
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol. , 08 July 2022
Sec. Virus and Host Immunity
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.916095
 Doty Ojwach1
Doty Ojwach1 Kamini Gounder1,2
Kamini Gounder1,2 Takalani Mulaudzi1
Takalani Mulaudzi1 Nombali Gumede1
Nombali Gumede1 Omolara O. Baiyegunhi1,2Kavidha Reddy1,2
Omolara O. Baiyegunhi1,2Kavidha Reddy1,2 Jennifer Giandhari3,4
Jennifer Giandhari3,4 Krista L. Dong5Zaza Ndhlovu1,2,5
Krista L. Dong5Zaza Ndhlovu1,2,5 Thumbi Ndung’u1,2,5,6
Thumbi Ndung’u1,2,5,6 Jaclyn K. Mann1*
Jaclyn K. Mann1*HIV variants present in the reservoir, particularly in tissues, may differ from those present in peripheral blood prior to therapy initiation, and characterisation of these reservoir variants could better inform immune-based interventions for HIV cure. In the present study, Gag sequence differences between variants derived from the lymph node and peripheral blood mononuclear cell (PBMC) reservoirs as well as those derived from pre-therapy plasma, were investigated in 24 HIV-1 subtype C-infected individuals. HIV gag amplification was successful for 20 individuals, where 4 were controls including one untreated individual and 3 early treated individuals with LN collection within 2 weeks of treatment initiation. The remaining 16 individuals with LN and PBMC collection > 3 months after treatment initiation (median = 665 days), were further characterised. Recombinant viruses encoding patient-derived Gag-protease sequences from the pre-therapy plasma, LN reservoir, and PBMC reservoir, were constructed and the replication-competent viruses that grew in vitro were used to further investigate whether there are specific features of Gag reservoir variants that may have relevance for strategies to cure HIV. Virus characteristics measured included replication capacity, interferon-alpha resistance, cell-to-cell spread ability, and induction of antiviral cytokines. A limited number of novel Gag mutations (median = 4) in the reservoir of 3/7 early treated participants and 9/9 late treated participants were observed, where the majority of these mutations were likely cytotoxic T lymphocyte (CTL)-driven and 48% were represented in the replication-competent viruses. The reservoir variants had very few unique potential CTL escape mutations (median = 3) in Gag compared to the number of these Gag mutations that were already present in the plasma-derived virus (median = 23) at the time of treatment initiation, which was similar whether treatment was initiated late or early. The data suggest that the extent of CTL escape in Gag overall is likely similar between early and late treated individuals as well as between the reservoir and pre-therapy variants. The sequence differences in Gag that were unique to the reservoir viruses did not result in significantly altered virus characteristics overall, and are therefore unlikely to affect effectiveness of immune-based interventions for virus eradication.
Antiretroviral therapy (ART) is highly effective in limiting HIV replication, prolonging the life span of infected individuals and reducing HIV transmission (1). However, ART is not curative due to the persistence of an infected reservoir of cells (2). Therefore, a viable cure requires that this HIV reservoir is eliminated.
The reservoir is in peripheral blood as well as in tissues (3), and any of these sites may contribute to the rebound virus (4). In animal models for AIDS it was shown that, during treatment, persisting viral DNA and RNA are highest in lymphoid tissues, in particular lymph nodes, spleen and gastrointestinal tissues (5, 6). There is also evidence of differences in the virus sequence (compartmentalisation) between different body compartments (3), including between lymph nodes and peripheral blood (7, 8). Therefore, although most previous studies of the reservoir focus on peripheral blood, it is relevant to study the reservoir in both peripheral blood and tissues, and particularly lymphoid tissues.
One approach being explored to achieve long-term remission or cure is early initiation of ART, followed by a combination approach of latency reversal agents and a cytotoxic T lymphocyte (CTL)-based therapeutic vaccine or other immunomodulatory agents to eliminate the reactivated viruses (2). Therapeutic vaccination will likely be important for virus eradication since previous studies have shown that cytopathic effect and non-stimulated CTLs are not sufficient to eliminate reactivated infected cells, while CTLs stimulated with Gag peptides that are unmutated in the reservoir are effective at killing reactivated infected cells (9). Early treatment is thought to be conducive to achieving cure as it not only reduces the viral reservoir and preserves the quality of immune responses (10, 11), but it may also limit CTL escape in the peripheral blood reservoir (12). It is relevant to CTL-based eradication efforts to investigate accumulation of CTL escape in the tissue reservoirs as well. Further, investigation of whether viral variants present in the reservoir differ from those circulating in peripheral blood prior to treatment initiation, and characterisation of the reservoir variants from peripheral blood and tissues could better inform immune-based interventions for HIV cure. It is also worth noting that most previous studies of the HIV reservoir have focussed on individuals infected with HIV subtype B. Given that HIV subtype C is the most predominant subtype world-wide, and that virus and host characteristics differ between populations infected with different subtypes, there is a need to study the HIV reservoir in non-subtype B regions as well.
In the present study, viral variants derived from plasma samples that were taken immediately prior to treatment initiation in HIV-1 subtype C-infected individuals from South Africa were sequenced, and these sequences were compared to those derived from lymph nodes (LN) and peripheral blood mononuclear cells (PBMCs) at a matched time-point collected at least 3 months after treatment initiation. We investigated whether there were unique mutations in the reservoir variants compared to the pre-therapy plasma variants and if these were represented in functional viral proteins (and could therefore have the potential to be in a rebound virus). We further investigated whether there were specific features of reservoir variants, in terms of replication capacity, interferon-alpha (IFN-α) resistance, cell-to-cell spread ability, and induction of antiviral cytokines, that may have relevance for strategies to cure HIV. Although studying the overall virus isolate to address these questions would be ideal, the present study focussed on HIV Gag, since it is a major target of CTL vaccine strategies (13), it influences antiviral cytokine production (14) and it contains determinants of cell-cell spread ability (15). Results showed limited novel mutations in the reservoir-derived variants compared to the variants derived from pre-therapy plasma, and there was a similar extent of CTL escape in the reservoir variants compared to the pre-therapy plasma variants. Further, reservoir-derived variants were not phenotypically distinct from pre-therapy plasma variants.
The study population included 24 HIV-infected individuals, where one was antiretroviral naïve, 14 initiated ART in early infection (Fiebig stage I-V) and 9 initiated ART in chronic infection (Table 1). Study participants were derived from the Females Rising through Education, Support and Health (FRESH) cohort (16) and HPP acute infection cohort (17) in Durban, South Africa. Lymph nodes were previously voluntarily excised as part of the lymph node study protocol (18). Stored LN mononuclear cells and PBMC samples closely matched to the time of LN excision were studied. In treated individuals, LN samples were obtained at a median of 553 days post-treatment initiation (inter-quartile range [IQR] 361-902 days). Plasma samples obtained at the nearest timepoint prior to treatment initiation were studied for comparison with the LN and PBMC samples obtained during treatment. Longitudinal viral loads and the CD4+ T cell counts, as well as HLA class I profiles, of these participants were available. With the exception of the untreated individual and the 3 individuals where the LN was excised within 2 weeks of treatment initiation, the viral load in all participants was <20 copies/ml at the time of LN excision and 4 participants had experienced viral blips in the interval following attainment of viral suppression and LN excision. In treated individuals, at the time of LN excision, the median CD4 count was 702 cells/mm3 (IQR 533-919 cells/mm3). The median age of participants was 24 years (IQR 22-25) and all were female. Written informed consent was obtained from all study participants and the study was approved by the Biomedical Research Ethics Committee of the University of KwaZulu-Natal.
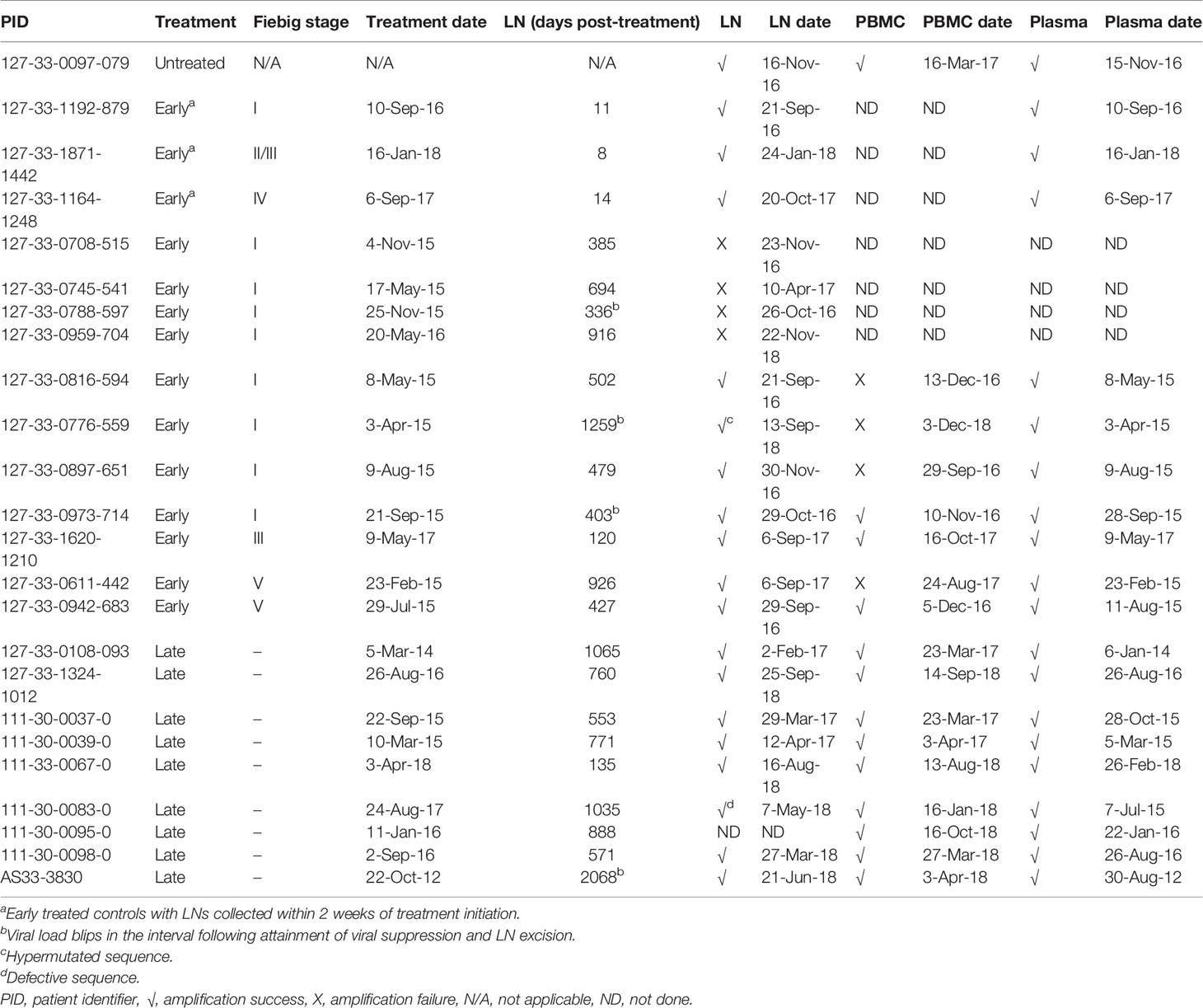
Table 1 HIV-1 gag amplification from lymph nodes (LN), peripheral blood mononuclear cells (PBMC) and plasma.
RNA was extracted from 140-500 µl plasma using the Qiagen Viral RNA Mini kit (Qiagen, Hilden, Germany), while DNA was extracted from 1.5-2 million frozen mononuclear cells obtained from LNs using the MasterPure Complete DNA and RNA Purification kit (Lucigen, Middleton, Wisconsin) and DNA was extracted from 10 million PBMCs using the QIAamp DNA blood Minikit (Qiagen), according to manufacturer’s instructions. HIV gag-protease was amplified by bulk PCR using previously described protocols (19). Single genome amplification (SGA) of gag was performed, as described previously (20), by serial dilution of cDNA to determine a dilution where ≤30% of wells were positive for amplification. The ABI Prism Big Dye Termination V 3.1 cycling sequencing kit (Applied Biosystems, Waltham, Massachussetts) was used to Sanger sequence bulk PCR products and single genome amplicons using previously published primers (19, 20). Sanger sequences were edited using Sequencher v5.1. Gag sequences were aligned using Genecutter and maximum likelihood phylogenetic trees were constructed using Phyml 3.0 (all available at http://www.hiv.lanl.gov) (21). Phylogenetic trees were visualized using Figtree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree). Deep sequencing of bulk PCR products was performed on a Miseq instrument (Illumina, San Diego, California), using the appropriate Illumina kits for preparation, and all variants present at a frequency of 2% and above, as detected using Geneious Prime version 2021.2.2, were considered. As previously (22), non-consensus mutations in or within 5 amino acids of defined CTL epitopes (Los Alamos HIV immunology database, https://www.hiv.lanl.gov/content/immunology/) that were restricted by HLA-I alleles expressed by that individual were considered to be potential escape mutations. Sanger sequences as well as consensus sequences derived from deep sequencing are available under Genbank accession numbers ON552272 - ON552477. Fastq files from deep sequencing are available under the Bioproject accession number PRJNA833316 in the sequence read archive (SRA) database.
Recombinant viruses encoding patient-derived gag-protease sequences were generated using gag-protease deleted NL4-3 backbone (23, 24). Briefly, the primers used during gag-protease amplification from plasma, LNs and PBMCs matched NL4-3 on either side of gag-protease to enable the homologous recombination of the gag-protease PCR product and the NL4-3 backbone. CEM-derived GXR25 green fluorescent protein (GFP)-reporter T cells (25) were transfected with gag-protease PCR product and linearised gag-protease deleted NL4-3 via electroporation. HIV-infected GXR cells produce GFP, allowing monitoring by flow cytometry of the percentage of infected cells in the culture. Supernatants containing the recombinant viruses were harvested when the culture had approximately 25-30% GFP-positive cells and aliquots of these virus stocks were stored at -80 °C.
Titration of virus stocks and replication capacity assays were performed as previously described (19). To determine the titre of virus stocks, 0.4 ml thawed virus stock was incubated with 1 million GXR cells for 2 days. The percentage GFP-expressing cells was then measured by flow cytometry and the volume of virus stock to infect GXR cells at a multiplicity of infection (MOI) of 0.003 for the replication capacity assays was calculated:
Volume of virus stock = (0.3/proportion GFP-expressing cells) X 400 μl.
GXR cells were then infected at a MOI of 0.003 and the mean slope of exponential growth in GFP-positive cells from days 3-6 post-infection was calculated using the semi-log method in Excel. This slope was normalized to the slope of growth of the wild-type NL4-3 control included in each assay. The NL4-3 normalized slope was used as the measure of replication capacity, where the replication capacity of NL4-3 was equal to 1.
To test for the IFN-α resistance using the replication capacity assay, the assay was performed in both the absence and presence of IFN-α in parallel (26, 27). GXR cells were pre-incubated with IFN-α at a concentration of 1000 U/ml before addition of virus and the assay was then performed in the same way, maintaining the cells in R10 medium containing IFN-α at 1000 U/ml. To calculate IFN-α resistance, the slope of growth in GFP-positive cells in the presence of IFN-α was divided by the slope of growth in GFP-positive cells in the absence of IFN-α. All replication capacity assays were performed independently in duplicate and the results averaged.
Cell-to-cell spread ability of recombinant viruses encoding gag-protease derived from plasma, LNs and PBMCs were tested by flow cytometry, as described previously (28). Briefly, one million Jurkat cells were infected with 100 ng p24 equivalent of virus stocks, as determined using HIV-1 p24 Antigen ELISA (Zeptomatrix Corporation Buffalo, New York). Infection of the Jurkat cells was performed by centrifugation at 1200 x g for 2 hours at room temperature, with a further incubation at 37 °C and 5% carbon dioxide for 4-5 days. HIV infection was detected by flow cytometry using intracellular Gag staining with a fluorescently-labelled antibody (anti-HIV-1 core antigen RD1, Beckman coulter, California). HIV-infected Jurkat cells were adjusted to 5% infected cells (donor cells), stained with eFlour450 (Thermofisher Scientific, Boston, Massachusetts), and then co-cultured at a ratio of 1:1 with CellTrace™-stained (CellTrace™ Far red cell proliferation kit, Thermofisher Scientific) uninfected Jurkat cells (target cells) for 48 hours. HIV cell-to-cell spread was thereafter measured by intracellular Gag staining in target cells (CellTrace™ stained cells). Zombie NIR (Biolegend, San Diego, California) was used to distinguish live cells from dead cells. Compensation was done using single stained cells and cells were acquired using an LSRFortessa (BD Biosciences, San Jose, California) with FACSDiva™ software. Results were analyzed using the FlowJo version 10.0.8 (Flowjo, LLC, Ashland, Oregon).
Cell culture supernatants were collected 24 hours after infection of GXRs at a MOI of 0.003 with recombinant viruses encoding gag-protease derived from plasma, LNs and PBMCs, and frozen at – 80°C. Infections and supernatant collections were done independently in duplicate. Cytokines were detected in the supernatants using a bead-based assay for detecting 27 human cytokines, chemokines, and growth factor biomarkers (Bio-Rad, Hercules, California) by following the manufacturer’s instructions. The 27 analytes detected by this panel are eotaxin, fibroblast growth factor (FGF)-basic, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-1β (IL-1β), IL-RA, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10,IL-12, IL-13,IL-15, IL-17, IFN-γ, IFN-γ-inducible protein 10 (IP-10), macrophage inflammatory protein 1α (MIP)-1α (CCL3), MIP-1β (CCL4), monocyte chemotactic protein 1 (MCP-1), platelet-derived growth factor (PDGF)-BB; regulated on activation normal T cell expressed and secreted (RANTES or CCL5), tumor necrosis factor alpha (TNF-α), and vascular endothelial growth factor (VEGF). The concentration of each analyte was determined from a standard curve derived from a human recombinant protein and therefore represents an estimate of the analyte concentration in each sample. The analyte measurements were done independently in duplicate for the duplicate supernatant collections. The multiplex plate was read with a Bio-Plex 200 suspension array Luminex system (Bio-Rad).
The Mann-Whitney U test was used to compare the number of unique Gag mutations in the reservoir of early treated and late treated individuals. Replication capacity, IFN-α resistance and cell-to-cell spread ability of the viruses encoding gag-protease derived from plasma, LNs and PBMCs, as well as cytokine levels induced by these viruses, were compared between matched compartments using repeated measures ANOVA. Paired T-tests were used to compare 2 compartments at a time. Pearson’s correlation was used to test the correlation between assay replicates. All analyses were done with GraphPad software (GraphPad Software Inc. Version 5.0) and p<0.05 was considered statistically significant. Q values, with a cutoff of q < 0.04, were used to account for multiple comparisons where appropriate.
HIV gag amplification was attempted from the LNs of 24 patients, and was successful for 20 (Table 1). Included in the 24 LNs were 4 controls for which gag was successfully amplified: LNs from an untreated patient as well as 3 early treated patients (Fiebig I, II/III, and IV) where LNs were collected within 2 weeks of treatment initiation. Of the remaining 20 patients from whom LNs were collected at least 120 days after therapy initiation, there was successful amplification for 16, where 7 initiated treatment in the acute phase (4 in Fiebig I, 1 in Fiebig III and 2 in Fiebig V) and 9 initiated treatment in the chronic phase. The 4 LNs from which gag failed to amplify were from patients who initiated treatment in Fiebig I. Overall, for LNs collected at least 3 months after therapy, there was 50% successful gag amplification from those who initiated therapy in Fiebig I compared to 100% successful amplification for those who initiated therapy in Fiebig III or later. This is consistent with data from our group showing less HIV-1 p24 antigen as well as HIV-1 RNA in LNs from Fiebig I treated patients compared to those initiating treatment later (Baiyegunhi et al., 2021, submitted).
For the 16 patients with LN collection at least 3 months after therapy initiation (median of 665 days; IQR of 466-953 days) and successful gag amplification, gag amplification was attempted for PBMCs at a closely matched timepoint. This was successful for 3 of 7 early treated patients and 9 of 9 late treated patients, while amplification from pre-therapy plasma was successful for all patients. Lower amplification success from PBMCs compared to LNs obtained during treatment is consistent with a previous report of lower HIV-1 copies in PBMCs than LNs in treated patients (29).
To investigate differences between the reservoir-derived variants and pre-therapy plasma variants, bulk Gag sequences were compared between compartments and amino acid differences between the pre-therapy plasma and either reservoir compartment were identified. For the early treated individuals, to investigate whether these mutations identified were unique to the reservoir (i.e. not present in the pre-therapy plasma), multiple amplicons from the pre-therapy plasma samples were pooled together for deep sequencing and SGA sequences from pre-therapy plasma were also interrogated for the presence of these mutations. In addition, where available (n=2), PBMCs obtained within one week of plasma virus suppression were also deep sequenced and SGAs performed to check for the presence of the new mutations observed in the reservoir viruses. For the late treated patients, only deep sequencing of the pre-therapy plasma was performed. Table S1 shows deep sequencing and SGA results for each novel mutation in each compartment for each patient, and also reports which mutations are in or within 5 amino acids of CTL epitopes that are restricted by the patient’s HLA-I alleles.
In early treated (Fiebig I-V treated) individuals, the bulk sequence comparison showed sequence differences between variants derived from the reservoir and pre-therapy plasma for 5 of 7 early treated individuals. Of these 5 early treated individuals where LN and PBMC samples were collected > 3 months (median of 427 days) after treatment initiation and novel mutations were detected in the reservoir by bulk sequencing, one had a hypermutated sequence in the LN, and of the remaining 4 individuals, 3 were confirmed by deep sequencing and SGA to have unique non-consensus mutations (1-6 amino acid mutations) in the PBMCs and/or LNs (Table 2). In contrast, 3 early treated individuals for whom LNs were obtained within two weeks of ART initiation showed no sequence differences between plasma and LN samples (Table 2). This may suggest that in the 3 early treated individuals where novel Gag mutations were detected in the reservoir, that mutations arose during treatment, however virus compartmentalisation prior to therapy initiation or selection of consensus variants in the pre-therapy plasma are alternative explanations. It should also be noted that one of these 3 individuals experienced a low-level virus blip of uncertain duration prior to LN collection.
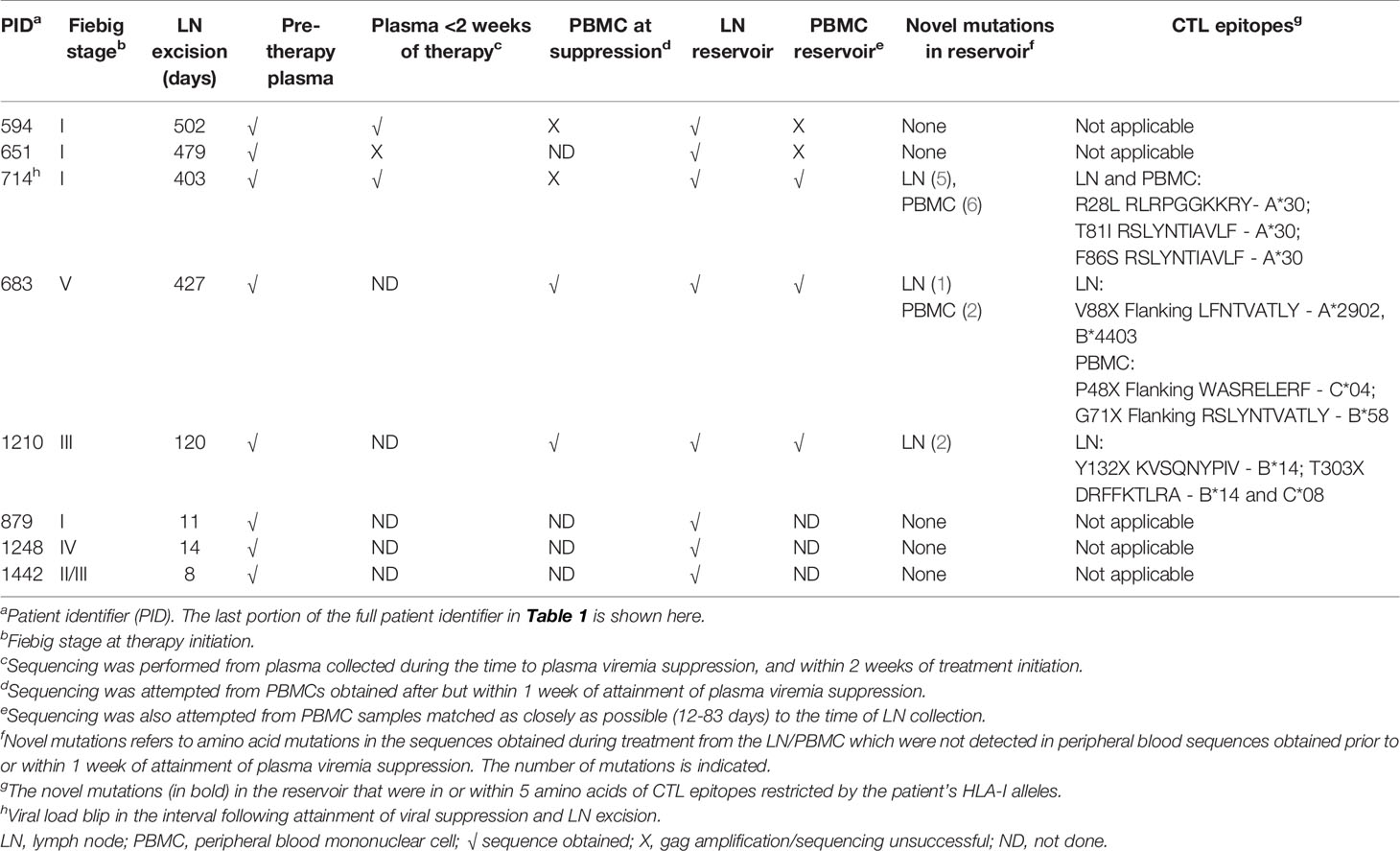
Table 2 Gag sequence differences between the reservoir-derived variants and pre-therapy plasma variants in early treated individuals.
Next, sequence differences between the reservoir-derived variants and those variants derived from plasma collected at the closest timepoint to treatment initiation were identified in late treated individuals, and compared to results obtained in the early treated individuals. Although only 3 of 7 early treated individuals had novel Gag mutations in the reservoir, all late treated individuals had unique mutations (2-19 amino acid mutations) in the PBMCs and/or LNs (Figures 1A–C). The median number of unique mutations in the reservoir overall was 4 (IQR, 2-5) and there was a trend for a higher number of unique mutations in the reservoir of late treated patients (median = 4) compared to early treated patients (median = 1), although this was not statistically significant (Mann-Whitney U test, p = 0.09) (Figures 1A–C). Interestingly, the majority (8/12 in early treated patients and 31/49 in late treated patients) of these novel Gag mutations were non-consensus variants in or within 5 amino acids of cytotoxic T lymphocyte (CTL) epitopes restricted by HLA-I alleles expressed by these individuals (Figures 1A–C; Table 2; Table S1), suggesting that these mutations were most likely selected by CTL. There was also a trend for a higher number of these potential CTL escape mutations unique to the reservoir in the late treated patients (median = 3) compared to early treated patients (median = 1) (Mann-Whitney U test, p = 0.08) (Figure 1A). In comparison, at the time of treatment initiation in the plasma, there were a median of 23 non-consensus mutations (IQR, 21-27) in or flanking CTL epitopes that could be targeted by the patient’s HLA-I alleles, and this number was similar in late and early treated participants (24 and 21, respectively) (Figure 1D). In summary, there were a limited number of new Gag mutations in the reservoir of some early treated participants, and in all late treated participants, where the majority of these mutations were likely CTL-driven. This limited number of novel potential CTL escape mutations in the reservoir was small in comparison to the number of potential CTL escape mutations already present in the pre-therapy plasma, which did not differ significantly between early treated and late treated individuals.
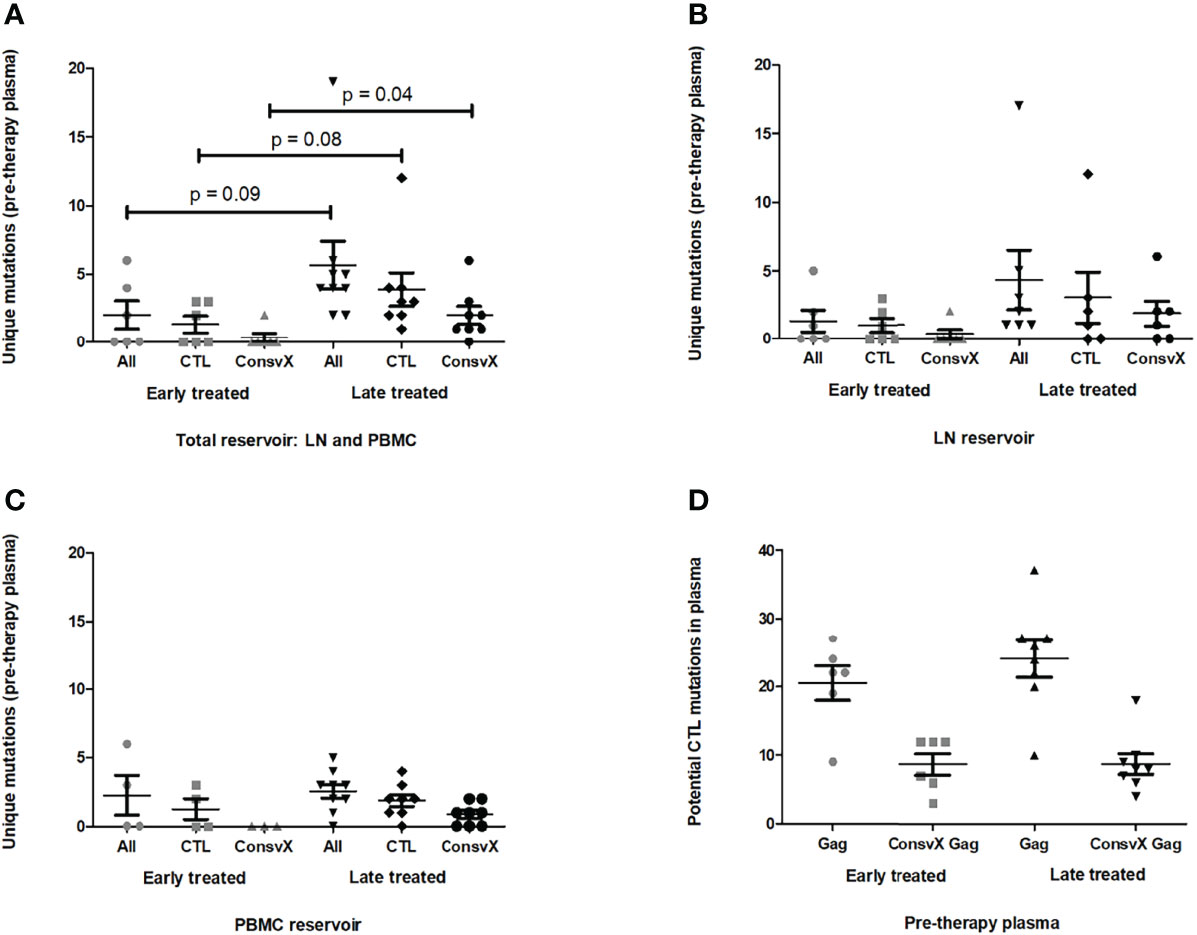
Figure 1 Gag sequence differences between the reservoir variants and pre-therapy plasma variants. (A–C) Panel (A) shows the number of unique amino acid mutations in the reservoir-derived variants with respect to the pre-therapy plasma variants for individuals treated early in infection (grey) or late in infection (black). The same is shown for the subset of these unique mutations that are non-consensus and in or flanking cytotoxic T lymphocyte (CTL) epitopes restricted by HLA-I alleles expressed by the patient i.e. potential CTL escape mutations. Also shown are potential CTL escape mutations in the HIVconsvX immunogen region only (HXB2 Gag amino acid positions 133-363 and 391-459). Panels (B) and (C) are specific to the lymph nodes (LN) and peripheral blood mononuclear cells (PBMC), respectively, while panel A represents both LN and PBMC. Unique mutations are those mutations in the reservoir viruses that are not detected by single genome amplicon (SGA) sequencing and/or deep sequencing of bulk amplicons in the pre-therapy plasma virus or virus derived from peripheral blood up to one week after attainment of plasma viremia suppression. (D) The number of potential CTL escape mutations in Gag, and in the HIVconsvX immunogen region only, is shown for the pre-therapy plasma for individuals treated early in infection (grey) or late in infection (black). The Mann-Whitney U test was used to compare the different mutation categories between the early and late treated groups. Only p values less than < 0.1 are shown.
Since the HIVconsvX is one of the most recent CTL-based vaccine immunogen designs under development and it contains a large portion of Gag (HXB2 Gag amino acid positions 133-363 and 391-459) (30), the amount of potential CTL-driven mutations unique to the reservoir that were in this region was assessed. Interestingly, approximately half (19/39) of the potential CTL-driven mutations unique to the reservoir were in the HIVconsvX region (Figures 1A–C, Table S1), and there was a trend for higher numbers of these mutations in the late treated individuals (median = 2) compared to the early treated individuals (median = 0) (Mann-Whitney U test, p = 0.04). Overall, there was a median of 9 (IQR, 6-12; data not shown) non-consensus mutations in or flanking CTL epitopes that were restricted by the patient’s HLA-I alleles and that were also in the HIVconsvX region in the plasma at the time of treatment initiation (Figure 1D), while in the reservoir there was a median of 1 (IQR, 0-2) unique potential CTL escape mutation in the HIVconsvX region (Figure 1A, Table S1). In summary, there were very few additional potential CTL escape mutations in the HIVconsvX region in the variants derived from the reservoir compared to those derived from pre-therapy plasma.
Since a large proportion of reservoir variants could be defective (31), recombinant viruses encoding the bulk gag-protease sequences from the LN and PBMC reservoirs, as well as from the pre-therapy plasma, were constructed and cultured to investigate whether the unique mutations identified in the reservoir were encoded in functional Gag proteins. Gag-protease recombinant viruses were constructed from 15 different treated individuals for whom gag amplification was successful (excluding the 1 patient with the hypermutated sequence). All recombinant virus stocks were resequenced and phylogenetic analysis confirmed participant origin in all cases (Figure S1). Of the 15 participants, 12 had mutations which were unique to the reservoirs and, overall, 48% of these unique mutations were present in the virus outgrowth from the reservoirs (Figure 2A). Of the mutations unique to the total reservoir that were present in replication competent Gag-protease recombinant viruses, 60% were non-consensus variants in (43%) or within 5 amino acids (17%) of CTL epitopes restricted by HLA alleles expressed by the patient, suggesting that these might represent CTL escape (Figure 2B). Similar proportions were observed when the same analyses were done separately for the LN reservoir (Figures 2C, D) and the PBMC reservoir (Figures 2E, F), although a larger percentage (72%) of the unique mutations in the PBMC reservoir were present in the virus outgrowth. Since each participant expressed HLA-I alleles that restrict epitopes covering 30 – 49% of the Gag protein (40% on average), no particular enrichment (or negative selection) of potential CTL escape in the functional Gag proteins from the reservoir is suggested by these data. In summary, approximately half of the unique Gag mutations (albeit limited in number) in the reservoir are represented in functional Gag proteins (which have the potential to be part of the rebound virus), and a significant portion of these functional unique mutations are potential CTL escape mutations.
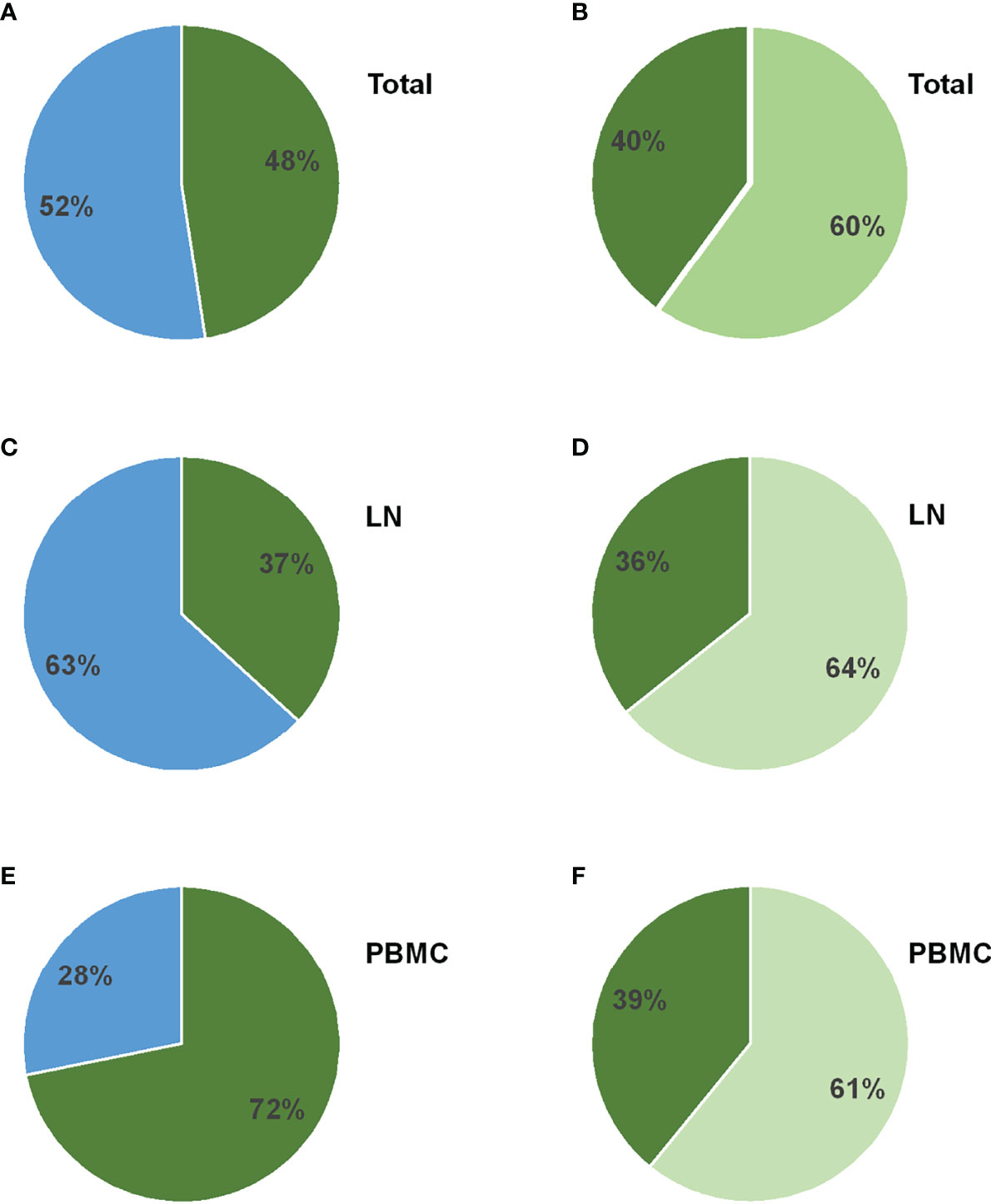
Figure 2 Gag mutations unique to the reservoir in replication-competent viruses encoding gag-protease from the reservoir. (A–B) The pie chart in panel (A) depicts the proportion of amino acid mutations unique to the reservoir, when compared to the pre-therapy plasma, that are present in the replication competent recombinant viruses encoding the patient-derived gag-protease from the lymph node (LN) and peripheral blood mononuclear cell (PBMC) reservoir. This proportion is shown in green, while the proportion of amino acid mutations unique to the reservoir that were not represented in the virus outgrowth are shown in blue. Of the unique mutations that were present in the virus outgrowth, panel (B) shows the proportion that are in or within 5 amino acids of CTL epitopes restricted by the patient HLA-I alleles (shown in light green), versus the proportion that were not in these epitopes (shown in darker green). (C–F) Panels (C) and (D) are specific to the LN reservoir only and panels E and F are specific to the PBMC reservoir only.
The replication capacities of the recombinant viruses encoding bulk Gag-protease sequences from the LN and PBMC reservoirs, as well as from the pre-therapy plasma, in the absence and presence of IFN-α was measured to assess whether or not viruses in the reservoir may be adapted to replicate more efficiently, especially in the presence of this key antiviral cytokine. In particular, the sequence of Gag is known to influence the virus sensitivity to IFN-α (32).
Duplicate replication capacity assay measurements were highly consistent (Pearson’s correlation, r > 0.94 and p < 0.0001) (data not shown), and the range of NL4-3 normalized Gag-protease replication capacities (range = 0.43 – 1.04; average = 0.72) was similar to that previously reported for subtype C Gag-protease (19, 33) (Figure 3A). The difference in replication capacity (in the absence of IFN-α) by compartment for each individual was not in a consistent direction, and therefore not significant overall (repeated measures ANOVA, p = 0.7) (Figure 3A). Similarly, two-way comparisons of replication capacity between compartments showed no significant differences (paired T test, p > 0.33) (Figure 3A). A sub-analysis including only individuals (n = 8) where the cultured virus outgrowth encoded unique mutations that were present in the reservoir virus with respect to the pre-therapy plasma virus, yielded similar results (paired T test, p > 0.41) (data not shown).
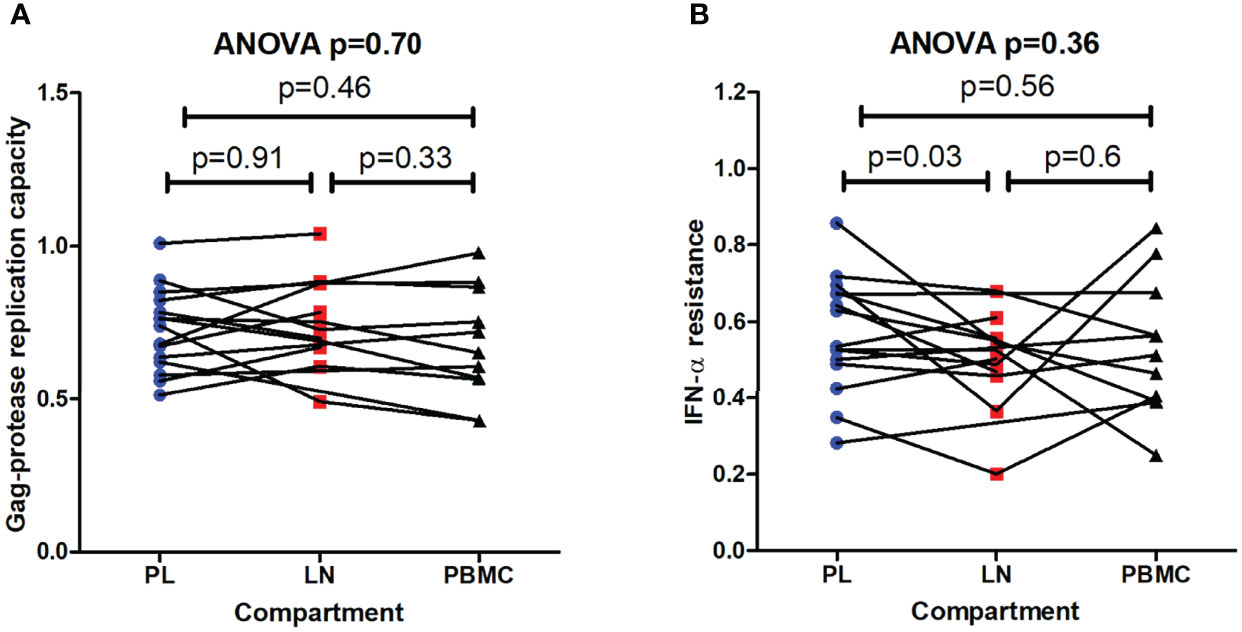
Figure 3 Replication capacities and interferon-alpha resistance of viruses encoding gag-protease from pre-therapy plasma and the reservoir. (A) The replication capacities of the recombinant viruses encoding gag-protease from the lymph node (LN) and peripheral blood mononuclear cell (PBMC) reservoirs, as well as from the pre-therapy plasma (PL), in the absence of type I interferon alpha (IFN-α) is shown. Replication capacity is expressed relative to the NL4-3 control (replication capacity = 1). (B) The IFN-α resistance (ratio of replication capacity in the presence and absence of IFN-α) of these viruses is shown. The overall repeated measures ANOVA p value is shown as well as the paired T test p values from two-way comparisons.
IFN-α resistance was calculated as the ratio of replication capacity in the presence and absence of IFN-α. Representative flow cytometry data used to calculate IFN-α resistance is shown in Figure S2. Overall, there was no significant difference in IFN-α resistance between the different compartments (repeated measures ANOVA, p =0.36), however two-way comparisons showed that there was tendency for lower IFN-α resistance in the viruses derived from the LN reservoir compared to those derived from pre-therapy plasma (paired T test, p = 0.03) (Figure 3B). A similar trend was observed when only the viruses with sequence differences between compartments were included in a sub-analysis (LN reservoir versus pre-therapy plasma, paired T test, p = 0.07) (data not shown).
In summary, Gag-driven replication capacity and IFN-α resistance were overall similar between reservoir viruses and pre-therapy plasma viruses, although there was a trend of lower IFN-α resistance of the viruses derived from the LN reservoir.
Cell-to-cell spread is an important mechanism of HIV-1 infection that may have higher relevance than cell-free spread in cell-dense regions (34), such as the lymph node, and may also be a mechanism to allow infection in the presence of antiretroviral drugs (35). Cell-to-cell spread ability of viruses encoding Gag-protease from the reservoirs and pre-therapy plasma was therefore compared.
Duplicate independent measurements of cell-to-cell spread ability were highly concordant (Pearson’s correlation, r = 0.96 and p < 0.0001) (data not shown). Representative flow cytometry plots used to calculate cell-to-cell spread are shown in Figure S3. Overall, there was no consistent direction of difference in viral cell-to-cell spread ability between compartments (repeated measures ANOVA, p = 0.32) (Figure 4). Similarly, two-way comparisons of virus cell-to-cell spread between compartments showed no significant differences (paired T test, p > 0.46). In summary, cell-to-cell spread ability was similar for viruses derived from the reservoir compartments and pre-therapy plasma.
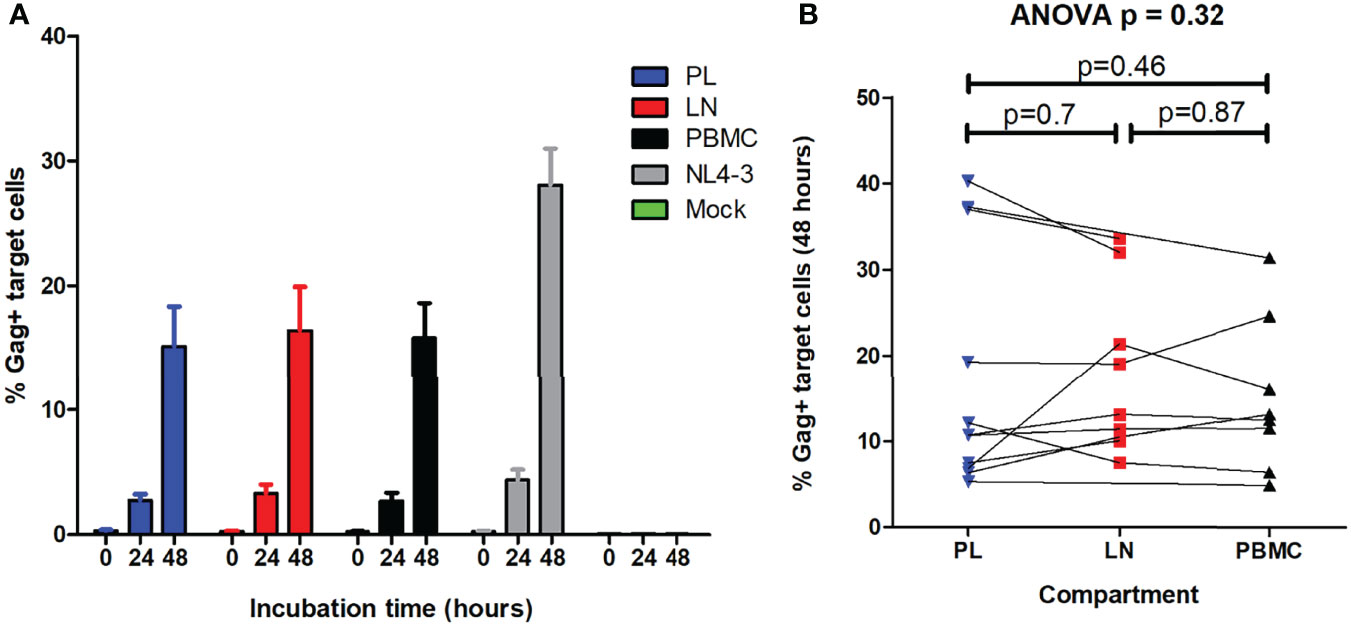
Figure 4 Cell-to-cell spread ability of viruses encoding gag-protease from pre-therapy plasma and the reservoir. HIV cell-to-cell spread of the recombinant viruses encoding gag-protease from the lymph node (LN) and peripheral blood mononuclear cell (PBMC) reservoirs, as well as from the pre-therapy plasma (PL), was measured. Cell-to-cell spread was measured by intracellular Gag staining in target cells, pre-stained with CellTrace™ far red, after incubation with HIV-infected donor cells (titrated to 5% infection) that were stained with eFlour450. (A) The percentage infected target cells at 0, 24 and 48 hours is shown for the viruses derived from the different compartments, as well as for the positive (NL4-3) and negative (mock) controls. (B) The percentage infected target cells at 48 hours was the measure of cell-cell spread ability. The overall repeated measures ANOVA p value is shown as well as the paired T test p values from two-way comparisons of viruses from the different compartments.
Cytokines are deployed in the initial stage of viral infections and most of the damage imposed on virus-infected cells is the result of cytokine responses (36). Mutations in the Gag protein have been reported to influence induction of antiviral cytokines (14). Therefore, cytokine induction in cellular supernatants following infection with viruses encoding Gag proteins from the reservoirs and pre-therapy plasma was investigated to assess whether viruses from these compartments differ in their ability to evade immune responses through altered induction of cytokines.
Overall, the 27 cytokines measured were consistent between compartments in the hierarchy of concentration in the cellular supernatants (Table 3). The levels of RANTES, FGF, PDGF-bb, and IL-4 differed significantly between compartments (repeated measures ANOVA, p < 0.05 and q < 0.4) (Table 3). To assess whether these cytokines were significantly altered by HIV infection or at similar levels in the supernatants of uninfected cell cultures, the cytokine level was compared between the supernatants of infected cell cultures (infected with viruses from the different compartments) and uninfected cell culture controls for these 4 cytokines. While FGF and IL-4 were at similar levels between the infected and uninfected cell cultures (Student’s T test, p = 0.96 and p = 0.21, respectively), RANTES and PDGF-bb were significantly higher in infected cell cultures than uninfected cell cultures (Student’s T test, p = 0.005 and p = 0.01, respectively). Thus, RANTES and PDGF-bb were both significantly different between the different compartments compared as well as significantly altered by HIV infection (Figure 5). Specifically, PBMC reservoir-derived viruses were associated with lower levels of RANTES when compared to LN reservoir-derived viruses (paired T test, p = 0.02). In addition, PDGF-bb was lower in supernatants of cultures infected with LN reservoir-derived viruses than pre-therapy plasma-derived viruses, although this was a trend and not statistically significant (paired T test, p = 0.07). A sub-analysis of only the 8 individuals for whom outgrowth viruses differed in sequence between compartments, supported the associations observed with the full dataset (data not shown).
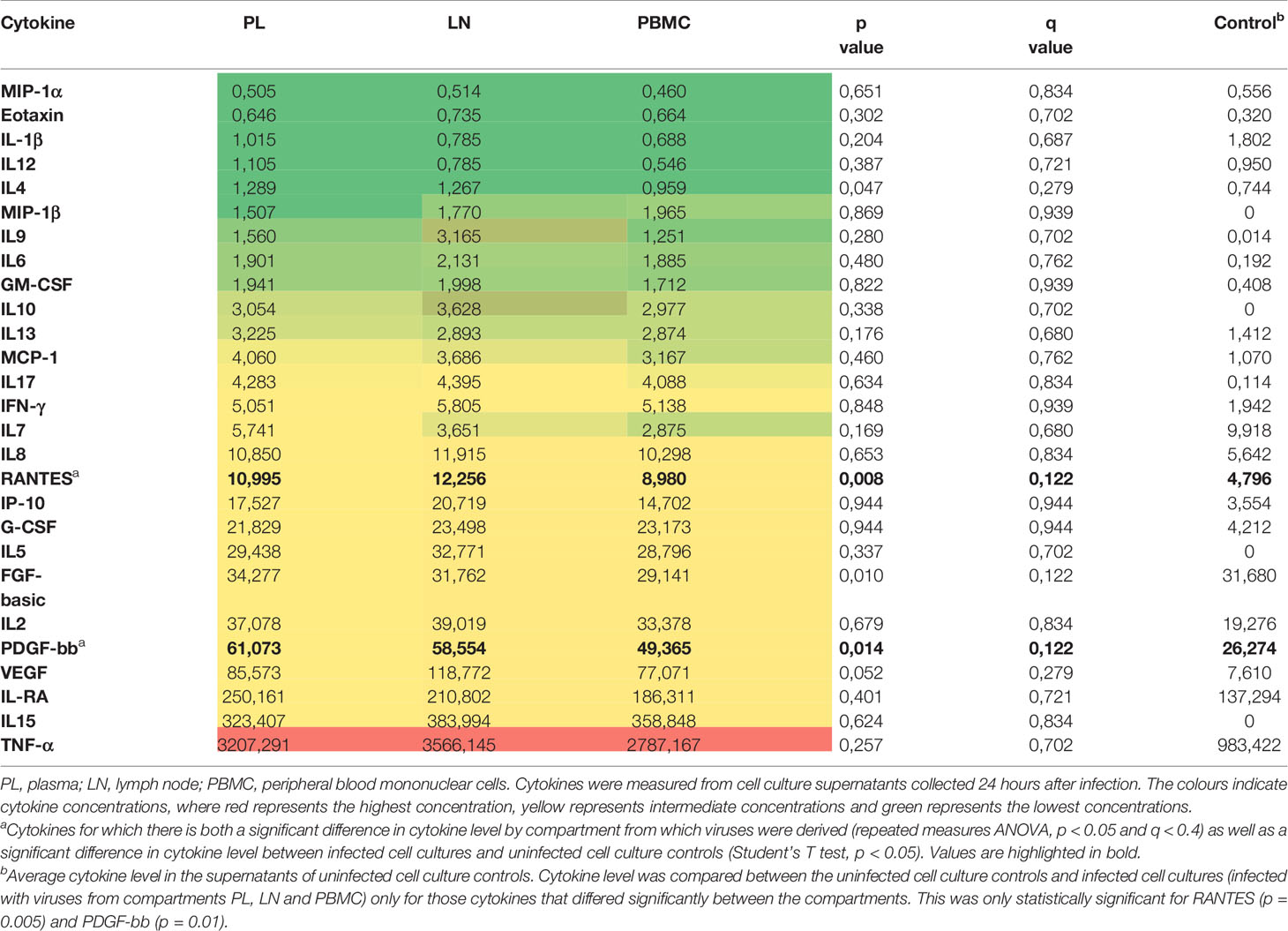
Table 3 Heat map of average cytokine concentrations (pg/ml) in cultures infected with Gag recombinant viruses derived from PL, LN and PBMC compartments.
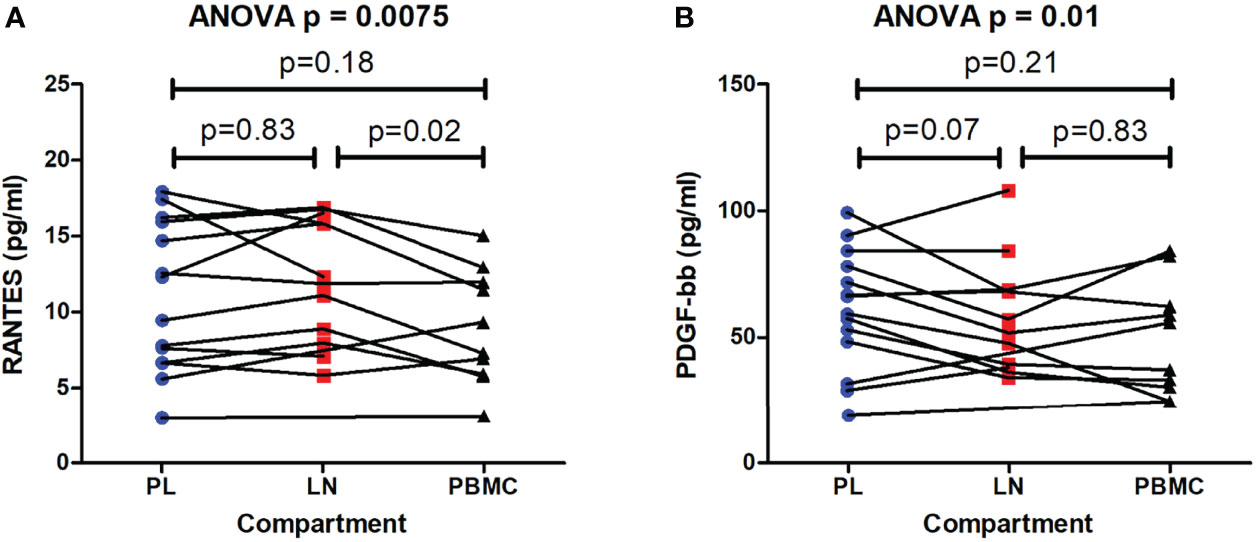
Figure 5 Differing cytokine levels induced by viruses encoding gag-protease from pre-therapy plasma and the reservoir. Cytokines were measured in the supernatants of cell cultures infected with recombinant viruses encoding gag-protease from the lymph node (LN) and peripheral blood mononuclear cell (PBMC) reservoirs, as well as from the pre-therapy plasma (PL). Cytokines that differed significantly between cultures infected with viruses from different compartments, and were also significantly higher in infected cell cultures compared to uninfected cell culture controls, are shown, namely RANTES (A), and PDGF-bb (B). The overall repeated measures ANOVA p value is shown as well as the paired T test p values from two-way comparisons of the PL, LN and PBMC compartments. The average cytokine level in the supernatants of the uninfected cell controls was 4.796 pg/ml and 26.274 pg/ml for RANTES and PDGF-bb, respectively, and differed significantly from the average cytokine level in the supernatants of cell cultures infected with viruses from the different compartments (Student’s T test, p = 0.005 and p = 0.01, respectively).
To further investigate whether there are differences in cytokine profiles induced by viruses from different compartments, cytokines were also grouped according to reported properties: pro-inflammatory (IL-1beta, IL-6, TNF-alpha, IL-8, MCP-1, MIP1-alpha, MIP1-beta), anti-inflammatory (IL-4, IL-10, IL-13, IL1-Ra), Th1 (IFN-gamma, IL-2), Th2 (IL-14, IL-10, IL-13, IL-5), pro-HIV replication (TNF-alpha, IL-1, IL-2, IL-6, IL-7, IL-15) and anti-HIV replication (IL-10, IL-13, MIP1-alpha/beta and RANTES) (37–40). There were no significant differences between compartments in these cytokine groups (data not shown).
HIV variants present in the reservoir, particularly in tissues, may differ from those present in peripheral blood prior to therapy initiation, and characterisation of these reservoir variants could better inform immune-based interventions for HIV cure. In the present study, Gag sequence differences as well as Gag phenotypic differences (in terms of replication capacity, IFN-α resistance, cell-to-cell spread ability, and induction of antiviral cytokines) between the viruses derived from the LN and PBMC reservoirs and those derived from pre-therapy plasma, were investigated.
In all late treated individuals, and 3 of 7 early treated individuals, a limited number of unique Gag mutations were identified in the LN and PBMC reservoir with respect to the pre-therapy plasma. The timing of the development of the novel mutations in the reservoir is not certain since longitudinal sampling of the LN reservoir spanning pre-treatment initiation and post-treatment initiation was not done. It is possible that the novel mutations in the reservoir were already present at the time of treatment initiation due to virus compartmentalization, which has been previously described for HIV variants when comparing LN and PBMCs (8), and when comparing PMBCs and plasma (41). Virus compartmentalization present at the time of treatment initiation likely explains in part the novel mutations observed in the reservoir, since there was an association between the timing of treatment initiation and identification of novel mutations in the reservoir, where all late treated individuals had novel mutations in the reservoir while this was observed for only half of the early treated individuals. Further, 2 of the 3 early treated individuals with novel mutations in the reservoir initiated treatment slightly later (in Fiebig stages III and V) than the other early treated individuals, allowing more time for development of mutations prior to treatment initiation. It is however important to note that, in these 2 individuals, it was confirmed that the novel mutations were not present in PBMCs shortly after viral suppression (Table 2 and Table S1). While LNs were not similarly collected around the time of ART initiation or virus suppression in these specific individuals, in another 3 early treated donors with LN collection within 2 weeks of ART initiation, no novel Gag mutations were observed in the LN, which may point to the possibility that novel mutations in the reservoir could also develop after treatment initiation.
One of the 3 early treated individuals with novel mutations in the reservoir, initiated treatment in Fiebig I and was infected with a single virus variant. However, the individual experienced a transient low-level plasma virus blip prior to LN collection, suggesting that non-optimal adherence to ART may have led to development novel mutations in the reservoir after ART initiation in this case. It is also possible that viral suppression in the LNs may take longer than in peripheral blood since drugs do not penetrate the LNs as readily (42), thereby allowing a limited opportunity for development of mutations in the LN after plasma virus suppression. Another possibility is that Gag evolution in the reservoir could occur via expression of Gag (yet without full virus replication) from infected cells followed by CTL-mediated deletion of cells with susceptible Gag, leading to selection of sequences in the reservoir that are resistant to CTL. A more controversial possibility is the development of mutations due to persisting low-level replication during therapy despite plasma virus suppression, which is supported by some studies to occur in a subset of individuals (42–46) while other studies did not find evidence of this (47–52). Various factors, including body compartment (50) and perhaps population differences involving virus and host factors (e.g. immune activation or co-infection status) (2), could potentially influence the answer to this question. Further research is required to address this controversial question in the setting of low- or middle-income countries. Furthermore, other reservoir sites for which some evidence of productive infection has been reported, in particular the gut ileum (43), should be considered in these studies. Resolving this controversy is critical to HIV cure efforts: if ongoing virus replication in sanctuary sites during ART contributes to virus persistence, then improving penetration of ART into these sites should be high priority in HIV cure strategies, and if not, then cure strategies should primarily focus on eliminating long-lived latently infected cells.
Interestingly, the majority of the novel Gag mutations in the reservoir were possible CTL escape, based on the HLA-I alleles expressed by the individuals. This suggests that the reservoir virus composition could be shaped by CTL pressure on Gag. As discussed above, the novel Gag CTL mutations may not necessarily have arisen uniquely in the reservoir after therapy initiation, but may be detected as unique in the reservoir due to virus compartmentalisation present at the time of therapy initiation, reversion of non-consensus variants to consensus in the pre-therapy plasma, or ongoing CTL-mediated deletion of cells in the reservoir that express susceptible Gag. The reservoir virus had very few unique potential CTL escape mutations in Gag (median = 3; and approximately half of these were in the outgrowth virus) compared to the number of potential CTL escape mutations that were already present in the plasma at the time of treatment initiation (median = 23). Although there was a tendency for less unique CTL mutations in the reservoir of early treated compared to late treated individuals, this number was very low in both groups. Interestingly, whether treatment was initiated late or early, there was a similar number of potential CTL escape mutations present in the plasma at the time of therapy initiation, and overall in the reservoir. This is in agreement with previous reports describing that most escape mutations are already present at transmission with a slow accumulation thereafter (53, 54) and with a previous study showing a similar depth and number of escaped epitopes in acute and chronic infection (55). These studies appear to conflict with another study which reports that the latent virus in peripheral blood has a greater dominance (or depth) of CTL escape mutations in individuals who initiated treatment in chronic infection compared to those who initiated treatment in acute infection (12). With the exception of that study (12), the data indicate that early or late treatment will not likely make a large difference to the extent of escape to a CTL-based vaccine targeting Gag. HIVconsvX, which is one of the most recent CTL-based vaccine immunogen designs, is a second generation conserved-region immunogen under development for use as an HIV therapeutic vaccine and it contains a large portion of Gag (30). In the present study, whether early treated or late treated, all individuals had a similar level of potential CTL escape mutations in the HIVconsvX region of Gag in the virus derived from pre-therapy plasma (median = 9), and very few unique mutations in the reservoir were in this HIVconsvX immunogen (median = 1). The data support that there is likely to be inherited escape in some of the epitopes within the HIVconsvX region that can be targeted by an individual’s HLA-I alleles, whether starting treatment early or late. It should be noted that the definition of potential CTL escape used in the present study is relaxed and is likely an overestimate of escape, particularly since a recent study showed that less than half of CTL epitopes containing non-consensus mutations actually resulted in a > 50% loss in CTL response (55). Taken together, the evidence presented here suggests that a CTL-based therapeutic vaccine targeting Gag is likely to have similar effectiveness against reservoir variants from the LN and PBMC, and against pre-therapy variants from peripheral blood, and that a similar quantity of escape mutations in Gag is likely to be present in early and late treated individuals.
Given the Gag sequence differences observed between reservoir viruses and pre-therapy plasma viruses, it was investigated whether these limited genetic differences resulted in phenotypic differences that could affect immune-based interventions proposed for HIV cure. Specifically, resistance to IFN-α, HIV cell-to-cell spread ability, and induction of cytokines, were measured, as the sequence of Gag is known to play a role in these outcomes (14, 15, 32). There was no evidence in the present study that the viruses in the PBMC or LN reservoir differed substantially in these characteristics, based on Gag alone, when compared to those in the pre-therapy plasma.
Although type I interferons are important antiviral cytokines and may have therapeutic benefits, the evidence largely shows that blockade of IFN-α and thereby dampening of chronic inflammation reduces the reservoir, enhances CD8+ T cell function, and delays viral rebound on ART interruption, and therefore interferon blockade as well as anti-inflammatory agents may be useful tools in virus eradication (2, 56). In the present study, IFN-α resistance level was overall similar between reservoir viruses and pre-therapy plasma virus, although there was a tendency for slightly lower IFN-α resistance of viruses derived from the LN reservoir when compared to pre-therapy plasma virus, which had similar IFN-α resistance to viruses derived from the PBMC reservoir. A recent study also reported similar IFN-α resistance of isolates derived from the PBMC reservoir when compared to isolates obtained from plasma just before ART initiation, however, in that study it was found that the rebound viruses following ART interruption had enhanced IFN-α resistance (57). This suggests that host innate pressures are high during virus rebound in vivo and that in vitro outgrowth virus may not be fully representative of the rebound virus, particularly in terms of IFN-α resistance. Cure strategies that rely on the IFN-α pathway may need to take into account IFN-α resistance ability of the rebound virus, while approaches that seek to block this pathway are not as likely to be affected by this characteristic.
Cell-to-cell spread is an important mechanism of HIV infection that may allow infection in the presence of antiretroviral drugs (35), hence cell-to-cell ability of pre-therapy plasma and reservoir-derived variants were compared. Moreover, broadly neutralising antibodies, an important component of immune-enhancing virus eradication strategies (2), have varying abilities in inhibiting viral cell-to-cell spread (58). In the present study, there was no evidence of a more efficient ability of reservoir-derived variants for this mode of cellular propagation. These are Gag focused studies and so a potential effect mediated by Env cannot be ruled out as both Gag and Env are determinants of cell-to-cell spread (15, 59).
Cytokines have dichotomous effects depending on the cell types present, timing of cytokine exposure and level of cytokine, therefore it is difficult to interpret in vitro results and translate them to the in vivo situation or proposed therapies (60, 61). Examples of cytokines proposed for use as part of an HIV eradication strategy include IL-15 (due to its effect of increasing cytotoxic T cells in LN follicles) (62) and IL-21 (reduces gut inflammation via promoting Th17 and Th22 cell restoration in the gut) (63). In the present study, the cytokine profile (considering 27 different cytokines) induced was very similar between virus variants from the reservoir and pre-therapy plasma. There were some differences between reservoir and pre-therapy variants for two cytokines only, namely PDGF-bb and RANTES. PDGF (64) has been implicated as a mediator of pathology (neuro-inflammation) in HIV-infected individuals, while RANTES (65–67) can have either antiviral effects or stimulate HIV replication depending on the conditions. However, when considering the effects of cytokines as groups with similar well-known effects there were no overall patterns that suggested selection for reservoir viruses with altered induction of antiviral cytokines, pro-inflammatory cytokines or anti-inflammatory cytokines. Therefore, based on these results, there is no strong evidence that the Gag sequence of viruses in different reservoir compartments is a factor influencing immune-based interventions that rely on induction of cytokines.
A major limitation of this study is that only Gag was studied, and other genes could contribute to phenotypic differences in reservoir viruses. Although replication-competent Gag was identified in the reservoir, it was not determined if the functional Gag protein was within a virus that was replication-competent. A large portion of proviral DNA is defective (31). However, the minority (40%) of defective viruses have an intact Gag while the majority (60%) of defective viruses have a defective Gag, as has been observed from single genome amplification within HIV-infected Durban cohorts (31). Future studies of other virus proteins, or the whole isolate, from the virus reservoir will be required for a more comprehensive picture. It should also be noted that the viruses studied encoded bulk Gag-protease sequences from the reservoir. The goal of using the bulk approach for functional analysis is to attempt to capture the virus pool such that it is closely representative of the quasispecies within the patient, however the disadvantage is that the function of precise sequences is not known. Another potential limitation of the bulk approach is that the variants in the quasispecies with the highest replication capacity may be selected in culture [although about 60% of the mixtures in bulk PCR products are represented in the virus stocks obtained using this assay and the assay is able to distinguish high and low replicating strains (19, 33, 68)]. Another study limitation is that viruses isolated in vitro may not necessarily represent the rebound virus in vivo (57), and further work to characterise rebound viruses is warranted. It is also important to note that the cytokine work was performed using a T cell line and results may differ in monocyte cell lines or in primary cells.
In conclusion, a limited number of novel Gag mutations in the reservoir of some early treated participants and all late treated participants was observed, where the majority were likely CTL-driven. The data suggest that partial CTL escape in conserved immunogen regions of Gag is present in the reservoir and that the extent of escape is likely similar between early and late treated individuals, as well as between the reservoir and pre-therapy virus variants. The unique sequence differences in Gag from the reservoir viruses did not result in reservoir viruses that were substantially different to pre-therapy plasma viruses, in terms of IFN-α resistance, HIV cell-to-cell spread ability and induction of cytokines, suggesting that these sequence differences are unlikely to significantly affect immune-based interventions for virus eradication.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, ON552272 — ON552477. https://www.ncbi.nlm.nih.gov/sra, PRJNA833316.
The studies involving human participants were reviewed and approved by Biomedical Research Ethics Committee of the University of KwaZulu-Natal. The patients/participants provided their written informed consent to participate in this study.
JM and TN conceived and designed the study. TN, ZN, and KD initiated the study cohorts. DO, JM, TM, NG, OB, KR, and JG performed experiments. JM, DO, KG, and JG performed data analysis. JM and DO drafted the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by the FLAIR Fellowship Programme, which is a partnership between the African Academy of Sciences and the Royal Society funded by the UK Government’s Global Challenges Research Fund (FLR\R1\201494 to JM). This work was supported in part by grants from the International AIDS Vaccine Initiative (IAVI) (UKZNRSA1001), Bill and Melinda Gates Foundation – OPP1212883, Gilead Sciences, Inc (Grant ID#00406), the NIAID (R37AI067073), the South African Research Chairs Initiative (grant # 64809) the Witten Family Foundation, Dan and Marjorie Sullivan, the Mark and Lisa Schwartz Foundation, Ursula Brunner, and the Howard Hughes Medical Institute. This work was also supported in part through the Sub-Saharan African Network for TB/HIV Research Excellence (SANTHE), a DELTAS Africa Initiative [grant # DEL-15-006]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [grant # 107752/Z/15/Z] and the United Kingdom (UK) government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust or the UK government.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.916095/full#supplementary-material
Supplementary Figure 1 | Phylogenetic relatedness of the gag-protease sequences from reservoirs, pre-therapy plasma and their matched recombinant viruses. A maximum likelihood tree showing the phylogenetic relatedness of HIV-1 subtype C gag-protease sequences from the pre-therapy plasma (in blue), peripheral blood mononuclear cells (in black), lymph nodes (in red) and respective recombinant viruses at a scale of 0.008. “PL” indicates pre-therapy plasma, “PB” indicates peripheral blood mononuclear cells and “LN” indicates lymph node mononuclear cells. The suffix “PCR” indicates the sequences of amplicons from the different body compartments, while “Virus” indicates the sequences from the matched cultured recombinant viruses. Highlighted in green are the HIV 1 subtype C consensus and NL4-3 laboratory strain virus sequences.
Supplementary Figure 2 | Representative flow cytometry plots measuring replication capacities of viruses in the presence and absence of interferon alpha. The replication capacities of the recombinant viruses encoding gag-protease from the lymph node (LN) and peripheral blood mononuclear cell (PBMC) reservoirs, as well as from the pre-therapy plasma, in the presence and absence of type I interferon alpha (IFN-α) was measured. Replication capacity was measured as the spread of infection from days 3 to 6 post-infection, where infected cells (expressing green fluorescent protein [GFP]) were detected by measurement of GFP by flow cytometry. Replication capacity was calculated as the natural log of the slope of exponential increase in GFP positive cells. IFN-α resistance was calculated as the ratio of the replication capacity in the presence and absence of IFN-α. Representative data from participant 111-33-0067-0 (PID 0067) is shown.
Supplementary Figure 3 | Flow cytometry gating strategies in the HIV cell-to-cell spread assay. (A) Gating of Jurkat cells based on forward scatter (FSC) and side scatter (SSC) plots is shown. Zombie NIR was used to distinguish live cells from dead cells. Plots show (i) uninfected Jurkat cells and (ii) Jurkat cells infected with 100ng p24 equivalent of virus derived from the plasma of participant 111-30-0098-0 (0098PL), after 4-5 days of incubation. (B) CellTrace™ far red-stained Jurkat cells (target cells - APC) and eFlour 450-stained Jurkat cells (donor cells – BV 450) are shown. (C) Plots show the spread of infection in both donor and target cells from 0-48 hours following co-culture of donor (5% infected) and uninfected target cells at a ratio of 1:1. Infected cells were detected by intracellular Gag p24 staining (p24-PE).
1. Pau AK, George JM. Antiretroviral Therapy: Current Drugs. Infect Dis Clin North Am (2014) 28(3):371–402. doi: 10.1016/j.idc.2014.06.001
2. Pitman MC, Lau JSY, McMahon JH, Lewin SR. Barriers and Strategies to Achieve a Cure for HIV. Lancet HIV (2018) 5(6):e317–e28. doi: 10.1016/S2352-3018(18)30039-0
3. Wong JK, Yukl SA. Tissue Reservoirs of HIV. Curr Opin HIV AIDS (2016) 11(4):362–70. doi: 10.1097/COH.0000000000000293
4. De Scheerder MA, Vrancken B, Dellicour S, Schlub T, Lee E, Shao W, et al. HIV Rebound Is Predominantly Fueled by Genetically Identical Viral Expansions From Diverse Reservoirs. Cell Host Microbe (2019) 26(3):347–58.e7. doi: 10.1016/j.chom.2019.08.003
5. North TW, Higgins J, Deere JD, Hayes TL, Villalobos A, Adamson L, et al. Viral Sanctuaries During Highly Active Antiretroviral Therapy in a Nonhuman Primate Model for AIDS. J Virol (2010) 84:2913–22. doi: 10.1128/JVI.02356-09
6. Kline C, Ndjomou J, Franks T, Kiser R, Coalter V, Smedley J, et al. Persistence of Viral Reservoirs in Multiple Tissues After Antiretroviral Therapy Suppression in a Macaque RT-SHIV Model. PloS One (2013) 8(12):e84275. doi: 10.1371/journal.pone.0084275
7. Günthard HF, Havlir DV, Fiscus S, Zhang ZQ, Eron J, Mellors J, et al. Residual Human Immunodeficiency Virus (HIV) Type 1 RNA and DNA in Lymph Nodes and HIV RNA in Genital Secretions and in Cerebrospinal Fluid After Suppression of Viremia for 2 Years. J Infect diseases (2001) 183:1318–27. doi: 10.1086/319864
8. Haddad DN, Birch C, Middleton T, Dwyer DE, Cunningham AL, Saksena NK. Evidence for Late Stage Compartmentalization of HIV-1 Resistance Mutations Between Lymph Node and Peripheral Blood Mononuclear Cells. AIDS (2000) 14(15):2273–81. doi: 10.1097/00002030-200010200-00008
9. Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-Specific Cytolytic T Lymphocytes Facilitates Elimination of Latent Viral Reservoir After Virus Reactivation. Immunity (2012) 36(3):491–501. doi: 10.1016/j.immuni.2012.01.014
10. Oxenius A, Price DA, Easterbrook PJ, O’Callaghan CA, Kelleher AD, Whelan JA, et al. Early Highly Active Antiretroviral Therapy for Acute HIV-1 Infection Preserves Immune Function of CD8+ and CD4+ T Lymphocytes. Proc Natl Acad Sci U S A (2000) 97:3382–7. doi: 10.1073/pnas.97.7.3382
11. Crowell TA, Fletcher JL, Sereti I, Pinyakorn S, Dewar R, Krebs SJ, et al. Initiation of Antiretroviral Therapy Before Detection of Colonic Infiltration by HIV Reduces Viral Reservoirs, Inflammation and Immune Activation. J Int AIDS Soc (2016) 19(1):21163. doi: 10.7448/IAS.19.1
12. Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, et al. Broad CTL Response is Required to Clear Latent HIV-1 Due to Dominance of Escape Mutations. Nature (2015) 517(7534):381–5. doi: 10.1038/nature14053
13. Mothe B, Hu X, Llano A, Rosati M, Olvera A, Kulkarni V, et al. A Human Immune Data-Informed Vaccine Concept Elicits Strong and Broad T-Cell Specificities Associated With HIV-1 Control in Mice and Macaques. J Transl Med (2015) 13(1):60. doi: 10.1186/s12967-015-0392-5
14. Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, et al. HIV-1 Evades Innate Immune Recognition Through Specific Cofactor Recruitment. Nature (2013) 503(7476):402–5. doi: 10.1038/nature12769
15. Gardiner JC, Mauer EJ, Sherer NM. HIV-1 Gag, Envelope, and Extracellular Determinants Cooperate To Regulate the Stability and Turnover of Virological Synapses. J Virol (2016) 90(14):6583–97. doi: 10.1128/JVI.00600-16
16. Ndhlovu ZM, Kamya P, Mewalal N, Kløverpris HN, Nkosi T, Pretorius K, et al. Magnitude and Kinetics of CD8+ T Cell Activation During Hyperacute HIV Infection Impact Viral Set Point. Immunity (2015) 43(3):591–604. doi: 10.1016/j.immuni.2015.08.012
17. Radebe M, Nair K, Chonco F, Bishop K, Wright JK, van der Stok M, et al. Limited Immunogenicity of HIV CD8+ T-Cell Epitopes in Acute Clade C Virus Infection. J Infect Diseases (2011) 204(5):768–76. doi: 10.1093/infdis/jir394
18. Ogunshola FJ, Smidt W, Naidoo AF, Nkosi TP, Ngubane T, Khaba PT, et al. Hypermethylation at the CXCR5 Gene Locus Limits Trafficking Potential of CD8+ T Cells Into B-Cell Follicles During HIV-1 Infection. Blood advances (2022) 6(6):1904–16. doi: 10.1182/bloodadvances.2021006001
19. Wright JK, Brumme ZL, Carlson JM, Heckerman D, Kadie CM, Brumme CJ, et al. Gag-Protease-Mediated Replication Capacity in HIV-1 Subtype C Chronic Infection: Associations With HLA Type and Clinical Parameters. J Virol (2010) 84(20):10820–31. doi: 10.1128/JVI.01084-10
20. Gounder K, Oyaro M, Padayachi N, Zulu TM, de Oliveira T, Wylie J, et al. Complex Subtype Diversity of HIV-1 Among Drug Users in Major Kenyan Cities. AIDS Res Hum Retroviruses (2017) 33(5):500–10. doi: 10.1089/AID.2016.0321
21. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst Biol (2010) 59(3):307–21. doi: 10.1093/sysbio/syq010
22. Ojwach DBA, MacMillan D, Reddy T, Novitsky V, Brumme ZL, Brockman MA, et al. Pol-Driven Replicative Capacity Impacts Disease Progression in HIV-1 Subtype C Infection. J Virol (2018) 92(19):e00811–18. doi: 10.1128/JVI.00811-18
23. Brumme ZL, Li C, Miura T, Sela J, Rosato PC, Brumme CJ, et al. Reduced Replication Capacity of NL4-3 Recombinant Viruses Encoding Reverse Transcriptase-Integrase Sequences From HIV-1 Elite Controllers. J Acquir Immune Defic Syndr (2011) 56(2):100–8. doi: 10.1097/QAI.0b013e3181fe9450
24. Wright JK, Naidoo VL, Brumme ZL, Prince JL, Claiborne DT, Goulder PJR, et al. Impact of HLA-B*81-Associated Mutations in HIV-1 Gag on Viral Replication Capacity. J Virol (2012) 86(6):3193–9. doi: 10.1128/JVI.06682-11
25. Brockman MA, Tanzi GO, Walker BD, Allen TM. Use of a Novel GFP Reporter Cell Line to Examine Replication Capacity of CXCR4-And CCR5-Tropic HIV-1 by Flow Cytometry. J Virological Methods (2006) 131(2):134–42. doi: 10.1016/j.jviromet.2005.08.003
26. Deymier MJ, Ende Z, Fenton-May AE, Dilernia DA, Kilembe W, Allen SA, et al. Heterosexual Transmission of Subtype C HIV-1 Selects Consensus-Like Variants Without Increased Replicative Capacity or Interferon-α Resistance. PloS Pathog (2015) 11(9):e1005154. doi: 10.1371/journal.ppat.1005154
27. Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, et al. Phenotypic Properties of Transmitted Founder HIV-1. Proc Natl Acad Sci U S A (2013) 110(17):6626–33. doi: 10.1073/pnas.1304288110
28. Jolly C, Booth NJ, Neil SJ. Cell-Cell Spread of Human Immunodeficiency Virus Type 1 Overcomes Tetherin/BST-2-Mediated Restriction in T Cells. J Virol (2010) 84(23):12185–99. doi: 10.1128/JVI.01447-10
29. Wong JK, Günthard HF, Havlir DV, Zhang Z-Q, Haase AT, Ignacio CC, et al. Reduction of HIV-1 in Blood and Lymph Nodes Following Potent Antiretroviral Therapy and the Virologic Correlates of Treatment Failure. Proc Natl Acad Sci (1997) 94(23):12574–9. doi: 10.1073/pnas.94.23.12574
30. Ondondo B, Murakoshi H, Clutton G, Abdul-Jawad S, Wee EG, Gatanaga H, et al. Novel Conserved-Region T-Cell Mosaic Vaccine With High Global HIV-1 Coverage Is Recognized by Protective Responses in Untreated Infection. Mol Ther (2016) 24(4):832–42. doi: 10.1038/mt.2016.3
31. Lee GQ, Reddy K, Einkauf KB, Gounder K, Chevalier JM, Dong KL, et al. HIV-1 DNA Sequence Diversity and Evolution During Acute Subtype C Infection. Nat Commun (2019) 10(1):2737. doi: 10.1038/s41467-019-10659-2
32. Sultana T, Mamede JI, Saito A, Ode H, Nohata K, Cohen R, et al. Multiple Pathways To Avoid Beta Interferon Sensitivity of HIV-1 by Mutations in Capsid. J Virol (2019) 93(23):e00986–19. doi: 10.1128/JVI.00986-19
33. Wright JK, Novitsky V, Brockman MA, Brumme ZL, Brumme CJ, Carlson JM, et al. Influence of Gag-Protease-Mediated Replication Capacity on Disease Progression in Individuals Recently Infected With HIV-1 Subtype C. J Virol (2011) 85(8):3996–4006. doi: 10.1128/JVI.02520-10
34. Iwami S, Takeuchi JS, Nakaoka S, Mammano F, Clavel F, Inaba H, et al. Cell-To-Cell Infection by HIV Contributes Over Half of Virus Infection. Elife (2015) 4:e08150. doi: 10.7554/eLife.08150
35. Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, et al. Cell-To-Cell Spread of HIV Permits Ongoing Replication Despite Antiretroviral Therapy. Nature (2011) 477(7362):95–8. doi: 10.1038/nature10347
36. Kenway-Lynch CS, Das A, Lackner AA, Pahar B. Cytokine/Chemokine Responses in Activated CD4+ and CD8+ T Cells Isolated From Peripheral Blood, Bone Marrow, and Axillary Lymph Nodes During Acute Simian Immunodeficiency Virus Infection. J Virol (2014) 88(16):9442–57. doi: 10.1128/JVI.00774-14
37. Dinarello CA. Proinflammatory Cytokines. Chest (2000) 118(2):503–8. doi: 10.1378/chest.118.2.503
38. Kedzierska K, Crowe SM. Cytokines and HIV-1: Interactions and Clinical Implications. Antivir Chem Chemother (2001) 12(3):133–50. doi: 10.1177/095632020101200301
39. Dinarello CA. Historical Insights Into Cytokines. Eur J Immunol (2007) 37(S1):S34–45. doi: 10.1002/eji.200737772
40. Ryabov VV, Kirgizova MA, Suslova TE, Karas SI, Markov VA, Karpov RS. Relationships of Growth Factors, Proinflammatory Cytokines, and Anti-Inflammatory Cytokines With Long-Term Clinical Results of Autologous Bone Marrow Mononuclear Cell Transplantation in STEMI. PloS One (2017) 12(5):e0176900. doi: 10.1371/journal.pone.0176900
41. Simmonds P, Zhang LQ, McOmish F, Balfe P, Ludlam CA, Brown AJ. Discontinuous Sequence Change of Human Immunodeficiency Virus (HIV) Type 1 Env Sequences in Plasma Viral and Lymphocyte-Associated Proviral Populations In Vivo: Implications for Models of HIV Pathogenesis. J Virol (1991) 65(11):6266–76. doi: 10.1128/jvi.65.11.6266-6276.1991
42. Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, et al. Persistent HIV-1 Replication is Associated With Lower Antiretroviral Drug Concentrations in Lymphatic Tissues. Proc Natl Acad Sci U S A (2014) 111(6):2307–12. doi: 10.1073/pnas.1318249111
43. Yukl SA, Shergill AK, McQuaid K, Gianella S, Lampiris H, Hare CB, et al. Effect of Raltegravir-Containing Intensification on HIV Burden and T-Cell Activation in Multiple Gut Sites of HIV-Positive Adults on Suppressive Antiretroviral Therapy. AIDS (2010) 24(16):2451–60. doi: 10.1097/QAD.0b013e32833ef7bb
44. Buzón MJ, Massanella M, Llibre JM, Esteve A, Dahl V, Puertas MC, et al. HIV-1 Replication and Immune Dynamics Are Affected by Raltegravir Intensification of HAART-Suppressed Subjects. Nat Med (2010) 16(4):460–5. doi: 10.1038/nm.2111
45. Joseph SB, Kincer LP, Bowman NM, Evans C, Vinikoor MJ, Lippincott CK, et al. Human Immunodeficiency Virus Type 1 RNA Detected in the Central Nervous System (CNS) After Years of Suppressive Antiretroviral Therapy Can Originate From a Replicating CNS Reservoir or Clonally Expanded Cells. Clin Infect Dis (2019) 69(8):1345–52. doi: 10.1093/cid/ciy1066
46. Real F, Capron C, Sennepin A, Arrigucci R, Zhu A, Sannier G, et al. Platelets From HIV-Infected Individuals on Antiretroviral Drug Therapy With Poor CD4(+) T Cell Recovery can Harbor Replication-Competent HIV Despite Viral Suppression. Sci Trans Med (2020) 12(535):eaat6263. doi: 10.1126/scitranslmed.aat6263
47. Bozzi G, Simonetti FR, Watters SA, Anderson EM, Gouzoulis M, Kearney MF, et al. No Evidence of Ongoing HIV Replication or Compartmentalization in Tissues During Combination Antiretroviral Therapy: Implications for HIV Eradication. Sci Adv (2019) 5(9):eaav2045. doi: 10.1126/sciadv.aav2045
48. Van Zyl GU, Katusiime MG, Wiegand A, McManus WR, Bale MJ, Halvas EK, et al. No Evidence of HIV Replication in Children on Antiretroviral Therapy. J Clin Invest (2017) 127(10):3827–34. doi: 10.1172/JCI94582
49. McManus WR, Bale MJ, Spindler J, Wiegand A, Musick A, Patro SC, et al. HIV-1 in Lymph Nodes Is Maintained by Cellular Proliferation During Antiretroviral Therapy. J Clin Invest (2019) 129(11):4629–42. doi: 10.1172/JCI126714
50. Rasmussen TA, McMahon JH, Chang JJ, Audsley J, Rhodes A, Tennakoon S, et al. The Effect of Antiretroviral Intensification With Dolutegravir on Residual Virus Replication in HIV-Infected Individuals: A Randomised, Placebo-Controlled, Double-Blind Trial. Lancet HIV (2018) 5(5):e221–e30. doi: 10.1016/S2352-3018(18)30040-7
51. Vancoillie L, Hebberecht L, Dauwe K, Demecheleer E, Dinakis S, Vaneechoutte D, et al. Longitudinal Sequencing of HIV-1 Infected Patients With Low-Level Viremia for Years While on ART Shows No Indications for Genetic Evolution of the Virus. Virology (2017) 510:185–93. doi: 10.1016/j.virol.2017.07.010
52. Colby DJ, Trautmann L, Pinyakorn S, Leyre L, Pagliuzza A, Kroon E, et al. Rapid HIV RNA Rebound After Antiretroviral Treatment Interruption in Persons Durably Suppressed in Fiebig I Acute HIV Infection. Nat Med (2018) 24(7):923–6. doi: 10.1038/s41591-018-0026-6
53. Gounder K, Padayachi N, Mann JK, Radebe M, Mokgoro M, van der Stok M, et al. High Frequency of Transmitted HIV-1 Gag HLA Class I-Driven Immune Escape Variants But Minimal Immune Selection Over the First Year of Clade C Infection. PloS One (2015) 10(3):e0119886. doi: 10.1371/journal.pone
54. Roberts HE, Hurst J, Robinson N, Brown H, Flanagan P, Vass L, et al. Structured Observations Reveal Slow HIV-1 CTL Escape. PloS Genet (2015) 11(2):e1004914. doi: 10.1371/journal.pgen.1004914
55. Warren JA, Zhou S, Xu Y, Moeser MJ, MacMillan DR, Council O, et al. The HIV-1 Latent Reservoir Is Largely Sensitive to Circulating T Cells. Elife (2020) 9:e57246. doi: 10.7554/eLife.57246
56. Deeks SG, Odorizzi PM, Sekaly R-P. The Interferon Paradox: Can Inhibiting an Antiviral Mechanism Advance an HIV Cure? J Clin Invest (2017) 127(1):103–5. doi: 10.1172/JCI91916
57. Gondim MVP, Sherrill-Mix S, Bibollet-Ruche F, Russell RM, Trimboli S, Smith AG, et al. Heightened Resistance to Host Type 1 Interferons Characterizes HIV-1 at Transmission and After Antiretroviral Therapy Interruption. Sci Trans Med (2021) 13(576):eabd8179. doi: 10.1126/scitranslmed.abd8179
58. Abela IA, Berlinger L, Schanz M, Reynell L, Günthard HF, Rusert P, et al. Cell-Cell Transmission Enables HIV-1 to Evade Inhibition by Potent CD4bs Directed Antibodies. PloS Pathog (2012) 8(4):e1002634. doi: 10.1371/journal.ppat.1002634
59. Durham ND, Chen BK. HIV-1 Cell-Free and Cell-To-Cell Infections Are Differentially Regulated by Distinct Determinants in the Env Gp41 Cytoplasmic Tail. J Virol (2015) 89(18):9324–37. doi: 10.1128/JVI.00655-15
60. Montaner LJ, Gordon S. Th2-Mediated HIV1 Virostatic State: Macrophage-Specific Regulation In Vitro. Res Immunol (1994) 145(8):583–8. doi: 10.1016/S0923-2494(05)80037-9
61. Kelly MD, Naif HM, Adams SL, Cunningham AL, Lloyd AR. Cutting Edge: Dichotomous Effects of β-Chemokines on HIV Replication in Monocytes and Monocyte-Derived Macrophages. J Immunol (1998) 160(7):3091–5. doi: 10.1126/scitranslmed.abd8179
62. Watson DC, Moysi E, Valentin A, Bergamaschi C, Devasundaram S, Fortis SP, et al. Treatment With Native Heterodimeric IL-15 Increases Cytotoxic Lymphocytes and Reduces SHIV RNA in Lymph Nodes. PloS Pathog (2018) 14(2):e1006902. doi: 10.1371/journal.ppat.1006902
63. Micci L, Ryan ES, Fromentin R, Bosinger SE, Harper JL, He T, et al. Interleukin-21 Combined With ART Reduces Inflammation and Viral Reservoir in SIV-Infected Macaques. J Clin Invest (2015) 125(12):4497–513. doi: 10.1172/JCI81400
64. Bethel-Brown C, Yao H, Hu G, Buch S. Platelet-Derived Growth Factor (PDGF)-BB-Mediated Induction of Monocyte Chemoattractant Protein 1 in Human Astrocytes: Implications for HIV-Associated Neuroinflammation. J Neuroinflammation (2012) 9:262. doi: 10.1186/1742-2094-9-262
65. Kinter A, Catanzaro A, Monaco J, Ruiz M, Justement J, Moir S, et al. CC-Chemokines Enhance the Replication of T-Tropic Strains of HIV-1 in CD4(+) T Cells: Role of Signal Transduction. Proc Natl Acad Sci USA (1998) 95(20):11880–5. doi: 10.1073/pnas.95.20.11880
66. Kelly MD, Naif HM, Adams SL, Cunningham AL, Lloyd AR. Dichotomous Effects of Beta-Chemokines on HIV Replication in Monocytes and Monocyte-Derived Macrophages. J Immunol (1998) 160(7):3091–5.
67. Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 Alpha, and MIP-1 Beta as the Major HIV-Suppressive Factors Produced by CD8+ T Cells. Science (1995) 270(5243):1811–5. doi: 10.1126/science.270.5243.1811
Keywords: HIV-1, reservoir, lymph nodes, peripheral blood mononuclear cells, HIV-1 gag, cytotoxic T lymphocyte-driven mutations, interferon-alpha resistance, cell-to-cell spread
Citation: Ojwach D, Gounder K, Mulaudzi T, Gumede N, Baiyegunhi OO, Reddy K, Giandhari J, Dong KL, Ndhlovu Z, Ndung’u T and Mann JK (2022) Limited Sequence Variation and Similar Phenotypic Characteristics of HIV-1 Subtype C Gag Variants Derived From the Reservoir and Pre-Therapy Plasma. Front.Virol. 2:916095. doi: 10.3389/fviro.2022.916095
Received: 08 April 2022; Accepted: 30 May 2022;
Published: 08 July 2022.
Edited by:
Caroline T. Tiemessen, National Institute of Communicable Diseases (NICD), South AfricaReviewed by:
Maarten Van De Klundert, Karolinska University Hospital, SwedenCopyright © 2022 Ojwach, Gounder, Mulaudzi, Gumede, Baiyegunhi, Reddy, Giandhari, Dong, Ndhlovu, Ndung’u and Mann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaclyn K. Mann, bWFubmpAdWt6bi5hYy56YQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.