2017.07.010
91. Shimizu S, Hong P, Arumugam B, Pokomo L, Boyer J, Koizumi N, et al. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood. (2010) 115(8):1534–44. doi: 10.1182/blood-2009-04-215855
92. Allers K, Hütter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, et al. Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood. (2011) 117(10):2791–9. doi: 10.1182/blood-2010-09-309591
93. Gupta RK, Peppa D, Hill AL, Gálvez C, Salgado M, Pace M, et al. Evidence for HIV-1 cure after CCR5Δ32/Δ32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: a case report. Lancet HIV. (2020) 7(5):e340–7. doi: 10.1016/S2352-3018(20)30069-2
94. Connell BJ, Hermans LE, Wensing AMJ, Schellens I, Schipper PJ, van Ham PM, et al. Immune activation correlates with and predicts CXCR4 co-receptor tropism switch in HIV-1 infection. Sci Rep (2020) 10(1):15866. doi: 10.1038/s41598-020-71699-z
95. Ledger S, Howe A, Turville S, Aggarwal A, Savkovic B, Ong A, et al. Analysis and dissociation of anti-HIV effects of shRNA to CCR5 and the fusion inhibitor C46. J Gene Med (2018) 20(2–3):e3006. doi: 10.1002/jgm.3006
96. Spanevello F, Calistri A, Del Vecchio C, Mantelli B, Frasson C, Basso G, et al. Development of lentiviral vectors simultaneously expressing multiple siRNAs against CCR5, vif and tat/rev genes for an HIV-1 gene therapy approach. Mol Ther Nucleic Acids (2016) 5:e312. doi: 10.1038/mtna.2016.24
97. Liu YP, von Eije KJ, Schopman NC, Westerink JT, ter BO, Haasnoot J, et al. Combinatorial RNAi against HIV-1 using extended short hairpin RNAs. Mol Ther (2009) 17(10):1712–23. doi: 10.1038/mt.2009.176
98. Walker JE, Chen RX, McGee J, Nacey C, Pollard RB, Abedi M, et al. Generation of an HIV-1-Resistant immune system with CD34 + hematopoietic stem cells transduced with a triple-combination anti-HIV lentiviral vector. J Virol (2012) 86(10):5719–29. doi: 10.1128/JVI.06300-11
99. Li MJ, Kim J, Li S, Zaia J, Yee JK, Anderson J, et al. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol Ther (2005) 12(5):900–9. doi: 10.1016/j.ymthe.2005.07.524
100. Choi JG, Bharaj P, Abraham S, Ma H, Yi G, Ye C, et al. Multiplexing seven miRNA-based shRNAs to suppress HIV replication. Mol Ther (2015) 23(2):310–20. doi: 10.1038/mt.2014.205
101. Herrera-Carrillo E, Berkhout B. Attacking HIV-1 RNA versus DNA by sequence-specific approaches: RNAi versus CRISPR-Cas. Biochem Soc Trans (2016) 44(5):1355–65. doi: 10.1042/BST20160060
102. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity (2012) 337(6096):816–21. doi: 10.1126/science.1225829
103. Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. (2014) 346(6213):1258096. doi: 10.1126/science.1258096
104. Ophinni Y, Inoue M, Kotaki T, Kameoka M. CRISPR/Cas9 system targeting regulatory genes of HIV-1 inhibits viral replication in infected T-cell cultures. Sci Rep (2018) 8(1):7784. doi: 10.1038/s41598-018-26190-1
105. Kaminski R, Chen Y, Fischer T, Tedaldi E, Napoli A, Zhang Y, et al. Elimination of HIV-1 genomes from human T-lymphoid cells by CRISPR/Cas9 gene editing. Sci Rep (2016) 6(1):22555. doi: 10.1038/srep22555
106. Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X, et al. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol Ther (2017) 25(5):1168–86. doi: 10.1016/j.ymthe.2017.03.012
107. Mancuso P, Chen C, Kaminski R, Gordon J, Liao S, Robinson JA, et al. CRISPR based editing of SIV proviral DNA in ART treated non-human primates. Nat Commun (2020) 11(1):6065. doi: 10.1038/s41467-020-19821-7
108. Schmidt JK, Strelchenko N, Park MA, Kim YH, Mean KD, Schotzko ML, et al. Genome editing of CCR5 by CRISPR-Cas9 in Mauritian cynomolgus macaque embryos. Sci Rep (2020) 10(1):18457. doi: 10.1038/s41598-020-75295-z
109. Wang G, Zhao N, Berkhout B, Das AT. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol Ther (2016) 24(3):522–6. doi: 10.1038/mt.2016.24
110. Yoder KE, Bundschuh R. Host double strand break repair generates HIV-1 strains resistant to CRISPR/Cas9. Sci Rep (2016) 6(1):29530. doi: 10.1038/srep29530
111. Lai M, Maori E, Quaranta P, Matteoli G, Maggi F, Sgarbanti M, et al. CRISPR/Cas9 ablation of integrated HIV-1 accumulates proviral DNA circles with reformed long terminal repeats. J Virol (2021) 95(23):e01358–21. doi: 10.1128/JVI.01358-21
112. Wang G, Zhao N, Berkhout B, Das AT. A combinatorial CRISPR-Cas9 attack on HIV-1 DNA extinguishes all infectious provirus in infected T cell cultures. Cell Rep (2016) 17(11):2819–26. doi: 10.1016/j.celrep.2016.11.057
113. Sullivan NT, Dampier W, Chung CH, Allen AG, Atkins A, Pirrone V, et al. Novel gRNA design pipeline to develop broad-spectrum CRISPR/Cas9 gRNAs for safe targeting of the HIV-1 quasispecies in patients. Sci Rep (2019) 9(1):17088. doi: 10.1038/s41598-019-52353-9
114. Chung CH, Allen AG, Atkins A, Link RW, Nonnemacher MR, Dampier W, et al. Computational design of gRNAs targeting genetic variants across HIV-1 subtypes for CRISPR-mediated antiviral therapy. Front Cell Infect Microbiol (2021) 11:593077. doi: 10.3389/fcimb.2021.593077
115. Gao Z, Fan M, Das AT, Herrera-Carrillo E, Berkhout B. Extinction of all infectious HIV in cell culture by the CRISPR-Cas12a system with only a single crRNA. Nucleic Acids Res (2020) 48(10):5527–39. doi: 10.1093/nar/gkaa226
116. Yin L, Zhao F, Sun H, Wang Z, Huang Y, Zhu W, et al. CRISPR-Cas13a inhibits HIV-1 infection. Mol Ther - Nucleic Acids (2020) 21:147–55. doi: 10.1016/j.omtn.2020.05.030
117. Qu X, Wang P, Ding D, Li L, Wang H, Ma L, et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res (2013) 41(16):7771–82. doi: 10.1093/nar/gkt571
118. Ebina H, Kanemura Y, Misawa N, Sakuma T, Kobayashi T, Yamamoto T, et al. A high excision potential of TALENs for integrated DNA of HIV-based lentiviral vector. PLoS One (2015) 10(3):e0120047. doi: 10.1371/journal.pone.0120047
119. De Silva Feelixge HS, Stone D, Pietz HL, Roychoudhury P, Greninger AL, Schiffer JT, et al. Detection of treatment-resistant infectious HIV after genome-directed antiviral endonuclease therapy. Antiviral Res (2016) 126:90–8. doi: 10.1016/j.antiviral.2015.12.007
120. Liang SQ, Liu P, Smith JL, Mintzer E, Maitland S, Dong X, et al. Genome-wide detection of CRISPR editing in vivo using GUIDE-tag. Nat Commun (2022) 13(1):437. doi: 10.1038/s41467-022-28135-9
121. Naeem M, Majeed S, Hoque MZ, Ahmad I. Latest developed strategies to minimize the off-target effects in CRISPR-Cas-Mediated genome editing. Cells. (2020) 9(7):E1608. doi: 10.3390/cells9071608
Keywords: HIV, drug resistance, viral escape, antiretroviral therapies, CRISPR-Cas, immunotherapies, shock and kill, RNAi
Citation: Magro G, Calistri A and Parolin C (2022) How to break free: HIV-1 escapes from innovative therapeutic approaches. Front. Virol. 2:933418. doi: 10.3389/fviro.2022.933418
Received: 30 April 2022; Accepted: 30 June 2022;
Published: 29 July 2022.
Edited by:
Mayte Coiras, Instituto de Salud Carlos III (ISCIII), SpainReviewed by:
Nancie M. Archin, University of North Carolina Hospitals, United StatesCopyright © 2022 Magro, Calistri and Parolin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Parolin, Y3Jpc3RpbmEucGFyb2xpbkB1bmlwZC5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.