 Morgan E. Meissner
Morgan E. Meissner Nathaniel Talledge
Nathaniel Talledge Louis M. Mansky
Louis M. Mansky- 1Institute for Molecular Virology, University of Minnesota – Twin Cities, Minneapolis, MN, United States
- 2Molecular, Cellular, Developmental Biology and Genetics Graduate Program, University of Minnesota – Twin Cities, Minneapolis, MN, United States
- 3Division of Basic Sciences, School of Dentistry, University of Minnesota – Twin Cities, Minneapolis, MN, United States
- 4Masonic Cancer Center, University of Minnesota – Twin Cities, Minneapolis, MN, United States
Studies of retroviruses have led to many extraordinary discoveries that have advanced our understanding of not only human diseases, but also molecular biology as a whole. The most recognizable human retrovirus, human immunodeficiency virus type 1 (HIV-1), is the causative agent of the global AIDS epidemic and has been extensively studied. Other human retroviruses, such as human immunodeficiency virus type 2 (HIV-2) and human T-cell leukemia virus type 1 (HTLV-1), have received less attention, and many of the assumptions about the replication and biology of these viruses are based on knowledge of HIV-1. Existing comparative studies on human retroviruses, however, have revealed that key differences between these viruses exist that affect evolution, diversification, and potentially pathogenicity. In this review, we examine current insights on disparities in the replication of pathogenic human retroviruses, with a particular focus on the determinants of structural and genetic diversity amongst HIVs and HTLV.
Pathogenic Human Retroviruses
Human Immunodeficiency Viruses
Human immunodeficiency virus (HIV) is the etiologic agent of acquired immunodeficiency syndrome (AIDS) (1, 2). Nearly 40 years after its discovery, HIV remains a chronic global public health concern. Since its emergence in the human population, HIV has infected approximately 75 million people and is responsible for the deaths of almost 32 million individuals (3). As of 2018, an estimated 38 million people are infected with HIV around the world (4). Despite advancements in the understanding of viral transmission and antiviral drug development, approximately 1.7 million new infections and 770,000 HIV-related deaths still occur annually (4).
Most HIV infections are caused by HIV type 1 (HIV-1), which is the primary driver of the pandemic. A much smaller subset of HIV infections are caused by HIV type 2 (HIV-2), which accounts for only about 1 to 2 million infections and is almost exclusively geographically constrained to West Africa (5). Both HIV-1 and HIV-2 belong to the Retroviridae family of viruses. Retroviruses are a unique class of viruses that challenge the central dogma of biology, such that their RNA genomes are first reverse transcribed into DNA. The DNA copy of the viral genome is then integrated into the host DNA, which contributes to lifelong, persistent infections. Messenger RNA (mRNA) and viral genomic RNA (vRNA) are both transcribed from the integrated genome, which allows for sustained propagation of viral progeny so long as the host cell survives.
Both HIV-1 and HIV-2 infect immune cells that express the CD4 molecule on their surface, namely CD4+ T cells and macrophages. During the acute stage, which represents the first weeks of infection, CD4+ T cells are robustly depleted from the lymphoid system. This is driven by both direct targeting of virally-infected cells as well as the activation-induced death of bystander T cells (6). Infected individuals may experience mononucleosis-like symptoms, such as fever, headache, and muscle pains. These symptoms may be so mild that many individuals do not realize they are infected. The acute stage of infection is followed by a long period of clinical latency, known as the asymptomatic phase, which typically lasts about 6 to 8 years (7). During this time, the concentration of CD4+ T cell continues to decline and viremia increases. When CD4+ T cell counts drop below 500 cells/µL, patients enter the symptomatic phase of infection, at which point viremia rapidly increases and progression to AIDS (defined as CD4+ T cell concentration <200 cells/µL) typically occurs within a couple of years (7). Left untreated, 52% of patients will die within 2 years of progression to AIDS and 82% will die within 6 years (8).
Compared with HIV-1, HIV-2 exhibits a significantly attenuated disease phenotype, characterized by lower viral loads within individuals, lower rates of both vertical and sexual transmissibility, and a slower progression to AIDS (9–14). In areas where HIV-2 has historically been endemic, rates of HIV-2 infection are decreasing while the prevalence of HIV-1 is on the rise (15–17). For example, in Guinea-Bissau, the prevalence of HIV-1 rose from 0.5% in 1990 to 3.6% in 2007, whereas HIV-2 prevalence dropped from 8.3% to 4.7% in the same timeframe (17). Even in the absence of antiretrovirals, the prognosis for HIV-2-infected individuals is better than for HIV-1-infected individuals. A study of women with HIV-1 and HIV-2 infections from 1985 to 1993 found that the 5-year AIDS-free survival rate for HIV-1-infected women was 67% compared with 100% for HIV-2-infected women (14).
The reason for the attenuated disease progression of HIV-2 compared with HIV-1 remains unclear. Humanized mice studies in HIV-2 are lacking, and no head-to-head comparisons have been done, but preliminary studies suggest that viral loads in HIV-2-infected humanized mice are an order of magnitude lower than those of HIV-1-infected humanized mice during the first several months of infection (18, 19). Studies of HIV-1 elite controllers and long-term non-progressors (i.e., those individuals who maintain high CD4+ T-cell counts in the absence of treatment) have found that these patients have lower levels of proviral DNA and smaller pools of latently infected cells compared with viremic individuals (20–22). However, results comparing HIV-1 and HIV-2 long-term non-progressors have been conflicting. One study found that differences in proviral loads between HIV-1- and HIV-2-infected individuals may explain disparities in viremia and disease progression (23). However, another group demonstrated that low plasma viral load was independent of proviral load in HIV-2-infected individuals, which were comparable with proviral loads for HIV-1-infected individuals at similar disease states (24).
Human T-Cell Leukemia Virus Type 1
Human T cell leukemia virus type 1 (HTLV-1) represents the other major pathogenic human retrovirus. HTLV-1 was the first human retrovirus discovered, laying the groundwork for much of the work that has been done to understand HIV-1 infection (25, 26). HTLV-1 is in the deltaretrovirus genus, whereas HIV-1 and HIV-2 are in the lentivirus genus. Like HIV-1 and HIV-2, HTLV-1 predominantly infects CD4+ T cells, but also infects a wide variety of other immune cells, endothelial cells, myeloid cells, and fibroblasts (27–31). Although HTLV-1 is also characterized by relatively low rates of transmission between individuals, an estimated 5 to 10 million people are infected worldwide, and the seroprevalence may reach as high as 40% in endemic regions such as Japan, the Caribbean, central Africa, Brazil, parts of the Middle East, and the Pacific Islands, including Australia (32, 33).
While most HTLV-1 infections remain asymptomatic, there is a 0.25% to 3% lifetime risk for pathogenicity for infected individuals (32). In these individuals, the disease may manifest as a lymphoproliferative adult T cell leukemia/lymphoma (ATL) or as a collection of neurological disorders, namely HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) (25, 34–36). HTLV-1 is also associated with a number of other inflammatory disorders, including bronchiectasis. A recent study found that in Indigenous adults in central Australia, HTLV-1 proviral loads are 8-fold higher in patients with bronchiectasis compared with healthy controls, and patients with an HTLV-1 proviral load greater than 1000 copies per 100,000 peripheral blood leukocytes were over 12-times more likely to develop bronchiectasis (37).
Compared with HIV-1, both HIV-2 and HTLV-1 have been significantly understudied. A search of the online PubMed database performed December 2021 yields over 100,000 results using the search terms “HIV-1” or “human immunodeficiency virus type-1”, compared with approximately 6,300 results for “HIV-2” or “human immunodeficiency virus type-2” and 6,600 to 8,500 results for “HTLV-1” or “human T cell leukemia virus type-1”. Although the viruses share many similarities, it is important to consider both HIV-2 and HTLV-1 independently, as there are key differences that distinguish these viruses from HIV-1 that have important implications for replication, disease pathogenesis, and treatment.
The Retroviral Lifecycle
HIV-1, HIV-2, and HTLV-1 Genomes
At the most basic level, retroviral genomes consist of at least 3 core genes which are encoded in a 5’-gag-pol-env-3’ structure. The proteins encoded by these genes facilitate the fundamental steps in viral replication: attachment/entry, genome replication, particle assembly, and egress. All 3 major human retroviruses also encode a set of auxiliary genes, known as accessory genes, which are dispensable for replication in certain in vitro cell culture systems (Figure 1). The proteins encoded by these genes play a variety of important roles within the host system and help facilitate transmission and replication. Two identical copies of the single-stranded RNA genome are packaged into viral particles and serve as the templates for the generation of a single proviral genome, which is integrated into the host DNA.
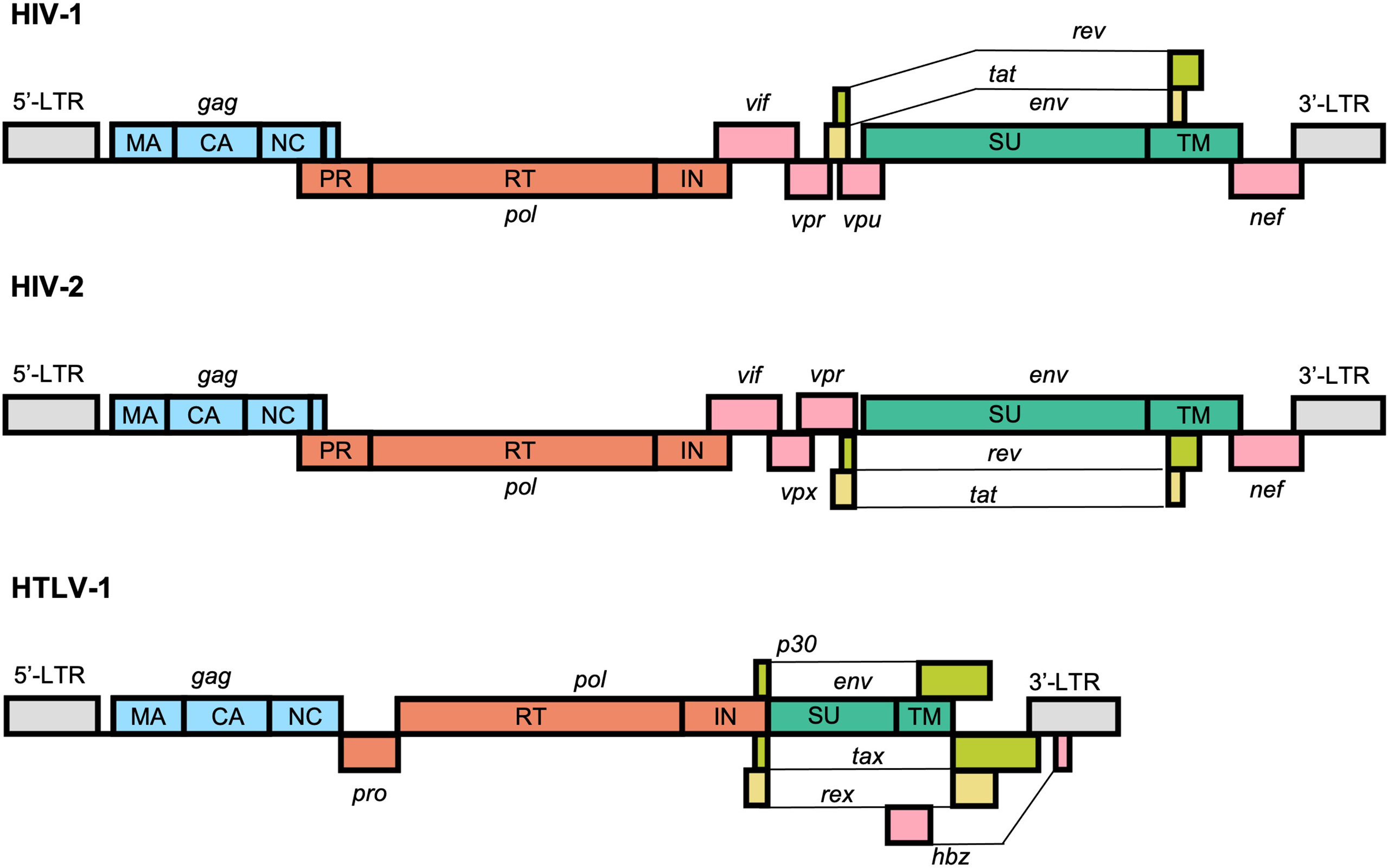
Figure 1 Schematic representation of the typical structure of the HIV-1, HIV-2, and HTLV-1 proviral genomes. All retroviruses encode a core set of genes in a 5’-gag-pol-env-3’ structure flanked by long-terminal repeats (LTRs) that facilitate replication and integration within the genome. HIV-1, HIV-2, and HTLV-1 also encode a unique set of accessory genes, shown in pink, lime green, and yellow, that help facilitate genomic replication and immune evasion and counteract cellular defense mechanisms against the virus.
The HIV-1 genome is approximately 9,200 to 9,600 nucleotides and is flanked at both ends by long terminal repeat (LTR) sequences. There are 9 protein-coding viral genes encoded by overlapping reading frames: gag, pol, vif, vpr, tat, rev, vpu, env, and nef. The RNA genome also contains a number of highly structured elements that regulate replication, including the 5’ trans-activation response (TAR) element, primer binding site (PBS), dimerization initiation sequence (DIS), and the Rev response element (RRE) (38).
The HIV-2 genome is structurally very similar to the HIV-1 genome and is approximately 9,800 nucleotides in length. The most prominent difference between them is the absence of vpu, which is replaced instead with vpx. At the sequence level, however, HIV-1 and HIV-2 are highly divergent. The viruses emerged in the human population via distinct zoonotic transmissions of simian immunodeficiency viruses (SIVs) from either chimpanzees or gorillas (HIV-1) or sooty mangabeys (HIV-2) (39). As result, HIV-1 and HIV-2 share approximately 60% identity at the amino acid level and just 48% identity at the nucleotide level (40).
The HTLV-1 genome is approximately 9,000 nucleotides in length and, similarly to HIV-1 and HIV-2, is flanked by LTR sequences. In addition to the 3 core genes, HTLV-1 also contains a regulatory pX region, which encodes the viral accessory proteins Tax-1, Rex, p8/p12, p13, p30, and HBZ (encoded in the antisense direction) (41). Several of these unique accessory factors, namely Tax and HBZ, have been implicated in oncogenesis and serve to partly explain the distinct disease pathogeneses of HTLV-1 from HIVs (42). HTLV-1 is genetically distinct from both HIV-1 and HIV-2 and is the result of multiple zoonotic transmissions of simian T-lymphotropic virus type 1 (STLV-1) from apes to humans. Whereas this transmission is thought to have occurred relatively recently for HIV-1 and HIV-2, likely sometime between 1920 and 1940 (43, 44), HTLV-1 is thought to be a much older human pathogen, with some transmission events estimated to have occurred as early as the Paleolithic period (45, 46). Indeed, preserved HTLV-1 proviral DNA has been identified in 1500-year-old Andean mummies (47). Even in the most conserved structural regions of the genome, such as the capsid (CA) domain of the Gag protein, HTLV-1 shares only about 50% amino acid identity with HIV-1 (48).
Attachment, Fusion, and Entry
Viral entry into target cells is facilitated by the envelope glycoprotein (Env) on the surface of the viral particle (Figure 2A) . Env is initially translated as a polyprotein precursor which is subsequently cleaved into the surface protein (SU) and a transmembrane protein (TM), the latter of which is anchored within the lipid membrane of the viral envelope. The mature Env protein consists of a trimer of the SU-TM heterodimers that is heavily glycosylated during protein trafficking to the plasma membrane of the host cell (49–51). The SU protein binds to a series of receptors on the host cell which facilitate viral uptake. For both HIV-1 and HIV-2, the primary cellular receptor is CD4. CD4 is abundantly expressed on the surface of CD4+ T cells but is also expressed on a number of other immune cell types, including hematopoietic progenitor cells (52). Subsequent interaction with a coreceptor on the cell surface, namely one of the chemokine receptors CCR5 or CXCR4, triggers a conformational change within Env which results in the fusion of the viral and cellular membranes mediated by the TM protein. The process is similar for HTLV-1, except that HTLV-1 SU first interacts with heparin sulfate proteoglycans (HSPGs), followed by neuropilin-1 (NRP-1), and finally glucose transporter 1 (GLUT1). Binding to NRP-1 induces a conformational change within SU which allows for interaction with GLUT1 which subsequently facilitates fusion of the viral and cellular membranes (53).
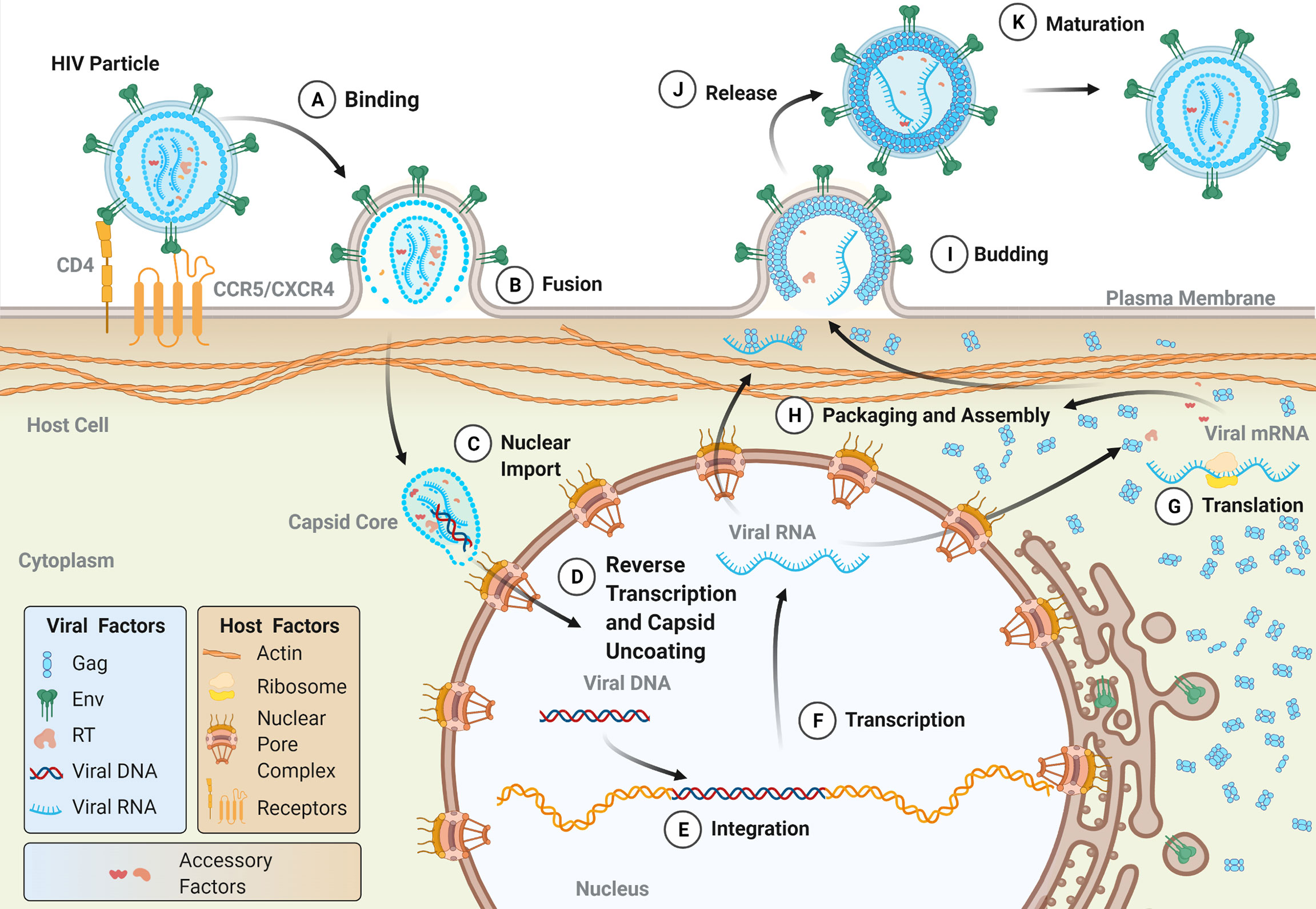
Figure 2 The lifecycle of human retroviruses. The major steps of the retroviral lifecycle are shown. Viral proteins are colored according to those used in the genomic structure diagram in Figure 1. (A) The retroviral lifecycle begins when the envelope proteins of a mature, infectious virion attach to the appropriate receptor (for HIV-1, CD4) and co-receptor (CCR5 or CXRC4) on the surface of the cell, which facilitates viral membrane fusion with the plasma membrane. Following release into the cytoplasm (B), the intact (or near intact) capsid core is trafficked to nuclear pore complexes (NPCs) on the nuclear envelope. During or shortly after nuclear import (C), capsid uncoating and reverse transcription occur (D) (see Figure 3 for more details on reverse transcription). The double-stranded DNA product is subsequently integrated into the host genome by integrase (E). Host machinery transcribes the proviral genome (F) into both the viral genomic RNA (vRNA) and mRNA, which templates the translation of viral proteins both in the cytosol and on the surface of the ER, with co-translational insertion of the transmembrane Env protein occurring in the ER (G). A milieu of viral and host proteins traffics along with vRNA to the plasma membrane where they are packaged into nascent budding particles (H). Following the budding (I) and release of the particle from the plasma membrane (J), the viral protease cleaves Gag resulting in the formation of a mature, infectious particle (K).
Reverse Transcription and Integration
Following membrane fusion, the viral capsid core is released into the cytoplasm of the cell along with a host of viral and cellular proteins, including reverse transcriptase (RT), integrase (IN), and cellular proviral and restriction factors (Figure 2B). The early stages of genomic replication within the cell have remained a topic of debate. Reverse transcription is intimately linked to the dissociation of the capsid core structure, known as uncoating, and perturbation of uncoating kinetics can disrupt reverse transcription events (54–58). While it was historically believed that capsid core uncoating and reverse transcription occurred in the cytoplasm, recent evidence suggests that capsid cores remain largely intact until they reach nuclear pore complexes (NPCs) on the nuclear envelope (59–63). Recent reports suggest that these cores are subsequently trafficked through the NPC in an intact state, and that both uncoating and reverse transcription occur in the nucleus (59–61). Others, however, suggest that capsid remodeling or partial uncoating occur at the NPC, and that the early formation of viral DNA products occurs at or near the nuclear envelope (Figures 2C, D) (62, 63). Such studies, which have reshaped our understanding of the timing of capsid uncoating and reverse transcription, have been aided by advancements in molecular imaging techniques. Further improvements in live cell imaging microscopy will allow for deeper insights into the relationship between uncoating and reverse transcription, as well as the timing of these processes.
The details of reverse transcription are shown in Figure 3. Cellular tRNAs serve as the primer for initiation of reverse transcription (tRNALys3 for HIV-1 and HIV-2, tRNAPro for HTLV-1), which bind to the PBS at the 5’ end of the RNA genome (64). From this primer, the RT enzyme begins synthesis of the negative-sense strand of single-stranded DNA (-ssDNA) in a 5’-to-3’ direction. As RT proceeds with DNA synthesis, the RNase H domain of the enzyme degrades the RNA genome behind it. RT proceeds through the unique 5’ (U5) and repeated (R) regions of the 5’ leader sequence of the RNA genome, at which point the first strand transfer occurs. The R region is thusly named as it is repeated at both ends of the genome, allowing for binding of the newly synthesized ssDNA to the 3’ end of the genome. DNA elongation continues through the unique 3’ (U3) region, generating the first of the LTRs. As synthesis of the -ssDNA continues, RNase H continues to degrade the viral RNA genome, until it reaches the centrally located polypurine tract (PPT). This portion of the RNA is resistant to RNase H cleavage and serves as the template for generation of the positive-sense single-stranded DNA (+ssDNA). As synthesis of the -ssDNA continues, +ssDNA elongation proceeds to the 3’ end of the genome through U3, R, and U5. The second strand transfer occurs, in which the PBS from both ssDNA molecules hybridize. Both strands are allowed to complete synthesis using one another as templates and the dsDNA genome is completed.
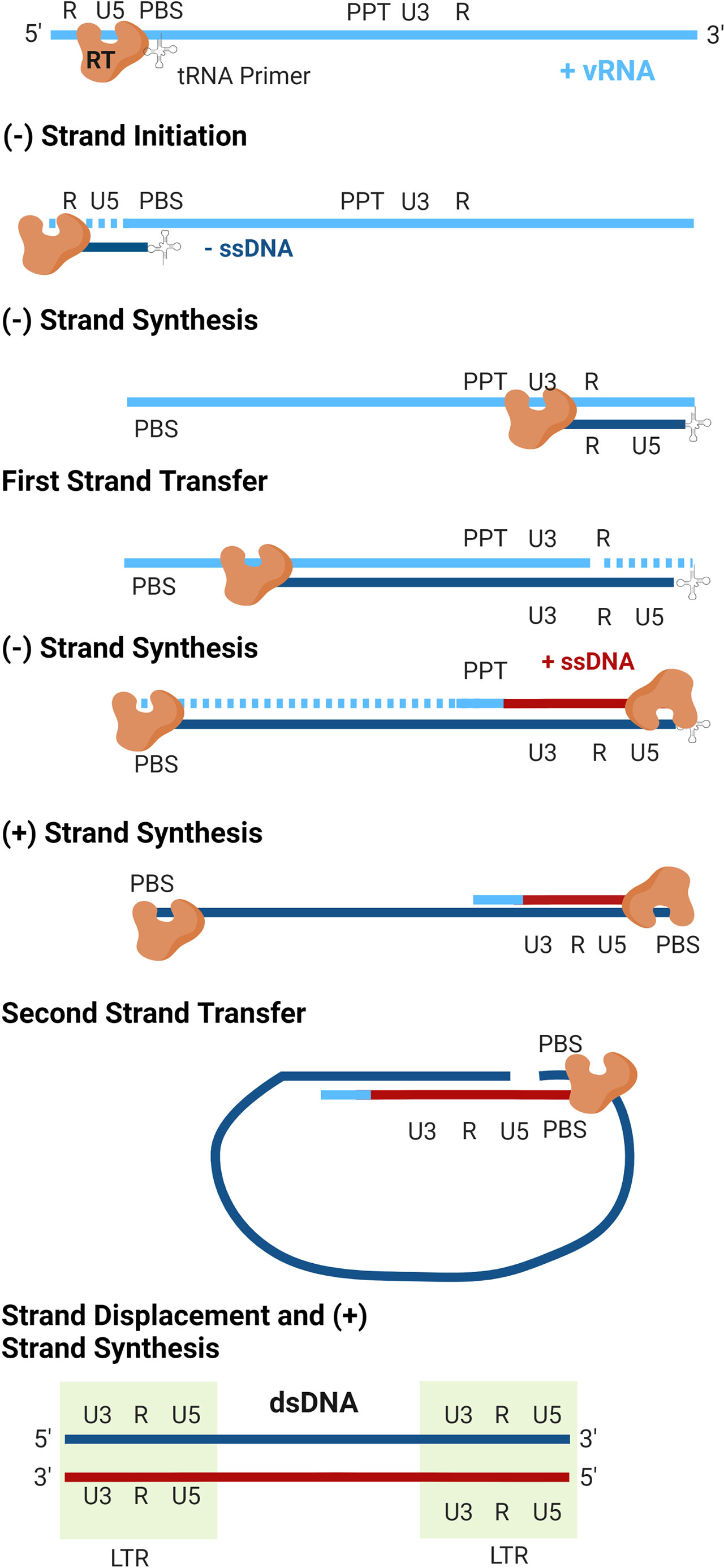
Figure 3 Reverse transcription. The virally encoded reverse transcriptase (RT) converts the viral genomic RNA (vRNA) into double-stranded DNA. As replication occurs, the RNaseH activity of RT degrades the vRNA template (dashed lines). Following completion of plus-strand synthesis, the double-stranded DNA product is integrated into the host genome by integrase to form a transcriptionally active provirus (not shown).
Within the nucleus, the dsDNA product exists as a ribonucleotide complex made up of viral and cellular proteins, known as the pre-integration complex (PIC). Included within the PIC is IN, which acts to catalyze the integration of the viral dsDNA into the host genome, forming a completed provirus (Figure 2E). Integration of retroviral genomes occurs in a semi-random fashion. Research suggests that both HIV-1 and HIV-2 preferentially integrate within or adjacent to protein-coding regions and in CpG-rich regions (65–70). One study found that during in vitro infection of peripheral blood mononuclear cells, 82% of HIV-2 integrations occurred within annotated genes (68). On the other hand, HTLV-1 does not exhibit a preference for integration within open reading frames (71, 72). According to one analysis of integration sites in cells from HTLV-1-infected individuals, HTLV-1 proviruses integrate within gene transcriptional units at a frequency similar to what is expected based on random chance (71). Although specific genomic hotspots have not been identified, research suggests that HTLV-1 may preferentially integrate at palindromic targets near transcription start sites (72).
Retroviral transcription from integrated proviruses is driven by promoter and enhancer elements within the LTR (73, 74). A number of structures and sequences are included within the LTRs that promote viral gene transcription, including the TATA box, the 5’ trans-activating response (TAR) element, and a collection of host transcription factor binding sites. Together, these factors recruit the cellular RNA polymerase II, which catalyzes the transcription of viral mRNAs as well as genomic RNA starting at the U3 region of the LTR (73). Because HIV-1 and HIV-2 integrate near or within transcriptionally active regions of the genome, enhancement of transcription from the proviral LTRs can also affect the gene expression profile of infected cells (75). Additionally, some of the cellular factors recruited to the proviral LTR, including NF-κB and CBF-1, also recruit histone deacetylases (HDACs) that suppress viral gene transcription and help establish viral latency (76–78).
Assembly, Budding, and Maturation
Viral particle assembly occurs as the vRNA and transcribed proteins (Figure 2G), including Gag, Gag-Pol, Env, and a collection of accessory proteins (Figure 2F), traffic to the plasma membrane (Figure 2H). A key step in this process is the oligomerization of Gag proteins, which promotes the formation of the immature Gag lattice of the viral particle. For HIV-1, Gag oligomerization is driven by interactions between the C-terminal domains within the CA region of different Gag proteins (79). In contrast, HTLV-1 Gag oligomerization is driven by a variety of Gag-Gag interactions across the full length of the protein, including in the N-terminal domain of CA as well as in the matrix (MA) and nucleocapsid (NC) domains (80, 81). In the case of HIV-1 infection, Gag oligomers form primarily in the cytoplasm before trafficking to the plasma membrane (82). HTLV-1 Gag is instead thought to traffic to the plasma membrane as a monomer before oligomerizing on the inner leaflet of the plasma membrane (82). The MA domain of Gag is responsible for the membrane-binding activity of the protein, which anchors the protein within lipid rafts at the plasma membrane (83–87). For both HIV-1 and HTLV-1, electrostatic interactions between the highly basic region of MA and phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] are key regulators of membrane binding, though recent research suggests that co-translational myristoylation of the G2 position in MA is the primary driver of HTLV-1 Gag membrane binding (88, 89).
Although NC is thought to be the main driver of Gag vRNA-binding activity, MA can bind to vRNA as well (90–93). It is currently unknown whether HIV-1 and HTLV-1 Gag proteins bind to vRNA within the cytoplasm of the cell and traffic to the membrane together, or whether vRNA reaches sites of particle assembly via transient diffusion (94–96). Preliminary evidence from in vitro biophysical studies suggests that ribonucleoprotein complexes, including Gag-RNA complexes, may be partially excluded from the plasma membrane-adjacent space via the actin cortex, which may act as a steric hinderance to particle assembly (97). Although the biological implications of these observations remain to be elucidated, these results suggest that the actin cortex may serve as a physical determinant of particle assembly sites on the plasma membrane.
The curvature of the Gag lattice at the plasma membrane causes the formation of a roughly spherical particle, the budding and release of which is catalyzed by host proteins including the ESCRT machinery (98–102). During or shortly after virus particle release, the viral PR cleaves Gag, triggering the maturation of the immature virus particle (Figures 2I–K). The mature particle contains MA embedded along the inner surface of the viral envelope; a CA core surrounding the viral genomic RNA, which is studded with NC, RT, and IN proteins; and a myriad of host and viral accessory proteins within the tegument of the particle. The maturation process is the final step in the development of an infectious viral particle that contains all the necessary factors to infect a naïve target cell (95, 103).
Morphological Diversity in Human Retrovirus Particles
Mature HIV-1 and HIV-2 particles exhibit an iconic conical capsid core structure (104, 105). Lentiviral core structure is unique among retroviruses, though, and the Retroviridae family exhibits a great deal of structural diversity, both among mature and immature particles (106, 107). Similar to other retroviruses, including murine leukemia virus (MLV), Rous sarcoma virus (RSV), and foamy viruses, HTLV-1 mature particles have been found to be roughly polygonal in nature (108–111). However, in analyses of HTLV-1 mature core morphology using chronically infected T cell lines, populations of particles with distinct morphologies have been identified (111, 112). In these analyses, particles with a distinct core-like structure accounted for just 10% to 15% of particles observed, and an additional 5% to 20% contained what were termed “partially mature” capsid cores, in which the capsid structure appeared incomplete. Still another 60% to 70% exhibited uniform electron density within the viral particle, with no discernable core-like structure observed (111, 112). The large proportion of HTLV-1 particles lacking CA organization and containing partially mature CA cores provides a plausible hypothesis to explain the decreased transmissibility and infectivity of HTLV-1 particles in a cell-free environment, though further research is needed to define this relationship.
Significant structural diversity has also been observed in immature particle structure across different human and animal retroviral species. In a comprehensive analysis of the structure of immature retroviral particles, it was determined that immature HTLV-1 particles are distinct among all retroviruses, characterized by discontinuous Gag lattices and flat regions of multimerized CA that appear disconnected from the plasma membrane (106, 113). The retroviral Gag lattice is the most prominent feature in both mature and immature particles, and the complex arrangement of these lattices is dictated by intermolecular protein interactions located primarily within the CA domain (79–81). While the structural determinants of HIV-1 Gag oligomerization have been extensively studied in authentic particles (114–116), virus-like particles (117–119), and in vitro assemblies of purified CA proteins (120), high resolution of the structure of the HTLV-1 has been limited to nuclear magnetic resonance (NMR) studies of the HTLV-1 CA domain (121). Comparative studies probing residues within CA have identified many of the key interaction interfaces required for maintaining replication and morphology of HIV-1 (122, 123), HIV-2 (124), and HTLV-1 (81), including those that dictate the formation of structural features such as the six-helix bundle of CA-SP1 and the two- and three-fold CA interfaces. Recent determination of the structure of the HIV-2 immature lattice demonstrates a similar hexameric organization to that of HIV-1 (125), further emphasizing the unique nature of HTLV-1 among human retroviruses.
Understanding structural diversity, and its impact on infectivity, in retroviruses is important, as form dictates function. As Moshe Safdie wrote in his 1970 novel Beyond Habitat, “nature makes form, and form is a by-product of evolution.” Using point mutations introduced into the HIV-1 CA protein that either increase or decrease the stability of the CA core structure, research has demonstrated that core stability and disassembly are finely tuned processes, and that structural abnormalities that disrupt their kinetics in either direction inhibit viral replication (56, 58). This is likely due to the observation that CA core uncoating is connected to reverse transcription kinetics; consequently, disruption of core disassembly may perturb replication of the viral genome (126, 127). There is therefore a need to better understand the structural diversity within retroviruses, and particularly within HTLV-1, and how this relates to viral replication and infectivity. In addition, more quantitative analyses identifying the differences in viral and host protein incorporation into particles, properties of Gag oligomerization, and maturation dynamics are critical to support the next generation of comparative studies of human retroviral particles.
Genetic Diversity in Human Retroviruses
A high rate of viral evolution is a hallmark feature of HIV-1 that drives the rapid diversification of viral genomes, both within and between hosts. The genetic diversity of HIV-1 within a single infected individual is comparable with the genetic diversity observed in influenza sequences circulating around the globe within a given year (128). The genetic diversity of the virus has led to the classification of HIV-1 into groups, subtypes, and sub-subtypes (Figure 4A). HIV-1 can be classified as group M, N, O, or P; group M consists of subtypes A, B, C, D, F, G, H, J, and K, and subtypes A and F can be further classified into sub-subtypes A1, A2, A3, A4, F1, and F2 (129). The genetic variability between subtypes generally ranges from about 25% to 35%, and within subtype variability can be as high as 20% (130). The various HIV-1 groups are thought to have originated via distinct zoonotic transmission events, with the further diversification of HIV-1 group M driven by a high rate of viral mutation.
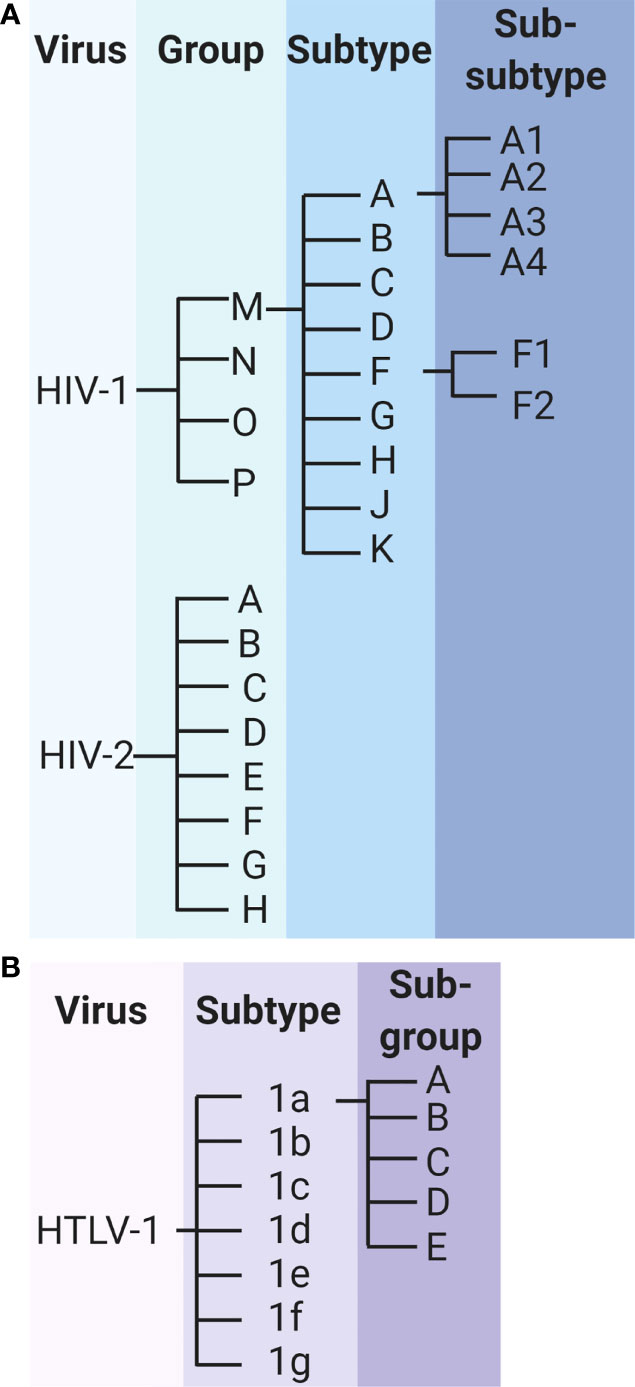
Figure 4 Classification of HIV-1, HIV-2 and HTLV-1. (A) Classification of HIV-1 and HIV-2 groups, subtypes, and sub-subtypes. (B) Classification of HTLV-1 subtypes and subgroups.
The genetic diversity of HIV-1 has important consequences for the clinical progression of disease. A diverse pool of viral genomes promotes immune evasion, cell tropism changes, and the emergence of antiretroviral drug resistance (131). The rapid evolution of HIV-1 has necessitated a shift in the HIV-1 treatment paradigm towards combination active antiretroviral therapy (cART, or simply ART), which uses a customized combination of different classes of antiretrovirals to suppress HIV replication. The use of ART has significantly increased the proportion of people living with HIV who remain progression-free and can delay the progression to AIDS by 85% and the risk for AIDS-related death by 98% (132, 133). However, resistance to ART has already started emerging globally. In 2019, the World Health Organization reported that more than 10% of adults infected with HIV in 12 countries in Africa, Asia, and Central and South America have developed drug resistance to efavirenz and nevirapine, two non-nucleoside analog reverse transcriptase inhibitors that form the backbone of many ART regimens. In sub-Saharan Africa, the prevalence of ART resistance is especially high, with approximately 50% of newly diagnosed infants within the region being infected with a strain of HIV that is resistant to one or both of these drugs (134).
Like HIV-1, HIV-2 is further characterized into groups A through H (Figure 4A). Preliminary research suggested that the genetic diversity between these groups may reach as high as 25% (135), but these data are nearly 3 decades old and do not reflect the potential genetic diversity in newly identified HIV-2 groups. In addition to the sparsity of research, a clear understanding of HIV-2 diversification has been confounded by conflicting results from in vivo studies of HIV-2 evolution. In a 2007 study of env gene evolution among 8 HIV-2-infected individuals from Senegal over approximately 10 years, HIV-2 evolution was found to be significantly slower than that of HIV-1 sequences over a similar time period (136). In contrast, a similar study published in 2010, which compared HIV-1 and HIV-2 env sequence evolution among 22 Swedish and Portuguese patients, reported significantly faster rates of genetic evolution for HIV-2 compared with HIV-1 over up to 13 years of follow up (137). In a more recent study exploring the selective pressure of the immune system on HIV-1 and HIV-2, HIV-2 was found to be under greater purifying selection than HIV-1, suggestive of stronger constraints on viral evolution (138). The discrepancies in results may be related to the regions of the env gene sequenced, as HIV-2 evolution has been found to vary significantly across various regions of env (139). Similarly incongruent results have been observed in in vitro studies of HIV-1 and HIV-2 RT fidelity and kinetics (140, 141). However, single-cycle replication studies suggest that HIV-2 accumulates significantly fewer mutations during a single round of replication than does HIV-1 (142, 143).
HTLV-1 is divided into 7 subtypes (1a through 1g), of which subtype 1a is further characterized into subgroups A through E (Figure 4B). The diversity between these subtypes is considerably lower than is observed for HIV-1 or HIV-2, with one study estimating less than 1% genetic divergence between isolates of HTLV-1 subtypes (144). This diversity represents the result of thousands of years of evolution within the human population, underscoring the extremely low rate of HTLV-1 evolution (45–47). Studies estimate that substitutions accumulate within the HTLV-1 genome at a frequency of approximately 1% per thousand years (145, 146).
Research on the determinants of viral mutagenesis and evolution suggest that the diversification of retroviruses is driven by a complex interplay of internal and external factors. For example, while the error rate of HTLV-1 RT has been found to be several orders of magnitude lower than that of HIV-1 RT (147, 148), studies analyzing virus replication suggest that the substitution rate of HTLV-1 is comparable with those observed in HIV-1 and HIV-2 (137, 139, 149, 150). These results suggest that human retroviral evolution is driven by multiple factors beyond intrinsic replication errors alone. Indeed, retroviral evolution is thought to be driven by a combination of viral mutation, the pace of viral replication, the longevity of infection, and the size of the replicating viral population (131). The relative contribution of these factors has been found to vary considerable between retroviruses. For example, while HTLV-1 diversification is thought to be driven primarily by clonal expansion of infected cells (33), a high viral mutation rate and rapid replication of the virus are thought to be key contributors to viral evolution of HIV-1. Historical studies have estimated that the HIV-1 mutation rate is between 1.4 and 3.4 x 10-5 mutations per base pair (mut/bp), although more recent studies have estimated that the mutation rate may be closer to 4.1 x 10-3 mut/bp per cell in vivo (151–155).
The occurrence of multiple zoonotic transmission events of SIVs are also thought to contribute to the genetic diversity of HIV-1 and HIV-2. While most strains of HIV-1 exhibit the highest level of similarity to SIVs from chimpanzees, group O viruses are more closely related to SIVs isolated from gorillas, indicating distinct transmission events that have contributed to the pool of circulating HIV-1. Additionally, phylogenetic analyses demonstrate that HIV-2 isolates are genetically interspersed with SIV strains from sooty mangabey monkeys, suggesting multiple cross-species transmissions (156). Zoonotic transmission events are undoubtedly an important earlier driver of the genetic diversity of human retroviruses, as adaptive mutations have been found to coincide with cross-species transmission events (157).
Molecular Determinants of Retroviral Mutagenesis
Reverse Transcriptase
The error-prone RT encoded by the virus is thought to be the primary driver of HIV-1 mutagenesis (158). In vitro studies suggest that the error rate of the HIV-1 group M RT (subtype B) is approximately 0.6 to 2.0 x 10-4 mut/bp, nearly 100- to 1000-times greater than cellular DNA polymerases (148). The extremely high error rates associated with HIV-1 and other retroviral polymerases are due in part to a lack of intrinsic proofreading ability, which corrects up to 99% of mistakes encoded by cellular DNA polymerases (159). Instead, polymerase errors become fixed within the retroviral genome and contribute to viral diversification.
A variety of intrinsic and extrinsic factors can modulate the fidelity of retroviral polymerases. Mutations within the pol gene that encode for drug resistance-associated mutations, for example, can significantly increase or decrease the fidelity of RT. The M184V mutation in HIV-1, which confers resistance to the nucleoside analogue reverse transcriptase inhibitors (NRTIs) lamivudine (3TC) and emtricitabine (FTC), is associated with a 20% reduction in the relative HIV-1 mutant frequency (160). In contrast, the K65R mutation, which confers resistance to the NRTIs 3TC, abacavir (ABC), and tenofovir disoproxil fumarate (TDF) in both HIV-1 and HIV-2, increases the mutant frequency of HIV-2 by approximately 10% compared with WT (141, 161). Small molecules and NRTIs themselves can also affect viral mutagenesis and can be used to increase the viral mutation rate above a theoretical error threshold to promote viral population collapse (a novel therapeutic strategy known as lethal mutagenesis) (160, 162–165). Retroviral polymerases are also prone to “slipping” and “jumping”, which often occur at runs of repeated nucleotides and can result in deletions and frameshift mutations (166).
The fidelity of retroviral polymerases can also be affected by cellular concentrations of deoxynucleoside triphosphates (dNTPs), which are needed for generation of viral genomic DNA products (167). The mutation rate of HIV-1 has been found to differ by up to 30% between macrophages and T cells, which have drastically different dNTP concentrations based on their dividing status (168). In the absence of dNTPs, ribonucleotides are frequently incorporated during HIV-1 replication in macrophages, which can result in viral mutation if unrepaired (169). Indeed, recent research suggests that perturbations of dNTP pools within macrophages results in a 20% to 30% increase in the viral mutant frequency (170). However, the misincorporation of ribonucleotides during retroviral replication in macrophages is reduced following expression of the HIV-2/SIV accessory protein viral protein X (Vpx) (171). Vpx counteracts the cellular HIV-restriction factor SAMHD1 (sterile alpha motif and HD domain-containing protein 1), which acts as a deoxynucleoside triphosphohydrolase in non-dividing myeloid-lineage cells and depletes cellular dNTP pools (172). Collectively, these results suggest that depletion of dNTPs within macrophages by SAMHD1 contributes to HIV mutagenesis, but direct evidence of this effect remains to be observed. However, in further support of this activity, SAMHD1 expression is associated with perturbations in dNTP levels similar to those observed with the ribonucleoside reductase inhibitors, which have been shown to increase the HIV-1 mutagenic properties of nucleoside analogs (173–175).
The retroviral polymerase, RT, is also highly prone to recombination, which further promotes diversification. Retroviruses, including HIV-1, HIV-2, and HTLV-1, are considered pseudodiploid, meaning that viral particles each contain two complete copies of the viral genomic RNA. During replication of the viral genome and synthesis of the DNA provirus, template switching may occur, in which the viral RT enzyme “jumps” between RNA copies to complete reverse transcription (176). When identical copies of the RNA genome are packaged, no effect is observed. However, mutations can arise when genetically distinct copies of the RNA genome are packaged, either as a result of superinfection (infection of the host cell with more than one virus) or, much less frequently, errors resulting from host DNA polymerases (176). Recombination itself can also lead to mutations. One study demonstrated that 15% to 20% of all mutations that occur during reverse transcription in cell culture are associated with recombination events (177).
In endemic regions where individuals may be repeatedly exposed to HIV, recombination can occur between HIV subtypes within the context of host superinfection, resulting in the formation of recombinant forms of the virus (178). As of June 2021, 102 circulating recombinant forms (CRFs) of HIV-1 have been identified in the Los Alamos National Laboratory HIV sequence database (179). Recombination between subtypes of HIV-2, however, is much less common, and only one HIV-2 CRF has been identified to date (179, 180). Recombination between HTLV-1 strains has been observed similarly infrequently (33, 181).
Recombination is an essential process in retroviral replication and occurs frequently during the course of infection. Early in vitro studies estimated that over 20% of proviruses within a population are the result of recombination events after as little as 15 days (182). Blocking recombination results in a reduction in HIV-1 viral titers and significant deletions within genomes that are able to propagate, indicating that recombination is essential for maintaining genomic integrity (183). Studies of HIV-1 suggest that retroviral recombination is dependent on regions of high sequence similarity between templates as well as RNA secondary structures (40, 184–186). Low levels of cellular dNTP pools may also promote recombination, as macrophages have been found to have recombination rates up to 4-fold higher than observed in T cells, and HIV-1 template switching in macrophages is reduced to T cell levels following degradation of SAMHD1 via Vpx (168, 187).
APOBEC3 Proteins
Host cellular factors, including the APOBEC3 (apolipoprotein B mRNA editing-enzyme catalytic polypeptide-like 3; A3) family of restriction factors, also contribute to viral mutagenesis (Figure 5). These proteins are a family of cytosine deaminases that catalyze the deamination of cytosine (C) to uracil (U) within ssDNA substrates (188, 189). During reverse transcription, these C-to-U mutations template the improper insertion of adenine (A) in place of guanine (G) in the complimentary DNA strand, effectively catalyzing G-to-A mutations within the positive-sense DNA strand (188, 190). Of the 7 APOBEC3 proteins encoded within the human genome (A3A-D, A3F-H), A3D, A3F, A3G, and A3H (haplotypes II, V, and VII) have been shown to restrict HIV-1 infection via the introduction of G-to-A mutations during reverse transcription (191–195). Editing by APOBEC3 proteins is dependent on both DNA structure and sequence, with A3G preferentially targeting cytosines within a 5’-CC-3’ (5’-GG-3’) context and all other A3 proteins more potently mutagenizing cytosines within a 5’-TC-3’ (5’-GA-3’) context (189, 196–200). APOBEC3 proteins, particularly A3G, can introduce several G-to-A mutations within an HIV-1 genome during a single round of replication, a phenomenon termed hypermutation. One study found that up to 10% of plus-strand guanines in the HIV-1 viral genome were converted to adenines by A3G during a single round of replication; some individual base pairs were especially prone to mutagenesis, being changed to adenines at frequencies ranging from 33% to 100% (196). APOBEC3-induced G-to-A mutations are often lethal, as a G-to-A transition within a 5’-TGG-3’ context results in the generation of any one of the 3 translational stop codons (TAG, TAA, or TGA). In an analysis of the HIV-1 gag gene, it was found that over 80% of proviruses contained stop codons introduced by A3G editing within a cell culture system (196).
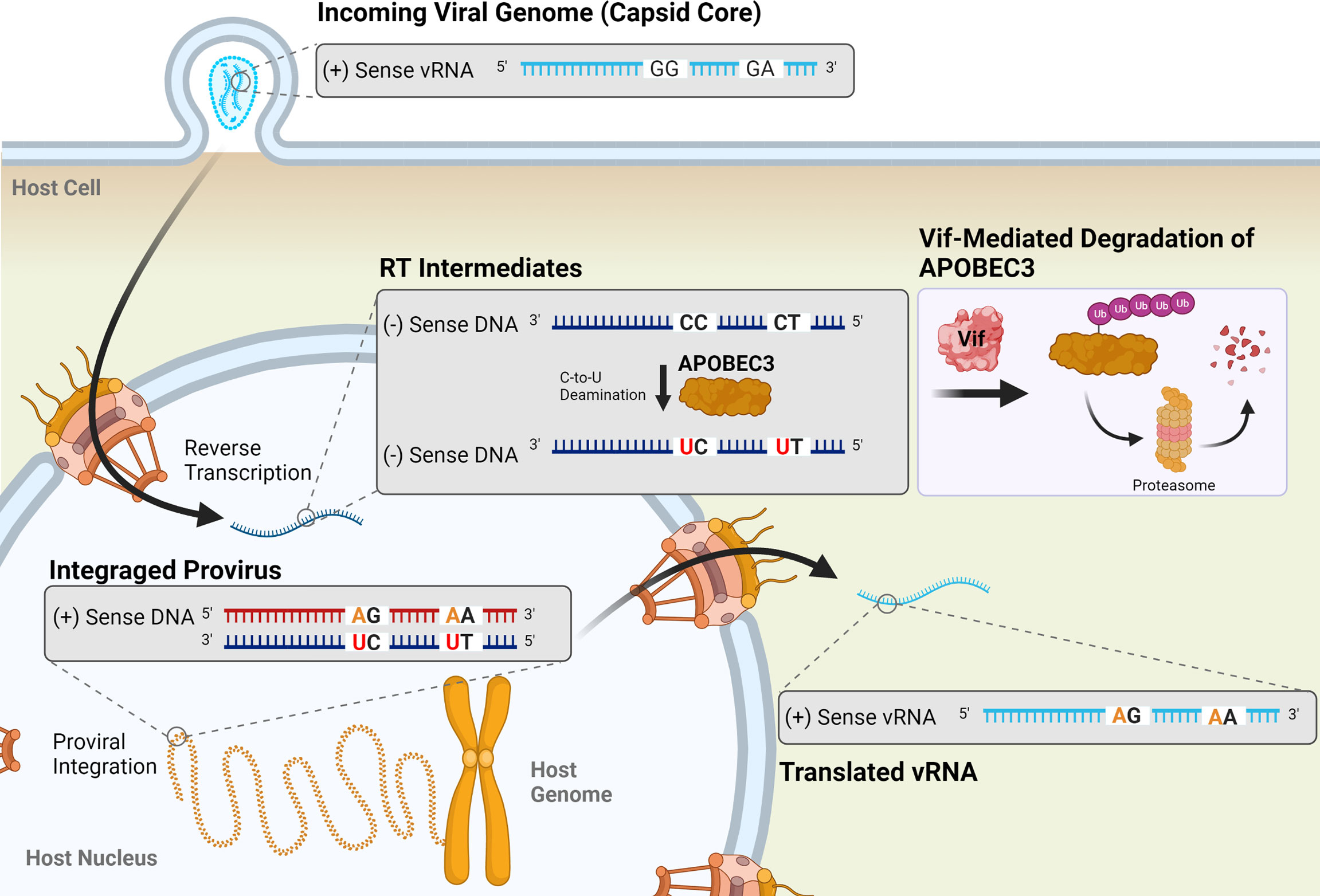
Figure 5 Mutagenesis by APOBEC3 proteins results in G-to-A transition mutations in HIV-1 proviruses. Four APOBEC3 proteins (A3D, A3F, A3G, and A3H) are known to restrict HIV-1 infection by catalyzing the deamination of cytosine in single-stranded DNA (ssDNA) into a uracil (red) during reverse transcription. This templates the improver insertion of an adenine (orange) in place of a guanine, resulting in a G-to-A mutation in the positive-sense DNA strand of the resulting provirus. These mutations preferentially occur in a 5’-GG-3’ or 5’-GA-3’ dinucleotide context. APOBEC3 activity is inhibited by the viral accessory protein Vif (shown in pink box), which targets APOBEC3 proteins for degradation by the proteasome. Vif suppression of APOBEC3 activity is incomplete, though, and sublethal levels of G-to-A mutations can be observed in HIV-1 proviruses.
HIV-1 escapes restriction by APOBEC3 proteins via expression of the Vif (viral infectivity factor) protein. In viral producer cells, Vif interacts with the cellular cofactor CBF-β to catalyze the formation of an active E3 ubiquitin ligase complex, comprised of CUL5, ELOB, ELOC, and RBX1, to promote the ubiquitination and subsequent proteasomal degradation of APOBEC3 proteins (188, 201–205). In the absence of Vif, APOBEC3 proteins are packaged into budding particles along with viral proteins and genomic RNA. Following infection of a target cell, they exert their antiviral activity by deaminating cytosines within the nascent ssDNA. Targeting of the APOBEC3 proteins for degradation within the producer cell by Vif prevents their incorporation into viral particles and allows the virus to circumvent APOBEC3-mediated restriction.
However, even in the presence of Vif expression, sublethal levels of G-to-A mutations are observed within the HIV-1 genome, indicating that suppression of APOBEC3-mediated restriction is incomplete (142, 143, 158, 206, 207). These mutations have important implications within the context of viral infection, and have been linked to changes in coreceptor usage, immune evasion, and the failure of ART (208–214). One study found that incomplete neutralization of A3G and A3F was associated with a significant increase in the presence of CXCR4-tropic viruses in the population, a shift that has previously been found to correlate with cell tropism changes, disease progression, and a more rapid decline in CD4+ T cell counts in HIV-1 infected individuals (209, 215). In a study of 88 Indian HIV-1-positive patients (87 infected with subtype C, 1 infected with a recombinant A1C strain), 11.4% exhibited evidence of A3G-mediated hypermutation within proviral sequences, and patients failing ART were nearly 5-times more likely to exhibit evidence of lethal G-to-A hypermutation than treatment naïve patients (214). Collectively with other in vitro and in vivo studies, these results suggest that, despite their relatively minor contribution to the HIV-1 mutation rate (158), G-to-A mutations resulting from sublethal APOBEC3-mediated mutagenesis are important drivers of HIV-1 diversification and evolution.
The restrictive activity of APOBEC3 proteins against other human retroviruses has remained largely uncharacterized relative to HIV-1. Studies on the anti-HIV-2 activity of APOBEC3 proteins have largely been conducted in a piecewise fashion, primarily as an evolutionary comparator with HIV-1 and SIV (216–218). However, one study demonstrated that HIV-1 and HIV-2 Vif proteins interact with A3G and A3F via disparate moieties in both the Vif and APOBEC3 proteins (218). Additionally, our group has demonstrated that HIV-2 is characterized by a relative lack of G-to-A mutations, and specifically hypermutations, relative to HIV-1 (142, 143). Consistent with these observations, restriction of HIV-2 was found to be mediated by a smaller subset of APOBEC3 proteins than HIV-1 (A3F, A3G, and A3H), with only A3F and A3G serving as potent mutagens of HIV-2 (219). Collectively, these results indicate that APOBEC3-mediated restriction of HIV-2 is distinct from that of HIV-1.
The effects of APOBEC3 proteins on HTLV-1 have been studied, but with somewhat inconclusive results. In a single cell culture study, A3A and A3B were found to restrict HTLV-1 infection in a deaminase-dependent manner (220). A3G and A3H (haplotype II) have also been found to restrict HTLV-1 infection in vitro, though these proteins are thought to act in a deaminase-independent manner and the exact mechanisms remain to be established (220, 221). However, two studies independently examined proviral sequences obtained from the blood of HTLV-1-infected individuals to determine the effects of APOBEC3 editing in vivo. While one study found no evidence of G-to-A hypermutation within HTLV-1 proviruses (222), the other reported extensive editing of proviral sequences within dinucleotide contexts suggestive of APOBEC3-mediated mutagenesis (220). A more recent study examining the evolutionary footprint of APOBEC3 proteins within the genome of human viruses found that the HBZ-encoding region of the HTLV-1 genome is relatively depleted of 5’-NTC-3’ codons, indicative of historic APOBEC3-mediated editing throughout the evolution of the virus (223). More research is needed to fully understand the effects of the HTLV-1-restrictive activity of APOBEC3, but studies have been confounded by difficulties propagating the virus in cell culture (224–226).
ADAR Proteins
Host ADAR (adenosine deaminases acting on RNA) proteins have also been implicated as potential sources of mutation during HIV-1 replication (Figure 6A). ADAR proteins are RNA-editing enzymes that catalyze the deamination of adenosine into the purine nucleoside inosine (I) in double-stranded RNA structures (227, 228). During splicing and translation events, cellular machinery treat inosine as guanosine, resulting in changes to both splicing patterns and protein amino acid sequences (228–230). This also results in the introduction of A-to-G mutations in proviral DNA when these editing events occur during reverse transcription (154, 231). A-to-G, and U-to-C, hypermutation have been observed in many negative-sense RNA viruses but are observed relatively infrequently in retroviruses such as HIV-1 and HIV-2 (142, 154, 207, 232, 233).
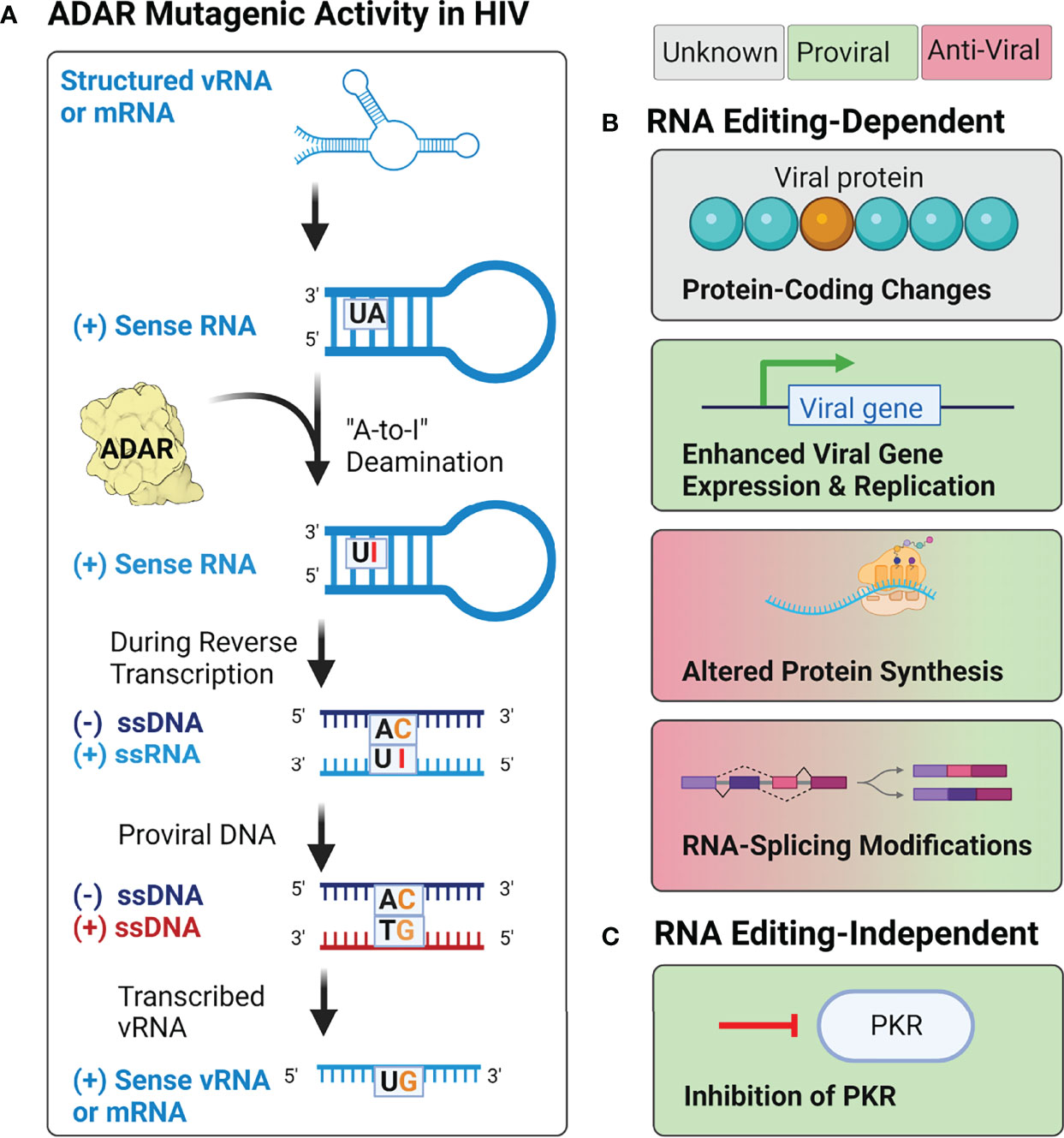
Figure 6 Potential effects of ADAR activity during HIV-1, HIV-2, and HTLV-1 infection. (A) Adenosine deamination reactions by ADAR proteins during retroviral replication are thought to induce A-to-I mutations within structured RNA elements from both the incoming vRNA genome and transcribed mRNA products. When these mutations occur during reverse transcription, it may lead to the introduction of A-to-G mutations in the integrated provirus. (B) Proposed RNA editing-dependent enzymatic activities of ADAR during retroviral infection have reported pro- and antiviral effects on protein synthesis, viral gene expression and replication, rates of protein synthesis, and RNA-splicing. (C) Proposed RNA editing-independent effects include inhibition of PKR.
Research on the effects of ADAR editing on HIV-1 infectivity has yielded conflicting results (Figure 6B). Several studies have demonstrated that ADAR1 and ADAR2, the two ADAR proteins expressed throughout the body, serve as proviral factors for HIV-1 (and HTLV-1) infection (234–238). Studies of ADAR1 overexpression in HIV-1 producer cells have demonstrated that editing by ADAR1 in the 5’UTR, as well as the env, rev, and tat genes, results in enhanced expression of the viral genome and increased protein production (234, 236). Experimental evidence using both HIV-1 and HTLV-1 suggest that the proviral activity of ADAR1 is also mediated by ADAR1-induced inhibition of PKR (protein kinase RNA activated), an antiviral protein that inhibits translational initiation of viral RNAs (Figure 6C) (235, 238). However, although ADAR1 has been shown to facilitate HIV-1 replication in CD4+ T cells (238), it has been found to inhibit HIV-1 replication in human alveolar macrophages (233). In these cells, overexpression of ADAR1 inhibited viral replication, but did not affect transcription of viral genes, suggesting the antiviral activity may be regulated post-transcriptionally (233). This hypothesis is supported by evidence from a variety of cell lines (293T, HeLa, Jurkat T, and primary CD4+ T cells), which found that ADAR1-induced A-to-G mutations in highly structured regions of the genome (i.e., rev, env, and the RRE) inhibited viral protein synthesis, but not viral RNA synthesis (239). The conflicting results observed in existing studies, as well as the lack of research on the effects of ADAR1 and ADAR2 on HIV-2 replication, highlight the need for further research on the cellular sources of genetic diversity within human retroviruses, and their effects on viral replication.
Conclusion
Much of our current knowledge on the biology of human retroviruses is based on studies of HIV-1, the most prominent and deadly of the human pathogenic Retroviridae. However, comparative studies demonstrate key genetic, morphological, and physiologic differences between HIV-1 and other retroviruses including HIV-2 and HTLV-1. Historically, assumptions about these viruses, particularly HIV-2, have been made based on what is known about HIV-1. Indeed, even clinical guidelines for the treatment of HIV-2-infected individuals are based on extrapolation of data from the treatment of HIV-1, as clinical trials and formal studies in HIV-2 patients are lacking (240). As indicated in this review, the available research demonstrates that significant diversity exists among human retroviruses, and each should be studied independently for a complete understanding of their biology, including the cellular and molecular determinants of replication and mutagenesis. Comparative analysis among the human retroviruses can be powerful in understanding their replication, evolution, persistence, and pathogenicity.
Author Contributions
Writing—original draft preparation, MM. Writing—reviewing and editing, MM, NT, and LM. Visualization, MM and NT. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institutes of Health grant R01 AI150468 (to LM). MM was supported by NIH grants T32 AI083196 and F31 AI 50487. NT was supported by NIH grants T32 DA007097, F32 AI150351, and American Cancer Society Postdoctoral Fellowship PF-21-189-01-MPC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Dr. Nikunj Somia for his thoughtful comments and advice on this manuscript. Figures were created with BioRender.com.
References
1. Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, et al. Isolation of a T-Lymphotropic Retrovirus From a Patient at Risk for Acquired Immune Deficiency Syndrome (AIDS). Science (1983) 220(4599):868–71. doi: 10.1126/science.6189183
2. Gallo RC, Sarin PS, Gelmann EP, Robert-Guroff M, Richardson E, Kalyanaraman VS, et al. Isolation of Human T-Cell Leukemia Virus in Acquired Immune Deficiency Syndrome (AIDS). Science (1983) 220(4599):865–7. doi: 10.1126/science.6601823
3. UNAIDS. Global HIV & AIDS Statistics — 2019 Fact Sheet (2020). Available at: https://www.unaids.org/en/resources/fact-sheet.
4. World Health Organization. HIV Data and Statistics (2020). Available at: https://www.who.int/hiv/data/en/.
5. Visseaux B, Damond F, Matheron S, Descamps D, Charpentier C. Hiv-2 Molecular Epidemiology. Infect Genet Evol (2016) 46:233–40. doi: 10.1016/j.meegid.2016.08.010
6. Walker B, McMichael A. The T-Cell Response to HIV. Cold Spring Harb Perspect Med (2012) 2(11). doi: 10.1101/cshperspect.a007054
7. Pantaleo G, Fauci AS. Immunopathogenesis of HIV Infection. Annu Rev Microbiol (1996) 50:825–54. doi: 10.1146/annurev.micro.50.1.825
8. Poorolajal J, Sarr AD, Travers KU, Guèye-Ndiaye A, Mboup S, Essex ME, et al. Survival Rate of AIDS Disease and Mortality in HIV-Infected Patients: A Meta-Analysis. Public Health (2016) 139:3–12. doi: 10.1016/j.puhe.2016.05.004
9. Popper SJ, Hooshmand E, Mahjub H, Esmailnasab N, Jenabi E. Lower Human Immunodeficiency Virus (HIV) Type 2 Viral Load Reflects the Difference in Pathogenicity of HIV-1 and HIV-2. J Infect Dis (1999) 180(4):1116–21. doi: 10.1086/315010
10. Andersson S, Norrgren H, da Silva Z, Biague A, Bamba S, Kwok S, et al. Plasma Viral Load in HIV-1 and HIV-2 Singly and Dually Infected Individuals in Guinea-Bissau, West Africa: Significantly Lower Plasma Virus Set Point in HIV-2 Infection Than in HIV-1 Infection. Arch Intern Med (2000) 160(21):3286–93. doi: 10.1001/archinte.160.21.3286
11. Kanki PJ, Travers KU, S MB, Hsieh CC, Marlink RG, Gueye NA, et al. Slower Heterosexual Spread of HIV-2 Than HIV-1. Lancet (1994) 343(8903):943–6. doi: 10.1016/S0140-6736(94)90065-5
12. Adjorlolo-Johnson G, Norrgren H, da Silva Z, Biague A, Bamba S, Kwok S, et al. Prospective Comparison of Mother-to-Child Transmission of HIV-1 and HIV-2 in Abidjan, Ivory Coast. JAMA (1994) 272(6):462–6. doi: 10.1001/jama.1994.03520060062033
13. O’Donovan D, Ariyoshi K, Milligan P, Ota M, Yamuah L, Sarge-Njie R, et al. Maternal Plasma Viral RNA Levels Determine Marked Differences in Mother-to-Child Transmission Rates of HIV-1 and HIV-2 in The Gambia. MRC/Gambia Government/University College London Medical School Working Group on Mother-Child Transmission of HIV. AIDS (2000) 14(4):441–8.
14. Marlink R, Kanki P, Thior I, Travers K, Marlink R, et al. Reduced Rate of Disease Development After HIV-2 Infection as Compared to HIV-1. Science (1994) 265(5178):1587–90. doi: 10.1126/science.7915856
15. van der Loeff MF, Awasana AA, Sarge-Njie R, van der Sande M, Jaye A, Sabally S, et al. Sixteen Years of HIV Surveillance in a West African Research Clinic Reveals Divergent Epidemic Trends of HIV-1 and HIV-2. Int J Epidemiol (2006) 35(5):1322–8. doi: 10.1093/ije/dyl037
16. da Silva ZJ, Oliveira I, Andersen A, Dias F, Rodrigues A, Holmgren B, et al. Changes in Prevalence and Incidence of HIV-1, HIV-2 and Dual Infections in Urban Areas of Bissau, Guinea-Bissau: Is HIV-2 Disappearing? AIDS (2008) 22(10):1195–202. doi: 10.1097/QAD.0b013e328300a33d
17. Tienen C, van der Loeff MS, Zaman SM, Vincent T, Sarge-Njie R, Peterson I, et al. Two Distinct Epidemics: The Rise of HIV-1 and Decline of HIV-2 Infection Between 1990 and 2007 in Rural Guinea-Bissau. J Acquir Immune Defic Syndr (2010) 53(5):640–7. doi: 10.1097/QAI.0b013e3181bf1a25
18. Berges BK, Akkina SR, Remling L, Akkina R. Humanized Rag2(-/-)Gammac(-/-) (RAG-Hu) Mice can Sustain Long-Term Chronic HIV-1 Infection Lasting More Than a Year. Virology (2010) 397(1):100–3. doi: 10.1016/j.virol.2009.10.034
19. Hu S, Neff CP, Kumar DM, Habu Y, Akkina SR, Seki T, et al. A Humanized Mouse Model for HIV-2 Infection and Efficacy Testing of a Single-Pill Triple-Drug Combination Anti-Retroviral Therapy. Virology (2017) 501:115–8. doi: 10.1016/j.virol.2016.11.013
20. Julg B, Pereyra F, Buzon MJ, Piechocka-Trocha A, Clark MJ, Baker BM, et al. Infrequent Recovery of HIV From But Robust Exogenous Infection of Activated CD4(+) T Cells in HIV Elite Controllers. Clin Infect Dis (2010) 51(2):233–8. doi: 10.1086/653677
21. Garcia M, Gorgolas M, Cabello A, Estrada V, Ligos JM, Fernandez-Guerrero M, et al. Peripheral T Follicular Helper Cells Make a Difference in HIV Reservoir Size Between Elite Controllers and Patients on Successful cART. Sci Rep (2017) 7(1):16799. doi: 10.1038/s41598-017-17057-y
22. Sharaf R, Lee GQ, Sun X, Etemad B, Aboukhater LM, Hu Z, et al. HIV-1 Proviral Landscapes Distinguish Posttreatment Controllers From Noncontrollers. J Clin Invest (2018) 128(9):4074–85. doi: 10.1172/JCI120549
23. Gueudin M, Damond F, Braun J, Taieb A, Lemee V, Plantier JC, et al. Differences in Proviral DNA Load Between HIV-1- and HIV-2-Infected Patients. AIDS (2008) 22(2):211–5. doi: 10.1097/QAD.0b013e3282f42429
24. Popper SJ, Sarr AD, Gueye-Ndiaye A, Mboup S, Essex ME, Kanki PJ. Low Plasma Human Immunodeficiency Virus Type 2 Viral Load is Independent of Proviral Load: Low Virus Production In Vivo. J Virol (2000) 74(3):1554–7. doi: 10.1128/JVI.74.3.1554-1557.2000
25. Yoshida M, Miyoshi I, Hinuma Y. Isolation and Characterization of Retrovirus From Cell Lines of Human Adult T-Cell Leukemia and its Implication in the Disease. Proc Natl Acad Sci USA (1982) 79(6):2031–5. doi: 10.1073/pnas.79.6.2031
26. Popovic M, Sarin PS, Robert-Gurroff M, Kalyanaraman VS, Mann D, Minowada J, et al. Isolation and Transmission of Human Retrovirus (Human T-Cell Leukemia Virus). Science (1983) 219(4586):856–9. doi: 10.1126/science.6600519
27. Yamamoto N, Matsumoto T, Koyanagi Y, Tanaka Y, Hinuma Y. Unique Cell Lines Harbouring Both Epstein-Barr Virus and Adult T-Cell Leukaemia Virus, Established From Leukaemia Patients. Nature (1982) 299(5881):367–9. doi: 10.1038/299367a0
28. Longo DL, Gelmann EP, Cossman J, Young RA, Gallo RC, O'Brien SJ, et al. Isolation of HTLV-Transformed B-Lymphocyte Clone From a Patient With HTLV-Associated Adult T-Cell Leukaemia. Nature (1984) 310(5977):505–6. doi: 10.1038/310505a0
29. Ho DD, Rota TR, Hirsch MS. Infection of Human Endothelial Cells by Human T-Lymphotropic Virus Type I. Proc Natl Acad Sci USA (1984) 81(23):7588–90. doi: 10.1073/pnas.81.23.7588
30. Yoshikura H, Nishida J, Yoshida M, Kitamura Y, Takaku F, Ikeda S, et al. Isolation of HTLV Derived From Japanese Adult T-Cell Leukemia Patients in Human Diploid Fibroblast Strain IMR90 and the Biological Characters of the Infected Cells. Int J Cancer (1984) 33(6):745–9. doi: 10.1002/ijc.2910330606
31. Koyanagi Y, Itoyama Y, Nakamura N, Takamatsu K, Kira J, Iwamasa T, et al. In Vivo Infection of Human T-Cell Leukemia Virus Type I in non-T Cells. Virology (1993) 196(1):25–33. doi: 10.1006/viro.1993.1451
32. Gessain A, Mahieux R. Tropical Spastic Paraparesis and HTLV-1 Associated Myelopathy: Clinical, Epidemiological, Virological and Therapeutic Aspects. Rev Neurol (Paris) (2012) 168(3):257–69. doi: 10.1016/j.neurol.2011.12.006
33. Afonso PV, Cassar O, Gessain A. Molecular Epidemiology, Genetic Variability and Evolution of HTLV-1 With Special Emphasis on African Genotypes. Retrovirology (2019) 16(1):39. doi: 10.1186/s12977-019-0504-z
34. Yoshida M, Seiki M, Yamaguchi K, Takatsuki K. Monoclonal Integration of Human T-Cell Leukemia Provirus in All Primary Tumors of Adult T-Cell Leukemia Suggests Causative Role of Human T-Cell Leukemia Virus in the Disease. Proc Natl Acad Sci USA (1984) 81(8):2534–7. doi: 10.1073/pnas.81.8.2534
35. Gessain A, Barin F, Vernant JC, Gout O, Maurs L, Calender A, et al. Antibodies to Human T-Lymphotropic Virus Type-I in Patients With Tropical Spastic Paraparesis. Lancet (1985) 2(8452):407–10. doi: 10.1016/S0140-6736(85)92734-5
36. Osame M, Usuku K, Izumo S, Ijichi N, Amitani H, Igata A, et al. HTLV-I Associated Myelopathy, a New Clinical Entity. Lancet (1986) 1(8488):1031–2. doi: 10.1016/S0140-6736(86)91298-5
37. Einsiedel L, Pham H, Au V, Hatami S, Wilson K, Spelman T, et al. Predictors of non-Cystic Fibrosis Bronchiectasis in Indigenous Adult Residents of Central Australia: Results of a Case-Control Study. ERJ Open Res (2019) 5(4). doi: 10.1183/23120541.00001-2019
38. Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, Swanstrom R, et al. Architecture and Secondary Structure of an Entire HIV-1 RNA Genome. Nature (2009) 460(7256):711–6. doi: 10.1038/nature08237
39. Tebit DM, Arts EJ. Tracking a Century of Global Expansion and Evolution of HIV to Drive Understanding and to Combat Disease. Lancet Infect Dis (2011) 11(1):45–56. doi: 10.1016/S1473-3099(10)70186-9
40. Dilley KA, Ni N, Nikolaitchik OA, Chen J, Galli A, Hu WS. Determining the Frequency and Mechanisms of HIV-1 and HIV-2 RNA Copackaging by Single-Virion Analysis. J Virol (2011) 85(20):10499–508. doi: 10.1128/JVI.05147-11
41. Harrod R. Silencers of HTLV-1 and HTLV-2: The pX-Encoded Latency-Maintenance Factors. Retrovirology (2019) 16(1):25. doi: 10.1186/s12977-019-0487-9
42. Bangham CR, Ratner L. How Does HTLV-1 Cause Adult T-Cell Leukaemia/Lymphoma (ATL)? Curr Opin Virol (2015) 14:93–100. doi: 10.1016/j.coviro.2015.09.004
43. Sharp PM, Hahn BH. Origins of HIV and the AIDS Pandemic. Cold Spring Harb Perspect Med (2011) 1(1):a006841. doi: 10.1101/cshperspect.a006841
44. German Advisory Committee Blood, S. A. o. P. T. b. B. Human Immunodeficiency Virus (HIV). Transfus Med Hemother (2016) 43(3):203–22.
45. Sonoda S, Li HC, Tajima K. Ethnoepidemiology of HTLV-1 Related Diseases: Ethnic Determinants of HTLV-1 Susceptibility and its Worldwide Dispersal. Cancer Sci (2011) 102(2):295–301. doi: 10.1111/j.1349-7006.2010.01820.x
46. Paiva A, Casseb J. Origin and Prevalence of Human T-Lymphotropic Virus Type 1 (HTLV-1) and Type 2 (HTLV-2) Among Indigenous Populations in the Americas. Rev Inst Med Trop Sao Paulo (2015) 57(1):1–13. doi: 10.1590/S0036-46652015000100001
47. Sonoda S, Li HC, Cartier L, Nunez L, Tajima K. Ancient HTLV Type 1 Provirus DNA of Andean Mummy. AIDS Res Hum Retroviruses (2000) 16(16):1753–6. doi: 10.1089/08892220050193263
48. Rayne F, Bouamr F, Lalanne J, Mamoun RZ. The NH2-Terminal Domain of the Human T-Cell Leukemia Virus Type 1 Capsid Protein is Involved in Particle Formation. J Virol (2001) 75(11):5277–87. doi: 10.1128/JVI.75.11.5277-5287.2001
49. Bernstein HB, Tucker SP, Hunter E, Schutzbach JS, Compans RW. Human Immunodeficiency Virus Type 1 Envelope Glycoprotein is Modified by O-Linked Oligosaccharides. J Virol (1994) 68(1):463–8. doi: 10.1128/jvi.68.1.463-468.1994
50. Allan JS, Coligan JE, Barin F, McLane MF, Sodroski JG, Rosen CA, et al. Major Glycoprotein Antigens That Induce Antibodies in AIDS Patients are Encoded by HTLV-III. Science (1985) 228(4703):1091–4. doi: 10.1126/science.2986290
51. Leonard CK, Spellman MW, Riddle L, Harris RJ, Thomas JN, Gregory TJ. Assignment of Intrachain Disulfide Bonds and Characterization of Potential Glycosylation Sites of the Type 1 Recombinant Human Immunodeficiency Virus Envelope Glycoprotein (Gp120) Expressed in Chinese Hamster Ovary Cells. J Biol Chem (1990) 265(18):10373–82. doi: 10.1016/S0021-9258(18)86956-3
52. Sebastian NT, Zaikos TD, Terry V, Taschuk F, McNamara LA, Onafuwa-Nuga A, et al. CD4 is Expressed on a Heterogeneous Subset of Hematopoietic Progenitors, Which Persistently Harbor CXCR4 and CCR5-Tropic HIV Proviral Genomes In Vivo. PloS Pathog (2017) 13(7):e1006509. doi: 10.1371/journal.ppat.1006509
53. Ghez D, Lepelletier Y, Jones KS, Pique C, Hermine O. Current Concepts Regarding the HTLV-1 Receptor Complex. Retrovirology (2010) 7:99. doi: 10.1186/1742-4690-7-99
54. Temin HM, Mizutani S. RNA-Dependent DNA Polymerase in Virions of Rous Sarcoma Virus. Nature (1970) 226(5252):1211–3. doi: 10.1038/2261211a0
55. Baltimore D. RNA-Dependent DNA Polymerase in Virions of RNA Tumour Viruses. Nature (1970) 226(5252):1209–11. doi: 10.1038/2261209a0
56. Forshey BM, von Schwedler U, Sundquist WI, Aiken C. Formation of a Human Immunodeficiency Virus Type 1 Core of Optimal Stability is Crucial for Viral Replication. J Virol (2002) 76(11):5667–77. doi: 10.1128/JVI.76.11.5667-5677.2002
57. Hulme AE, Perez O, Hope TJ. Complementary Assays Reveal a Relationship Between HIV-1 Uncoating and Reverse Transcription. Proc Natl Acad Sci USA (2011) 108(24):9975–80. doi: 10.1073/pnas.1014522108
58. Hulme AE, Kelley Z, Okocha EA, Hope TJ. Identification of Capsid Mutations That Alter the Rate of HIV-1 Uncoating in Infected Cells. J Virol (2015) 89(1):643–51. doi: 10.1128/JVI.03043-14
59. Dharan A, Bachmann N, Talley S, Zwikelmaier V, Campbell EM. Nuclear Pore Blockade Reveals That HIV-1 Completes Reverse Transcription and Uncoating in the Nucleus. Nat Microbiol (2020) 5(9):1088–95. doi: 10.1038/s41564-020-0735-8
60. Li C, Burdick RC, Nagashima K, Hu WS, Pathak VK. HIV-1 Cores Retain Their Integrity Until Minutes Before Uncoating in the Nucleus. Proc Natl Acad Sci USA (2021) 118(10). doi: 10.1073/pnas.2019467118
61. Zila V, Margiotta E, Turonova B, Muller TG, Zimmerli CE, Mattei S, et al. Cone-Shaped HIV-1 Capsids are Transported Through Intact Nuclear Pores. Cell (2021) 184(4):1032–1046.e1018. doi: 10.1016/j.cell.2021.01.025
62. Blanco-Rodriguez G, Gazi A, Monel B, Frabetti S, Scoca V, Mueller F, et al. Remodeling of the Core Leads HIV-1 Preintegration Complex Into the Nucleus of Human Lymphocytes. J Virol (2020) 94(11). doi: 10.1128/JVI.00135-20
63. Guedan A, Donaldson CD, Caroe ER, Cosnefroy O, Taylor IA, Bishop KN. HIV-1 Requires Capsid Remodelling at the Nuclear Pore for Nuclear Entry and Integration. PloS Pathog (2021) 17(9):e1009484. doi: 10.1371/journal.ppat.1009484
64. Jin D, Musier-Forsyth K. Role of Host tRNAs and aminoacyl-tRNA Synthetases in Retroviral Replication. J Biol Chem (2019) 294(14):5352–64. doi: 10.1074/jbc.REV118.002957
65. Kitamura Y, Lee YM, Coffin JM. Nonrandom Integration of Retroviral DNA In Vitro: Effect of CpG Methylation. Proc Natl Acad Sci USA (1992) 89(12):5532–6. doi: 10.1073/pnas.89.12.5532
66. Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell (2002) 110(4):521–9. doi: 10.1016/S0092-8674(02)00864-4
67. Holman AG, Coffin JM. Symmetrical Base Preferences Surrounding HIV-1, Avian Sarcoma/Leukosis Virus, and Murine Leukemia Virus Integration Sites. Proc Natl Acad Sci USA (2005) 102(17):6103–7. doi: 10.1073/pnas.0501646102
68. MacNeil A, Sankale JL, Meloni ST, Sarr AD, Mboup S, Kanki P. Genomic Sites of Human Immunodeficiency Virus Type 2 (HIV-2) Integration: Similarities to HIV-1 In Vitro and Possible Differences In Vivo. J Virol (2006) 80(15):7316–21. doi: 10.1128/JVI.00604-06
69. Soto MJ, Pena A, Vallejo FG. A Genomic and Bioinformatics Analysis of the Integration of HIV in Peripheral Blood Mononuclear Cells. AIDS Res Hum Retroviruses (2011) 27(5):547–55. doi: 10.1089/aid.2010.0182
70. Kok YL, Vongrad V, Chaudron SE, Shilaih M, Leemann C, Neumann K, et al. HIV-1 Integration Sites in CD4+ T-Cells During Primary, Chronic, and Late Presentation of HIV-1 Infection. JCI Insight (2021). doi: 10.1172/jci.insight.143940
71. Doi K, Wu X, Taniguchi Y, Yasunaga J, Satou Y, Okayama A, et al. Preferential Selection of Human T-Cell Leukemia Virus Type I Provirus Integration Sites in Leukemic Versus Carrier States. Blood (2005) 106(3):1048–53. doi: 10.1182/blood-2004-11-4350
72. Derse D, Crise B, Li Y, Princler G, Lum N, Stewart C, et al. Human T-Cell Leukemia Virus Type 1 Integration Target Sites in the Human Genome: Comparison With Those of Other Retroviruses. J Virol (2007) 81(12):6731–41. doi: 10.1128/JVI.02752-06
73. Pluta A, Jaworski JP, Cortes-Rubio CN. Balance Between Retroviral Latency and Transcription: Based on HIV Model. Pathogens (2020) 10(1). doi: 10.3390/pathogens10010016
74. Pluta A, Jaworski JP, Douville RN. Regulation of Expression and Latency in BLV and HTLV. Viruses (2020) 12(10). doi: 10.3390/v12101079
75. Ciuffi A, Bushman FD. Retroviral DNA Integration: HIV and the Role of LEDGF/P75. Trends Genet (2006) 22(7):388–95. doi: 10.1016/j.tig.2006.05.006
76. He G, Margolis DM. Counterregulation of Chromatin Deacetylation and Histone Deacetylase Occupancy at the Integrated Promoter of Human Immunodeficiency Virus Type 1 (HIV-1) by the HIV-1 Repressor YY1 and HIV-1 Activator Tat. Mol Cell Biol (2002) 22(9):2965–73. doi: 10.1128/MCB.22.9.2965-2973.2002
77. Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB P50 Promotes HIV Latency Through HDAC Recruitment and Repression of Transcriptional Initiation. EMBO J (2006) 25(1):139–49. doi: 10.1038/sj.emboj.7600900
78. Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. A Limited Group of Class I Histone Deacetylases Acts to Repress Human Immunodeficiency Virus Type 1 Expression. J Virol (2009) 83(10):4749–56. doi: 10.1128/JVI.02585-08
79. Lanman J, Lam TT, Barnes S, Sakalian M, Emmett MR, Marshall AG, et al. Identification of Novel Interactions in HIV-1 Capsid Protein Assembly by High-Resolution Mass Spectrometry. J Mol Biol (2003) 325(4):759–72. doi: 10.1016/S0022-2836(02)01245-7
80. Martin JL, Mendonca LM, Angert I, Mueller JD, Zhang W, Mansky LM. Disparate Contributions of Human Retrovirus Capsid Subdomains to Gag-Gag Oligomerization, Virus Morphology, and Particle Biogenesis. J Virol (2017) 91(14). doi: 10.1128/JVI.00298-17
81. Martin JL, Mendonca LM, Marusinec R, Zuczek J, Angert I, Blower RJ, et al. Critical Role of the Human T-Cell Leukemia Virus Type 1 Capsid N-Terminal Domain for Gag-Gag Interactions and Virus Particle Assembly. J Virol (2018) 92(14). doi: 10.1128/JVI.00333-18
82. Eichorst JP, Chen Y, Mueller JD, Mansky LM. Distinct Pathway of Human T-Cell Leukemia Virus Type 1 Gag Punctum Biogenesis Provides New Insights Into Enveloped Virus Assembly. mBio (2018) 9(5). doi: 10.1128/mBio.00758-18
83. Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. Interaction Between the Human Immunodeficiency Virus Type 1 Gag Matrix Domain and Phosphatidylinositol-(4,5)-Bisphosphate is Essential for Efficient Gag Membrane Binding. J Virol (2008) 82(5):2405–17. doi: 10.1128/JVI.01614-07
84. Ono A. HIV-1 Assembly at the Plasma Membrane: Gag Trafficking and Localization. Future Virol (2009) 4(3):241–57. doi: 10.2217/fvl.09.4
85. Inlora J, Collins DR, Trubin ME, Chung JY, Ono A. Membrane Binding and Subcellular Localization of Retroviral Gag Proteins are Differentially Regulated by MA Interactions With Phosphatidylinositol-(4,5)-Bisphosphate and RNA. mBio (2014) 5(6):e02202. doi: 10.1128/mBio.02202-14
86. Maldonado JO, Martin JL, Mueller JD, Zhang W, Mansky LM. New Insights Into Retroviral Gag-Gag and Gag-Membrane Interactions. Front Microbiol (2014) 5:302. doi: 10.3389/fmicb.2014.00302
87. Thornhill D, Murakami T, Ono A. Rendezvous at Plasma Membrane: Cellular Lipids and tRNA Set Up Sites of HIV-1 Particle Assembly and Incorporation of Host Transmembrane Proteins. Viruses (2020) 12(8). doi: 10.3390/v12080842
88. Herrmann D, Zhou LW, Hanson HM, Willkomm NA, Mansky LM, Saad JS. Structural Insights Into the Mechanism of Human T-Cell Leukemia Virus Type 1 Gag Targeting to the Plasma Membrane for Assembly. J Mol Biol (2021) 433(19):167161. doi: 10.1016/j.jmb.2021.167161
89. Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) Bisphosphate Regulates HIV-1 Gag Targeting to the Plasma Membrane. Proc Natl Acad Sci USA (2004) 101(41):14889–94. doi: 10.1073/pnas.0405596101
90. Sun M, Grigsby IF, Gorelick RJ, Mansky LM, Musier-Forsyth K. Retrovirus-Specific Differences in Matrix and Nucleocapsid Protein-Nucleic Acid Interactions: Implications for Genomic RNA Packaging. J Virol (2014) 88(2):1271–80. doi: 10.1128/JVI.02151-13
91. Kroupa T, Datta SAK, Rein A. Distinct Contributions of Different Domains Within the HIV-1 Gag Polyprotein to Specific and Nonspecific Interactions With RNA. Viruses (2020) 12(4). doi: 10.3390/v12040394
92. Mouhand A, Pasi M, Catala M, Zargarian L, Belfetmi A, Barraud P, et al. Overview of the Nucleic-Acid Binding Properties of the HIV-1 Nucleocapsid Protein in Its Different Maturation States. Viruses (2020) 12(10). doi: 10.3390/v12101109
93. Wu W, Hatterschide J, Syu YC, Cantara WA, Blower RJ, Hanson HM, et al. Human T-Cell Leukemia Virus Type 1 Gag Domains Have Distinct RNA-Binding Specificities With Implications for RNA Packaging and Dimerization. J Biol Chem (2018) 293(42):16261–76. doi: 10.1074/jbc.RA118.005531
94. Chen J, Grunwald D, Sardo L, Galli A, Plisov S, Nikolaitchik OA, et al. Cytoplasmic HIV-1 RNA is Mainly Transported by Diffusion in the Presence or Absence of Gag Protein. Proc Natl Acad Sci USA (2014) 111(48):E5205-5213. doi: 10.1073/pnas.1413169111
95. Martin JL, Maldonado JO, Mueller JD, Zhang W, Mansky LM. Molecular Studies of HTLV-1 Replication: An Update. Viruses (2016) 8(2). doi: 10.3390/v8020031
96. Comas-Garcia M, Davis SR, Rein A. On the Selective Packaging of Genomic RNA by HIV-1. Viruses (2016) 8(9). doi: 10.3390/v8090246
97. Angert CI, Kakura SR, Mansky LM, Mueller JD. Partitioning of Ribonucleoprotein Complexes From the Cellular Actin Cortex. bioRxiv (2021). doi: 10.1101/2021.10.01.462753
98. Demirov DG, Freed EO. Retrovirus Budding. Virus Res (2004) 106(2):87–102. doi: 10.1016/j.virusres.2004.08.007
99. Morita E, Sundquist WI. Retrovirus Budding. Annu Rev Cell Dev Biol (2004) 20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350
100. Weiss ER, Gottlinger H. The Role of Cellular Factors in Promoting HIV Budding. J Mol Biol (2011) 410(4):525–33. doi: 10.1016/j.jmb.2011.04.055
101. Sundquist WI, Krausslich HG. HIV-1 Assembly, Budding, and Maturation. Cold Spring Harb Perspect Med (2012) 2(7):a006924.
102. Ehrlich LS, Carter CA. HIV Assembly and Budding: Ca(2+) Signaling and Non-ESCRT Proteins Set the Stage. Mol Biol Int (2012) 2012:851670. doi: 10.1155/2012/851670
103. Konvalinka J, Krausslich HG, Muller B. Retroviral Proteases and Their Roles in Virion Maturation. Virology (2015) 479-480:403–17. doi: 10.1016/j.virol.2015.03.021
104. Goto T, Ashina T, Fujiyoshi Y, Kume N, Yamagishi H, Nakai M. Projection Structures of Human Immunodeficiency Virus Type 1 (HIV-1) Observed With High Resolution Electron Cryo-Microscopy. J Electron Microsc (Tokyo) (1994) 43(1):16–9.
105. Kewalramani VN, Emerman M. Vpx Association With Mature Core Structures of HIV-2. Virology (1996) 218(1):159–68. doi: 10.1006/viro.1996.0176
106. Martin JL, Cao S, Maldonado JO, Zhang W, Mansky LM. Distinct Particle Morphologies Revealed Through Comparative Parallel Analyses of Retrovirus-Like Particles. J Virol (2016) 90(18):8074–84. doi: 10.1128/JVI.00666-16
107. Zhang W, Cao S, Martin JL, Mueller JD, Mansky LM. Morphology and Ultrastructure of Retrovirus Particles. AIMS Biophys (2015) 2(3):343–69. doi: 10.3934/biophy.2015.3.343
108. Yeager M, Wilson-Kubalek EM, Weiner SG, Brown PO, Rein A. Supramolecular Organization of Immature and Mature Murine Leukemia Virus Revealed by Electron Cryo-Microscopy: Implications for Retroviral Assembly Mechanisms. Proc Natl Acad Sci USA (1998) 95(13):7299–304. doi: 10.1073/pnas.95.13.7299
109. Butan C, Winkler DC, Heymann JB, Craven RC, Steven AC. RSV Capsid Polymorphism Correlates With Polymerization Efficiency and Envelope Glycoprotein Content: Implications That Nucleation Controls Morphogenesis. J Mol Biol (2008) 376(4):1168–81. doi: 10.1016/j.jmb.2007.12.003
110. Effantin G, Estrozi LF, Aschman N, Renesto P, Stanke N, Lindemann D, et al. Cryo-Electron Microscopy Structure of the Native Prototype Foamy Virus Glycoprotein and Virus Architecture. PloS Pathog (2016) 12(7):e1005721. doi: 10.1371/journal.ppat.1005721
111. Cao S, Maldonado JO, Grigsby IF, Mansky LM, Zhang W. Analysis of Human T-Cell Leukemia Virus Type 1 Particles by Using Cryo-Electron Tomography. J Virol (2015) 89(4):2430–5. doi: 10.1128/JVI.02358-14
112. Meissner ME, Mendonca LM, Zhang W, Mansky LM. Polymorphic Nature of Human T-Cell Leukemia Virus Type 1 Particle Cores as Revealed Through Characterization of a Chronically Infected Cell Line. J Virol (2017) 91(16). doi: 10.1128/JVI.00369-17
113. Maldonado JO, Cao S, Zhang W, Mansky LM. Distinct Morphology of Human T-Cell Leukemia Virus Type 1-Like Particles. Viruses (2016) 8(5). doi: 10.3390/v8050132
114. Mattei S, Glass B, Hagen WJ, Krausslich HG, Briggs JA. The Structure and Flexibility of Conical HIV-1 Capsids Determined Within Intact Virions. Science (2016) 354(6318):1434–7. doi: 10.1126/science.aah4972
115. Schur FK, Hagen WJ, Rumlova M, Ruml T, Muller B, Krausslich HG, et al. Structure of the Immature HIV-1 Capsid in Intact Virus Particles at 8.8 A Resolution. Nature (2015) 517(7535):505–8. doi: 10.1038/nature13838
116. Ni T, Zhu Y, Yang Z, Xu C, Chaban Y, Nesterova T, et al. Structure of Native HIV-1 Cores and Their Interactions With IP6 and CypA. Sci Adv (2021) 7(47):eabj5715. doi: 10.1126/sciadv.abj5715
117. Mattei S, Tan A, Glass B, Muller B, Krausslich HG, Briggs JAG. High-Resolution Structures of HIV-1 Gag Cleavage Mutants Determine Structural Switch for Virus Maturation. Proc Natl Acad Sci USA (2018) 115(40):E9401–10. doi: 10.1073/pnas.1811237115
118. Schur FK, Obr M, Hagen WJ, Wan W, Jakobi AJ, Kirkpatrick JM, et al. An Atomic Model of HIV-1 Capsid-SP1 Reveals Structures Regulating Assembly and Maturation. Science (2016) 353(6298):506–8. doi: 10.1126/science.aaf9620
119. Mendonca L, Sun D, Ning J, Liu J, Kotecha A, Olek M, et al. CryoET Structures of Immature HIV Gag Reveal Six-Helix Bundle. Commun Biol (2021) 4(1):481. doi: 10.1038/s42003-021-01999-1
120. Li S, Hill CP, Sundquist WI, Finch JT. Image Reconstructions of Helical Assemblies of the HIV-1 CA Protein. Nature (2000) 407(6802):409–13. doi: 10.1038/35030177
121. Khorasanizadeh S, Campos-Olivas R, Clark CA, Summers MF. Sequence-Specific 1H, 13C and 15N Chemical Shift Assignment and Secondary Structure of the HTLV-I Capsid Protein. J Biomol NMR (1999) 14(2):199–200. doi: 10.1023/A:1008307507462
122. Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. Assembly Properties of the Human Immunodeficiency Virus Type 1 CA Protein. J Virol (2004) 78(5):2545–52. doi: 10.1128/JVI.78.5.2545-2552.2004
123. von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. Functional Surfaces of the Human Immunodeficiency Virus Type 1 Capsid Protein. J Virol (2003) 77(9):5439–50. doi: 10.1128/JVI.77.9.5439-5450.2003
124. Yang H, Talledge N, Arndt WG, Zhang W, Mansky LM. Human Immunodeficiency Virus Type 2 Capsid Protein Mutagenesis Defines the Determinants for Gag-Gag Interactions. BioRxiv (2022). doi: 10.1101/2022.01.31.478542
125. Talledge N, Yang H, Shi K, Yu G, Arndt WG, Mendonca LM, et al. HIV-2 Immature Particle Morphology Provides Insights Into Gag Lattice Stability and Virus Maturation. BioRxiv (2022). doi: 10.1101/2022.02.01.478508
126. Rankovic S, Deshpande A, Harel S, Aiken C, Rousso I. HIV-1 Uncoating Occurs via a Series of Rapid Biomechanical Changes in the Core Related to Individual Stages of Reverse Transcription. J Virol (2021). doi: 10.1101/2021.01.16.426924
127. Christensen DE, Ganser-Pornillos BK, Johnson JS, Pornillos O, Sundquist WI. Reconstitution and Visualization of HIV-1 Capsid-Dependent Replication and Integration In Vitro. Science (2020) 370(6513). doi: 10.1126/science.abc8420
128. Korber B, Gaschen B, Yusim K, Thakallapally R, Kesmir C, Detours V. Evolutionary and Immunological Implications of Contemporary HIV-1 Variation. Br Med Bull (2001) 58:19–42. doi: 10.1093/bmb/58.1.19
129. Taylor BS, Sobieszczyk ME, McCutchan FE, Hammer SM. The Challenge of HIV-1 Subtype Diversity. N Engl J Med (2008) 358(15):1590–602. doi: 10.1056/NEJMra0706737
130. Hemelaar J, Gouws E, Ghys PD, Osmanov S. Global and Regional Distribution of HIV-1 Genetic Subtypes and Recombinants in 2004. AIDS (2006) 20(16):W13–23. doi: 10.1097/01.aids.0000247564.73009.bc
131. Coffin J, Swanstrom R. HIV Pathogenesis: Dynamics and Genetics of Viral Populations and Infected Cells. Cold Spring Harb Perspect Med (2013) 3(1):a012526. doi: 10.1101/cshperspect.a012526
132. Tancredi MV, Waldman EA. Predictors of Progression to AIDS After HIV Infection Diagnosis in the Pre- and Post-HAART Eras in a Brazilian AIDS-Free Cohort. Trans R Soc Trop Med Hyg (2014) 108(7):408–14. doi: 10.1093/trstmh/tru078
133. Jiang H, Xie N, Cao B, Tan L, Fan Y, Zhang F, et al. Determinants of Progression to AIDS and Death Following HIV Diagnosis: A Retrospective Cohort Study in Wuhan, China. PloS One (2013) 8(12):e83078. doi: 10.1371/journal.pone.0083078
134. Mega ER. Alarming Surge in Drug-Resistant HIV Uncovered. Nature (2019). doi: 10.1038/d41586-019-02316-x
135. Gao F, Yue L, Robertson DL, Hill SC, Hui H, Biggar RJ, et al. Genetic Diversity of Human Immunodeficiency Virus Type 2: Evidence for Distinct Sequence Subtypes With Differences in Virus Biology. J Virol (1994) 68(11):7433–47. doi: 10.1128/jvi.68.11.7433-7447.1994
136. MacNeil A, Sankale JL, Meloni ST, Sarr AD, Mboup S, Kanki P. Long-Term Intrapatient Viral Evolution During HIV-2 Infection. J Infect Dis (2007) 195(5):726–33. doi: 10.1086/511308
137. Skar H, Borrego P, Wallstrom TC, Mild M, Marcelino JM, Barroso H, et al. HIV-2 Genetic Evolution in Patients With Advanced Disease is Faster Than That in Matched HIV-1 Patients. J Virol (2010) 84(14):7412–5. doi: 10.1128/JVI.02548-09
138. de Silva TI, Leligdowicz A, Carlson J, Garcia-Knight M, Onyango C, Miller N, et al. HLA-Associated Polymorphisms in the HIV-2 Capsid Highlight Key Differences Between HIV-1 and HIV-2 Immune Adaptation. AIDS (2018) 32(6):709–14. doi: 10.1097/QAD.0000000000001753
139. Rocha C, Calado R, Borrego P, Marcelino JM, Bartolo I, Rosado L, et al. Evolution of the Human Immunodeficiency Virus Type 2 Envelope in the First Years of Infection is Associated With the Dynamics of the Neutralizing Antibody Response. Retrovirology (2013) 10:110. doi: 10.1186/1742-4690-10-110
140. Lenzi GM, Domaoal RA, Kim DH, Schinazi RF, Kim B. Mechanistic and Kinetic Differences Between Reverse Transcriptases of Vpx Coding and Non-Coding Lentiviruses. J Biol Chem (2015) 290(50):30078–86. doi: 10.1074/jbc.M115.691576
141. Alvarez M, Sebastian-Martin A, Garcia-Marquina G, Menendez-Arias L. Fidelity of Classwide-Resistant HIV-2 Reverse Transcriptase and Differential Contribution of K65R to the Accuracy of HIV-1 and HIV-2 Reverse Transcriptases. Sci Rep (2017) 7:44834. doi: 10.1038/srep44834
142. Rawson JM, Gohl DM, Reilly CS, Mansky LM. HIV-1 and HIV-2 Exhibit Similar Mutation Frequencies and Spectra in the Absence of G-To-A Hypermutation. Retrovirology (2015) 12:60. doi: 10.1186/s12977-015-0180-6
143. Rawson JMO, Gohl DM, Landman SR, Roth ME, Meissner ME, Peterson TS, et al. Single-Strand Consensus Sequencing Reveals That HIV Type But Not Subtype Significantly Impacts Viral Mutation Frequencies and Spectra. J Mol Biol (2017). doi: 10.1016/j.jmb.2017.05.010
144. Nozuma S, Matsuura E, Kodama D, Tashiro Y, Matsuzaki T, Kubota R, et al. Effects of Host Restriction Factors and the HTLV-1 Subtype on Susceptibility to HTLV-1-Associated Myelopathy/Tropical Spastic Paraparesis. Retrovirology (2017) 14(1):26. doi: 10.1186/s12977-017-0350-9
145. Sherman MP, Saksena NK, Dube DK, Yanagihara R, Poiesz BJ. Evolutionary Insights on the Origin of Human T-Cell Lymphoma/Leukemia Virus Type I (HTLV-I) Derived From Sequence Analysis of a New HTLV-I Variant From Papua New Guinea. J Virol (1992) 66(4):2556–63. doi: 10.1128/jvi.66.4.2556-2563.1992
146. Gessain A, Mahieux R, de The G. Genetic Variability and Molecular Epidemiology of Human and Simian T Cell Leukemia/Lymphoma Virus Type I. J Acquir Immune Defic Syndr Hum Retrovirol (1996) 13 Suppl 1:S132–145. doi: 10.1097/00042560-199600001-00022
147 . Mansky LM. In Vivo Analysis of Human T-Cell Leukemia Virus Type 1 Reverse Transcription Accuracy. J Virol (2000) 74(20):9525–31. doi: 10.1128/JVI.74.20.9525-9531.2000
148. Menendez-Arias L. Mutation Rates and Intrinsic Fidelity of Retroviral Reverse Transcriptases. Viruses (2009) 1(3):1137–65. doi: 10.3390/v1031137
149. Nobre AFS, Almeida DS, Ferreira LC, Ferreira DL, Junior ECS, Viana M, et al. Low Genetic Diversity of the Human T-Cell Lymphotropic Virus (HTLV-1) in an Endemic Area of the Brazilian Amazon Basin. PloS One (2018) 13(3):e0194184. doi: 10.1371/journal.pone.0194184
150. Patino-Galindo JA, Gonzalez-Candelas F. The Substitution Rate of HIV-1 Subtypes: A Genomic Approach. Virus Evol (2017) 3(2):vex029. doi: 10.1093/ve/vex029
151. Mansky LM, Temin HM. Lower In Vivo Mutation Rate of Human Immunodeficiency Virus Type 1 Than That Predicted From the Fidelity of Purified Reverse Transcriptase. J Virol (1995) 69(8):5087–94. doi: 10.1128/jvi.69.8.5087-5094.1995
152. Mansky LM. Forward Mutation Rate of Human Immunodeficiency Virus Type 1 in a T Lymphoid Cell Line. AIDS Res Hum Retroviruses (1996) 12(4):307–14. doi: 10.1089/aid.1996.12.307
153. Kim T, Mudry RA Jr, Rexrode CA 2nd, Pathak VK. Retroviral Mutation Rates and A-To-G Hypermutations During Different Stages of Retroviral Replication. J Virol (1996) 70(11):7594–602. doi: 10.1128/jvi.70.11.7594-7602.1996
154. Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. Nature, Position, and Frequency of Mutations Made in a Single Cycle of HIV-1 Replication. J Virol (2010) 84(19):9864–78. doi: 10.1128/JVI.00915-10
155. Cuevas JM, Geller R, Garijo R, Lopez-Aldeguer J, Sanjuan R. Extremely High Mutation Rate of HIV-1 In Vivo. PloS Biol (2015) 13(9):e1002251. doi: 10.1371/journal.pbio.1002251
156. Sharp PM, Hahn BH. The Evolution of HIV-1 and the Origin of AIDS. Philos Trans R Soc Lond B Biol Sci (2010) 365(1552):2487–94. doi: 10.1098/rstb.2010.0031
157. Mansky LM, Cunningham KS. Virus Mutators and Antimutators: Roles in Evolution, Pathogenesis and Emergence. Trends Genet (2000) 16(11):512–7. doi: 10.1016/S0168-9525(00)02125-9
158. Delviks-Frankenberry KA, Nikolaitchik OA, Burdick RC, Gorelick RJ, Keele BF, Hu WS. Minimal Contribution of APOBEC3-Induced G-To-A Hypermutation to HIV-1 Recombination and Genetic Variation. PloS Pathog (2016) 12(5):e1005646. doi: 10.1371/journal.ppat.1005646
160. Mansky LM, Bernard LC. 3’-Azido-3’-Deoxythymidine (AZT) and AZT-Resistant Reverse Transcriptase can Increase the In Vivo Mutation Rate of Human Immunodeficiency Virus Type 1. J Virol (2000) 74(20):9532–9. doi: 10.1128/JVI.74.20.9532-9539.2000
161. Brenner BG, Coutsinos D. The K65R Mutation in HIV-1 Reverse Transcriptase: Genetic Barriers, Resistance Profile and Clinical Implications. HIV Ther (2009) 3(6):583–94. doi: 10.2217/hiv.09.40
162. Domingo E, Sheldon J, Perales C. Viral Quasispecies Evolution. Microbiol Mol Biol Rev (2012) 76(2):159–216. doi: 10.1128/MMBR.05023-11
163. Dapp MJ, Patterson SE, Mansky LM. Back to the Future: Revisiting HIV-1 Lethal Mutagenesis. Trends Microbiol (2013) 21(2):56–62. doi: 10.1016/j.tim.2012.10.006
164. Domingo E, Perales C. Viral Quasispecies. PloS Genet (2019) 15(10):e1008271. doi: 10.1371/journal.pgen.1008271
165. Rawson JM, Daly MB, Xie J, Clouser CL, Landman SR, Reilly CS, et al. 5-Azacytidine Enhances the Mutagenesis of HIV-1 by Reduction to 5-Aza-2’-Deoxycytidine. Antimicrob Agents Chemother (2016) 60(4):2318–25. doi: 10.1128/AAC.03084-15
166. Pathak VK, Temin HM. Broad Spectrum of In Vivo Forward Mutations, Hypermutations, and Mutational Hotspots in a Retroviral Shuttle Vector After a Single Replication Cycle: Substitutions, Frameshifts, and Hypermutations. Proc Natl Acad Sci USA (1990) 87(16):6019–23. doi: 10.1073/pnas.87.16.6019
167. Julias JG, Pathak VK. Deoxyribonucleoside Triphosphate Pool Imbalances In Vivo are Associated With an Increased Retroviral Mutation Rate. J Virol (1998) 72(10):7941–9. doi: 10.1128/JVI.72.10.7941-7949.1998
168. Cromer D, Schlub TE, Smyth RP, Grimm AJ, Chopra A, Mallal S, et al. HIV-1 Mutation and Recombination Rates Are Different in Macrophages and T-Cells. Viruses (2016) 8(4):118. doi: 10.3390/v8040118
169. Kennedy EM, Amie SM, Bambara RA, Kim B. Frequent Incorporation of Ribonucleotides During HIV-1 Reverse Transcription and Their Attenuated Repair in Macrophages. J Biol Chem (2012) 287(17):14280–8. doi: 10.1074/jbc.M112.348482
170. Shepard C, Xu J, Holler J, Kim DH, Mansky LM, Schinazi RF, et al. Effect of Induced dNTP Pool Imbalance on HIV-1 Reverse Transcription in Macrophages. Retrovirology (2019) 16(1):29. doi: 10.1186/s12977-019-0491-0
171. Oo A, Kim DH, Schinazi RF, Kim B. Viral Protein X Reduces the Incorporation of Mutagenic Noncanonical rNTPs During Lentivirus Reverse Transcription in Macrophages. J Biol Chem (2020) 295(2):657–66. doi: 10.1074/jbc.RA119.011466
172. St Gelais C, Wu L. SAMHD1: A New Insight Into HIV-1 Restriction in Myeloid Cells. Retrovirology (2011) 8:55. doi: 10.1186/1742-4690-8-55
173. Hollenbaugh JA, Tao S, Lenzi GM, Ryu S, Kim DH, Diaz-Griffero F, et al. dNTP Pool Modulation Dynamics by SAMHD1 Protein in Monocyte-Derived Macrophages. Retrovirology (2014) 11:63. doi: 10.1186/s12977-014-0063-2
174. Rawson JM, Roth ME, Xie J, Daly MB, Clouser CL, Landman SR, et al. Synergistic Reduction of HIV-1 Infectivity by 5-Azacytidine and Inhibitors of Ribonucleotide Reductase. Bioorg Med Chem (2016) 24(11):2410–22. doi: 10.1016/j.bmc.2016.03.052
175. Rawson JM, Roth ME, Xie J, Daly MB, Clouser CL, Landman SR, et al. 5,6-Dihydro-5-Aza-2’-Deoxycytidine Potentiates the Anti-HIV-1 Activity of Ribonucleotide Reductase Inhibitors. Bioorg Med Chem (2013) 21(22):7222–8. doi: 10.1016/j.bmc.2013.08.023
176. Onafuwa-Nuga A, Telesnitsky A. The Remarkable Frequency of Human Immunodeficiency Virus Type 1 Genetic Recombination. Microbiol Mol Biol Rev (2009) 73(3):451–80. doi: 10.1128/MMBR.00012-09
177. Schlub TE, Grimm AJ, Smyth RP, Cromer D, Chopra A, Mallal S, et al. Fifteen to Twenty Percent of HIV Substitution Mutations are Associated With Recombination. J Virol (2014) 88(7):3837–49. doi: 10.1128/JVI.03136-13
178. Bbosa N, Kaleebu P, Ssemwanga D. HIV Subtype Diversity Worldwide. Curr Opin HIV AIDS (2019) 14(3):153–60. doi: 10.1097/COH.0000000000000534
179. LANL HIV Sequence Database. HIV Circulating Recombinant Forms (CRFs) . Available at: https://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html.
180. Ibe S, Yokomaku Y, Shiino T, Tanaka R, Hattori J, Fujisaki S, et al. HIV-2 CRF01_AB: First Circulating Recombinant Form of HIV-2. J Acquir Immune Defic Syndr (2010) 54(3):241–7. doi: 10.1097/QAI.0b013e3181dc98c1
181. Desrames A, Cassar O, Gout O, Hermine O, Taylor GP, Afonso PV, et al. Northern African Strains of Human T-Lymphotropic Virus Type 1 Arose From a Recombination Event. J Virol (2014) 88(17):9782–8. doi: 10.1128/JVI.01591-14
182. Kuwata T, Miyazaki Y, Igarashi T, Takehisa J, Hayami M. The Rapid Spread of Recombinants During a Natural In Vitro Infection With Two Human Immunodeficiency Virus Type 1 Strains. J Virol (1997) 71(9):7088–91. doi: 10.1128/jvi.71.9.7088-7091.1997
183. Rawson JMO, Nikolaitchik OA, Keele BF, Pathak VK, Hu WS. Recombination is Required for Efficient HIV-1 Replication and the Maintenance of Viral Genome Integrity. Nucleic Acids Res (2018) 46(20):10535–45. doi: 10.1093/nar/gky910
184. An W, Telesnitsky A. Effects of Varying Sequence Similarity on the Frequency of Repeat Deletion During Reverse Transcription of a Human Immunodeficiency Virus Type 1 Vector. J Virol (2002) 76(15):7897–902. doi: 10.1128/JVI.76.15.7897-7902.2002
185. Motomura K, Chen J, Hu W. Genetic Recombination Between Human Immunodeficiency Virus Type 1 (HIV-1) and HIV-2, Two Distinct Human Lentiviruses. J Virol (2008) 82(4):1923–33. doi: 10.1128/JVI.01937-07
186. Simon-Loriere E, Martin DP, Weeks KM, Negroni M. RNA Structures Facilitate Recombination-Mediated Gene Swapping in HIV-1. J Virol (2010) 84(24):12675–82. doi: 10.1128/JVI.01302-10
187. Nguyen LA, Kim DH, Daly MB, Allan KC, Kim B. Host SAMHD1 Protein Promotes HIV-1 Recombination in Macrophages. J Biol Chem (2014) 289(5):2489–96. doi: 10.1074/jbc.C113.522326
188. Harris RS, Dudley JP. APOBECs and Virus Restriction. Virology (2015) 479-480:131–45. doi: 10.1016/j.virol.2015.03.012
189. McDaniel YZ, Wang D, Love RP, Adolph MB, Mohammadzadeh N, Chelico L, et al. Deamination Hotspots Among APOBEC3 Family Members are Defined by Both Target Site Sequence Context and ssDNA Secondary Structure. Nucleic Acids Res (2020) 48(3):1353–71. doi: 10.1093/nar/gkz1164
190. Okada A, Iwatani Y. APOBEC3G-Mediated G-To-A Hypermutation of the HIV-1 Genome: The Missing Link in Antiviral Molecular Mechanisms. Front Microbiol (2016) 7:2027. doi: 10.3389/fmicb.2016.02027
191. Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, et al. Human and Rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H Demonstrate a Conserved Capacity to Restrict Vif-Deficient HIV-1. J Virol (2011) 85(21):11220–34. doi: 10.1128/JVI.05238-11
192. Chaipan C, Smith JL, Hu WS, Pathak VK. APOBEC3G Restricts HIV-1 to a Greater Extent Than APOBEC3F and APOBEC3DE in Human Primary CD4+ T Cells and Macrophages. J Virol (2013) 87(1):444–53. doi: 10.1128/JVI.00676-12
193. Ooms M, Brayton B, Letko M, Maio SM, Pilcher CD, Hecht FM, et al. HIV-1 Vif Adaptation to Human APOBEC3H Haplotypes. Cell Host Microbe (2013) 14(4):411–21. doi: 10.1016/j.chom.2013.09.006
194. Refsland EW, Hultquist JF, Luengas EM, Ikeda T, Shaban NM, Law EK, et al. Natural Polymorphisms in Human APOBEC3H and HIV-1 Vif Combine in Primary T Lymphocytes to Affect Viral G-To-A Mutation Levels and Infectivity. PloS Genet (2014) 10(11):e1004761. doi: 10.1371/journal.pgen.1004761
195. Krisko JF, Begum N, Baker CE, Foster JL, Garcia JV. APOBEC3G and APOBEC3F Act in Concert To Extinguish HIV-1 Replication. J Virol (2016) 90(9):4681–95. doi: 10.1128/JVI.03275-15
196. Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, et al. Single-Strand Specificity of APOBEC3G Accounts for Minus-Strand Deamination of the HIV Genome. Nat Struct Mol Biol (2004) 11(5):435–42. doi: 10.1038/nsmb758
197. Chelico L, Pham P, Calabrese P, Goodman MF. APOBEC3G DNA Deaminase Acts Processively 3’ –> 5’ on Single-Stranded DNA. Nat Struct Mol Biol (2006) 13(5):392–9. doi: 10.1038/nsmb1086
198. Hache G, Liddament MT, Harris RS. The Retroviral Hypermutation Specificity of APOBEC3F and APOBEC3G is Governed by the C-Terminal DNA Cytosine Deaminase Domain. J Biol Chem (2005) 280(12):10920–4. doi: 10.1074/jbc.M500382200
199. Rathore A, Carpenter MA, Demir O, Ikeda T, Li M, Shaban NM, et al. The Local Dinucleotide Preference of APOBEC3G can be Altered From 5’-CC to 5’-TC by a Single Amino Acid Substitution. J Mol Biol (2013) 425(22):4442–54. doi: 10.1016/j.jmb.2013.07.040
200. Holtz CM, Sadler HA, Mansky LM. APOBEC3G Cytosine Deamination Hotspots are Defined by Both Sequence Context and Single-Stranded DNA Secondary Structure. Nucleic Acids Res (2013) 41(12):6139–48. doi: 10.1093/nar/gkt246
201. Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, et al. Induction of APOBEC3G Ubiquitination and Degradation by an HIV-1 Vif-Cul5-SCF Complex. Science (2003) 302(5647):1056–60. doi: 10.1126/science.1089591
202. Shirakawa K, Takaori-Kondo A, Kobayashi M, Tomonaga M, Izumi T, Fukunaga K, et al. Ubiquitination of APOBEC3 Proteins by the Vif-Cullin5-ElonginB-ElonginC Complex. Virology (2006) 344(2):263–6. doi: 10.1016/j.virol.2005.10.028
203. Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, et al. Vif Hijacks CBF-Beta to Degrade APOBEC3G and Promote HIV-1 Infection. Nature (2011) 481(7381):371–5.
204. Zhang W, Du J, Evans SL, Yu Y, Yu XF. T-Cell Differentiation Factor CBF-Beta Regulates HIV-1 Vif-Mediated Evasion of Host Restriction. Nature (2011) 481(7381):376–9.
205. Guo Y, Dong L, Qiu X, Wang Y, Zhang B, Liu H, et al. Structural Basis for Hijacking CBF-Beta and CUL5 E3 Ligase Complex by HIV-1 Vif. Nature (2014) 505(7482):229–33. doi: 10.1038/nature12884
206. Sadler HA, Stenglein MD, Harris RS, Mansky LM. APOBEC3G Contributes to HIV-1 Variation Through Sublethal Mutagenesis. J Virol (2010) 84(14):7396–404. doi: 10.1128/JVI.00056-10
207. Meissner ME, Julik EJ, Badalamenti JP, Arndt WG, Mills LJ, Mansky LM. Development of a User-Friendly Pipeline for Mutational Analyses of HIV Using Ultra-Accurate Maximum-Depth Sequencing. Viruses (2021) 13(7). doi: 10.3390/v13071338
208. Sato K, Takeuchi JS, Misawa N, Izumi T, Kobayashi T, Kimura Y, et al. APOBEC3D and APOBEC3F Potently Promote HIV-1 Diversification and Evolution in Humanized Mouse Model. PloS Pathog (2014) 10(10):e1004453. doi: 10.1371/journal.ppat.1004453
209. Alteri C, Surdo M, Bellocchi MC, Saccomandi P, Continenza F, Armenia D, et al. Incomplete APOBEC3G/F Neutralization by HIV-1 Vif Mutants Facilitates the Genetic Evolution From CCR5 to CXCR4 Usage. Antimicrob Agents Chemother (2015) 59(8):4870–81. doi: 10.1128/AAC.00137-15
210. Monajemi M, Woodworth CF, Zipperlen K, Gallant M, Grant MD, Larijani M. Positioning of APOBEC3G/F Mutational Hotspots in the Human Immunodeficiency Virus Genome Favors Reduced Recognition by CD8+ T Cells. PloS One (2014) 9(4):e93428. doi: 10.1371/journal.pone.0093428
211. Squires KD, Monajemi M, Woodworth CF, Grant MD, Larijani M. Impact of APOBEC Mutations on CD8+ T Cell Recognition of HIV Epitopes Varies Depending on the Restricting HLA. J Acquir Immune Defic Syndr (2015) 70(2):172–8. doi: 10.1097/QAI.0000000000000689
212. Mulder LC, Harari A, Simon V. Cytidine Deamination Induced HIV-1 Drug Resistance. Proc Natl Acad Sci USA (2008) 105(14):5501–6. doi: 10.1073/pnas.0710190105
213. Kim EY, Bhattacharya T, Kunstman K, Swantek P, Koning FA, Malim MH, et al. Human APOBEC3G-Mediated Editing can Promote HIV-1 Sequence Diversification and Accelerate Adaptation to Selective Pressure. J Virol (2010) 84(19):10402–5. doi: 10.1128/JVI.01223-10
214. Neogi U, Shet A, Sahoo PN, Bontell I, Ekstrand ML, Banerjea AC, et al. Human APOBEC3G-Mediated Hypermutation is Associated With Antiretroviral Therapy Failure in HIV-1 Subtype C-Infected Individuals. J Int AIDS Soc (2013) 16:18472. doi: 10.7448/IAS.16.1.18472
215. Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in Coreceptor Use Correlates With Disease Progression in HIV-1–Infected Individuals. J Exp Med (1997) 185(4):621–8. doi: 10.1084/jem.185.4.621
216. Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A Second Human Antiretroviral Factor, APOBEC3F, is Suppressed by the HIV-1 and HIV-2 Vif Proteins. EMBO J (2004) 23(12):2451–8. doi: 10.1038/sj.emboj.7600246
217. Ribeiro AC, Maia e Silva A, Santa-Marta M, Pombo A, Moniz-Pereira J, Goncalves J, et al. Functional Analysis of Vif Protein Shows Less Restriction of Human Immunodeficiency Virus Type 2 by APOBEC3G. J Virol (2005) 79(2):823–33. doi: 10.1128/JVI.79.2.823-833.2005
218. Smith JL, Izumi T, Borbet TC, Hagedorn AN, Pathak VK. HIV-1 and HIV-2 Vif Interact With Human APOBEC3 Proteins Using Completely Different Determinants. J Virol (2014) 88(17):9893–908. doi: 10.1128/JVI.01318-14
219. Meissner ME, Willkomm NA, Lucas J, Arndt WG, Aitken SF, Julik EJ, et al. Differential Activity of APOBEC3F, APOBEC3G, and APOBEC3H in the Restriction of HIV-2. J Mol Biol (2021), 167355.
220. Ooms M, Krikoni A, Kress AK, Simon V, Munk C. APOBEC3A, APOBEC3B, and APOBEC3H Haplotype 2 Restrict Human T-Lymphotropic Virus Type 1. J Virol (2012) 86(11):6097–108. doi: 10.1128/JVI.06570-11
221. Sasada A, Takaori-Kondo A, Shirakawa K, Kobayashi M, Abudu A, Hishizawa M, et al. APOBEC3G Targets Human T-Cell Leukemia Virus Type 1. Retrovirology (2005) 2:32. doi: 10.1186/1742-4690-2-32
222. Mahieux R, Suspene R, Delebecque F, Henry M, Schwartz O, Wain-Hobson S, et al. Extensive Editing of a Small Fraction of Human T-Cell Leukemia Virus Type 1 Genomes by Four APOBEC3 Cytidine Deaminases. J Gen Virol (2005) 86(Pt 9):2489–94. doi: 10.1099/vir.0.80973-0
223. Poulain F, Lejeune N, Willemart K, Gillet NA. Footprint of the Host Restriction Factors APOBEC3 on the Genome of Human Viruses. PloS Pathog (2020) 16(8):e1008718. doi: 10.1371/journal.ppat.1008718
224. Kimata JT, Wong FH, Wang JJ, Ratner L. Construction and Characterization of Infectious Human T-Cell Leukemia Virus Type 1 Molecular Clones. Virology (1994) 204(2):656–64. doi: 10.1006/viro.1994.1581
225. Derse D, Hill SA, Lloyd PA, Chung H, Morse BA. Examining Human T-Lymphotropic Virus Type 1 Infection and Replication by Cell-Free Infection With Recombinant Virus Vectors. J Virol (2001) 75(18):8461–8. doi: 10.1128/JVI.75.18.8461-8468.2001
226. Ohsugi T, Kumasaka T, Ohsugi T. Construction of a Full-Length Human T Cell Leukemia Virus Type I Genome From MT-2 Cells Containing Multiple Defective Proviruses Using Overlapping Polymerase Chain Reaction. Anal Biochem (2004) 329(2):281–8. doi: 10.1016/j.ab.2004.02.036
227. Bass BL. RNA Editing by Adenosine Deaminases That Act on RNA. Annu Rev Biochem (2002) 71:817–46. doi: 10.1146/annurev.biochem.71.110601.135501
228. Nishikura K. Editor Meets Silencer: Crosstalk Between RNA Editing and RNA Interference. Nat Rev Mol Cell Biol (2006) 7(12):919–31. doi: 10.1038/nrm2061
229. Basilio C, Wahba AJ, Lengyel P, Speyer JF, Ochoa S. Synthetic Polynucleotides and the Amino Acid Code. V. Proc Natl Acad Sci USA (1962) 48:613–6.
230. Rueter SM, Dawson TR, Emeson RB. Regulation of Alternative Splicing by RNA Editing. Nature (1999) 399(6731):75–80. doi: 10.1038/19992
231. Steele EJ, Lindley RA, Wen J, Weiller GF. Computational Analyses Show A-To-G Mutations Correlate With Nascent mRNA Hairpins at Somatic Hypermutation Hotspots. DNA Repair (Amst) (2006) 5(11):1346–63. doi: 10.1016/j.dnarep.2006.06.002
232. Cattaneo R. Biased (A–>I) Hypermutation of Animal RNA Virus Genomes. Curr Opin Genet Dev (1994) 4(6):895–900. doi: 10.1016/0959-437X(94)90076-0
233. Weiden MD, Hoshino S, Levy DN, Li Y, Kumar R, Burke SA , et al. Adenosine Deaminase Acting on RNA-1 (ADAR1) Inhibits HIV-1 Replication in Human Alveolar Macrophages. PloS One (2014) 9(10):e108476. doi: 10.1371/journal.pone.0108476
234. Phuphuakrat A, Kraiwong R, Boonarkart C, Lauhakirti D, Lee TH, Auewarakul P. Double-Stranded RNA Adenosine Deaminases Enhance Expression of Human Immunodeficiency Virus Type 1 Proteins. J Virol (2008) 82(21):10864–72. doi: 10.1128/JVI.00238-08
235. Clerzius G, Gelinas JF, Daher A, Bonnet M, Meurs EF, Gatignol A. ADAR1 Interacts With PKR During Human Immunodeficiency Virus Infection of Lymphocytes and Contributes to Viral Replication. J Virol (2009) 83(19):10119–28. doi: 10.1128/JVI.02457-08
236. Doria M, Neri F, Gallo M, Farace MG, Michienzi A. Editing of HIV-1 RNA by the Double-Stranded RNA Deaminase ADAR1 Stimulates Viral Infection. Nucleic Acids Res (2009) 37(17):5848–58. doi: 10.1093/nar/gkp604
237. Cuadrado E, Booiman T, van Hamme JL, Jansen MH, van Dort KA, Vanderver A, et al. ADAR1 Facilitates HIV-1 Replication in Primary CD4+ T Cells. PloS One (2015) 10(12):e0143613. doi: 10.1371/journal.pone.0143613
238. Cachat A, Alais S, Chevalier SA, Journo C, Fusil F, Dutartre H, et al. ADAR1 Enhances HTLV-1 and HTLV-2 Replication Through Inhibition of PKR Activity. Retrovirology (2014) 11:93. doi: 10.1186/s12977-014-0093-9
239. Biswas N, Wang T, Ding M, Tumne A, Chen Y, Wang Q, et al. ADAR1 is a Novel Multi Targeted Anti-HIV-1 Cellular Protein. Virology (2012) 422(2):265–77. doi: 10.1016/j.virol.2011.10.024
240. DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents With HIV: HIV-2 Infection . Available at: https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/AdultARV_GL_HIV-2.pdf.
Keywords: retrovirus, molecular biology, diversification, human T-cell leukemia virus, human immunodeficiency virus
Citation: Meissner ME, Talledge N and Mansky LM (2022) Molecular Biology and Diversification of Human Retroviruses. Front.Virol. 2:872599. doi: 10.3389/fviro.2022.872599
Received: 09 February 2022; Accepted: 07 March 2022;
Published: 02 June 2022.
Edited by:
Akio Adachi, Kansai Medical University, JapanReviewed by:
Fatah Kashanchi, George Mason University, United StatesMikako Fujita, Kumamoto University, Japan
Eric O. Freed, National Cancer Institute at Frederick (NIH), United States
Copyright © 2022 Meissner, Talledge and Mansky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Louis M. Mansky, bWFuc2t5QHVtbi5lZHU=