 Riccardo Delli Ponti
Riccardo Delli Ponti Jiaxu Wang2
Jiaxu Wang2 Roland G. Huber
Roland G. Huber
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Virol. , 27 June 2022
Sec. Bioinformatic and Predictive Virology
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.878679
This article is part of the Research Topic Insights In Bioinformatic and Predictive Virology:2021 View all 4 articles
Identifying structural elements in long and complex RNAs, such as long non-coding and RNA viruses, can shed light on the functionality and mechanisms of such RNAs. Here we present RNAvigator, a tool able to identify elements of structural importance by using experimental SHAPE data or SHAPE-like predictions in conjunction with stability and entropy assessments. RNAvigator recognizes regions that are the most stable, unambiguous, and structured on RNA molecules, and thus potentially functional. When relying on predictions, RNAvigator uses the CROSS algorithm, a neural network trained on experimental data that achieved an AUC of 0.74 on hepatitis C virus SHAPE-MaP data and which was able to improve the predictive power of Superfold. By using RNAvigator, we can identify known functional regions on the complete hepatitis C virus genome, including the regulatory regions CRE and IRES, and the 3’ UTR of dengue virus, a region known for the presence of structural elements essential for its replication, and functional regions of long non-coding RNAs such as XIST and HOTAIR. We envision that RNAvigator will be a useful tool for studying long and complex RNA molecules using known chemical probing data or, if they are not available, by employing predicted profiles.
Recently, significant advances have been made in the elucidation of RNA structures. It has been understood for some time that the RNA structure can play a critical role in gene regulation. Structural motifs in RNA can influence various biological processes, ranging from translation initiation and termination (1), resistance to degradation (2, 3), splicing (4), or ligand-dependent conformational switching (5, 6). Unfortunately, determining the RNA structure has proven challenging (7), as most RNAs are not readily amenable to conventional structural biology approaches such as X-ray crystallography or cryo-electron microscopy, mainly due to their conformational flexibility and low propensity to crystallize (8, 9). While some progress has been made using nuclear magnetic resonance spectroscopy, challenges regarding isotope labeling and size restrictions remain, especially for large and complex RNA molecules (10, 11). The advent of high-throughput sequencing has enabled the application of the previously developed chemical structure probing techniques on a larger scale. Several protocols based on selective 2’-hydroxyl acylation analysed by primer extension (SHAPE) have been developed, allowing the extraction of secondary structure information of particular RNAs, among them SHAPE-MaP (7) and in vivo click SHAPE (icSHAPE) (12, 13). It has been shown that the incorporation of chemical probing data with traditional computational structure prediction methods allows for the accurate determination of RNA secondary structure in large RNAs, e.g., long non-coding RNAs (lncRNAs) or viral genomes of RNA viruses, while also improving the predictive power of thermodynamics-based algorithms (7, 14).
While significant progress has been made in RNA structure determination, the challenge of interpreting these structures and identifying functional elements remain. Generally, structural surveys readily produce a plethora of structural elements, but a significant proportion of these elements are likely opportunistic folds without much functional relevance (7, 15, 16). Other structural elements play a key role in the biological processing of these RNAs, and a number of examples are well described. In particular, elements in the 5’- and 3’-untranslated regions (UTRs) of coding RNA segments, as well as lncRNAs, contain structural elements crucial for their function (1, 17–20). However, examples of functional segments have also been found in coding segments (18, 21, 22). Such functional elements are likely to be conserved as evolutionary selection pressures apply to them. Moreover, such elements generally have a well-defined, unambiguous, and stable fold to exert their function (20, 23). Using these criteria, we can survey structural probing data and structural models of RNA to identify candidate functional elements.
We developed the RNAvigator pipeline to aid in the identification of functionally relevant RNA structures. RNAvigator takes aa input a target sequence and chemical probing data to search for the most stable, unambiguous, and structured segments as candidates for functional elements. In the absence of chemical probing data, RNAvigator uses Computational Recognition of Secondary Structure (CROSS), an AI-based tool that has previously shown good performance when compared with SHAPE data (24), to impute SHAPE-like data to feed the pipeline. Here, we demonstrate how RNAvigator allows us to recover known functional elements in viral RNAs, and we demonstrate that we can retrieve accurate structure models even in the absence of SHAPE data. We created a simple tool that processes the inputs and provides illustrative charts highlighting the different criteria and consensus regions for functional candidates along the length of the query RNA. RNAvigator is available on GitHub (https://github.com/RiccardoDP/RNAvigator) under the terms of the GNU public license v3 (GPLv3).
SHAPE data and the relative viral sequence for the complete genomes of hepatitis C virus (HCV; hcv 77) and dengue virus (DENV; DENV-1) were downloaded/extracted from the original papers (21, 25). The exact sequences were selected from the SHAPE-MaP files to keep consistency with SHAPE reactivities.
We used Scanfold (26) to discover structurally stable RNA regions. Specifically, we used the ScanFold-Scan module with a step of one nucleotide and a window of 150. We also studied different windows, and the results showed a high degree of consistency across window sizes ranging from 150 to 500 (Supplementary Figure 1). RNAvigator selects the most stable regions, corresponding to the lowest 20% of regions for block-averaged z-score regions.
The secondary structure predictions were computed using the CROSS (Computational Recognition of Secondary Structure) algorithm. CROSS is a machine learning approach trained on experimental data (SHAPE, PARS, NMR/X-Ray, and icSHAPE), which was applied to large and complex molecules such as viral genomes. We used the Global Score model, considering nucleotides with a score of >0 as double-stranded, and <0 as single-stranded. For integrating CROSS as a SHAPE-like constraint inside the Superfold, we applied a normalization (check Shannon Entropy Calculation With Superfold; Supplementary Figure 2).
To assess the secondary structure of RNA molecules, we used Superfold (7). The tool is optimized to employ SHAPE data as constraints for RNAstructure (27) and to merge sliding windows of local structure models to obtain the full-length structure of large RNAs. We used Superfold using both SHAPE-MaP data and CROSS predictions. For both the pipelines, Superfold was launched using standard settings for the SHAPE slope and intercept. CROSS predictions were normalized to match a SHAPE-like distribution processable by Superfold, using the following formula:
The three algorithms comprising the RNAvigator pipeline were independently used to select regions of the following: 1) high double-stranded propensity (SHAPE and/or CROSS), 2) structural stability (Scanfold), and 3) low entropy, indicating unambiguous structures (Superfold). For the output from each algorithm, we ranked the averaged regions of the selected metric (reactivity/propensity for SHAPE/CROSS; z-score for Scanfold; entropy value for Superfold), and we selected the bottom 20% regions for each metric, thus identifying the most structured, most stable, and least ambiguous structures along the RNA of interest. Unambiguous, stable and structured regions are often encountered in known functional elements, e.g., the flavivirus 5’ and 3’ UTRs (21, 28). We used a window of 150 nucleotides to average the different scores.
The pipeline was built using the previously described methods in order to identify regions of importance according to structural stability, secondary structure profiles, and entropy. Searching for the structural stability by using Scanfold is an independent step, while the secondary structure profiles, obtained using SHAPE-MaP data or normalized CROSS predictions, are also used as input to compute the entropy with Superfold. After obtaining the three profiles from the different metrics, regions of interest are selected following the methodology in Selecting Regions of Structural Importance. The main output of the pipeline is a plot showing the highlighted regions for each metric and a file containing information not only on how many regions are identified by each metric but also the exact secondary structure of those regions (dots and brackets notation extracted from Superfold, ct output).
To generate a p-value to determine how significant it is to identify specific regions of known RNA molecules, such as for XIST and HOTAIR, we randomly shuffled the RNA regions (150 nucleotides for XIST, 50 nucleotides for HOTAIR). We then selected the bottom 20% following the RNAvigator pipeline, and we used this information to check how many times we could have more or equal random regions following inside our domain of interest compared with our data. We repeated this 10,000 times, and we used the information to build a p-value.
For the statistics, metrics, and figures, we used the most up-to-date version of Statistical Software R (29) and the associated libraries.
Particularly for novel or complex RNA, identifying regions of structural importance is a fundamental step toward their characterization and functional discovery (Figure 1A). The idea behind RNAvigator is to create a comprehensive pipeline to identify regions of interest in complex RNA molecules. To identify the aforementioned regions, the pipeline implements three levels of data: 1) structural stability; 2) secondary structure profiles, and 3) entropy. For this purpose, we developed two parallel pipelines, one for use when experimental data are available, specifically SHAPE-MaP chemical probing profiles, and the other based on secondary structure predictions (Figure 1B). A combination of these data sets and criteria has proven successful in highlighting functionally important regions, e.g., in Flaviviruses, HIV, HCV, or most recently in SARS-CoV-2 genomes (21, 30–33).

Figure 1 (A) Graphical representation of how understanding the RNA secondary structure, by using experiments or predictions, is crucial to assess the functionality of a RNA molecule. (B) Workflow of RNAvigator explaining the differences in the two pipelines while using experimental data (SHAPE) or predicted secondary structure profiles (CROSS) as input.
To analyze the structural stability, we used Scanfold (26). Scanfold assesses the stability of secondary structure motifs in a particular region and hence allows the ranking of regions by their propensity to fold into stable conformations. RNAvigator generates Scanfold profiles for each nucleotide in a window of 150 nucleotides, as suggested in previous publications (21, 33, 34). Nucleotides were ranked based on the reported z-score, a metric that assesses structural stability. We studied the different z-scores reported by Scanfold on the complete DENV-1 when using a different window (from 150 to 500), and we found a satisfying correlation between the values (Supplementary Figure 1), thus indicating that the algorithm is robust for various window sizes.
We built RNAvigator to work when experimental data are available, specifically SHAPE chemical probing data, or in the absence of such data, to rely on predictions using the CROSS algorithm. Secondary structure data are a key element to understanding regions of structural importance, especially for identifying nucleotides with low SHAPE reactivity, indicating a high propensity for double-stranded and thus structured conformations. Structured regions are essential to identifying regions of potential functionality, especially in long and complex RNAs such as lncRNAs or viral genomes. For this reason, RNAvigator uses SHAPE data to identify RNA regions enriched in high-propensity double-stranded nucleotides, but it can also include predictions in cases where experimental data are not available.
We used Superfold, an algorithm specifically developed to improve the predictions of RNA secondary structure by using SHAPE data (7, 19, 35), and the underlying RNAstructure (14, 27) software package, to derive pairing probability, which allows us to calculate structure models and Shannon entropy at specific positions. A Shannon entropy indicates the degree of uncertainty about a specific base pair, so a position that is uniquely paired with another nucleotide will have a Shannon entropy of 0, while a nucleotide that has various predicted interaction partners will exhibit a higher Shannon entropy. Superfold generates not only the complete list of secondary structure interactions but also the positional Shannon entropy based on the matching probabilities generated by the partition function (7, 14). It is worth specifying that nucleotides with low entropy are more prone to being part of unambiguous structures, which can be important for assessing the functionality of RNA elements (22, 33, 34, 36).
The experimental- and predictive-based pipelines of RNAvigator differ specifically in the handling of secondary structure data, while the steps encoding for structure stability and entropy are similar (Figure 1B).
We used the CROSS algorithm to build the predicting side of RNAvigator, allowing the identification of regions of structural importance even when SHAPE data are not available. We selected CROSS since the algorithm already showed good similarity with SHAPE data compared with RNA crystal structures, e.g., of the E. coli ribosome, and it has already been successfully employed as SHAPE-like constraints inside thermodynamics-based modeling tools (24). Moreover, CROSS showed high performance in terms of area under the ROC curve (AUC) compared with SHAPE data in viral genomes, such as HIV, SARS-CoV-2, and DENV-1 (24, 37). To further validate the approach, we also applied CROSS to predict SHAPE data on the HCV genome, reaching an AUC of 0.74 (Figure 2A). As previously reported, CROSS has already been tested on DENV-1 data, achieving an AUC of 0.85 (37).
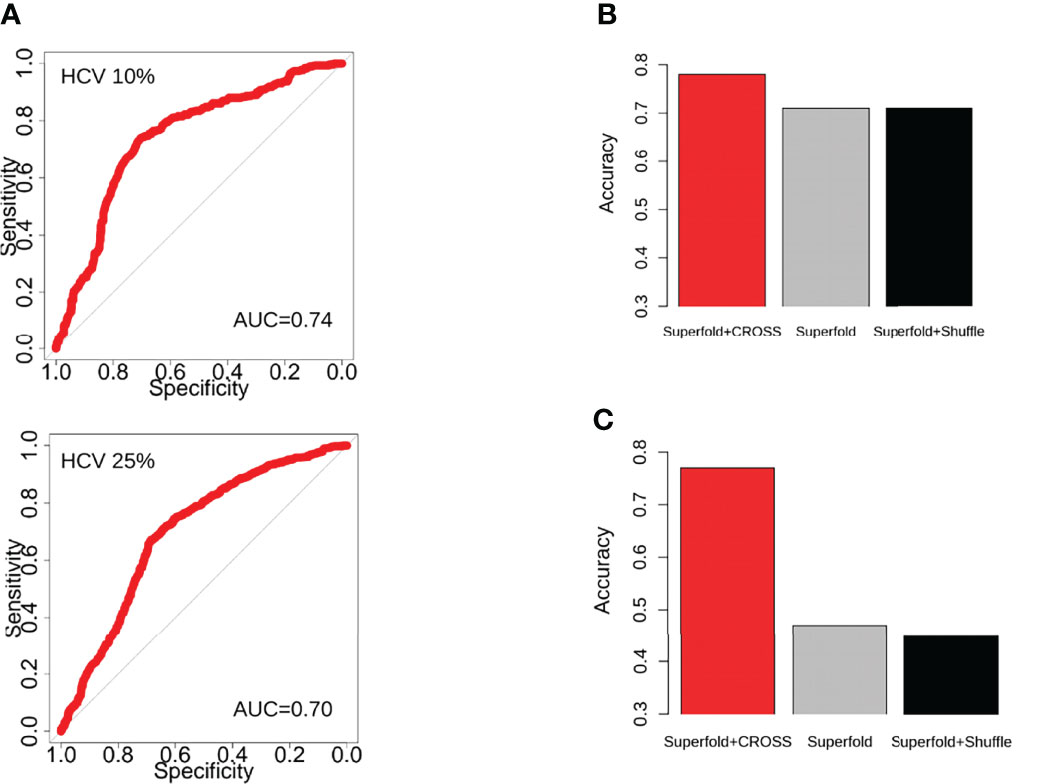
Figure 2 (A) ROC curves and AUCs of the CROSS algorithm to predict the top and bottom (10–25%) nucleotides ranked for their SHAPE reactivity for HCV. The 25% corresponds to half of the dataset. (B) Barplot showing the performance (Accuracy) of Superfold while using the best SHAPE-like normalisation of CROSS (red), an empty Shape-MaP file (gray), and the shuffled CROSS profile (black), compared with the structure (same interactions for paired nucleotides) obtained with Superfold using SHAPE-MaP for HCV (first 1,500 nt) as input. (C) Barplot showing the performance (Accuracy) of Superfold while using the best SHAPE-like normalization of CROSS (red), an empty Shape-MaP file (gray), and the shuffled CROSS profile (black), compared with the structure (same interactions for paired nucleotides) obtained with Superfold using SHAPE-MaP for DENV-1 (last 1,000 nt) as input.
Furthermore, we studied how the predicted data are processed by Superfold as SHAPE-like constraints. We used Superfold to compute the Shannon entropy and to identify structurally unambiguous regions. Since Superfold receives a SHAPE map file as the input, CROSS predictions were normalized to follow a SHAPE-like distribution while keeping the same level of information about the secondary structure (see Shannon Entropy Calculation with Superfold). To do that, we studied >90 different normalization approaches to convert the predicted data into SHAPE-like reactivities, and we compared the structures obtained using Superfold as input SHAPE data with those ones obtained using normalized predicted data as input (Shannon Entropy Calculation With Superfold; Supplementary Figure 2). We applied this procedure both for HCV and DENV-1 and, to speed-up the computational time, we used as reference well-known structured regions, specifically the internal ribosome entry site (IRES)-contained region for HCV (i.e., first 1,500 nt) and the 3’ UTR for DENV-1 (i.e., last 1,000 nt). The best normalization (i.e., the highest number of identical nucleotides matching the SHAPE-obtained structure) is different between the two viruses (Supplementary Figure 2), but we found that the best normalization for DENV-1 is the third top ranking for HCV (Supplementary Figure 2).
Afterward, we analyzed how well Superfold works with and without constraints. Interestingly, Superfold generates better structures, compared with the ones obtained with SHAPE data, when using normalized predicted data coming from CROSS than when using an empty SHAPE map file (i.e., all reactivities = −999; Figures 2B, C), both for HCV and DENV-1. Moreover, CROSS data also show a consistent signal when implemented as constraints, and the performance of Superfold decreases when CROSS data are shuffled before being used as constraints (Figures 2B, C). Together, these results show how predicted CROSS profiles can be used inside RNAvigator as an in silico alternative when SHAPE experiments are not available.
To show the potential of our approach, we applied RNAvigator to the complete HCV and DENV-1 genomes, using SHAPE data as input (21, 30). The profiles generated using SHAPE data, Shannon entropy (Superfold using SHAPE data), and the predicted stable regions (Scanfold z-score), are used together to identify the most interesting regions for their secondary structure (Figure 3). To analyze only the most important regions, each metric was individually ranked, and only the bottom 20% of regions were selected for each score (SHAPE reactivity, entropy, and Scanfold z-score), but we also noticed similar results when selecting only the bottom 10% and 30% (Supplementary Figure 3). After studying multiple normalizing windows (from 50 to 300 nt), we decided to use a window of 150 nucleotides since it was already employed in a similar study (33) (Supplementary Figure 4). By selecting only the lowest scores, we are sure to analyze only the regions that are most highly enriched in double-stranded nucleotides (low SHAPE reactivity), structurally stable (low Scanfold z-score), and unambiguous (low Shannon entropy). Interestingly, the region encoding the IRES (735–1,185 nt) is identified in the bottom 20% for stability both by Scanfold and entropy, highlighting the importance of this structure for HCV (Figure 3A, 25; p-value = 0.02 under best normalization). Interestingly, the region 3,750–3,900 nucleotides (nt) is in agreement between the three methodologies and is associated with a high-propensity double-stranded, energetically and structurally stable region. This could be explained by the fact that this region is partially corresponds to a well-conserved structured region across three strains of HCV (30).
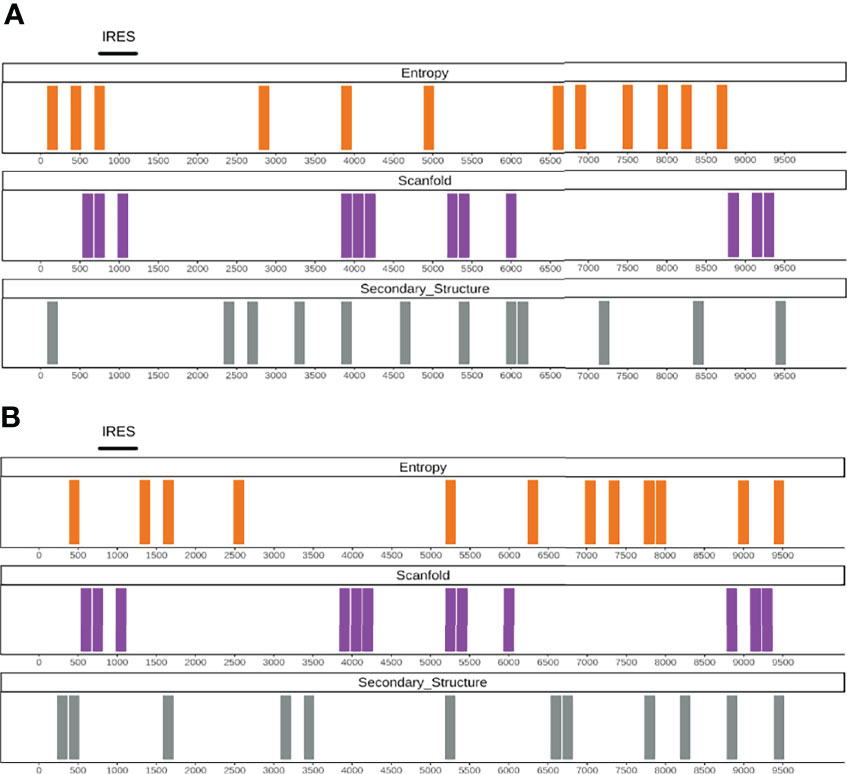
Figure 3 (A) Output example of the RNAvigator pipeline for the complete HCV genome showing the bottom 20% regions for SHAPE reactivity (Secondary_Structure; gray), Scanfold z-score (Scanfold; purple), and the entropy obtained by using SHAPE data as input inside Superfold (Entropy; orange). The notorious functional element IRES is highlighted at the top. (B) Output example of the RNAvigator pipeline for the complete HCV genome showing the bottom 20% regions for CROSS predictions (Secondary_Structure; gray), Scanfold z-score (Scanfold; purple), and the entropy obtained by using CROSS SHAPE-like data as input inside Superfold (Entropy; orange). The notorious functional element IRES is highlighted at the top.
The predicted data for HCV highlight similar regions (Figure 3B). Considering secondary structure profiles, between the bottom 20% of most structured regions, ~50% are identical or in close proximity (i.e., +-one shift of the window, in this case 150 nt) when considering experimental and predicted data. The entropy profiles also different between predictions and experimental data since Superfold uses the predicted data as SHAPE-like constraints, but also in this case, the regions are quite similar at ~50% when using the third best normalization of HCV in common with DENV-1 (see Shannon Entropy Calculation With Superfold; Figure 3B and Supplementary Figure 2), and reach 75% of identity or close proximity when using the best normalization window of HCV data (Supplementary Figure 5). Surprisingly, predictions are identifying more structural elements in the terminal 3’ regions, including conserved elements between three HCV strains around 8,800–9,000 (30; identified by secondary structure predictions and Scanfold), and the region around 9,300 nt, which includes the well-known NS5B cis-regulatory element (CRE; 38).
The same approach was used to study regions of structural importance in DENV-1 using SHAPE data and predictions. The experimental data highlight how the 3’ UTR, a known regulatory structural region essential for DENV replication (39, 40), is important both for its structure and entropy (bottom 20% both for SHAPE and Shannon entropy; Figure 4A) and is highly stable for Scanfold in multiple consecutive regions. Moreover, the region encoding for NS2A (~3,500–4,000 nt), one of the most conserved and with a low mutation rate between DENV and Zika virus (ZIKV; 21), is also identified as a highly-structured and low-entropy region.
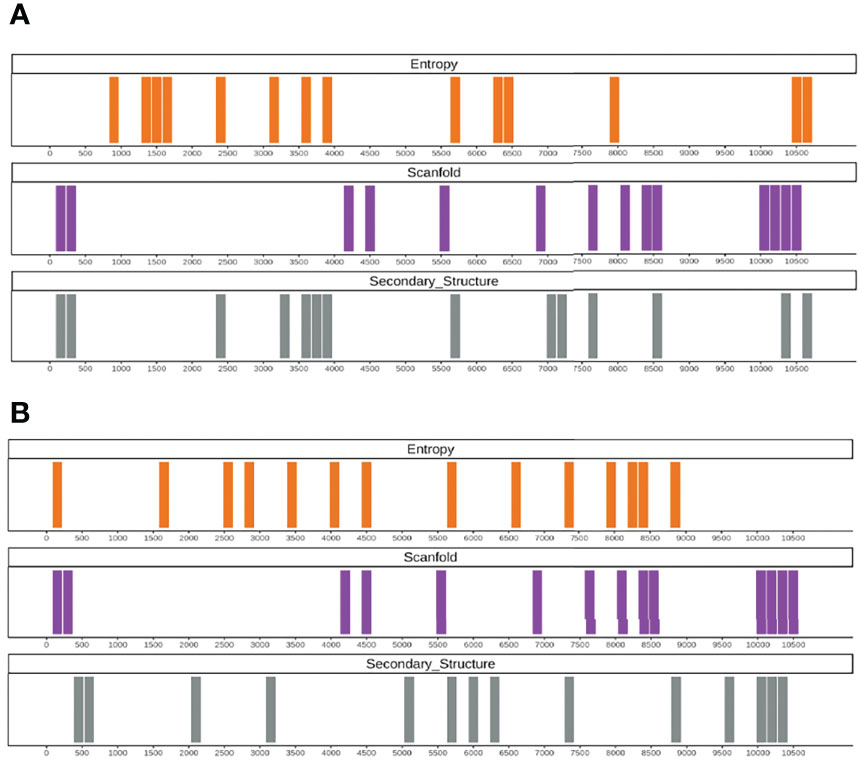
Figure 4 (A) Output example of the RNAvigator pipeline for the complete DENV-1 genome showing the bottom 20% regions for SHAPE reactivity (Secondary_Structure; gray), Scanfold z-score (Scanfold; purple), and the entropy obtained by using SHAPE data as input inside Superfold (Entropy; orange). (B) Output example of the RNAvigator pipeline for the complete DENV-1 genome showing the bottom 20% of regions for CROSS predictions (Secondary_Structure; gray), Scanfold z-score (Scanfold; purple), and the entropy obtained by using CROSS SHAPE-like data as input inside Superfold (Entropy; orange).
Here, considering secondary-structure profiles between experimental and predicted data, the identical or closely proximate regions in the bottom 20% are ~35% (Figure 4B). With regard to Shannon entropy, the results are similar compared to HCV, reaching ~50% identity. Interestingly, the secondary-structure predicted profiles identify the 3’ UTR accurately as a key element, matching the consecutive regions identified as structurally stable by Scanfold.
Long non-coding RNAs (lncRNAs) are an important class of large and complex RNAs in which the structure is of crucial importance for their functionality (41). To further validate our approach, we tested RNAvigator on the complete murine X inactive specific transcript (XIST), a lncRNA responsible for the inactivation of the X chromosome in mammals and characterized by several structural and repetitive domains (19, 41). As in previous analysis, we employed both SHAPE-MaP data and the predictive pipeline based on CROSS. With the exception of the repetitive region (Rep) F, every Rep region is identified by at least one metric (Entropy, Secondary Structure, or Scanfold; Figure 5). Interestingly, the predictive pipeline can also correctly identify the repetitive elements, and while domains with only one region predicted as structurally important, such as Rep H, are not very significant (p-value = 0.18; see P-Value for Regions of Structural Importance), other repetitive domains containing multiple predicted regions of structural importance, for example, Rep C for Scanfold and entropy, are quite significant (p-values of 5 × 10−4 and 5 × 10−2, respectively). Indeed, Rep C is a region crucial for the activity of XIST, which, when disrupted, prevents the binding of the lncRNA with chromatin (42). It is also worth mentioning that Rep C is often partially missing from the experiments due to the complex mappability of this region (19), thus reinforcing the RNAvigator approach of also implementing a predictive pipeline.
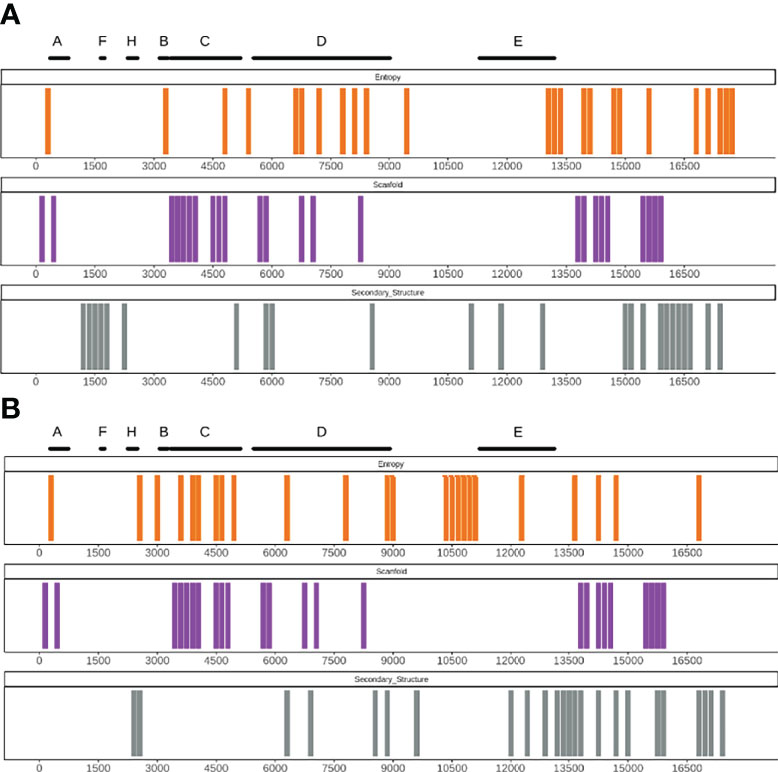
Figure 5 (A) Output example of the RNAvigator pipeline for the complete XIST showing the bottom 20% of regions for SHAPE reactivity (Secondary_Structure; gray), Scanfold z-score (Scanfold; purple), and the entropy obtained by using SHAPE data as input inside Superfold (Entropy; orange). Notorious repetitive regions (Rep) are highlighted on the top. (B) Output example of the RNAvigator pipeline for the complete XIST genome showing the bottom 20% of regions for CROSS predictions (Secondary_Structure; gray), Scanfold z-score (Scanfold; purple), and the entropy obtained by using CROSS SHAPE-like data as input inside Superfold (Entropy; orange). Notorious repetitive regions (Rep) are highlighted on the top (A-H).
HOX transcript antisense RNA (HOTAIR) is another well-studied lncRNA with a known secondary structure involved in cancer development and metastasis (43, 44). We also applied the complete RNAvigator pipeline to this lncRNA, using SHAPE data and predictions (Supplementary Figure 6). Interestingly, the 5’ and 3’ terminal regions, which are essential for the chromatin remodeling activation by interacting with different proteins including PRC2 and LSD1, are well-identified both using SHAPE data and CROSS predictions (Supplementary Figure 6). This result could suggest how our approach could also be extended into identifying possible protein-interacting regions that indeed follow some common rules, such as a tendency to have specific structures and accessibility (45). Moreover, the conserved helices H7, part of the PCR2-interacting region, and H10, part of the well-studied D1 domain, are correctly identified by CROSS Entropy (p-value = 0.01) (44).
We also analyzed coding RNAs with SHAPE data available on embryonic stem cells (ESC; 46). To perform the RNAvigator analyses, we selected only coding RNAs for which SHAPE data were available with a coverage of >90%, with complete extremities, and a total length in the range of 400–600 nucleotides (RPL24, RPS13, TMSB10, RPLP2, UBA52, and RPL26). The selected RNAs are mainly associated with ribosomal proteins, showing similar content (between 33 and 40%) of high-propensity double-stranded nucleotides (i.e., SHAPE reactivity <0.2) and a comparable distribution of SHAPE data (Supplementary Figure 7). We then used SHAPE data as input for RNAvigator to predict regions of structural importance in windows of 20 nucleotides. By studying the distribution of the identified regions of 20 nt, it is clear that the 5’ UTR (0–100 nt) is indeed more often recognized as a region of functionality, in agreement with literature [Supplementary Figure 8; (46)].
These results support the importance of our pipeline not only to identify important regions in RNA viruses but also in other classes of long and complex RNAs such as lncRNAs. Moreover, by employing predictions we can also identify regions that are difficult to profile with experimental techniques such as the Rep C of XIST, but which are indeed crucial for the RNA functionality.
The progress made using genome-wide chemical probing techniques, such as icSHAPE and SHAPE-MaP, paved the way for a further understanding of the RNA secondary structure, especially in large and complex RNA molecules, for example, viral RNA genomes and lncRNAs (12, 19, 33, 47). In this context, structural elements play an important role since they are often crucial for the function of these complex RNAs, thus the need to characterize structural elements to understand RNA functionality (19, 25, 40). We developed RNAvigator to contribute to achieving this task by creating a comprehensive pipeline to identify regions of structural importance in long RNAs such as viral genomes. RNAvigator uses chemical probing data or predictions to identify the most stable, unambiguous, and structured regions of RNA as potential functional elements.
RNAvigator works with and without experimental data, allowing the user to employ predictions to analyze novel RNAs for which experimental data are not available. For this reason, we used the CROSS algorithm to work as predictive SHAPE-like constraints. CROSS already showed in previous publications good performance on SHAPE data, and in this study, it reached an AUC of 0.74 on HCV SHAPE-MaP data (Figure 2A), while also improving the predictive power of Superfold to build a structure similar to the one obtained using SHAPE data (Figure 2B).
The experimental- and predictive-based pipelines comprising RNAvigator were applied to identify regions of structural interest, and thus of potential functionality, in the complete genomes of HCV and DENV-1, and in the lncRNAs XIST and HOTAIR. RNAvigator works by ranking the different signals to select the most important regions (default: bottom 20%) for each metric, and then visualizes the common regions and provides their secondary structure. We also studied more selective (bottom 10%) and permissive (bottom 30%) thresholds, while also facilitating the user to select their personal case-specific values (Supplementary Figure 3). RNAvigator can identify known functional elements, such as CRE or IRES on HCV and the 3’ UTR of DENV-1, by using SHAPE-MaP data and CROSS predictions, highlighting how the pipeline can work with both types of data. Moreover, the identification through predictions of the functional Rep C in XIST, a region otherwise difficult to experimentally map, supports our use of a predictive pipeline inside the RNAvigator.
Since RNAvigator needs only a single RNA as input to identify elements of structural importance, we employ three levels of information that can be extracted from the primary sequence. However, further knowledge of the RNA in the analysis, including multiple strains in the case of viruses, could be helpful to add additional levels of structural characterization, including conservation and mutation rate. We understand that our pipeline could benefit from information coming from multiple sequences, which is a feature that can be implemented in a future version of the tool. We believe that the layered way in which RNAvigator is built will allow the implementation of additional features, for example, including sequence or structural conservation. Moreover, other chemical-probing data, such as icSHAPE, can be easily implemented inside RNAvigator since the reactivities have a similar ranking to SHAPE-MaP data. Enzyme-based techniques, such as Parallel Analysis of RNA Structure (PARS), could also be used as the main input for the pipeline, for example, by considering the distribution of the S1/V1 ratio.
Considering the good performance of our pipeline for both experimental and predicted data in terms of AUC and when used as constraints inside Superfold and as a basis for structure prediction, especially for RNA viruses and lncRNAs, we posit that RNAvigator will be a valuable tool for future research into functional RNA domains.
Publicly available datasets were analyzed in this study. This data can be found at https://www.ncbi.nlm.nih.gov/ in the GEO series with the accession number: GSE106483.
RH and RDP designed the study. RDP performed the analysis. YW and JW provided and helped with ESC SHAPE data. RDP and RH wrote the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by A*STAR BMRC through CDF Grant 192D8050 to RH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.878679/full#supplementary-material
CROSS, Computational Recognition of Secondary Structure; SHAPE, Selective 2′ Hydroxyl Acylation analyzed by Primer Extension; icSHAPE, In vivo click SHAPE; PARS, Parallel Analysis of RNA Structure; HCV, Hepatitis C virus; UTR, Untranslated region; lncRNA, Long non-coding RNA; AUC, Area under the ROC curve; ss-RNA, Single-stranded RNA; nt, Nucleotide; IRES, Internal ribosome entry site; CRE, Cis-regulatory element; ESC, Embryonic stem cell; XIST X Inactive Specific Transcript; Repm Repetitive region; HOTAIR, HOX Transcript Antisense RNA; DENV, Dengue virus; ZIKV, Zika virus.
1. Gray NK, Hentze MW. Regulation of Protein Synthesis by mRNA Structure. Mol Biol Rep (1994) 19, 195–200. doi: 10.1007/BF00986961
2. Chapman EG, Moon SL, Wilusz J, Kieft JS. RNA Structures That Resist Degradation by Xrn1 Produce a Pathogenic Dengue Virus RNA. Elife (2014) eLife3:e01892. doi: 10.7554/eLife.01892
3. Pandey NB, Marzluff WF. The Stem-Loop Structure at the 3’ End of Histone mRNA is Necessary and Sufficient for Regulation of Histone mRNA Stability. Mol Cell Biol (1987). 7(12):4557–9. doi: 10.1128/mcb.7.12.4557-4559.1987
4. Shepard PJ, Hertel KJ. Conserved RNA Secondary Structures Promote Alternative Splicing. RNA (2008) 14:1463–9. doi: 10.1261/rna.1069408
5. Montange RK, Batey RT. Riboswitches: Emerging Themes in RNA Structure and Function. Annu Rev Biophys (2008) 37:117–33. doi: 10.1146/annurev.biophys.37.032807.130000
6. Ganser LR, Kelly ML, Herschlag D, Al-Hashimi HM. The Roles of Structural Dynamics in the Cellular Functions of RNAs. Nat Rev Mol Cell Biol (2019) 20:474–89. doi: 10.1038/s41580-019-0136-0
7. Siegfried NA, Busan S, Rice GM, Nelson JAE, Weeks KM. RNA Motif Discovery by SHAPE and Mutational Profiling (SHAPE-MaP). Nat Methods (2014) 11:959–65. doi: 10.1038/nmeth.3029
8. Reddy T, Sansom MSP. Computational Virology: From the Inside Out. Biochim Biophys Acta - Biomembr (2016) 1858:1610–8. doi: 10.1016/j.bbamem.2016.02.007
9. McCaskill JS. The Equilibrium Partition Function and Base Pair Binding Probabilities for RNA Secondary Structure. Biopolymers (2018) 29:1105–19. doi: 10.1002/bip.360290621
10. Wunderlich CH, Huber RG, Spitzer R, Liedl KR, Kloiber K, Kreutz C. A Novel Paramagnetic Relaxation Enhancement Tag for Nucleic Acids: A Tool to Study Structure and Dynamics of RNA. ACS Chem Biol (2013) 8:2697–706. doi: 10.1021/cb400589q
11. Schroeder SJ. Challenges and Approaches to Predicting RNA With Multiple Functional Structures. RNA (2018) 24:1615–24. doi: 10.1261/rna.067827.118
12. Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, et al. Structural Imprints In Vivo Decode RNA Regulatory Mechanisms. Nature (2015) 519:86–490. doi: 10.1038/nature14263
13. Flynn RA, Zhang QC, Spitale RC, Lee B, Mumbach MR, Chang HY. Transcriptome-Wide Interrogation of RNA Secondary Structure in Living Cells With icSHAPE. Nat Protoc (2016) 11:273–90. doi: 10.1038/nprot.2016.011
14. Low JT, Weeks KM. SHAPE-Directed RNA Secondary Structure Prediction. Methods (2010) 52:150–8. doi: 10.1016/j.ymeth.2010.06.007
15. Darlix JL, Lapadattapolsky M, Derocquigny H, Roques BP. First Glimpses At Structure-Function-Relationships Of The Nucleocapsid Protein Of Retroviruses. J Mol Biol (1995) 254:523–37. doi: 10.1006/jmbi.1995.0635
16. Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, et al. Genome-Wide Measurement of RNA Secondary Structure in Yeast. Nature (2010) 467:103–7. doi: 10.1038/nature09322
17. Funk A, Truong K, Nagasaki T, Torres S, Floden N, Balmori Melian E, et al. RNA Structures Required for Production of Subgenomic Flavivirus RNA. J Virol (2010) 84:11407–17. doi: 10.1128/jvi.01159-10
18. Gebhard LG, Filomatori CV, Gamarnik AV. Functional RNA Elements in the Dengue Virus Genome. Viruses (2011) 3(9):1739–1736. doi: 10.3390/v3091739
19. Smola MJ, Christy TW, Inoue K, Nicholson CO, Friedersdorf M, Keene JD, et al. SHAPE Reveals Transcript-Wide Interactions, Complex Structural Domains, and Protein Interactions Across the Xist lncRNA in Living Cells. Proc Natl Acad Sci (2016) 113:10322–7. doi: 10.1073/pnas.1600008113
20. Sharma S, Varani G. NMR Structure of Dengue West Nile Viruses Stem-Loop B: A Key Cis-Acting Element for Flavivirus Replication. Biochem Biophys Res Commun (2020) 531:522–7. doi: 10.1016/j.bbrc.2020.07.115
21. Huber RG, Lim XN, Ng WC, Sim AYL, Poh HX, Shen Y, et al. Structure Mapping of Dengue and Zika Viruses Reveals Functional Long-Range Interactions. Nat Commun (2019) 10, 1408. doi: 10.1038/s41467-019-09391-8
22. Ziv O, Price J, Shalamova L, Kamenova T, Goodfellow I, Weber F, et al. The Short-and Long-Range RNA-RNA Interactome of SARS-CoV-2. Mol Cell (2020) 80:1067–77. doi: 10.1016/j.molcel.2020.11.004
23. Selisko B, Potisopon S, Agred R, Priet S, Varlet I, Thillier Y, et al. Molecular Basis for Nucleotide Conservation at the Ends of the Dengue Virus Genome. PloS Pathog (2012) 8:e1002912. doi: 10.1371/journal.ppat.1002912
24. Delli Ponti R, Marti S, Armaos A, Tartaglia GG. A High-Throughput Approach to Profile RNA Structure. Nucleic Acids Res (2017) 45:e35–:e35. doi: 10.1093/nar/gkw1094
25. Lukavsky PJ. Structure and Function of HCV IRES Domains. Virus Res (2009) 139:166–71. doi: 10.1016/j.virusres.2008.06.004
26. Andrews RJ, Roche J, Moss WN. ScanFold: An Approach for Genome-Wide Discovery of Local RNA Structural Elements—Applications to Zika Virus and HIV. PeerJ (2018) 6:e6136. doi: 10.7717/peerj.6136
27. Reuter JS, Mathews DH. RNAstructure: Software for RNA Secondary Structure Prediction and Analysis. BMC Bioinf (2010) 11:129. doi: 10.1186/1471-2105-11-129
28. De Falco L, Silva NM, Santos NC, Huber RG, Martins IC. The Pseudo-Circular Genomes of Flaviviruses: Structures, Mechanisms, and Functions of Circularization. Cells (2021) 10:642. doi: 10.3390/cells10030642
29. Team RC. R: A Language and Environment for Statistical Computing. (2013). R Foundation for Statistical Computing, Vienna, Austria
30. Mauger DM, Golden M, Yamane D, Williford S, Lemon SM, Martin DP, et al. Functionally Conserved Architecture of Hepatitis C Virus RNA Genomes. Proc Natl Acad Sci U.S.A (2015) 112(12):3692–3697. doi: 10.1073/pnas.1416266112
31. Li P, Wei Y, Mei M, Tang L, Sun L, Huang W, et al. Integrative Analysis of Zika Virus Genome RNA Structure Reveals Critical Determinants of Viral Infectivity. Cell Host Microbe (2018) 24:875–86. doi: 10.1016/j.chom.2018.10.011
32. Ziv O, Gabryelska MM, Lun ATL, Gebert LFR, Sheu-Gruttadauria J, Meredith LW, et al. COMRADES Determines In Vivo RNA Structures and Interactions. Nat Methods (2018) 15:785–8. doi: 10.1038/s41592-018-0121-0
33. Yang SL, DeFalco L, Anderson DE, Zhang Y, Aw JGA, Lim SY, et al. Comprehensive Mapping of SARS-CoV-2 Interactions In Vivo Reveals Functional Virus-Host Interactions. Nat Commun (2021) 12:1–15. doi: 10.1038/s41467-021-25357-1
34. Huston NC, Wan H, Strine MS, de Cesaris Araujo Tavares R, Wilen CB, et al. Comprehensive In Vivo Secondary Structure of the SARS-CoV-2 Genome Reveals Novel Regulatory Motifs and Mechanisms. Mol Cell (2021) 81:584–98. doi: 10.1016/j.molcel.2020.12.041
35. Smola MJ, Rice GM, Busan S, Siegfried NA, Weeks KM. Selective 2’-Hydroxyl Acylation Analyzed by Primer Extension and Mutational Profiling (SHAPE-MaP) for Direct, Versatile and Accurate RNA Structure Analysis. Nat Protoc (2015) 10:1643–69. doi: 10.1038/nprot.2015.103
36. Sun L, Li P, Ju X, Rao J, Huang W, Zhang S, et al. In Vivo Structural Characterization of the Whole SARS-CoV-2 RNA Genome Identifies Host Cell Target Proteins Vulnerable to Re-Purposed Drugs. Cell. J.cell. (2021). 184(7):1865–1883.e20 doi: 10.1101/2020.07.07.192732
37. Delli Ponti R, Mutwil M. Structural Landscape of the Complete Genomes of Dengue Virus Serotypes and Other Viral Hemorrhagic Fevers. BMC Genomics (2021) 22:1–14. doi: 10.1186/s12864-021-07638-7
38. Lee H, Shin H, Wimmer E, Paul AV. Cis-Acting RNA Signals in the NS5B C-Terminal Coding Sequence of the Hepatitis C Virus Genome. J Virol (2004) 78:10865–77. doi: 10.1128/JVI.78.20.10865-10877.2004
39. Reid DW, Campos RK, Child JR, Zheng T, Chan KWK, Bradrick SS, et al. Dengue Virus Selectively Annexes Endoplasmic Reticulum-Associated Translation Machinery as a Strategy for Co-Opting Host Cell Protein Synthesis. J Virol (2018) 92:e01766–17. doi: 10.1128/JVI.01766-17
40. de Borba L, Villordo SM, Marsico FL, Carballeda JM, Filomatori CV, Gebhard LG, et al. RNA Structure Duplication in the Dengue Virus 3´ UTR: Redundancy or Host Specificity? MBio (2019) 10:1–18. doi: 10.1128/mBio.02506-18
41. Nesterova TB, Slobodyanyuk SY, Elisaphenko EA, Shevchenko AI, Johnston C, Pavlova ME, et al. Characterization of the Genomic Xist Locus in Rodents Reveals Conservation of Overall Gene Structure and Tandem Repeats But Rapid Evolution of Unique Sequence. Genome Res (2001) 11:833–49. doi: 10.1101/gr.174901
42. Sarma K, Levasseur P, Aristarkhov A, Lee JT. Locked Nucleic Acids (LNAs) Reveal Sequence Requirements and Kinetics of Xist RNA Localization to the X Chromosome. Proc Natl Acad Sci (2010) 107:22196–201. doi: 10.1073/pnas.1009785107
43. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell (2007) 129:1311–23. doi: 10.1016/j.cell.2007.05.022
44. Somarowthu S, Legiewicz M, Chillón I, Marcia M, Liu F, Pyle AM. HOTAIR Forms an Intricate and Modular Secondary Structure. Mol Cell (2015) 58:353–61. doi: 10.1016/j.molcel.2015.03.006
45. Li X, Quon G, Lipshitz HD, Morris Q. Predicting In Vivo Binding Sites of RNA-Binding Proteins Using mRNA Secondary Structure. Rna (2010) 16:1096–107. doi: 10.1261/rna.2017210
46. Wang J, Zhang T, Yu Z, Tan WT, Wen M, Shen Y, et al. Genome-Wide RNA Structure Changes During Human Neurogenesis Modulate Gene Regulatory Networks. Mol Cell (2021) 81:4942–4953.e8. doi: 10.1016/j.molcel.2021.09.027
Keywords: RNA structure, structure prediction, chemical probing, pipeline, RNA virus
Citation: Delli Ponti R, Wang J, Wan Y and Huber RG (2022) RNAvigator: A Pipeline to Identify Candidates for Functional RNA Structure Elements. Front.Virol. 2:878679. doi: 10.3389/fviro.2022.878679
Received: 18 February 2022; Accepted: 19 May 2022;
Published: 27 June 2022.
Edited by:
Diego Forni, Eugenio Medea (IRCCS), ItalyReviewed by:
Caroline C. Friedel, Ludwig Maximilian University of Munich, GermanyCopyright © 2022 Delli Ponti, Wang, Wan and Huber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roland G. Huber, cmdodWJlckBiaWkuYS1zdGFyLmVkdS5zZw==; Riccardo Delli Ponti, cmljY2FyZG9fcG9udGlAYmlpLmEtc3Rhci5lZHUuc2c=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.