
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Virol., 29 April 2022
Sec. Translational Virology
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.863202
This article is part of the Research TopicTranslational Virology in PregnancyView all 12 articles
 Sukanta Jash
Sukanta Jash Surendra Sharma*
Surendra Sharma*Recent reports have suggested a tight relationship between viral infections and neurodevelopmental disorders. In this regard, fetal brain damage can be caused by direct viral infection or induced immune responses and cytokine storm. Although recent years have seen phenomenal progress in diagnosing autism spectrum disorders (ASD) and identifying genetic and epigenetic causative factors contributing to this group of neurodevelopmental disorders, almost 60% cases in children remain of unknown etiology. Little is known about the collective pathophysiology of ASD. In this regard, epidemiological data suggest that viral infections during pregnancy are associated with high risk of having an autistic child. Although SARS-CoV-2 infections have been documented in pregnant women, we do not yet know whether COVID-19 pandemic will contribute to the onset of autism-like features in the offspring or impact autistic individuals. We hypothesize that ASD are programmed in the mother's womb and that uterine, not peripheral, immune activation is the initial trigger to induce fetal brain developmental anomalies. We further hypothesize that exposure to infections only during a temporal window of pregnancy impact the onset of ASD-like pathology, particularly in the male fetus/offspring. We will discuss the role of uterine regulatory T cells and their inflammatory trans-differentiation in the pathophysiology of ASD and comment on possible therapeutic intervention options.
Obstetric infections are particularly dangerous to both the mother and the developing fetus as they may orchestrate events that interfere with normal fetal developmental programs (1–4). The placenta is now considered a specialized immune organ, and in this regard, intrauterine infections may adversely affect the immune balance regulated by the placenta in concert with the maternal immune system (5, 6). Viruses have evolved contemporary ways to evade the immune system and to cause diseases (7). Local cytokine storm and functionally or proportionally altered immune cell profiles have been the main consequences of viral infections (8, 9). Other co-infections by bacteria or parasites may further compound these immune responses. In the case of fetal growth and survival, these infections will have enormous deleterious effects. The well-known congenitally acquired infections/pathogens that cause morbidity and mortality in newborns are called TORCH (Toxoplasma gondii, Others like Treponema pallidum, Rubella, Cytomegalovirus, Herpes Simplex Virus) infections. The TORCH group has been expanded to include Parvovirus B19, human immunodeficiency virus (HIV), Varicella zoster virus, Hepatitis C virus (HCV), Zika virus (ZIKV), and Plasmodium falciparum among others (5, 10–14). These maternal infections are passed either transplacentally or during the birth process. These pathogens may act independently or in concert to cause neonatal morbidities. More importantly, an array of literature now suggests an association between viral infections and poor fetal brain development and childhood diseases such as autism, schizophrenia, bipolar disorders, microcephaly and other serious brain disorders (14–16). As a matter of fact, several of these adverse neurobehavioral outcomes share the perinatal inflammation pathogenesis.
Autism spectrum disorders (ASDs) are characterized by symptoms such as early social impairment, repetitive behavior, communication challenges, and learning and speech impairments, among other social traits (17, 18). Children and adults with autism have impaired social cognitive ability and perception, common executive dysfunction, and delayed information processing. Genetic causes have a crucial role in the development of ASD-like disorders, but early exposure to environmental factors during brain development has been shown to significantly increase the severity of the disorder (19, 20). According to the World Health Organization (WHO), ASDs affect around 1 in every 160 children worldwide. However, in the United States, the prevalence is astonishingly high, at one in every 55 children. The male offspring show a prevalence of ASD that is four to five times higher than that of their female counterparts (21).
Since inflammation, cytokine imbalance, and viral neurotropism could have an impact on fetal brain development, the potential role of immunological dysregulation in autism has garnered particular attention (22, 23). Viruses may cause ASD by directly infecting the brain, by inducing local and/or systemic cytokine storm, or by altering maternal or offspring localized immune responses. Several prior studies have linked ASD to various viral infections (24, 25).
In this review, we will focus on the evidence that uterine infection/specialized inflammation during a narrow gestational window (second trimester) has deleterious effects on fetal neurodevelopment. How viruses can access cellular components of the placenta and the pathogenic mechanisms that facilitate the process of specialized inflammation at the maternal–fetal interface is a subject of intense discussion. We also discuss resistance mechanisms used by the fetus and maternal–fetal interface that protect viral infections and inhibit the onset of ASD-like diseases. We summarize the emerging body of evidence in both humans and animals that links viral infection with increased incidence of autism. We propose a mechanism of inflammatory transformation of a uterine immune cell type that could be the basis of the early onset of brain developmental defects associated with ASD.
The decidual lining of the uterus, which is high in leukocytes, is one of the first lines of defense for the mother and fetus at the maternal–fetal interface. The decidua is replete with effector T lymphocytes, regulatory T cells (Tregs), NK cells, innate lymphoid cells, and macrophages (26). As the trophoblast invades and establishes the placental vascular bed and interacts with decidual immune cells, a milieu of immune tolerance is established. In order to orchestrate immune-tolerance, decidual cells remain in close contact with invading extravillous trophoblasts (EVTs) during pregnancy, particularly first and second trimesters. Using cell-to-cell fusion and the production of interferons (IFNs), exosomes, and antimicrobial peptides, trophoblasts create a functional barrier that confers antiviral resistance (3, 27). Fetal macrophages, also known as Hofbauer cells, proliferate rapidly upon virus infection. Hofbauer cells are targets of many viruses, including CMV and ZIKV. It is not clear if Hofbauer cells act as a check on viral propagation or more as a reservoir for the virus itself (28, 29). However, despite the fact that decidual immune cells generally have a more anti-inflammatory profile than their blood-borne counterparts, there is evidence of inflammatory transformation of decidual natural killer cells (dNKs), macrophages, and T cells in response to viral and bacterial products (30, 31). When infected with the influenza RNA virus during pregnancy, pDC exhibited an increased response and type I IFN production (32, 33). This is in contrast to required physiological inflammation during embryo implantation. Post implantation, the maternal–fetal interface is dominated by an “anti-inflammatory” phenotype, which is associated with preponderance of regulatory NK cells, M2 macrophages, and regulatory T cells (Tregs) that are critical for fetal protection. Tregs appear to play a vital role in regulating inflammation in early pregnancy and developing a responsive uteroplacental environment through their potent anti-inflammatory regulation (34, 35). When encountered with pathogenic assault, Tregs may switch to Th17 phenotype and suffer from plasticity or acquire a dual phenotype of Treg-Th17 cells (36, 37). Depending on the pathogen and the gestational age, these cells may promote overlap (plasticity) with Th17 cells and reprogram their conventional role as suppressive T cells (37). Our unreported findings suggest that uterine immune activation during pregnancy may lead Tregs to trans-differentiate into Th17 cells, altering fetal brain development and causing an ASD-like behavioral phenotype in the male offspring (38) (Figure 1).
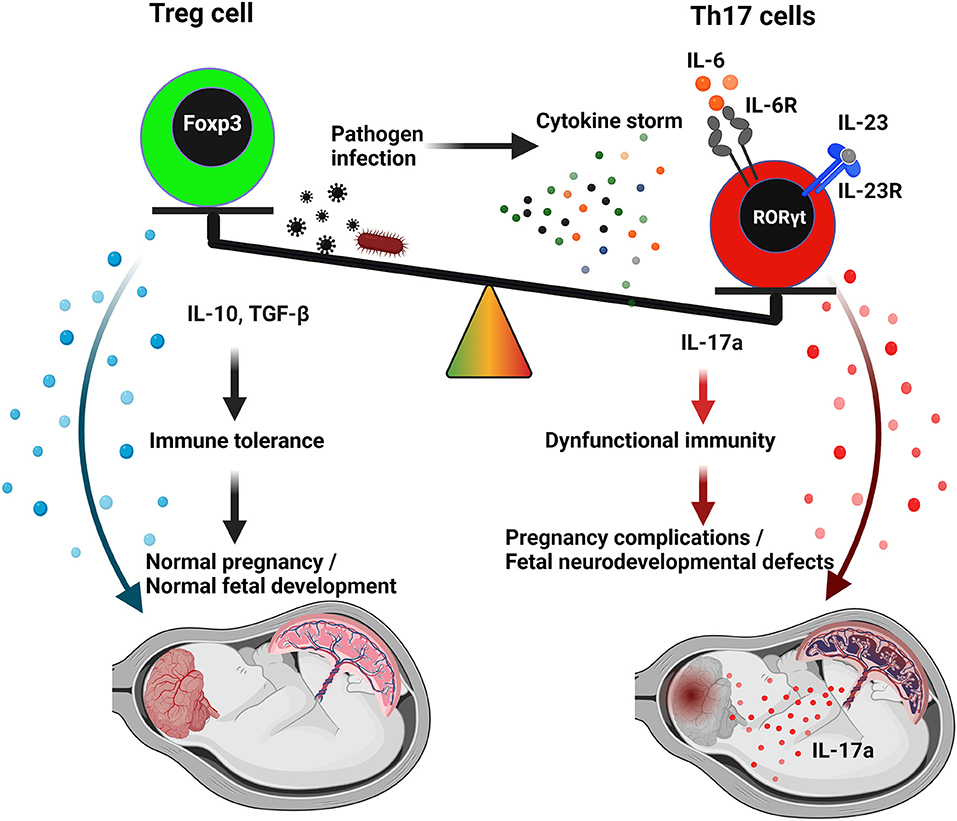
Figure 1. A proposed model for Th17/Treg imbalance and IL-17a in viral infection mediating autism spectrum disorders (ASD). Uncontrolled systemic or uterine inflammation during pregnancy by pathogenic infection might generate cytokine storm effect. The principal component of cytokine storm, IL-6, could modify the uterine cytokine microenvironment and facilitate alteration of the Th17/Treg imbalance in favor of the Th17-population. Th17 cells, in an exacerbated proinflammatory response, can promote ASD through IL-17a. In contrast, Treg cells maintain self-tolerance and maintain normal fetal development through an anti-inflammatory response (IL-10, TGFβ). Th17 cells and Tregs, despite their seemingly diametrically opposed functions, share comparable features and differentiation mechanisms. IL-17 released by Th17 cells traverses the placenta and could potentially reach the fetal brain in the presence of an incomplete blood–brain barrier.
The placenta is a temporary reproductive organ that allows the intermediation between the mother and the fetus during pregnancy. Because of its ability to modulate maternal immune responses, it is also called a potent immune organ. The syncytiotrophoblast (STB) forms and maintains the outer layer of villi by fusing the inner layer of proliferative progenitor cells known as villous cytotrophoblasts (CTBs) (39). CTBs also differentiate into invading trophoblasts primarily residing in the anchoring villi. These invading trophoblasts further differentiate into endovascular trophoblasts that are involved in spiral artery remodeling in the decidua. Invading extravillous trophoblast (EVTs) are involved in the cross-talk with decidual immune cells to regulate immune tolerance, angiogenesis, and fetal development (39, 40). Within the maternal–fetal contact, viruses can infect a variety of cell types and also traverse from one cell to another through caveolin-dependent endocytosis, macropinocytosis, or uptake processes, without the need for cellular receptors (41).
In the case of ZIKV, evidence of viral replication was found in proliferating villus and Hofbauer cells in the villous core. Through infected maternal blood macrophages, ZIKV or HIV can get into placental trophoblast cells and infect them (42, 43). Significant maternal viremia is required for placental infection to occur. By bathing terminal villi in maternal blood, the STB barrier serves as a common entry point for infectious agents to enter fetal blood and other organs later in the gestation. Viruses such as CMV, ZIKV, and SARS-CoV-2 may all infect the placenta directly by attaching to viral receptors on the maternal side of STBs, while other viruses can use antibody-dependent enhancement (ADE) to get through the STB barrier (44–46). Complex immune evasion approaches are employed by members of the HSV and CMV families. These viral infections may be curtailed by robust IFN responses and anti-inflammatory activities of hormones and cytokines. In normal pregnancy, the placenta is equipped to protect the mother and baby from low viremia cases. However, overwhelming viremia and compromised local immunity may lead to infections in the placenta. CMV, ZIKV, and other infections have been shown to maintain vigorous viral replication in the placenta (47, 48). The evasion mechanisms employed by the viruses often target components in the type I interferon pathway. Congenital infections such as ZIKV, HIV, CMV, SARS-CoV-2, and others have been shown to suppress this pathway (49–52). Consequently, proinflammatory cytokines are normally induced, leading to the downstream events of tissue inflammation and damage (49, 50). The ZIKV protein NS5 antagonizes type I interferon-mediated induction of RIG-I pathway to neutralize the placental defense. Similar to ZIKV, CMV utilizes its IE1 protein to weaken the host's natural defense mechanisms (53, 54). When it comes to mimicking congenital pathogens, CMV and ZIKV are particularly effective since they both target the host interferon response while producing widespread infections during early pregnancy when innate defense in the placenta is challenged by immune evading mechanisms (53, 54).
In the context of the new COVID-19 pandemic and placenta infection, SARS-CoV-2 has been screened in placental sections, amniotic fluid, and cord blood. SARS-CoV-2 was found in the STB and villous fibroblasts of a COVID-19 patient with severe disease using transmission electron microscopy (TEM) (55). In the case of SARS-CoV-2, the spike protein must attach to angiotensin-converting enzyme 2 (ACE2) in order for the virus to enter the cells. Molecular analysis has revealed the presence of ACE2 in various components of the placenta, including syncytiotrophoblasts, endothelium, and vascular smooth muscle (56). Transmembrane protease serine 2 (TMPRSS2) and molecules, such as cathepsin B/L7 and furin, are required for active viral infection. Researchers have discovered the substantial expression of ACE2 in the placenta by single-cell RNAseq, but not TMPRSS2 in the placenta (57, 58). Recent studies using single cell RNAseq during early gestation indicated that ACE2 was expressed in the placenta, but TMPRSS2 expression was either absent or extremely low (58). It is then possible that other proteases that may contribute to viral replication in the placenta remain to be identified. There are a number of interesting aspects of placentation that ACE2 is involved in, including trophoblast migration and maternal vasodilation (59, 60). Uterine arterial malfunction in pregnant mice lacking the ACE2 gene was linked to reduced umbilical blood flow and placental hypoxia. Adverse pregnancy outcomes such as miscarriage, ectopic pregnancy, and hypertension have also been linked to ACE2 (60). There is therefore the possibility of placental anomalies and pregnancy consequences if SARS-CoV-2 affects the expression of ACE2 in the placenta. The presence of ACE2 in the placenta suggests that SARS-CoV-2 may be able to attach to it, resulting in the initiation of viral infection. Another way for the virus to breach the placental barrier is for it to be carried by blood cells. However, there is controversy regarding whether SARS-CoV-2 efficiently infects CTBs and/or EVTs (61, 62).
Although there are case reports of vertical transmission, evidence, so far, suggests that SARSCoV-2 does not transmit vertically (63). SARS-CoV-2 has not been discovered in cord blood, throat and nasopharyngeal swabs, urine, or feces from many neonates screened for the virus at birth. SARS-CoV-2-negative amniotic fluid samples have also been collected from COVID-positive pregnancies (64, 65). These observations suggest that SARS-CoV-2-mediated effects on fetal development must be regulated by local cytokine storm and/or uterine immune activation.
Cytokine storm disorders are caused by a complex interwoven network of cells, signaling pathways, and cytokines. It is considered that cytokines, such as interferon-γ, interleukin-1, interleukin-6, tumor necrosis factor (TNF)-α, IL17, and interleukin-18, play critical roles in immunopathology when their levels are increased during a cytokine storm (8, 9). The microbiome, genetic traits, and underlying conditions all influence the cytokine patterns. Innate immune cells recognize and respond to a wide variety of microbes by releasing cytokines that activate cells of the adaptive immune system via pattern recognition receptors that are not antigen specific (8, 66). Cytokine storms frequently result in an inflammatory response that is more of a Th1 type. To protect the body from intracellular infections, effector T cells release significant amounts of interferon-γ that induces delayed hypersensitivity reactions and activate macrophages (8, 67). There is evidence that Th17 cells can be the driving force behind a cytokine storm that is not dependent on interferon-γ (68).
The deleterious cytokine storm has been reported in response to several viral infections, including influenza H5N1 virus, influenza H1N1 virus, and SARS-CoV-2. MERS-CoV infection was also reported to induce increased concentrations of proinflammatory cytokines, IFN-γ, TNF-α, IL15, and IL17. A significant number of COVID-19-associated deaths have been linked to acute cytokine storm (67). Plasma cytokine levels of 41 COVID-19 confirmed cases in China revealed elevated levels of an array of cytokines (68). A recent study with a large cohort of COVID-19 patients showed that serum IL-6 levels could be used to predict the patient's prognosis. These studies reported that serum IL-6 level was significantly high in mortality cases compared with recovery cases. Additional studies also confirmed the significance of plasma IL-6 as a measure of COVID-19 severity. Even in pediatric COVID-19 patients, ranging from 2 months to 15 years, significant increase in the levels of IL-6, IL-10, and IFN-γ was reported (67, 69).
Systemic “cytokine storm” can potentially cause secondary immune activation at the placental niche (70, 71). In animal studies, cytokines have been found to have a critical role in mediating the effects of uterine immune activation on the developing embryo. Pregnant women who have been infected with certain virus release proinflammatory cytokines at the maternal–fetal interface, amniotic fluid, and the fetal brain, which results in altered fetal behavioral outcomes. In animal models, IL-6, IL-17, TNF-α, and IL-1β have been identified as significant mediators of the fetal response to local inflammation, leading to ASD (72–74).
UIA-induced cytokine storm plays a central role in modulating the local immune system and possibly the brain immune responses. Cytokines act as messengers between the brain and the hypothalamus during infection, influencing the brain's response to fever and sickness (75). Increasing evidence points to the role of cytokines in higher-level brain processes, such as memory and cognition (76). Consequently, dysregulation of the immune system's cytokine signaling and/or regulation can result in a wide variety of neurological effects and complications. The importance of these interactions varies depending on when, how long, and how intense they are. For example, cytokines have varied effects on the developing and adult brains (76). For brain immune responses, neuronal stem cells (NSPCs) and immune cells communicate via cytokines, which can have both protective and harmful effects depending on the cytokine profile of each cell type (77, 78). The effects of cytokine and chemokine signaling on brain cell activity, proliferation, and survival are highly variable. Viral infections frequently change NSPCs, either directly through viral infection or indirectly through immune cell activity or cytokine/chemokine signaling (77). Multiple changes in the behavior of infected NSPCs can be induced by cytokines/chemokines that affect NSPC numbers, differentiation into other neural cells, migration to areas of injury, and eventually the development and repair of the human brain (79).
Individuals with autism are shown to have dysregulated interleukin-6 (IL-6). Children and adults with the disease have elevated amounts of IL-6 in their blood compared with healthy individuals. Postmortem brain tissues from children with ASD also show elevated levels of IL-6 (80, 81). According to immunohistochemistry examination of cerebellar sections, autistic postmortem brain specimens had considerably greater IL-6 staining (82). Autistic brain exhibits significantly higher IL-6 and its receptors in the brain (83, 84). In adults, circulating IL-6 from the peripheral tissues can pass the blood–brain barrier and affect a wide range of activities in the brain (85, 86). Multiple studies have shown that UIA triggers inflammatory response through proinflammatory cytokines in the embryonic brain. IL-6 mRNA and protein levels in the fetal brain have been found to increase in the wake of an UIA (87). This molecular interaction supports the feed-forward inflammatory cycle. The elevated expression of IL-6 has been seen in a number of central nervous system (CNS) illnesses (88), including those caused by HIV, CMV, and Zika viral infections (89–91). IL-6 induction appears to be responsible for many of the long-lasting behavioral changes seen in UIA-born offspring. The presence of IL-6 in the brain throughout neural development affects avoidance learning and causes autism-like behavior, whereas mice lacking IL-6 are more susceptible to infection and have impairments in fear conditioning (92, 93). Exposure to IL-6 in the womb alters the offspring's NSPC pools for the rest of their lives. In the fetus, maternal IL-6 treatment increases the number of cortical and forebrain neural precursors (94). Neuropathology, GABA dysregulation, and immune system alterations are all linked to IL-6 during the course of a child's life (95). Infection during pregnancy has the same effect. IL-6 has a wide range of effects on the developing brain. Neuronal self-renewal, migration, cell survival, and neurite outgrowth are all influenced by IL-6 and its family members (96–98). It is also possible that exposure to IL-6 during critical periods of pregnancy will change the synaptic networks of neurons in offspring. IL-6 overexpression causes a reduction in glutamate receptor expression in vitro and in vivo, as well as an increase in the ratio of excitatory to inhibitory synapses in the brain (99, 100). This is especially relevant in the context of autism, where a disproportionate excitatory-to-inhibitory neuronal ratio is thought to play a role in the development of the disorder. In this way, maternal IL-6 not only affects the pre-natal NSC pool but also has an effect on the post-natal NSPC pool. As a result of the changes in fetal NSPC activity, the SVZ pools of adult NSC display increased proliferation and neurogenesis (97, 98). Instead of increasing NSC proliferation and astrogliogenesis, when localized IL-6 was expressed from ZIKV infected microglia, there was a decrease in neurogenesis. This is in contrast to circulating maternal IL-6 (101). IL-6 has also been reported to have some protective effects on neuronal stem cells. There is a reduction in both the stem and immature neuron populations in vitro after infection with HSV1. When an active infection is present, however, IL-6 from microglia prevents these effects from occurring (102). It is possible that the setting of the inflammatory milieu, together with the presence of other cytokines, will have an impact on the effects of IL-6. However, subsequent human studies have cast doubt on the role of gestational IL-6 alone in the development of autism. Increased IL-6 levels in mid-pregnancy maternal serum and amniotic fluid were found to be linked with developmental problems but not autism in a longitudinal analysis.
Additionally, IL-6 plays a critical role in regulating the balance of proinflammatory Th17 cells and Tregs (103). Similar to IL-6, this upregulation of IL-17a in the pre-natal environment can also lead to abnormal fetal neurodevelopment and has been consistently associated with ASD (104). Furthermore, expression of IL-17RA in the brain dramatically increases during UIA. Cortical dysplasia is a prevalent characteristic in children with ASD (105, 106). The subtle pathogenic changes in the fetal brain that may occur in response to viral infections during pregnancy changes are depicted in Figure 2. This has been confirmed in mouse models. Interestingly, the architectural organization of neurons in the cortex was disrupted in embryonic mice injected IV with IL-17a at an early stage of development (E14.5). Indirect evidence supports the role of IL-17 in direct neuronal damage. Different neuronal populations express IL-17 receptor (104). IL-17 disrupts blood–brain barrier (BBB) tight junctions in vitro and in vivo and promotes CNS inflammation (107). IL-17 may contribute to CNS tissue damage by affecting the cells that express IL-17 receptor, such as microglia, endothelial cells, astrocytes, and neurons (108, 109). IL-17a was found to be sufficient to generate ASD-like traits in male offspring early and persistently. This is consistent with the male preponderance, and behavioral findings in ASD, which suggests that persistent maternal IL-17 contributes to the pathogenesis of ASD in offspring (104, 110). Persistent IL-17 during pregnancy may have lingering effects even during adulthood where it may cause defects in adult glia, inhibitory synapses, and behavior (111). It is also necessary to examine if maternal IL-17 are directly responsible for the cytokine responses reported in the fetal brain. Contrary to its embryonic effect on ASD development, IL-17 may have opposite effects during high fever scenario in adults (112, 113). This behavioral recovery was followed by a reduction in neuronal activity in the primary somatosensory cortex dysgranular zone (S1DZ). This finding tends to support the hypothesis that certain children with autism spectrum disorder (ASD) demonstrate behavioral gains when experiencing inflammation followed by fever (113).
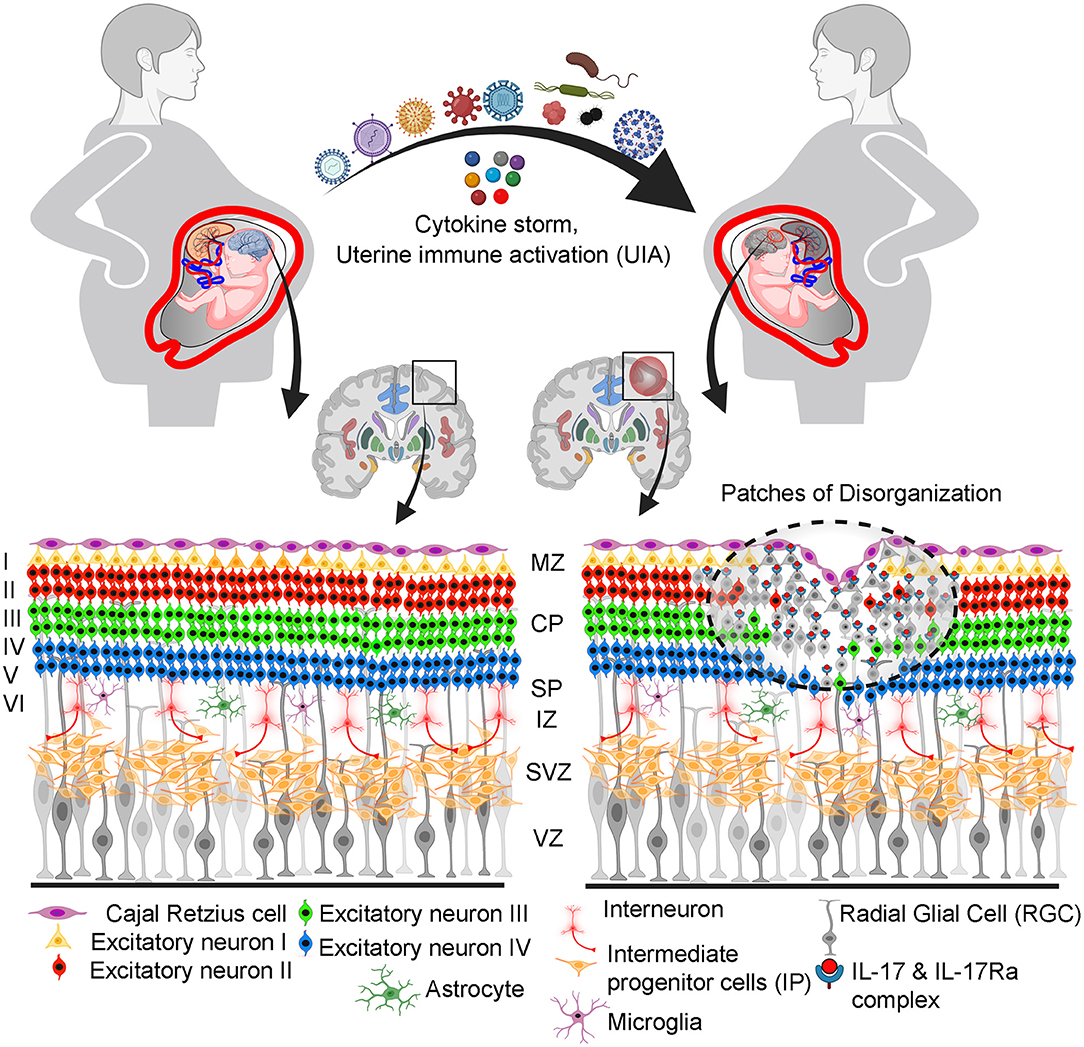
Figure 2. Proposed pathophysiological pathway illustrating how prenatal maternal viral infection and associated cytokine storm disrupted fetal brain programming leading to increased risk of ASD. Among cytokines, IL-17a binds to receptors in the upper layer neuronal cells and alters their identity and plasticity. This can lead to changes in the cortical layer organization and “patchy disorganization” in some regions of the somatosensory cortex. This disorganization potentially affects fine orchestration, proliferation, neurogenesis, migration, and gliogenesis (astrogliogenesis and oligodendrogenesis, and synaptogenesis). Cortical layering during embryonic development: MZ, marginal zone; CP, cortical plate; SP, subplate; IZ, intermediate zone; SVZ, subventricular zone; VZ, ventricular zone.
Pregnant women are more vulnerable to infections. During pregnancy, viral infections may or may not develop clinical indications in the mother. Indirect or direct consequences on the fetal development are inevitable. Infections will trigger immune responses, particularly in the placental microenvironment. However, immune responses are not always pregnancy compatible. The observations from human and animal studies suggest that pregnancy when challenged with viral infections is more likely to have an offspring with ASD-like features and other neurodevelopmental anomalies (114, 115). Neurodevelopmental problems in the fetus can be caused by both DNA and RNA viruses that cross the maternal–fetal interface (116). Here we have reviewed a few of the viral infections during pregnancy that have been associated with the onset of ASD. We also suggest mechanisms encompassing neurological immunological pathways that could play a role in the programming of ASD during fetal neurogenesis.
ZIKV is a single-stranded RNA virus of the Flaviviridae family. In general, in utero exposure to ZIKV is associated with birth defects such as microcephaly (117). Although ZIKV-associated neuro-immunological effects have been linked to the development of neurological diseases (117, 118), it is still not clear whether ZIKV infection during pregnancy significantly contributes to the incidence of ASD during the early years of infants (119). In a study of 216 infants during the Rio de Janeiro ZIKV epidemic of 2015–2016, the prematurity rate was very high (13%). Microcephaly was identified in 8 of 216 infants (120). With neurological Bayley-III and other assessments, ASD was diagnosed in the second year of life in previously healthy children. This suggests that ZIKV during pregnancy may primarily affect gestational age of birth and congenital defects such as microcephaly. It is noteworthy to state that improved neurodevelopmental outcomes were observed in female children, term babies, and maternal infection later in pregnancy (121). ASD developed in six of the 18 children with very low average performance. In another study, the findings in 156 infants from ZIKV infection during pregnancy and 79 infants without ZIKV infection suggested that there were minimal differences in neurodevelopmental outcomes at 24 months of age (122), suggesting that ZIKV infection if not timed for a temporal window of pregnancy may not lead to increased incidence of ASD. Although association between ZIKV infection during pregnancy and ASD is inclusive, more studies with larger cohorts are needed, particularly in the context of ZIKV infection during second trimester. Brazilian ZIKV strain may pass the placental barrier, infect progenitor cortical cells, and drive cell death by apoptosis and autophagy (123). Infection of cytotrophoblasts or the transmigration of infected primary human placental macrophages allowed this virus to infiltrate the embryonic neural cells (124). In newborns with congenital ZIKV infection, neuroimmune modulation may have a role in the development of autism. Proinflammatory cytokines, such as TNF-α- and IL-6, are generated at high levels in response to ZIKV infection and so predispose patients for this condition (125). Interestingly, ZIKV has been found to be highly specific for oRGs (radial glial cells) (126). Centrosomal abnormalities and early differentiation were observed in neural precursor cells (NPCs) infected with the virus. Early differentiation and maybe abnormal radial fiber migration of newborn neurons may have resulted from the breakdown of adherens junctions in the vRGs. The general view is that the ZIKV inhibits the proliferation of NPCs, triggers selective cell death, and shrinks the size of the brain in humans. Subsequently, detailed analysis of the structure of the brain of Zika-infected mice indicated a reduction in the number of VZ-like areas as well as the number of SOX2-positive RGs and TBR1-positive layer VI neurons (127, 128). Neurological problems have been documented in experimental studies in vivo and in vitro as a result of ZIKV neurotropism and the molecular signatures left by infection. Even after birth, infected nerve cells generate and release proinflammatory cytokines that are significantly linked to neuropsychological disorder. Moreover, ZIKV-infected human mesenchymal stem cells were also discovered to display ASD molecular markers in a distinct manner (129). Although there is no concrete evidence of ASD in newborns with maternal ZIKV infection, the altered neuro-immune axis may contribute to ASD.
Rubella virus is a single-stranded, plus-sense RNA virus belonging to the Togaviridae family. Congenital rubella syndrome (CRS), which includes sensorineural hearing impairment, cataracts, heart problems, and/or brain and nervous system damage, can arise from rubella infection during early pregnancy, which is normally a self-limiting condition (130). In the 1960s, the prevalence of intellectual disability and autism in CRS patients in the United States was substantially greater than in the general population, with 42 and 7.4% incidence rate, respectively (131). The virus enters the bloodstream by infected cells and alveolar macrophages, which goes to the lymph nodes in the affected area and cause lymphadenopathy (132). Exactly how Rubella reaches the maternal-fetal interface has not been thoroughly investigated. According to one theory, monocytes in the basal plate diffuse into the intervillous space and/or lymphatic arteries as a result of persistent infection. CTBs, endothelial cells of villous capillaries, amniotic epithelium, and different cells of the basal plate were found to contain detectable virions in placentas with CRS (133). Of the various side effects of CRS is ASD. In the 1970s, 200 times higher incidence of ASD was reported in children with CRS (134). CRS and autistic children show similar traits of hyperactivity and spasticity. Another similarity is that in both autism and CRS, certain changes in the brain are thought to be the result of dysregulated immune system (134, 135). After 3 to 5 years following the exposure, 95% of the children with CRS were suspected of having developmental issues and sensory dysfunction, and 41% were suspected of having autism (134, 135). At molecular level, viral replication in the host cell impacts the expression of genes involved in the development of sensory organs in a direct and indirect manner. Consequently, the long-term impacts of Rubella on the developing embryo are compounded by the host–virus interactions. Another important similarity between CRS and autism is that children with these disorders lack antibodies to rubella. Thus, introduction of rubella vaccines (RCVs) may result in significant reduction in CRS as well as neurodevelopmental and sensory issues in young children (136). Evidence for this supposition is available in small cohort studies.
Influenza is a contagious respiratory infection caused by influenza viruses A and B. Both influenza A and influenza B viruses are enveloped negative-sense RNA viruses belonging to the Orthomyxoviridae family. Pregnant women were among the most at risk during the 2009 H1N1 pandemic of influenza A (H1N1) (137). Third-trimester pregnant women with high temperature were more likely to develop severe disease during the 2009 H1N1 pandemic and in inter-pandemic periods, compared with those in earlier stages of pregnancy. Influenza virus infection can be more severe in pregnant women and their offspring, according to both animal research and clinical findings (137, 138). During pregnancy, influenza virus can have detrimental consequences on the fetus because of hormonal signaling imbalance, inflammation, or activation of the immune system against fetal tissue. As with other common viral pathogens, influenza during pregnancy has been linked to a range of neurodevelopmental issues, including ASD, bipolar disorder, and schizophrenia (139–141). In spite of the conflicting evidence, some epidemiological evidence suggests that pre-natal maternal influenza virus infection increases the likelihood of ASD in offspring. Autistic children were found among the offspring of ~8% of pregnant women who had influenza or had been exposed to it during their pregnancy. Conversely, analysis from a large cohort (196–929) of infants delivered at Kaiser Permanente Northern California between January 1, 2000 and December 31, 2010 at a gestational age of at least 24 weeks) found no link between maternal influenza infection during pregnancy and an elevated risk of ASD (142). When it comes to ASD, a new study found that influenza infection during pregnancy was not related with an increased risk. There is a pertinent debate on the maternal influenza A immunization during pregnancy and risk for autism in the offspring (143). In a large study of 39,726 infants from pre-natally exposed H1N1 vaccine with 13,845 in the first trimester and 29,293 infants from unexposed group, the authors found no correlation between H1N1 immunization during pregnancy and autism (144).
CMV and HSVs (HSV1 and HSV2) are double-stranded DNA viruses, which belong to the herpesviruses class. Like other TORCH pathogens, CMV, HSV1, and HSV2 may cause pregnancy complications, including spontaneous abortion, intrauterine growth restriction, preterm birth, brain anomalies, or visual impairment (145). Although infections with herpesviruses may not be primary infections, these viruses are associated with persistent or latent infections and may impact pregnancy if reactivated. What is important is gestational age at the time of infection which may control the risk of vertical transmission to the fetus and disease level (145, 146). There is a 40% chance that a fetus will be infected if the mother is infected. CMV and HSV2 sero-positivity has been used to examine a relationship between infections by these viruses and the incidence of ASD (147). One study involving 442 mothers of children with ASD suggested that high levels of HSV-2 IgG antibodies in maternal mid pregnancy were associated with increased risk of ASD in male offspring. In this study, no association was found between ASD and the sero-positivity for Toxoplasma gondii, rubella virus, CMV, or HSV-1 (148). However, in another study, CMV sero-positivity was found to be a more potent trigger than HSV-2 to influence the onset of ASD (149). These observations suggest that this association with ASD needs to be further evaluated in prospective studies with larger cohorts of pregnant women, particularly keeping the gestational age of infection in mind. It is not clear what immune changes occur during primary or reactivated CMV or HSV infections. The ability of CMV to infect trophoblasts has been demonstrated (150, 151). Several investigations have documented the inflammatory pathology that results from the placental immune response against CMV. Paracrine apoptosis of uninfected cells occurs during CMV infection of CTB and SYN possibly as a result of excessive production of TNF-α. This inflammatory reaction at the maternal–fetal interface has adverse consequences on the fetal neurodevelopment. Children with congenital CMV infection who were previously asymptomatic at birth have been found to have ASD (152, 153). In children with neurological disabilities and cerebral cortical abnormalities, teratogenic consequences of CMV infection were found by neuroimaging. Forty-five fetuses from women with a positive pre-natal diagnosis of CMV infection were examined for neuronal damage. This virus had been detected in the brain's cortex as well as its white and gray tissues as well as the germinal matrix and the leptomeninges (154). Neurons, neuroblasts, glia, endothelium, ependymal, and meningeal cells were among the CMV-positive cells identified. In the third layer of the cerebral cortex, there was significant laminar necrosis, with numerous macrophages replacing the growing neurons (155, 156). Multifocal aggregates of CD8+ T-lymphocytes and granzyme B+ T-lymphocytes were seen in the necrotic and CMV-positive portions of the inflammatory infiltrate. Severely brain-damaged fetuses had a noticeable invasion of fetal activated CD8+ T-cells (157–159).
According to the World Health Organization, around 1.3 million women living with HIV became pregnant each year. Transplacental transmission of HIV can occur during delivery and/or post-natal breastfeeding. Vertical transmission in utero is estimated to occur at a rate of around 1%−2% and is associated to maternal CD4 levels and viral load (160). Lymphocytes infected with HIV endocytose or transcytose the virion particle if they come into contact with trophoblast cells. Aside from that, CTB, SYN, and Hofbauer cells have been found to contain HIV genetic material (161). These findings are in line with epidemiological studies, which show that although HIV can be transmitted transplacentally, this is a rare occurrence (1%) (162, 163). On the other hand, the number of HIV-uninfected children who were exposed to HIV while in the womb or while breastfeeding is rising, and these children are referred to as HIV exposed uninfected (HEU). Many children who have been exposed to HIV but have not become infected have been shown to have ASD. Congenital infections, such as CMV and toxoplasmosis, are more common in HEU. As compared with uninfected individuals, those exposed to the HEU developed weaker cognitive functioning and poorer motor, mental, and language development. Researchers found that HEU diagnosed with an ASD-like pathology had higher leukocyte mitochondrial DNA content than controls, suggesting that mitochondrial malfunction may play a role in HEU's risk of developing ASD (164–166).
Although it is too early to examine an association between SARS-CoV-2 infection during pregnancy and the incidence of ASD or other neuropsychiatric disorders, infection-associated cytokine storm has been thought to be a possible risk factor for neurodevelopmental disorders in infants (167, 168). The common notion is that poor placental infection or vertical transmission, lack of trans-placental transfer of SARS-CoV-2 antibodies, and reduced co-expression of ACE-2 and TMPRSS2 may provide protection against placental infection and vertical transmission. However, cytokine storm induced by SARS-CoV-2 may reach the placenta and the fetus. In view of recent results indicating that pregnant women with SARS-CoV-2 infection have elevated levels of IL-6, IL-17, and other inflammatory cytokines (169, 170), fetal neurodevelopmental may be at risk. Although SARS-CoV-2 is rarely transferred to the fetus, it can breach the blood-cerebrospinal fluid barrier (BCSFB) by infecting epithelial cells expressing the ACE2 receptor (171). In light of maternal immune activation, it appears plausible to assume that infection with SARS-CoV-2 during the early stages of pregnancy could similarly result in serious fetal neurodevelopmental defects.
Although male fetuses are exposed to the same in utero environment, they have preponderance for generalized complications of pregnancy, showing higher vulnerability to placental inflammation, hypoxia, placental abruption, preeclampsia, eclampsia, and preterm birth (172, 173). These adverse pregnancy outcomes are invariably associated with increased risk of poor neurodevelopmental outcome. Emerging research points to a complicated relationship between infant's sex and maternal immune surveillance (174, 175). Differential crosstalk between male and female fetuses with the placenta might arise from the varying effects on innate immunity. Sex-specific differences in viral pathogenesis stem from gene dosage effect of X and Y chromosomes and specific sex hormone gradient (176, 177). There is evidence that sex-linked genes regulate innate, cellular, and humoral immune surveillance, significantly increasing the male susceptibility to adverse immune response (Figure 3A). The female placenta shares the same X-linked gene dosage as of the fetus as it is derived from the extra embryonic tissues. Gene dosage of X-linked genes, such as IL-13, IL-4, IL-10, XIST, TLR7, FOXP3, and Sox9 in female placenta could potentially contribute to female protection against viral infection compared with male through gene dosage and epigenetic modification. Taken together, sex-specific placental stress signaling and gene expression significantly impact the fetal adaptation in a sexually dimorphic manner to the in utero environment upon maternal immune response and stress (178–180). Epidemiological data reveal a higher mortality rate in male compared with female embryos due to viral infection. This unique immunological advantage to female is attributed to efficient humoral and cellular antiviral immune responses. Recent SARS pandemics (SARS-CoV-1 and SARS-CoV-2) also indicated sexual dimorphic IFN signaling, in blood and lungs, resulting in high morbidity in male patients (177, 181). Placental Type I and II IFN signaling at the maternal-fetal interface dampens viral pathogenicity. The propensity of placental Type I and Type II IFN signaling at the maternal–fetal interface during viral pathogenicity depends largely on sexual identity of the placenta. Sexual dimorphic antiviral response also arises from sex specific expression of TLR and IFN pathway genes (182, 183). Female immune cells show 10-fold higher expression of TLRs in comparison with its male counterpart (184). Furthermore, higher innate immune cell abundance in female confers higher immune protection against viruses. Interestingly, Treg abundance and proliferation rate is also higher in females. Interestingly, human female naive CD4+ T cells preferentially produce IFN-γ upon activation, but human male naive T cells produce more IL-17 than their female counterparts. Additional sexual dimorphism in IL-17 expression from T cells also exists (185–188). This sexual dimorphism in immune responses originates from X- and Y- linked chromatin remodeling. In addition to that, sex-specific microRNA expression, such as miR-124 and miR-202-5p/3p, could be attributed to sex-specific immune responses. For example, gestational expression of miR-124 in XX cells leads to induction of Foxp3+ Tregs by suppressing STAT3 signaling (189).
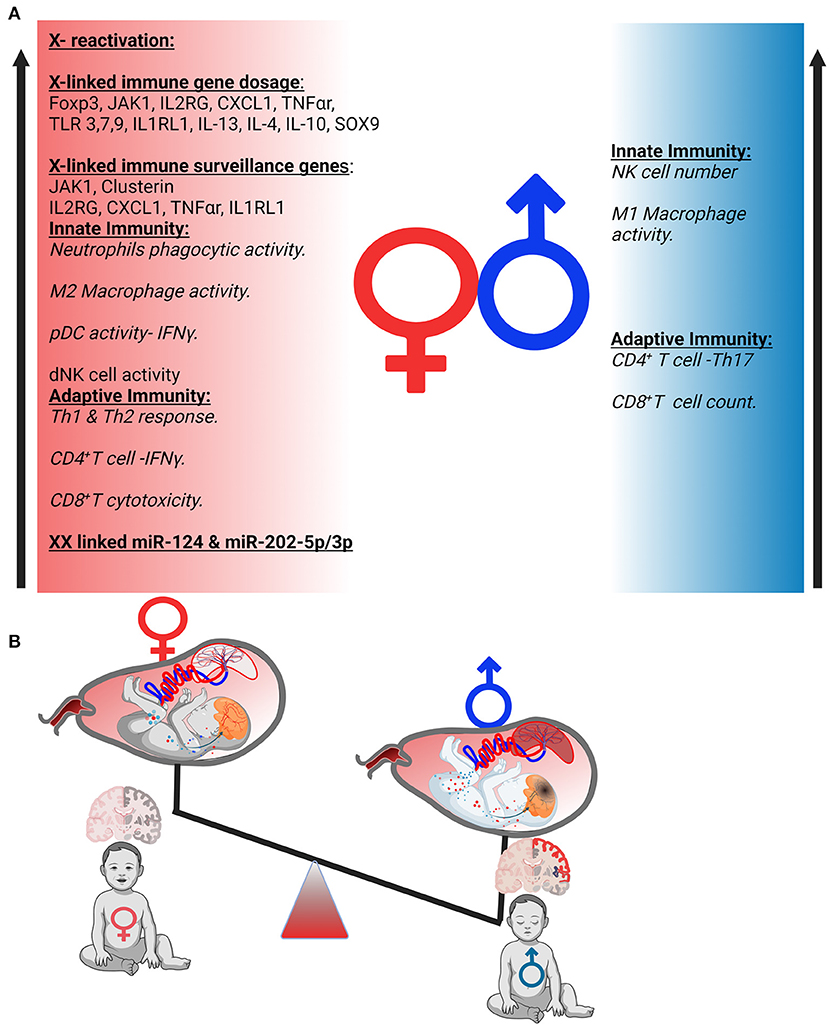
Figure 3. Possible mechanism of sexual dimorphism in uterine immunity and autism spectrum disorders (ASD) development. (A) Sexual dimorphism of the immune responses. Immune components of both innate and adaptive immunity are differently regulated in females and males. (B) Sex-specific placental response to inflammation generated a differential gradient in both sexes that affects fetal brain differentially.
A recent study highlighted a unique feature of the human placenta's ability to reactivate inactive X-chromosome at very early or late gestational period (190, 191). Unlike other tissues, these unique molecular features of the human female placenta highlight the possibility of inducing random X reactivation in an inimical intrauterine environment in the early stages of embryo development. This X-reactivation could potentially increase the gene dosage of certain X-linked genes as described above, leading to improved protection from viral infection. Sex-specific placental transcriptional analysis also reveals unique molecular adaptation under the same maternal in utero environment. The study highlighted the changes not only in the X- and Y-linked genes but also in immune regulatory pathways within autosomal gene cluster. The female placenta showed higher expression of immune surveillance genes including JAK1, IL2RB, Clusterin, LTBP, CXCL1, IL1RL1, and TNF-α receptor (179, 192).
Sexual dimorphism has been observed in a range of neurodevelopmental disorders including ASD, schizophrenia, attention deficit hyperactivity disorder, and intellectual disabilities (193–195). The brain is highly vulnerable to environmental insults during early gestational period, as it undergoes rapid developmental processes, including neurogenesis, neuronal migration, and synaptogenesis. Systemic and uterine immune activation during pregnancy is associated with disruption in fetal neurogenesis and predisposes to neuropsychiatric disease in male offspring (196–198) (Figure 3B). These sex-specific responses serve as additional mechanisms in which to consider male ASD predominance (199). The male preponderance does not appear to be directly linked to genetic factors, as sex-skewed expression of neurodevelopmental risk genes has not been discovered. Although male and female littermates are exposed to the same maternal in utero inflammation, it has been reported that behavioral shortfalls manifest mainly in the male offspring, mirroring the sex bias observed in placental immune response to viral infection (200). Because the placenta is the immunologic hub and the first site of fetal exposure to maternal inflammation, we propose that sex-specific reactions to uterine immune activation (UIA) that have deleterious impacts on fetal neurodevelopment may originate in the placenta.
Knowledge of placental molecular and immunologic pathways and their role in the transmission or protection from infection is critical to the care of pregnant women and the health of their newborn children. Infections during pregnancy can have serious implications. A variety of host–pathogen interactions specific to the maternal–fetal niche have been discovered as a result of the complexity and distinctive characteristics between the two hosts. Yet, the molecular mechanisms underlying infection-associated pathologies remain largely unknown, in part, due to the difficulties inherent in defining the interactions between the pathogen and the maternal and/or fetal hosts during pregnancy (201). There are significant differences in the placental architecture between humans and mice that prevent direct correlations of these findings to humans, despite the fact that mouse models have been useful for gaining valuable insights into numerous aspects of pregnancy. Even though pregnant women appear to be less susceptible to early infection than non-pregnant women, immunologic modifications with advanced pregnancy may hamper pathogen clearance, resulting in an increased severity of diseases caused by particular pathogens.
Neuronal development is a complex process that depends on the interaction between genetic and environmental factors. Various risk factors might affect embryonic development. Despite this, studies have shown that the inflammatory response to infections is a widespread and important feature. The identification of target cells for infection and study of possible neurodevelopmental effects require a better understanding of normal neurodevelopment and its comparison with the pathways that disrupt it.
Future research is critical to the development of tailored treatments that take into account the complex relationships between maternal and fetal tissues and how infections influence these interactions. Overall, research suggests that these pregnancy-related problems are linked to neurodevelopmental abnormalities, particularly ASD. The uterine immune system is affected by hormonal changes. Thus, therapeutic approaches to minimize the spread of infectious and other diseases by modifying the hormonal environment should be considered. In order to prevent and treat diseases, new preventive and therapeutic avenues that may interfere with the pathogen-placenta deleterious cross-talk are a possible prophylactic or therapeutic avenues. With the view of curtailing maternal and fetal inflammation during pregnancy, maternal immunizations may have long-term advantages for the offspring as well.
Both authors contributed equally to all aspects of the article.
This work was supported, in part, by the NIH grant P20 GM121298 to SS and a pilot project funding from this grant to SJ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the members of the Sharma Laboratory for critical reading of the manuscript and the Department of Pediatrics for continued support of the research.
ASD, autism spectrum disorders; UIA, uterine immune activation; Tregs, regulatory T cells; Th17, T-helper 17 cells; IL17a, interleukin 17a isoform; LPS, lipopolysaccharide; Poly I:C, polyinosinic:polycytidylic acid; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; COVID-19, coronavirus disease 2019; ZKV, Zika virus; CMV, Cytomegalovirus; TNF-α, tumor necrosis factor alpha; STB, syncytiotrophoblast.
1. Gibbs RS. The relationship between infections and adverse pregnancy outcomes: an overview. Ann Periodontol. (2001) 6:153–63. doi: 10.1902/annals.2001.6.1.153
2. Silasi M, Cardenas I, Kwon J-Y, Racicot K, Aldo P, Mor G. Viral infections during pregnancy. Am J Reprod Immunol. (2015) 73:199–213. doi: 10.1111/aji.12355
3. Megli CJ, Coyne CB. Infections at the maternal–fetal interface: an overview of pathogenesis and defence. Nat Rev Microbiol. (2021) 20:67–82. doi: 10.1038/s41579-021-00610-y
4. Slutsky R, Romero R, Xu Y, Galaz J, Miller D, Done B, et al. Exhausted and senescent T cells at the maternal-fetal interface in preterm and term labor. J Immunol Res. (2019) 2019:1–16. doi: 10.1155/2019/3128010
5. Chudnovets A, Liu J, Narasimhan H, Liu Y, Burd I. Role of inflammation in virus pathogenesis during pregnancy. J Virol. (2020) 95:e01381–19. doi: 10.1128/JVI.01381-19
6. Than NG, Hahn S, Rossi SW, Szekeres-Bartho J. Editorial: fetal-maternal immune interactions in pregnancy. Front Immunol. (2019) 10:2729. doi: 10.3389/fimmu.2019.02729
7. Alcami A, Ghazal P, Yewdell JW. Viruses in control of the immune system. EMBO Rep. (2002) 3:927–32. doi: 10.1093/embo-reports/kvf200
8. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
9. Teijaro JR. Cytokine storms in infectious diseases. Semin Immunopathol. (2017) 39:501–3. doi: 10.1007/s00281-017-0640-2
10. Jamieson DJ, Honein MA, Rasmussen SA, Williams JL, Swerdlow DL, Biggerstaff MS, et al. H1N1 2009 influenza virus infection during pregnancy in the USA. Lancet. (2009) 374:451–8. doi: 10.1016/S0140-6736(09)61304-0
11. Yockey LJ, Varela L, Rakib T, Khoury-Hanold W, Fink SL, Stutz B, et al. Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell. (2016) 166:1247–56.e4. doi: 10.1016/j.cell.2016.08.004
12. Fried M, Duffy PE. Malaria during Pregnancy. Cold Spring Harb Perspect Med. (2017) 7:a025551. doi: 10.1101/cshperspect.a025551
13. Leyser M, Marques FJP, do Nascimento OJM. potential risk of brain damage and poor developmental outcomes in children prenatally exposed to sars-cov-2: a systematic review. Rev Paul Pediatr. (2021) 40:e2020415. doi: 10.1590/1984-0462/2022/40/2020415
14. Frenkel LD, Gomez F, Sabahi F. The pathogenesis of microcephaly resulting from congenital infections: why is my baby's head so small? Eur J Clin Microbiol Infect Dis. (2018) 37:209–26. doi: 10.1007/s10096-017-3111-8
15. de Vries LS. Viral infections and the neonatal brain. Semin Pediatr Neurol. (2019) 32:100769. doi: 10.1016/j.spen.2019.08.005
16. Cordeiro CN, Tsimis M, Burd I. Infections and brain development. Obstet Gynecol Surv. (2015) 70:644–55. doi: 10.1097/OGX.0000000000000236
17. Courchesne E, Gazestani VH, Lewis NE. Prenatal origins of ASD: the when, what, and how of ASD development. Trends Neurosci. (2020) 43:326–42. doi: 10.1016/j.tins.2020.03.005
18. Howlin P, Magiati I. Autism spectrum disorder: outcomes in adulthood. Curr Opin Psychiatry. (2017) 30:69–76. doi: 10.1097/YCO.0000000000000308
19. Wiśniowiecka-Kowalnik B, Nowakowska BA. Genetics and epigenetics of autism spectrum disorder-current evidence in the field. J Appl Genet. (2019) 60:37–47. doi: 10.1007/s13353-018-00480-w
20. Trambacz-Oleszak S, Nosulia T. Genetic factors in autism spectrum disorders (ASD). Postepy Biochem. (2021) 67:28–33. doi: 10.18388/pb.2021_377
21. Beggiato A, Peyre H, Maruani A, Scheid I, Rastam M, Amsellem F, et al. Gender differences in autism spectrum disorders: divergence among specific core symptoms. Autism Res. (2017) 10:680–9. doi: 10.1002/aur.1715
22. Meltzer A, Van de Water J. The role of the immune system in autism spectrum disorder. Neuropsychopharmacology. (2017) 42:284–98. doi: 10.1038/npp.2016.158
23. Careaga M, Murai T, Bauman MD. Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol Psychiatry. (2017) 81:391–401. doi: 10.1016/j.biopsych.2016.10.020
24. Al-Haddad BJS, Oler E, Armistead B, Elsayed NA, Weinberger DR, Bernier R, et al. The fetal origins of mental illness. Am J Obstet Gynecol. (2019) 221:549–62. doi: 10.1016/j.ajog.2019.06.013
25. Shuid AN, Jayusman PA, Shuid N, Ismail J, Kamal Nor N, Mohamed IN. Association between viral infections and risk of autistic disorder: an overview. Int J Environ Res Public Health. (2021) 18:2817. doi: 10.3390/ijerph18062817
26. Erlebacher A. Immunology of the maternal-fetal interface. Annu Rev Immunol. (2013) 31:387–411. doi: 10.1146/annurev-immunol-032712-100003
27. Zeldovich VB, Clausen CH, Bradford E, Fletcher DA, Maltepe E, Robbins JR, et al. Placental syncytium forms a biophysical barrier against pathogen invasion. PLoS Pathog. (2013) 9:e1003821. doi: 10.1371/journal.ppat.1003821
28. Parker EL, Silverstein RB, Verma S, Mysorekar IU. Viral-immune cell interactions at the maternal-fetal interface in human pregnancy. Front Immunol. (2020) 11:2561. doi: 10.3389/fimmu.2020.522047
29. Thomas JR, Appios A, Zhao X, Dutkiewicz R, Donde M, Lee CYC, et al. Phenotypic and functional characterization of first-trimester human placental macrophages, Hofbauer cells. J Exp Med. (2021) 218:e20200891. doi: 10.1084/jem.20200891
30. Murphy SP, Fast LD, Hanna NN, Sharma S. Uterine NK Cells mediate inflammation-induced fetal demise in IL-10-null mice. J Immunol. (2005) 175:4084–90. doi: 10.4049/jimmunol.175.6.4084
31. Thaxton JE, Nevers T, Lippe EO, Blois SM, Saito S, Sharma S. NKG2D blockade inhibits poly(I:C)-triggered fetal loss in wild type but not IL-10–/– mice. J Immunol. (2013) 190:3639–47. doi: 10.4049/jimmunol.1203488
32. Shmeleva EV, Colucci F. Maternal natural killer cells at the intersection between reproduction and mucosal immunity. Mucosal Immunol. (2021) 14:991–1005. doi: 10.1038/s41385-020-00374-3
33. Jabrane-Ferrat N. Features of human decidual NK cells in healthy pregnancy and during viral infection. Front Immunol. (2019) 10:1397. doi: 10.3389/fimmu.2019.01397
34. Nevers T, Kalkunte S, Sharma S. Uterine regulatory T cells, IL-10 and hypertension. Am J Reprod Immunol. (2011) 66(Suppl. 1):88–92. doi: 10.1111/j.1600-0897.2011.01040.x
35. Sharma S. Natural killer cells and regulatory T cells in early pregnancy loss. Int J Dev Biol. (2014) 58:219–29. doi: 10.1387/ijdb.140109ss
36. Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced regulatory T cells: their development, stability, and applications. Trends Immunol. (2016) 37:803–11. doi: 10.1016/j.it.2016.08.012
37. Wan Z, Zhou Z, Liu Y, Lai Y, Luo Y, Peng X, et al. Regulatory T cells and T helper 17 cells in viral infection. Scand J Immunol. (2020) 91:e12873. doi: 10.1111/sji.12873
38. Jash S, Sharma S. In utero immune programming of autism spectrum disorder (ASD). Hum Immunol. (2021) 82:379–84. doi: 10.1016/j.humimm.2021.02.002
39. Maltepe E, Bakardjiev AI, Fisher SJ. The placenta: transcriptional, epigenetic, and physiological integration during development. J Clin Invest. (2010) 120:1016–25. doi: 10.1172/JCI41211
40. Ander SE, Diamond MS, Coyne CB. Immune responses at the maternal-fetal interface. Sci Immunol. (2019) 4:eaat6114. doi: 10.1126/sciimmunol.aat6114
41. Delorme-Axford E, Sadovsky Y, Coyne CB. The placenta as a barrier to viral infections. Annu Rev Virol. (2014) 1:133–46. doi: 10.1146/annurev-virology-031413-085524
42. Schwartz DA. Viral infection, proliferation, and hyperplasia of Hofbauer cells and absence of inflammation characterize the placental pathology of fetuses with congenital Zika virus infection. Arch Gynecol Obstet. (2017) 295:1361–8. doi: 10.1007/s00404-017-4361-5
43. Dorsamy V, Vallen C, Haffejee F, Moodley J, Naicker T. The role of trophoblast cell receptor expression in HIV-1 passage across the placenta in pre-eclampsia: an observational study. BJOG. (2017) 124:920–8. doi: 10.1111/1471-0528.14311
44. Schleiss MR, Aronow BJ, Handwerger S. Cytomegalovirus infection of human syncytiotrophoblast cells strongly interferes with expression of genes involved in placental differentiation and tissue integrity. Pediatr Res. (2007) 61:565–71. doi: 10.1203/pdr.0b013e318045be6d
45. Taglauer E, Benarroch Y, Rop K, Barnett E, Sabharwal V, Yarrington C, et al. Consistent localization of SARS-CoV-2 spike glycoprotein and ACE2 over TMPRSS2 predominance in placental villi of 15 COVID-19 positive maternal-fetal dyads. Placenta. (2020) 100:69–74. doi: 10.1016/j.placenta.2020.08.015
46. Brown JA, Singh G, Acklin JA, Lee S, Duehr JE, Chokola AN, et al. Dengue Virus Immunity Increases Zika virus-induced damage during pregnancy. Immunity. (2019) 50:751–62.e5. doi: 10.1057/978-1-137-55247-1
47. Jagger BW, Miner JJ, Cao B, Arora N, Smith AM, Kovacs A, et al. Gestational stage and IFN-λ signaling regulate ZIKV infection in utero. Cell Host Microbe. (2017) 22:366–76.e3. doi: 10.1016/j.chom.2017.08.012
48. Tabata T, Petitt M, Zydek M, Fang-Hoover J, Larocque N, Tsuge et al. Human cytomegalovirus infection interferes with the maintenance and differentiation of trophoblast progenitor cells of the human placenta. J Virol. (2015) 89:5134–47. doi: 10.1128/JVI.03674-14
49. Yockey LJ, Jurado KA, Arora N, Millet A, Rakib T, Milano KM, et al. Type I interferons instigate fetal demise after Zika virus infection. Sci Immunol. (2018) 3:eaao1680. doi: 10.1126/sciimmunol.aao1680
50. Johnson EL, Swieboda D, Olivier A, Enninga EAL, Chakraborty R. Robust innate immune responses at the placenta during early gestation may limit in utero HIV transmission. PLoS Pathog. (2021) 17:e1009860. doi: 10.1371/journal.ppat.1009860
51. Johnson EL, Swieboda D, Chakraborty R. Human cytomegalovirus preferentially infects early gestation placental macrophages and evades their antiviral immunogenicity through evasion of the type I IFN response. J Immunol. (2020) 204:171.10.
52. Fahmi A, Brügger M, Démoulins T, Zumkehr B, Oliveira Esteves BI, Bracher L, et al. SARS-CoV-2 can infect and propagate in human placenta explants. Cell Rep Med. (2021) 2:100456. doi: 10.1016/j.xcrm.2021.100456
53. Hertzog J, Dias Junior AG, Rigby RE, Donald CL, Mayer A, Sezgin E, et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur J Immunol. (2018) 48:1120–36. doi: 10.1002/eji.201847483
54. Paulus C, Krauss S, Nevels M. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc Natl Acad Sci USA. (2006) 103:3840–5. doi: 10.1073/pnas.0600007103
55. Brien M-E, Bouron-Dal Soglio D, Dal Soglio S, Couture C, Boucoiran I, Nasr Y, et al. Pandemic stress and SARS-CoV-2 infection are associated with pathological changes at the maternal-fetal interface. Placenta. (2021) 115:37–44. doi: 10.1016/j.placenta.2021.09.007
56. Moresi S, Dell'Aquila M, Salvi S, Rullo R, Fruci S, Stollagli F, et al. SARS-CoV-2 Infection in pregnancy: clinical signs, placental pathology, and neonatal outcome-implications for clinical care. Front Med. (2021) 8:676870. doi: 10.3389/fmed.2021.676870
57. Constantino FB, Cury SS, Nogueira CR, Carvalho RF, Justulin LA. prediction of non-canonical routes for SARS-CoV-2 infection in human placenta cells. Front Mol Biosci. (2021) 8:614728. doi: 10.3389/fmolb.2021.614728
58. Pique-Regi R, Romero R, Tarca AL, Luca F, Xu Y, Alazizi A, et al. Does the human placenta express the canonical cell entry mediators for SARS-CoV-2? eLife. 9:e58716. doi: 10.7554/eLife.58716
59. Azinheira Nobrega Cruz N, Stoll D, Casarini DE, Bertagnolli M. Role of ACE2 in pregnancy and potential implications for COVID-19 susceptibility. Clin Sci. (2021) 135:1805–24. doi: 10.1042/CS20210284
60. Valdés G, Neves LA, Anton L, Corthorn J, Chacón C, Germain AM, et al. Distribution of angiotensin-(1-7) and ACE2 in human placentas of normal and pathological pregnancies. Placenta. (2006) 27:200–7. doi: 10.1016/j.placenta.2005.02.015
61. Egloff C, Vauloup-Fellous C, Picone O, Mandelbrot L, Roques P. Evidence and possible mechanisms of rare maternal-fetal transmission of SARS-CoV-2. J Clin Virol. (2020) 128:104447. doi: 10.1016/j.jcv.2020.104447
62. To KF, Tong JHM, Chan PKS, Au FWL, Chim SSC, Chan KCA, et al. Tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: an in-situ hybridization study of fatal cases. J Pathol. (2004) 202:157–63. doi: 10.1002/path.1510
63. Kotlyar AM, Grechukhina O, Chen A, Popkhadze S, Grimshaw A, Tal O, et al. Vertical transmission of coronavirus disease 2019: a systematic review and meta-analysis. Am J Obstet Gynecol. (2021) 224:35–53.e3. doi: 10.1016/j.ajog.2020.07.049
64. Tolu LB, Ezeh A, Feyissa GT. Vertical transmission of severe acute respiratory syndrome coronavirus 2: a scoping review. PLoS ONE. (2021) 16:e0250196. doi: 10.1371/journal.pone.0250196
65. Edlow AG, Li JZ, Collier AY, Atyeo C, James KE, Boatin AA, et al. Assessment of maternal and neonatal SARS-CoV-2 viral load, transplacental antibody transfer, and placental pathology in pregnancies during the COVID-19 pandemic. JAMA Netw Open. (2020) 3:e2030455. doi: 10.1001/jamanetworkopen.2020.30455
66. Forbester JL, Humphreys IR. Genetic influences on viral-induced cytokine responses in the lung. Mucosal Immunol. (2020) 14:14–25. doi: 10.1038/s41385-020-00355-6
67. Mahmudpour M, Roozbeh J, Keshavarz M, Farrokhi S, Nabipour I. COVID-19 cytokine storm: the anger of inflammation. Cytokine. (2020) 133:155151. doi: 10.1016/j.cyto.2020.155151
68. Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect. (2020) 53:368–70. doi: 10.1016/j.jmii.2020.03.005
69. Pavel AB, Glickman JW, Michels JR, Kim-Schulze S, Miller RL, Guttman-Yassky E. Th2/Th1 cytokine imbalance is associated with higher COVID-19 risk mortality. Front Genet. (2021) 12:706902. doi: 10.3389/fgene.2021.706902
70. Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. (2020) 26:1070–6. doi: 10.1038/s41591-020-0944-y
71. Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. (2020) 369:eabc8511. doi: 10.1126/science.abc8511
72. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. (2012) 76:16–32. doi: 10.1128/MMBR.05015-11
73. Guo J, Wang S, Xia H, Shi D, Chen Y, Zheng S, et al. Cytokine signature associated with disease severity in COVID-19. Front Immunol. (2021) 12:681516. doi: 10.3389/fimmu.2021.681516
74. Paraschivescu C, Barbosa S, Lorivel T, Glaichenhaus N, Davidovic L. Cytokine changes associated with the maternal immune activation (MIA) model of autism: a penalized regression approach. PLoS ONE. (2020) 15:e0231609. doi: 10.1371/journal.pone.0231609
75. Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci. (2007) 8:221–32. doi: 10.1038/nrn2054
76. Asslih S, Damri O, Agam G. Neuroinflammation as a common denominator of complex diseases (cancer, diabetes type 2, and neuropsychiatric disorders). Int J Mol Sci. (2021) 22:6138. doi: 10.3390/ijms22116138
77. Chen L, Coleman R, Leang R, Tran H, Kopf A, Walsh CM, et al. Human neural precursor cells promote neurologic recovery in a viral model of multiple sclerosis. Stem Cell Rep. (2014) 2:825–37. doi: 10.1016/j.stemcr.2014.04.005
78. Hu S, Rotschafer JH, Lokensgard JR, Cheeran MC-J. Activated CD8+ T lymphocytes inhibit neural stem/progenitor cell proliferation: role of interferon-gamma. PLoS ONE. (2014) 9:e105219. doi: 10.1371/journal.pone.0105219
79. Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. (2009) 64:61–78. doi: 10.1016/j.neuron.2009.09.002
80. Emanuele E, Orsi P, Boso M, Broglia D, Brondino N, Barale F, et al. Low-grade endotoxemia in patients with severe autism. Neurosci Lett. (2010) 471:162–5. doi: 10.1016/j.neulet.2010.01.033
81. Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. (2011) 25:40–5. doi: 10.1016/j.bbi.2010.08.003
82. Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, Van de Water J. Altered T cell responses in children with autism. Brain Behav Immun. (2011) 25:840–9. doi: 10.1016/j.bbi.2010.09.002
83. Wei H, Zou H, Sheikh AM, Malik M, Dobkin C, Brown WT, et al. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflamm. (2011) 8:52. doi: 10.1186/1742-2094-8-52
84. Gadient RA, Otten U. Expression of interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) mRNAs in rat brain during postnatal development. Brain Res. (1994) 637:10–4. doi: 10.1016/0006-8993(94)91211-4
85. Gadient RA, Otten U. Differential expression of interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) mRNAs in rat hypothalamus. Neurosci Lett. (1993) 153:13–6. doi: 10.1016/0304-3940(93)90065-S
86. Banks WA, Kastin AJ, Gutierrez EG. Penetration of interleukin-6 across the murine blood-brain barrier. Neurosci Lett. (1994) 179:53–6. doi: 10.1016/0304-3940(94)90933-4
87. Zefferino R, Di Gioia S, Conese M. Molecular links between endocrine, nervous and immune system during chronic stress. Brain Behav. (2021) 11:e01960. doi: 10.1002/brb3.1960
88. Mandal M, Marzouk AC, Donnelly R, Ponzio NM. Maternal immune stimulation during pregnancy affects adaptive immunity in offspring to promote development of TH17 cells. Brain Behav Immun. (2011) 25:863–71. doi: 10.1016/j.bbi.2010.09.011
89. Alcendor DJ, Charest AM, Zhu WQ, Vigil HE, Knobel SM. Infection and upregulation of proinflammatory cytokines in human brain vascular pericytes by human cytomegalovirus. J Neuroinflamm. (2012) 9:95. doi: 10.1186/1742-2094-9-95
90. Bayless NL, Greenberg RS, Swigut T, Wysocka J, Blish CA. Zika virus infection induces cranial neural crest cells to produce cytokines at levels detrimental for neurogenesis. Cell Host Microbe. (2016) 20:423–8. doi: 10.1016/j.chom.2016.09.006
91. Nitkiewicz J, Borjabad A, Morgello S, Murray J, Chao W, Emdad L, et al. HIV induces expression of complement component C3 in astrocytes by NF-κB-dependent activation of interleukin-6 synthesis. J Neuroinflamm. (2017) 14:23. doi: 10.1186/s12974-017-0794-9
92. Wei H, Chadman KK, McCloskey DP, Sheikh AM, Malik M, Brown WT, et al. Brain IL-6 elevation causes neuronal circuitry imbalances and mediates autism-like behaviors. Biochim Biophys Acta. (2012) 1822:831–42. doi: 10.1016/j.bbadis.2012.01.011
93. Heyser CJ, Masliah E, Samimi A, Campbell IL, Gold LH. Progressive decline in avoidance learning paralleled by inflammatory neurodegeneration in transgenic mice expressing interleukin 6 in the brain. Proc Natl Acad Sci USA. (1997) 94:1500–5. doi: 10.1073/pnas.94.4.1500
94. Gallagher D, Norman AA, Woodard CL, Yang G, Gauthier-Fisher A, Fujitani M, et al. Transient maternal IL-6 mediates long-lasting changes in neural stem cell pools by deregulating an endogenous self-renewal pathway. Cell Stem Cell. (2013) 13:564–76. doi: 10.1016/j.stem.2013.10.002
95. Samuelsson A-M, Jennische E, Hansson H-A, Holmäng A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am J Physiol Regul Integr Comp Physiol. (2006) 290:R1345–56. doi: 10.1152/ajpregu.00268.2005
96. Yoshimatsu T, Kawaguchi D, Oishi K, Takeda K, Akira S, Masuyama N, et al. Non-cell-autonomous action of STAT3 in maintenance of neural precursor cells in the mouse neocortex. Development. (2006) 133:2553–63. doi: 10.1242/dev.02419
97. Mirabella F, Desiato G, Mancinelli S, Fossati G, Rasile M, Morini R, et al. Transient Maternal IL-6 boosts glutamatergic synapses and disrupts hippocampal connectivity in the offspring. BioRxiv [Preprint]. (2020). doi: 10.1101/2020.11.02.364356
98. Kumari E, Velloso F, Nasuhidehnavi A, Somasundaram A, Savanur VH, Buono K, et al. Developmental IL-6 exposure favors production of PDGF-responsive multipotential progenitors at the expense of neural stem cells and other progenitors. Stem Cell Rep. (2020) 14:861–75. doi: 10.1016/j.stemcr.2020.03.019
99. Vereyken EJF, Bajova H, Chow S, de Graan PNE, Gruol DL. Chronic interleukin-6 alters the level of synaptic proteins in hippocampus in culture and in vivo. Eur J Neurosci. (2007) 25:3605–16. doi: 10.1111/j.1460-9568.2007.05615.x
100. Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocrinol. (2012) 33:116–25. doi: 10.1016/j.yfrne.2011.12.002
101. Wang J, Liu J, Zhou R, Ding X, Zhang Q, Zhang C, et al. Zika virus infected primary microglia impairs NPCs proliferation and differentiation. Biochem Biophys Res Commun. (2018) 497:619–25. doi: 10.1016/j.bbrc.2018.02.118
102. Chucair-Elliott AJ, Conrady C, Zheng M, Kroll CM, Lane TE, Carr DJJ. Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia. (2014) 62:1418–34. doi: 10.1002/glia.22689
103. Kuwabara T, Ishikawa F, Kondo M, Kakiuchi T. The role of IL-17 and related cytokines in inflammatory autoimmune diseases. Mediat Inflamm. (2017) 2017:e3908061. doi: 10.1155/2017/3908061
104. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. (2016) 351:933–9. doi: 10.1126/science.aad0314
105. Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med. (2014) 370:1209–19. doi: 10.1056/NEJMoa1307491
106. Casanova MF, El-Baz AS, Kamat SS, Dombroski BA, Khalifa F, Elnakib A, et al. Focal cortical dysplasias in autism spectrum disorders. Acta Neuropathol Commun. (2013) 1:67. doi: 10.1186/2051-5960-1-67
107. Setiadi AF, Abbas AR, Jeet S, Wong K, Bischof A, Peng I, et al. IL-17A is associated with the breakdown of the blood-brain barrier in relapsing-remitting multiple sclerosis. J Neuroimmunol. (2019) 332:147–54. doi: 10.1016/j.jneuroim.2019.04.011
108. Milovanovic J, Arsenijevic A, Stojanovic B, Kanjevac T, Arsenijevic D, Radosavljevic G, et al. Interleukin-17 in chronic inflammatory neurological diseases. Front Immunol. (2020) 11:947. doi: 10.3389/fimmu.2020.00947
109. Cipollini V, Anrather J, Orzi F, Iadecola C. Th17 and cognitive impairment: possible mechanisms of action. Front Neuroanat. (2019) 13:95. doi: 10.3389/fnana.2019.00095
110. Shin Yim Y, Park A, Berrios J, Lafourcade M, Pascual LM, Soares N, et al. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature. (2017) 549:482–7. doi: 10.1038/nature23909
111. Barbosa S, Khalfallah O, Forhan A, Galera C, Heude B, Glaichenhaus N, et al. Immune activity at birth and later psychopathology in childhood. Brain Behav Immun Health. (2020) 8:100141. doi: 10.1016/j.bbih.2020.100141
112. Reed MD, Yim YS, Wimmer RD, Kim H, Ryu C, Welch GM, et al. IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature. (2020) 577:249–53. doi: 10.1038/s41586-019-1843-6
113. Hoogenraad CC, Riol-Blanco L. Interleukin-17: a social cytokine. Cell. (2020) 181:517–9. doi: 10.1016/j.cell.2020.03.060
114. Chen G, Liao Q, Ai J, Yang B, Bai H, Chen J, et al. Immune response to COVID-19 during pregnancy. Front Immunol. (2021) 12:675476. doi: 10.3389/fimmu.2021.675476
115. Boulanger-Bertolus J, Pancaro C, Mashour GA. Increasing role of maternal immune activation in neurodevelopmental disorders. Front Behav Neurosci. (2018) 12:230. doi: 10.3389/fnbeh.2018.00230
116. Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science. (2016) 353:772–7. doi: 10.1126/science.aag3194
117. Antoniou E, Orovou E, Sarella A, Iliadou M, Rigas N, Palaska E, et al. Zika virus and the risk of developing microcephaly in infants: a systematic review. Int J Environ Res Public Health. (2020) 17:3806. doi: 10.3390/ijerph17113806
118. Ganguli S, Chavali PL. Intrauterine viral infections: impact of inflammation on fetal neurodevelopment. Front Neurosci. (2021) 15:771557. doi: 10.3389/fnins.2021.771557
119. Carvalho A, Sales HF, Ventura P, Gnoatto-Medeiros M, Brites C, Lucena R. The neurodevelopmental spectrum of congenital Zika infection: a scoping review. Dev Med Child Neurol. (2020) 62:1356–62. doi: 10.1111/dmcn.14675
120. Mlakar J, Korva M, Tul N, Popović M, Poljšak-Prijatelj M, Mraz J, et al. Zika virus associated with microcephaly. N Engl J Med. (2016) 374:951–8. doi: 10.1056/NEJMoa1600651
121. Nielsen-Saines K, Brasil P, Kerin T, Vasconcelos Z, Gabaglia CR, Damasceno L, et al. Delayed childhood neurodevelopment and neurosensory alterations in the second year of life in a prospective cohort of ZIKV-exposed children. Nat Med. (2019) 25:1213–7. doi: 10.1038/s41591-019-0496-1
122. Abtibol-Bernardino MR, de Almeida Peixoto LdFA, de Oliveira GA, de Almeida TF, Rodrigues GRI, Otani RH, et al. Neurological findings in children without congenital microcephaly exposed to Zika virus in utero: a case series study. Viruses. (2020) 12:E1335. doi: 10.3390/v12111335
123. Grant R, Fléchelles O, Tressières B, Dialo M, Elenga N, Mediamolle N, et al. In utero Zika virus exposure and neurodevelopment at 24 months in toddler's normocephalic at birth: a cohort study. BMC Med. (2021) 19:12. doi: 10.1186/s12916-020-01888-0
124. Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JLM, Guimarães KP, et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature. (2016) 534:267–71. doi: 10.1038/nature18296
125. Vianna P, Gomes JdA, Boquett JA, Fraga LR, Schuch JB, Vianna FSL, et al. Zika virus as a possible risk factor for autism spectrum disorder: neuroimmunological aspects. Neuroimmunomodulation. (2018) 25:320–7. doi: 10.1159/000495660
126. Onorati M, Li Z, Liu F, Sousa AMM, Nakagawa N, Li M, et al. Zika virus disrupts phospho-TBK1 localization and mitosis in human neuroepithelial stem cells and radial glia. Cell Rep. (2016) 16:2576–92. doi: 10.1016/j.celrep.2016.08.038
127. Buchwalter RA, Ogden SC, York SB, Sun L, Zheng C, Hammack C, et al. Coordination of Zika virus infection and viroplasm organization by microtubules and microtubule-organizing centers. Cells. (2021) 10:3335. doi: 10.3390/cells10123335
128. Zhu Z, Mesci P, Bernatchez JA, Gimple RC, Wang X, Schafer ST, et al. Zika virus targets glioblastoma stem cells through a SOX2-integrin αvβ5 axis. Cell Stem Cell. (2020) 26:187–204.e10. doi: 10.1016/j.stem.2019.11.016
129. Beys-da-Silva WO, Rosa RL, Santi L, Berger M, Park SK, Campos AR, et al. Zika virus infection of human mesenchymal stem cells promotes differential expression of proteins linked to several neurological diseases. Mol Neurobiol. (2019) 56:4708–17. doi: 10.1007/s12035-018-1417-x
130. Vernon M, Hicks D. Relationship of rubella, herpes simplex, cytomegalovirus, and certain other viral disabilities. Am Ann Deaf. (1980) 125:529–34. doi: 10.1353/aad.2012.1506
131. Edlich RF, Winters KL, Long WB, Gubler KD. Rubella and congenital rubella (German measles). J Long Term Eff Med Implants. (2005) 15:319–28. doi: 10.1615/JLongTermEffMedImplants.v15.i3.80
132. Mawson AR, Croft AM. Rubella virus infection, the congenital rubella syndrome, and the link to autism. Int J Environ Res Public Health. (2019) 16:E3543. doi: 10.3390/ijerph16193543
133. Miller E, Cradock-Watson JE, Pollock TM. Consequences of confirmed maternal rubella at successive stages of pregnancy. Lancet. (1982) 2:781–4. doi: 10.1016/S0140-6736(82)92677-0
134. Chess S. Autism in children with congenital rubella. J Autism Child Schizophr. (1971) 1:33–47. doi: 10.1007/BF01537741
135. Hutton J. Does rubella cause autism: a 2015 reappraisal? Front Hum Neurosci. (2016) 10:25. doi: 10.3389/fnhum.2016.00025
136. Stubbs EG. Autistic children exhibit undetectable hemagglutination-inhibition antibody titers despite previous rubella vaccination. J Autism Child Schizophr. (1976) 6:269–74. doi: 10.1007/BF01543467
137. Louie JK, Acosta M, Jamieson DJ, Honein MA, California Pandemic (H1N1) Working Group. Severe 2009 H1N1 influenza in pregnant and postpartum women in California. N Engl J Med. (2010) 362:27–35. doi: 10.1056/NEJMoa0910444
138. Misra RS, Nayak JL. The importance of vaccinating children and pregnant women against influenza virus infection. Pathogens. (2019) 8:E265. doi: 10.3390/pathogens8040265
139. Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. (2003) 23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003
140. Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, et al. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry. (2004) 61:774–80. doi: 10.1001/archpsyc.61.8.774
141. Parboosing R, Bao Y, Shen L, Schaefer CA, Brown AS. Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiatry. (2013) 70:677–85. doi: 10.1001/jamapsychiatry.2013.896
142. Zerbo O, Qian Y, Yoshida C, Fireman BH, Klein NP, Croen LA. Association between influenza infection and vaccination during pregnancy and risk of autism spectrum disorder. JAMA Pediatr. (2017) 171:e163609. doi: 10.1001/jamapediatrics.2016.3609
143. Antoun S, Ellul P, Peyre H, Rosenzwajg M, Gressens P, Klatzmann D, Delorme R. Fever during pregnancy as a risk factor for neurodevelopmental disorders: results from a systematic review and meta-analysis. Mol Autism. (2021) 12:60. doi: 10.1186/s13229-021-00464-4
144. Ludvigsson JF, Winell H, Sandin S, Cnattingius S, Stephansson O, Pasternak B. Maternal influenza A(H1N1) immunization during pregnancy and risk for autism spectrum disorder in offspring : a cohort study. Ann Intern Med. (2020) 173:597–604. doi: 10.7326/M20-0167
145. Shet A. Congenital and perinatal infections: throwing new light with an old TORCH. Indian J Pediatr. (2011) 78:88–95. doi: 10.1007/s12098-010-0254-3
146. Neu N, Duchon J, Zachariah P. TORCH infections. Clin Perinatol. (2015) 42:77–103. doi: 10.1016/j.clp.2014.11.001
147. Straface G, Selmin A, Zanardo V, De Santis M, Ercoli A, Scambia G. Herpes simplex virus infection in pregnancy. Infect Dis Obstet Gynecol. (2012) 2012:385697. doi: 10.1155/2012/385697
148. Mahic M, Mjaaland S, Bøvelstad HM, Gunnes N, Susser E, Bresnahan M, et al. Maternal immunoreactivity to herpes simplex virus 2 and risk of autism spectrum disorder in male offspring. mSphere. (2017) 2:e00016–17. doi: 10.1128/mSphere.00016-17
149. Slawinski BL, Talge N, Ingersoll B, Smith A, Glazier A, Kerver J, et al. Maternal cytomegalovirus sero-positivity and autism symptoms in children. Am J Reprod Immunol. (2018) 79:e12840. doi: 10.1111/aji.12840
150. Kong Q, Li J, Zhao L, Shi P, Liu X, Bian C, et al. Human cytomegalovirus inhibits the proliferation and invasion of extravillous cytotrophoblasts via Hippo-YAP pathway. Virol J. (2021) 18:214. doi: 10.1186/s12985-021-01681-2
151. Liu T, Zheng X, Li Q, Chen J, Yin Z, Xiao J, et al. Role of human cytomegalovirus in the proliferation and invasion of extravillous cytotrophoblasts isolated from early placentae. Int J Clin Exp Med. (2015) 8:17248–60.
152. Stubbs EG, Ash E, Williams CP. Autism and congenital cytomegalovirus. J Autism Dev Disord. (1984) 14:183–9. doi: 10.1007/BF02409660
153. Booss J, Dann PR, Griffith BP, Kim JH. Host defense response to cytomegalovirus in the central nervous system. Predominance of the monocyte. Am J Pathol. (1989) 134:71–78.
154. Maeyama K, Tomioka K, Nagase H, Yoshioka M, Takagi Y, Kato T, et al. Congenital cytomegalovirus infection in children with autism spectrum disorder: systematic review and meta-analysis. J Autism Dev Disord. (2018) 48:1483–91. doi: 10.1007/s10803-017-3412-x
155. Gabrielli L, Bonasoni MP, Santini D, Piccirilli G, Chiereghin A, Petrisli E, et al. Congenital cytomegalovirus infection: patterns of fetal brain damage. Clin Microbiol Infect. (2012) 18:E419–27. doi: 10.1111/j.1469-0691.2012.03983.x
156. Krstanović F, Britt WJ, Jonjić S, Brizić I. Cytomegalovirus infection and inflammation in developing brain. Viruses. (2021) 13:1078. doi: 10.3390/v13061078
157. Cicin-Sain L, Brien JD, Uhrlaub JL, Drabig A, Marandu TF, Nikolich-Zugich J. Cytomegalovirus infection impairs immune responses and accentuates T-cell pool changes observed in mice with aging. PLoS Pathog. (2012) 8:e1002849. doi: 10.1371/journal.ppat.1002849
158. Lei J, Xie L, Zhao H, Gard C, Clemens JL, McLane MW, et al. Maternal CD8+ T-cell depletion alleviates intrauterine inflammation-induced perinatal brain injury. Am J Reprod Immunol. (2018) 79:e12798. doi: 10.1111/aji.12798
159. Lin X, Chen Y, Fang Z, Chen Q, Chen L, Han Q, et al. Effects of cytomegalovirus infection on extravillous trophoblast cells invasion and immune function of NK cells at the maternal-fetal interface. J Cell Mol Med. (2020) 24:11170–6. doi: 10.1111/jcmm.15638
160. Bernstein HB, Wegman AD. HIV Infection: antepartum Treatment and Management. Clin Obstet Gynecol. (2018) 61:122–36. doi: 10.1097/GRF.0000000000000330
161. Maury W, Potts BJ, Rabson AB. HIV-1 infection of first-trimester and term human placental tissue: a possible mode of maternal-fetal transmission. J Infect Dis. (1989) 160:583–8. doi: 10.1093/infdis/160.4.583
162. Townsend CL, Cortina-Borja M, Peckham CS, de Ruiter A, Lyall H, Tookey PA. Low rates of mother-to-child transmission of HIV following effective pregnancy interventions in the United Kingdom and Ireland, 2000-2006. AIDS. (2008) 22:973–81. doi: 10.1097/QAD.0b013e3282f9b67a
163. Johnson EL, Chakraborty R. HIV-1 at the placenta: immune correlates of protection and infection. Curr Opin Infect Dis. (2016) 29:248–55. doi: 10.1097/QCO.0000000000000267
164. Budd MA, Calli K, Samson L, Bowes J, Hsieh AYY, Forbes JC, et al. Blood mitochondrial DNA content in HIV-exposed uninfected children with autism spectrum disorder. Viruses. (2018) 10:E77. doi: 10.3390/v10020077
165. Jao J, Jacobson DL, Russell JS, Wang J, Yu W, Gojanovich GS, et al. Perinatally acquired HIV infection is associated with abnormal blood mitochondrial function during childhood/adolescence. AIDS. (2021) 35:1385–94. doi: 10.1097/QAD.0000000000002884
166. Singh K, Singh IN, Diggins E, Connors SL, Karim MA, Lee D, et al. Developmental regression and mitochondrial function in children with autism. Ann Clin Transl Neurol. (2020) 7:683–94. doi: 10.1002/acn3.51034
167. Mahallawi WH, Khabour OF, Zhang Q, Makhdoum HM, Suliman BA. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine. (2018) 104:8–13. doi: 10.1016/j.cyto.2018.01.025
168. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘Cytokine Storm' in COVID-19. J Infect. (2020) 80:607–13. doi: 10.1016/j.jinf.2020.03.037
169. Mendoza VMM. Interleukin-17: a potential therapeutic target in COVID-19. J Infect. (2020) 81:e136–8. doi: 10.1016/j.jinf.2020.05.072
170. Cunningham L, Kimber I, Basketter DA, McFadden JP. Why judiciously timed anti-IL 6 therapy may be of benefit in severe COVID-19 infection. Autoimmun Rev. (2020) 19:102563. doi: 10.1016/j.autrev.2020.102563
171. Pellegrini L, Albecka A, Mallery DL, Kellner MJ, Paul D, Carter AP, et al. SARS-CoV-2 infects the brain choroid plexus and disrupts the blood-csf barrier in human brain organoids. Cell Stem Cell. (2020) 27:951–61.e5. doi: 10.1016/j.stem.2020.10.001
172. Clifton VL. Review: sex and the human placenta: mediating differential strategies of fetal growth and survival. Placenta. (2010) 31(Suppl.):S33–9. doi: 10.1016/j.placenta.2009.11.010
173. Varì R, Scazzocchio B, Filardi T, Citarella A, Bellenghi M, Masella R, et al. Significance of sex differences in ncRNAs expression and function in pregnancy and related complications. Biomedicines. (2021) 9:1509. doi: 10.3390/biomedicines9111509
174. Wang J-Q, Hu Y-B, Gao H, Sheng J, Huang K, Zhang Y-W, et al. Sex-specific difference in placental inflammatory transcriptional biomarkers of maternal phthalate exposure: a prospective cohort study. J Expo Sci Environ Epidemiol. (2020) 30:835–44. doi: 10.1038/s41370-020-0200-z
175. Osei-Kumah A, Smith R, Jurisica I, Caniggia I, Clifton VL. Sex-specific differences in placental global gene expression in pregnancies complicated by asthma. Placenta. (2011) 32:570–8. doi: 10.1016/j.placenta.2011.05.005
176. Jacobsen H, Klein SL. Sex differences in immunity to viral infections. Front Immunol. (2021) 12:720952. doi: 10.3389/fimmu.2021.720952
177. Spiering AE, de Vries TJ. Why females do better: the X chromosomal TLR7 gene-dose effect in COVID-19. Front Immunol. (2021) 12:756262. doi: 10.3389/fimmu.2021.756262
178. Dubois A, Deuve JL, Navarro P, Merzouk S, Pichard S, Commere P-H, et al. Spontaneous reactivation of clusters of X-linked genes is associated with the plasticity of X-inactivation in mouse trophoblast stem cells. Stem Cells. (2014) 32:377–90. doi: 10.1002/stem.1557
179. Nugent BM, O'Donnell CM, Epperson CN, Bale TL. Placental H3K27me3 establishes female resilience to prenatal insults. Nat Commun. (2018) 9:2555. doi: 10.1038/s41467-018-04992-1
180. Nugent BM, Bale TL. The omniscient placenta: metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol. (2015) 39:28–37. doi: 10.1016/j.yfrne.2015.09.001
181. Bordt EA, Shook LL, Atyeo C, Pullen KM, De Guzman RM, Meinsohn M-C, et al. Maternal SARS-CoV-2 infection elicits sexually dimorphic placental immune responses. Sci Transl Med. (2021) 13:eabi7428. doi: 10.1126/scitranslmed.abi7428
182. Yockey LJ, Lucas C, Iwasaki A. Contributions of maternal and fetal antiviral immunity in congenital disease. Science. (2020) 368:608–12. doi: 10.1126/science.aaz1960
183. Braun AE, Carpentier PA, Babineau BA, Narayan AR, Kielhold ML, Moon HM, et al. “Females Are Not Just ‘Protected' Males”: sex-specific vulnerabilities in placenta and brain after prenatal immune disruption. eNeuro. (2019) 6:ENEURO.0358-19.2019. doi: 10.1523/ENEURO.0358-19.2019
184. Roberts BJ, Moussawi M, Huber SA. Sex differences in TLR2 and TLR4 expression and their effect on coxsackievirus-induced autoimmune myocarditis. Exp Mol Pathol. (2013) 94:58–64. doi: 10.1016/j.yexmp.2012.06.005
185. Sankaran-Walters S, Macal M, Grishina I, Nagy L, Goulart L, Coolidge K, et al. Sex differences matter in the gut: effect on mucosal immune activation and inflammation. Biol Sex Differ. (2013) 4:10. doi: 10.1186/2042-6410-4-10
186. Hewagama A, Patel D, Yarlagadda S, Strickland FM, Richardson BC. Stronger inflammatory/cytotoxic T-cell response in women identified by microarray analysis. Genes Immun. (2009) 10:509–16. doi: 10.1038/gene.2009.12
187. Zhang MA, Rego D, Moshkova M, Kebir H, Chruscinski A, Nguyen H. Peroxisome proliferator-activated receptor (PPAR)α and -γ regulate IFNγ and IL-17A production by human T cells in a sex-specific way. Proc Natl Acad Sci USA. (2012) 109:9505–10. doi: 10.1073/pnas.1118458109
188. Afshan G, Afzal N, Qureshi S. CD4+CD25(hi) regulatory T cells in healthy males and females mediate gender difference in the prevalence of autoimmune diseases. Clin Lab. (2012) 58:567–571.
189. Wei J, Wang F, Kong L-Y, Xu S, Doucette T, Ferguson SD, et al. miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res. (2013) 73:3913–26. doi: 10.1158/0008-5472.CAN-12-4318
190. Ohata T, Wutz A. Reactivation of the inactive X chromosome in development and reprogramming. Cell Mol Life Sci. (2013) 70:2443–61. doi: 10.1007/s00018-012-1174-3
191. Migeon BR, Axelman J, Jeppesen P. Differential X reactivation in human placental cells: implications for reversal of X inactivation. Am J Hum Genet. (2005) 77:355–64. doi: 10.1086/432815
192. Cissé YM, Chan JC, Nugent BM, Banducci C, Bale TL. Brain and placental transcriptional responses as a readout of maternal and paternal preconception stress are fetal sex specific. Placenta. (2020) 100:164–70. doi: 10.1016/j.placenta.2020.06.019
193. Dillon EF, Kanne S, Landa RJ, Annett R, Bernier R, Bradley C, et al. Sex differences in autism: examining intrinsic and extrinsic factors in children and adolescents enrolled in a national ASD cohort. J Autism Dev Disord. (2021). doi: 10.1007/s10803-021-05385-y. [Epub ahead of print].
194. Lai M-C, Lerch JP, Floris DL, Ruigrok ANV, Pohl A, Lombardo MV, et al. Imaging sex/gender and autism in the brain: etiological implications. J Neurosci Res. (2017) 95:380–97. doi: 10.1002/jnr.23948
195. Schubert C. Male biological clock possibly linked to autism, other disorders. Nat Med. (2008) 14:1170–0. doi: 10.1038/nm1108-1170a
196. Merikangas AK, Almasy L. Using the tools of genetic epidemiology to understand sex differences in neuropsychiatric disorders. Genes Brain Behav. (2020) 19:e12660. doi: 10.1111/gbb.12660
197. Xia Y, Xiao J, Yu Y, Tseng W-L, Lebowitz E, DeWan AT, et al. Rates of neuropsychiatric disorders and gestational age at birth in a danish population. JAMA Netw Open. (2021) 4:e2114913. doi: 10.1001/jamanetworkopen.2021.14913
198. Lai M-C, Lombardo MV, Auyeung B, Chakrabarti B, Baron-Cohen S. Sex/gender differences and autism: setting the scene for future research. J Am Acad Child Adolesc Psychiatry. (2015) 54:11–24. doi: 10.1016/j.jaac.2014.10.003
199. Pfaff DW, Rapin I, Goldman S. Male predominance in autism: neuroendocrine influences on arousal and social anxiety. Autism Res. (2011) 4:163–76. doi: 10.1002/aur.191
200. Han VX, Patel S, Jones HF, Dale RC. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat Rev Neurol. (2021) 17:564–579. doi: 10.1038/s41582-021-00530-8
Keywords: ASD autismspectrumdisorders, UIA uterine immune activation, Tregs regulatory T cells, Th17 T helper 17 cells, IL17a interleukin 17a isoform, poly I, C polyinosinic, polycytidylic acid
Citation: Jash S and Sharma S (2022) Viral Infections and Temporal Programming of Autism Spectrum Disorders in the Mother's Womb. Front. Virol. 2:863202. doi: 10.3389/fviro.2022.863202
Received: 26 January 2022; Accepted: 22 February 2022;
Published: 29 April 2022.
Edited by:
Vikki M. Abrahams, Yale University, United StatesReviewed by:
Irina Burd, Johns Hopkins Medicine, United StatesCopyright © 2022 Jash and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Surendra Sharma, c3NoYXJtYUB3aWhyaS5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.