
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol. , 13 April 2022
Sec. Bioinformatic and Predictive Virology
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.840952
This article is part of the Research Topic Insights In Bioinformatic and Predictive Virology:2021 View all 4 articles
 Loïc Borcard
Loïc Borcard Sonja GempelerMiguel A. Terrazos MianiChristian BaumannCarole GrädelRonald DijkmanFranziska Suter-Riniker
Sonja GempelerMiguel A. Terrazos MianiChristian BaumannCarole GrädelRonald DijkmanFranziska Suter-Riniker Stephen L. LeibPascal BittelStefan Neuenschwander
Stephen L. LeibPascal BittelStefan Neuenschwander Alban Ramette*
Alban Ramette*The SARS-CoV-2 Delta variant, corresponding to the Pangolin lineage B.1.617.2, was first detected in India in July 2020 and rapidly became dominant worldwide. The ARTIC v3 protocol for SARS-CoV-2 whole-genome sequencing, which relies on a large number of PCR primers, was among the first available early in the pandemic, but may be prone to coverage dropouts that result in incomplete genome sequences. A new set of primers (v4) was designed to circumvent this issue in June 2021. In this study, we investigated whether the sequencing community adopted the new sets of primers, especially in the context of the spread of the Delta lineage, in July 2021. Because information about protocols from individual laboratories is generally difficult to obtain, the aims of the study were to identify whether large under-sequenced regions were present in deposited Delta variant genome sequences (from April to August 2021), to investigate the extent of the coverage dropout among all the currently available Delta sequences in six countries, and to propose simple PCR primer modifications to sequence the missing region, especially for the first circulating Delta variants observed in 2021 in Switzerland. Candidate primers were tested on few clinical samples, highlighting the need to further pursue primer optimization and validation on a larger and diverse set of samples.
In late 2019, SARS-CoV-2 was detected in Wuhan (Hubei province, China). This discovery rapidly evolved in the global COVID-19 pandemic. SARS-CoV-2 is a positive-strand RNA virus of the Betacoronavirus genus from Coronaviridae family. The spike (S) protein, encoded by the S gene, has a key role in receptor recognition and cell membrane fusion process, and the emergence of new variants often relies on the appearance of mutations within this region, the predominant antigen of the virus (1, 2). There is a worldwide effort to monitor the emergence and evolution of SARS-CoV-2 genotypes due to the implications these new variants have for public health, society, and scientific research. Tools such as Nextstrain or Pangolin were developed to characterize emerging variants (3, 4). Such efforts have permitted the discovery of emerging variants, such as Alpha, Beta, and Delta, and to follow their progression in the human population. In October 2020, the Delta variant, corresponding to the Pangolin lineage B.1.617.2, was first detected in India. Since then, this variant has been detected in most countries and has rapidly become dominant worldwide (5–7). The Delta variant lineage harbors 29 mutations as compared to the Wuhan-Hu-1 reference virus sequence (e.g., S:P681R and S:L452R; Table 1). As of 12 August 2021, it was present in 115 countries. The Delta variant comprises several descendants of B.1.617.2, such as AY.4 and AY.12.
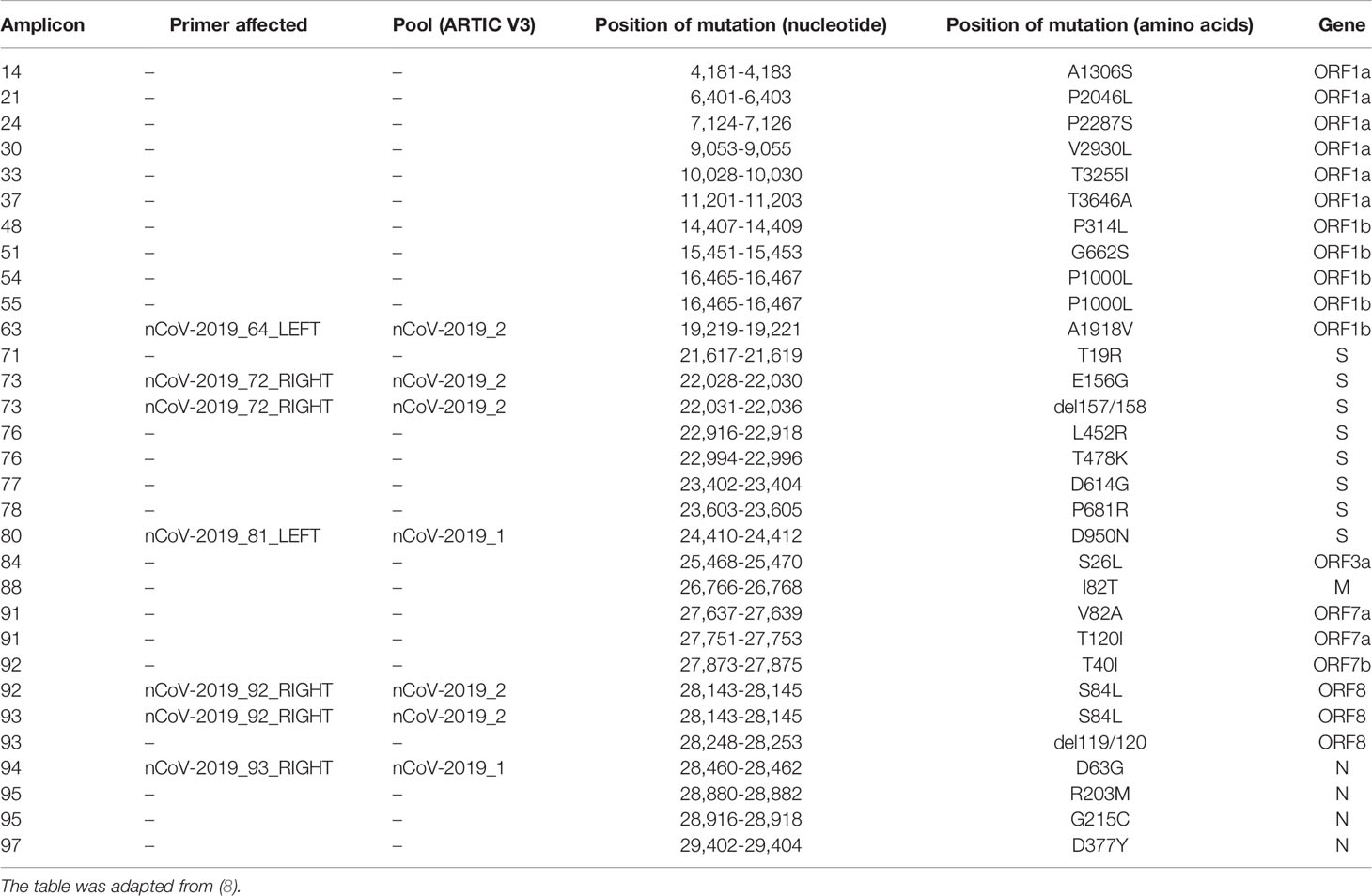
Table 1 Notable mutations of the Delta variant.
Whole-genome sequencing (WGS) currently represents the methodology of choice for lineage classification and for providing fine genomic comparisons among viral isolates from local to global scales, allowing their surveillance in an unprecedented close-to-real-time manner. The rapid near-full-length sequencing of the virus genome has been enabled by the combination of, on the one hand, protocols that used amplicon tilling strategies, whereby allowing the fast and reliable production of complete genome sequences based on sets of carefully chosen amplicons and on the other hand, the generalized use of Next-Generation sequencing (NGS) technologies to massively sequence the resulting amplicons via short-read or long-read NGS technologies (9, 10). Among the often-used approaches, the ARTIC protocol has received much attention, as it was among the first available early in the pandemic (12 February 2020). The ARTIC network pipeline comprises a set of materials to assist sequencing by providing laboratory and bioinformatic protocols (11). Each of the 98 pairs of proposed PCR primers in the protocol produces an insert of approximately 400 bases. The bioinformatic analysis of the data is performed by first filtering the reads, trimming primer sequences, and normalizing read coverage. Then, variant calling and consensus-building are performed. The consensus sequences are then taxonomically classified using NextClade (3) or the dynamic approach of Pangolin (4).
The reliance on a PCR-based strategy to rapidly sequence viral genomes may, however, present drawbacks: Amplicon sequencing is prone to coverage dropout that results in incomplete genome sequences. This phenomenon is identified by the presence of unknown bases (represented by “N”) in consensus sequences, which may prevent correct lineage classification. In addition, there is an inherent danger when relying on genome-specific primers, as the template used to create these primers is bound to rapidly become obsolete as the viral population evolves. Thus, there should be a constant effort to limit the emergence of under-sequenced regions (USR) due to mismatching primers resulting from viral mutations. In June 2021, a new set of primers (v4) (12) was designed to circumvent the mismatching of ARTIC v3 primers due to the emergence of SARS-CoV-2 Beta and Delta variants.
In this study, we investigated whether the sequencing community adopted the new sets of primers, especially in the context of the emergence and spread of the Delta lineage. Because information about protocols from individual laboratories, especially concerning amplification strategy, is difficult to obtain, the aims of the study were to identify whether large USRs were present in deposited Delta variant genome sequences, to investigate the extent of the coverage dropout among all the currently available Delta sequences from six countries, and to propose primer modifications in the ARTIC v3 protocol to allow the sequencing of the missing regions in circulating Delta variants observed in May-July 2021 in Switzerland.
Viral RNA samples analyzed in the study were obtained during the routine sequencing of SARS-CoV-2 viral genomes done at the Institute for Infectious Diseases (IFIK, Bern, Switzerland) for diagnostic or surveillance purposes. Ethics approval was granted by the Cantonal ethical commission for Research, Canton of Bern, Switzerland, on 17 February 2020 (GSI-KEK, BASEC-Nr Req-2020-00167) to sequence and genomically compare SARS-CoV-2 isolates starting from previously screened samples sent to the IFIK for viral diagnostics by treating physicians. Representative RNA samples were collected from 17 to 25 July 2021 from Lambda and Delta variants. All test results were anonymized prior to statistical analysis. Genome sequence information of all isolates is already available in the GISAID database [https://www.gisaid.org/; (13)]. Total nucleic acids were extracted using the STARMag 96 X4 Universal Cartridge Kit on a Seegene STARlet liquid handling platform (Seegene Inc., Seoul, South Korea). Briefly, 200 µl of original material was extracted in 100 µl elution buffer according to the manufacturer’s instructions. Nucleic acid eluates were immediately stored at -80°C after processing until further use. To investigate the situation in other countries, we obtained a total of 436,820 sequences from GISAID on 12 August 2021, consisting of all Delta variants (B1.167.2, and sub-lineages AY.1; AY.2; AY.3; AY.3.1) deposited since April 24 2021. The GISAID database offers the most exhaustive collection of curated sequences of SARS-CoV-2 originating from patient samples.
The ROI (Region of Interest, i.e., positions 21,357-22,246 of SARS-CoV-2 reference genome MN908947.3) was selected in the FASTA sequences by performing in silico PCR with seqkit [v0.16; (14)] using the ROI flanking primers. The forward primer binds between positions 21,357-21,386 (Table 2) and the reverse primer to positions 22,324-22,346 of the reference sequence. The proportion of Ns in the ROI sequences was calculated with the alphabetfrequency function of the Biostrings package (15). Next the total frequency of Ns was computed using the fx2tab function of seqkit. Simultaneously, we analyzed the presence of segments of N within the entire genome of SARS-CoV-2 by using the locate function of seqkit. All of this information was merged into one single dataset.

Table 2 Original primers (ARTIC v3 protocol) and alternative primers binding to the ROI defined in this study.
The RT-PCR mix contained for each sample 16 µl RNA and 4 µl LunaScript RT Supermix (5x) (New England BioLabs, Ipswich, USA). The RT-PCR conditions were set to 2 min 25°C, 10 min 55°C, 1 min 95°C on a S1000 Thermal Cycler (Bio-Rad, Cressier, Switzerland). A PCR Mastermix with 12.5 µl Q5 Hot Start High-Fidelity 2x Master Mix (New England BioLabs), 1.85 µl of each 10 nM primers L and R (Microsynth AG, Balgach, Switzerland) and 3.8 µl nuclease-free water (New England BioLabs) was prepared per sample. A total of 20 µl of the PCR Mastermix was added to 5 µl of the RT sample. The PCR was set on a Bio-Rad S1000 Thermal Cycler with initial denaturation at 98°C (30s), followed by 35 amplification cycles of 98°C for 15 s and 65°C for 5 min. Gel electrophoresis was performed for 1 h at 120 volts with 2% agarose gels prepared in 1x TAE buffer with 2.5 µl RedSafe (20,000x) (iNtRON Biotechnology, Burlington, USA). A total of 10 µl PCR reaction with DNA loading buffer (ROTI Load DNA with Saccharose, Carl Roth GmbH) was loaded per well, alongside a benchtop 100-bp DNA Ladder (Promega, Dübendorf, Switzerland). Gel images were acquired with the Fusio Fx (Vilber; Witec, Sursee, Switzerland) gel-documentation system. Purified PCR products were sent for Sanger sequencing at Microsynth. Forward and reverse strands were assembled into consensus sequences using SeqMan Pro (DNAStar, Madison, WI, USA).
SARS-CoV-2 RNA genomes were reverse-transcribed and amplified following the sequencing strategy of the ARTIC v3 protocol (https://artic.network/ncov-2019), which generates 400-bp amplicons that overlap by approximately 20 bp and covers the whole target genomes. Nanopore library preparation was performed with SQK-LSK109 (Oxford Nanopore Technologies, Oxford, UK) according to the ONT “PCR tiling of COVID-19 virus” (version: PTC_9096_v109_revE_06Feb2020, last update: 26/03/2020). Reagents, quality control and flow cell preparation were done as described previously (16, 17). Sequencing was performed on GridION X5 (Oxford Nanopore Technologies) with real-time basecalling enabled (ont-guppy-for-gridion v.4.2.3; high-accuracy basecalling mode). Bioinformatic analyses followed the ARTIC workflow (https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html) using version 1.1.3. Consensus sequences were generated using medaka (https://github.com/nanoporetech/medaka) and bcftools (18). The genome assembly sequences were deposited to the European Nucleotide Archive (ENA), under project accession PRJEB50374, and sample accessions ERS10420666-ERS10420673.
During routine sequencing of SARS-CoV-2 genomes at the Institute for Infectious Diseases in May 2021, we initially observed that the region surrounding the start of the S gene of SARS-CoV-2 Delta variant genome sequences (Figure 1) was systematically under-sequenced (Figure 2A), as compared to non-Delta variant sequences (Figure 2B). The consensus sequences displayed a large region containing mostly N (unidentified) bases from positions 21,357 to 22,346 of the genome sequence, region that is flanked by primers 71L and 73R of the ARTIC v3 protocol (11). We refer to this region as region of interest (ROI).
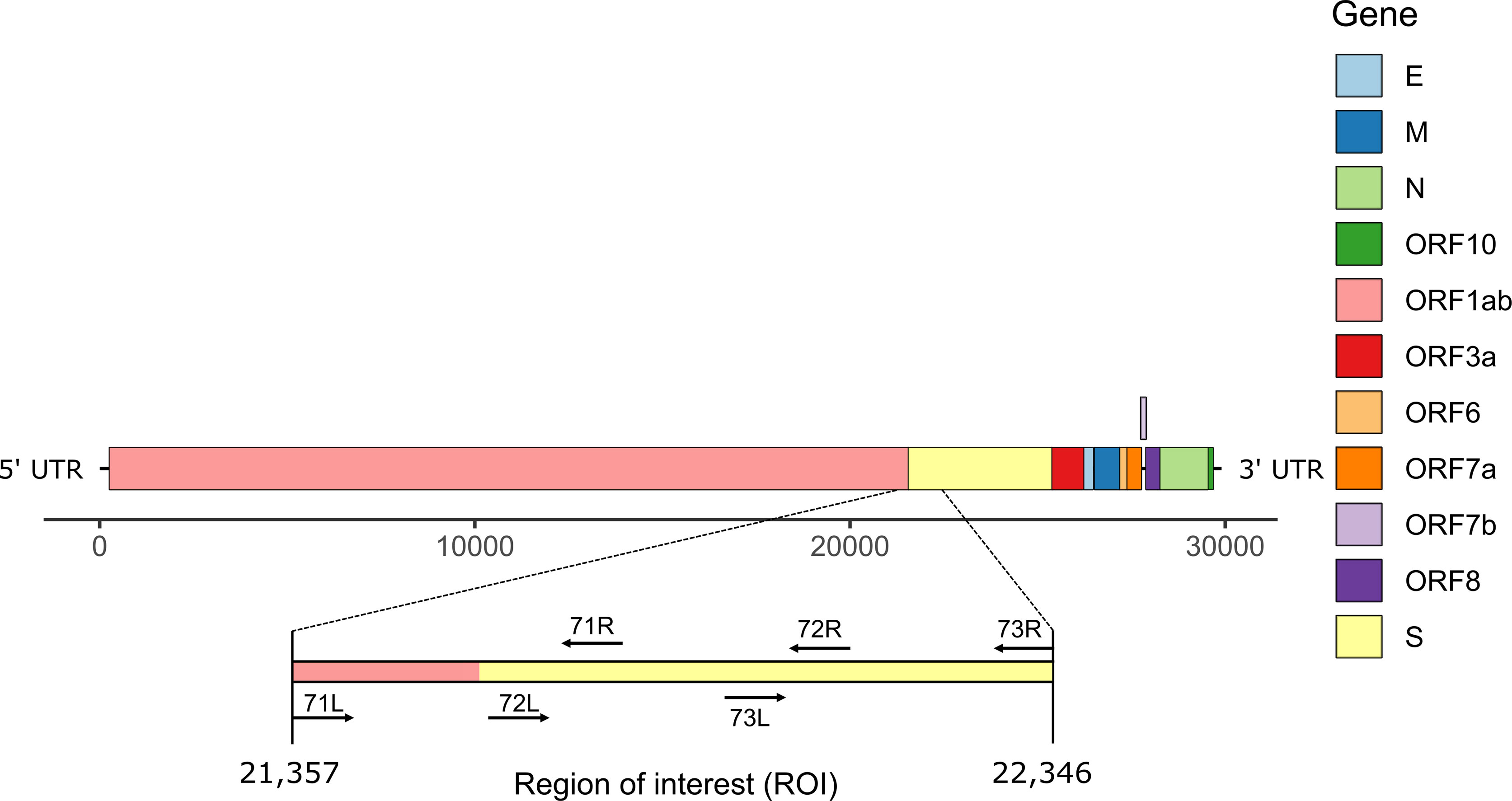
Figure 1 Schematic representation of SARS-CoV-2 genome structure. The region of interest (ROI) at positions 21,357-22,346 corresponds to the region between primers 71L and 73R (ARTIC primers v3).
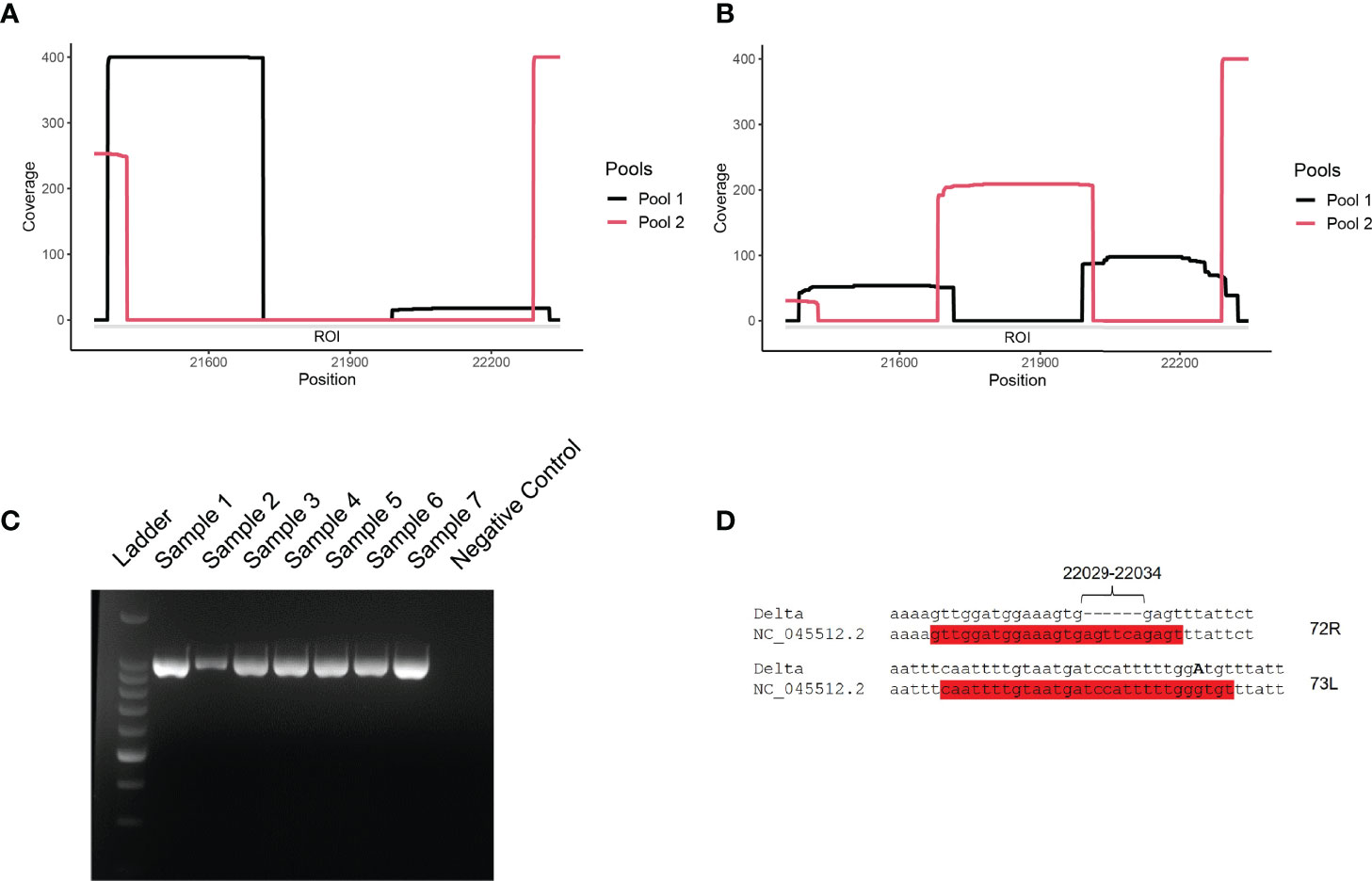
Figure 2 (A, B) Coverage analysis of the ROI (21,357-22,346 nt) for representative Delta variant (A) and lambda variant (B) clinical samples. Snapshots were taken directly from the visualization of the BAM files with IGV software. Pools 1 and 2 correspond to ARTIC v3 primer sets. (C) Agarose gel electrophoresis of PCR amplicons generated with primers 71L and 73R on Lambda (samples 1-2), Delta (samples 3-7), and negative (well 8; non-template control) samples. Ladder: 1-kb ladder. (D) Observed mutations at primer binding sites for primers 72R and 73L. The two primers are highlighted in red and the substitution G21987A is indicated in bold, capital letter.
The coverage of the ROI ranged from 0 to less than 50x (Figure 2A). The cause of the USR in the S gene was examined by PCR amplifying the ROI using USR-flanking primers 71L and 73R (Figure 2C). PCR results confirmed the absence of a major deletion in this region, as the expected ca. 990-nt long region was amplified for all Delta and Lambda samples tested. To test the hypothesis of primer mismatch, PCR amplicons were further sequenced, using Sanger sequencing, over positions 21,357-22,346 using the same two primers (71L and 73R) (Figure 2D). We could identify two non-matching primers: First, primer 72R (22,013-22,038) displayed a truncated binding site due to a deletion between positions 22,031-22,036 (Table 1). Second, a substitution (G21987A, S:G142D) (Table 1) was detected in the primer binding site of 73L, which binds between position 21,961 to 21,990. These two primer sequence mismatches were found in all five consensus sequences considered here, supporting the hypothesis that the low coverage of the ROI was due to the compromised binding of certain primers. These results are in accordance with previous reports indicating a mismatching of primer 72R in this region of the S gene (12).
We further investigated SARS-CoV-2 sequences originating from six countries (England, France, Germany, India, Switzerland, USA). First, we assessed the presence of N-containing sequences in the ROI of Delta variants by looking at all sequences available in the GISAID database until 12 August 2021 (i.e., B.1.167.2 and its descendants). We determined the proportion of Ns present in the ROI for each sequence by extracting the subsequence between primers 71L and 73R and compared it to the rest of the sequence. We were interested in longer USRs originating from PCR mis-amplification than in small segments containing N bases. Therefore, we filtered out all N-containing segments smaller than 200 nt and removed sequences with low coverage (overall N > 5%), following the criteria used by GISAID. The mapping of large N-containing segments for each genome clearly indicated that many of these segments were located in the ROI of sequences originating from all six countries considered (Figure 3A), especially when retaining N-containing segments larger than 300-nt (Figure 3B). Another striking observation was the large amounts of N-containing segments outside the ROI corresponding to other USR regions, particularly in countries contributing large amounts of sequencing data (e.g. USA, England). We also observed that the contribution of the USR in the S region to the total number of Ns in each genome sequence was disproportionally high (> 50%). This trend started from the early reported cases of Delta sequences in April-May 2021 across all countries considered here and continued to fluctuate markedly in all countries (Figure 4). Altogether, our observations indicate a clear coverage bias in this ROI specifically and that the issue was generalized geographically and persisted over time. This suggests that, although previous reports revealed the existence of primer mismatches, the sequencing community might not have adopted the new sets of primer sequences.
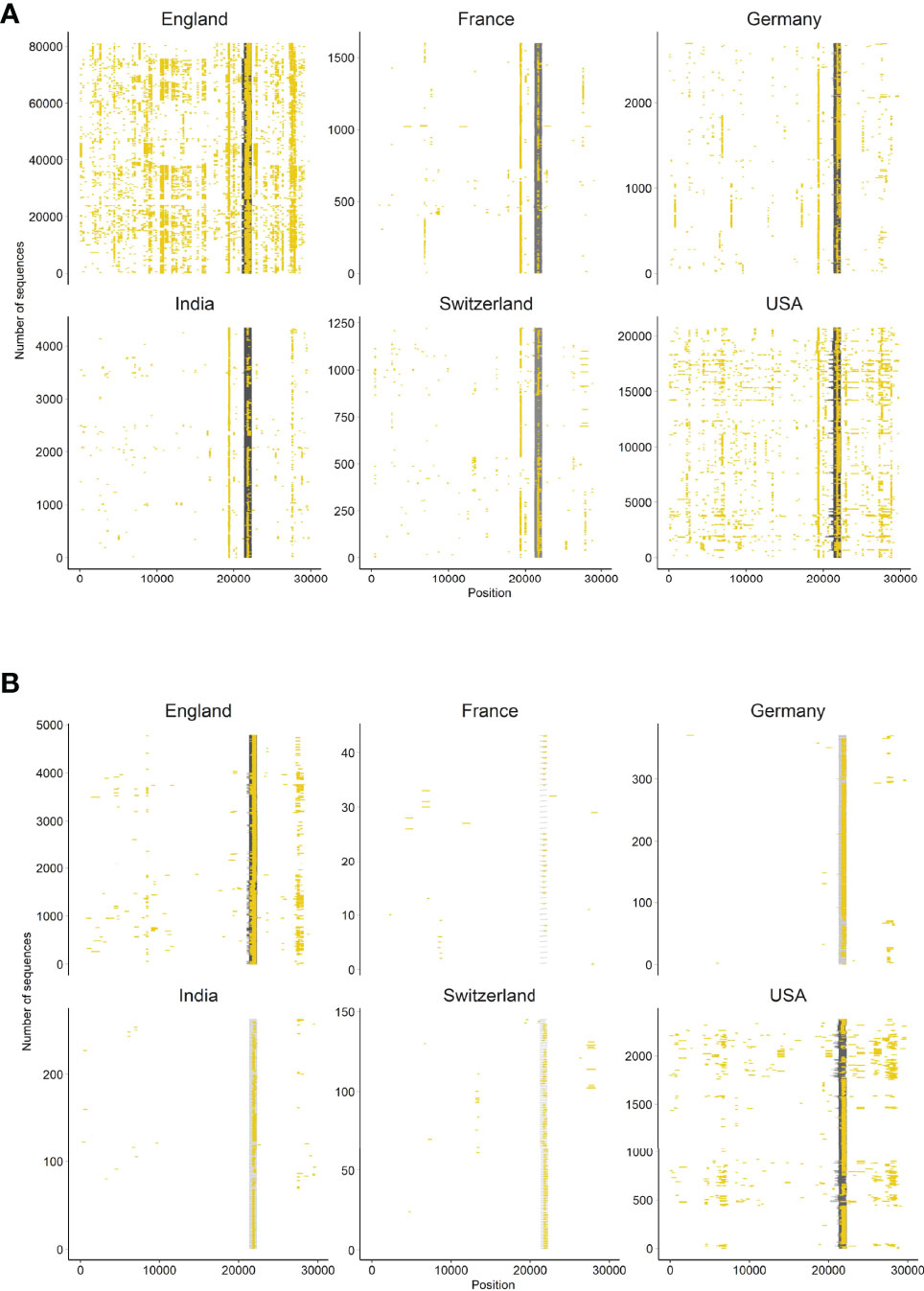
Figure 3 Occurrence of N-containing segments in SARS-CoV-2 genome sequences. Each N-containing segment is depicted in yellow and is positioned on the horizontal axis according to its position within the sequence. The vertical axis represents the number of sequences analyzed among six countries representative of the global situation. Only sequences with overall less than 5% Ns and (A) N-containing segments of lengths >200 nt, and (B) >300 nt are depicted.
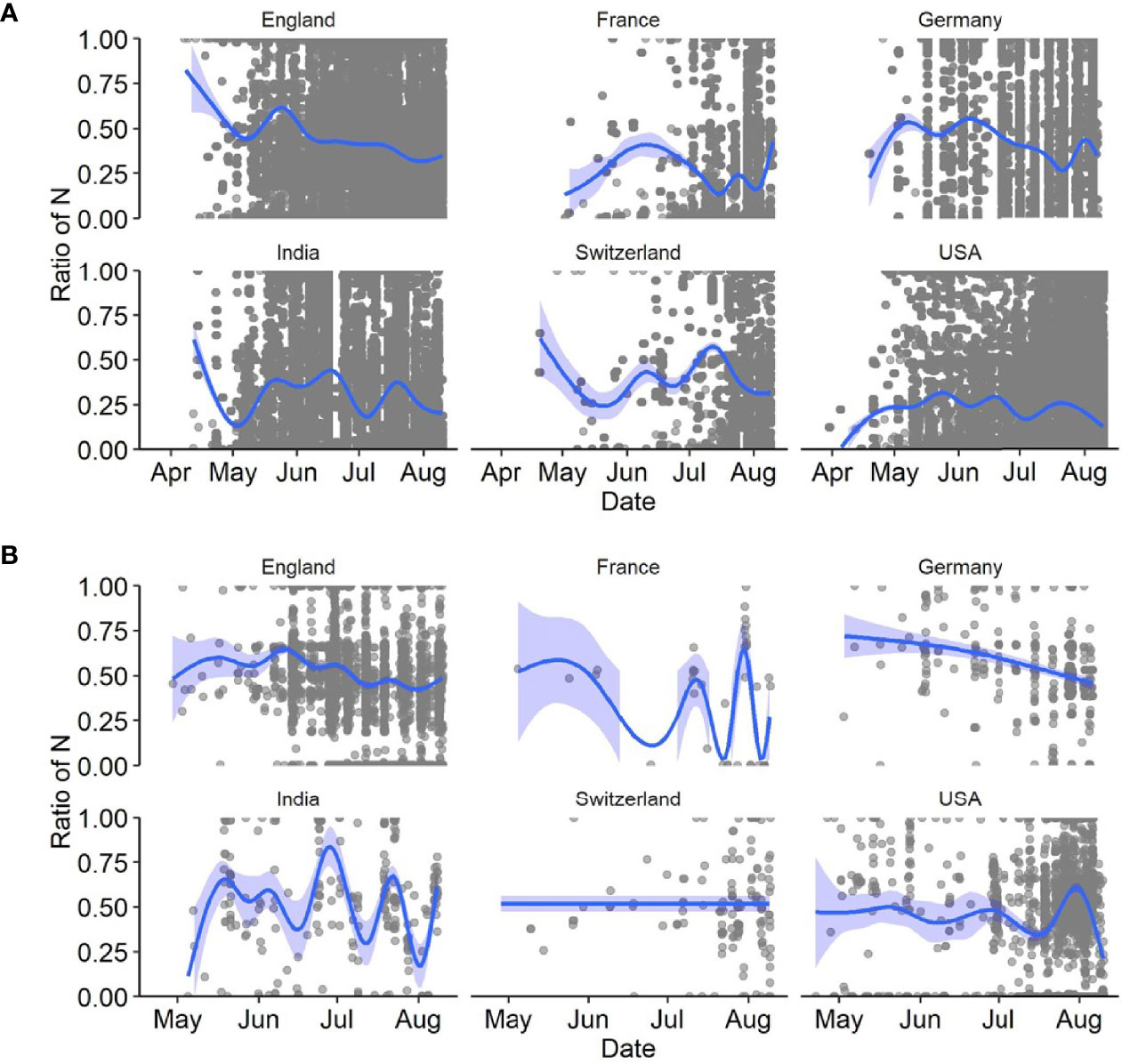
Figure 4 Ratio of number of Ns present in the ROI to the total number of Ns present in the entire genome of SARS-CoV-2 sequences for six countries in 2021. Considered here are (A) all sequences with a total Ns <5%, and (B) those with N-containing segments of length > 200 nt. The dark blue lines depict the best fitting lines of generalized additive models (GAM) with 95% confidence intervals (light blue areas).
Because primer mismatches can lead to lineage miss-classification and missing mutations of interest in the spike region, we modified the primer sequences (72R and 73L) that displayed matching issues. For primer 72R, we generated three alternative primers (72R_alt1, 72R_alt2 and 72R_alt3), and for 73L, another three primers (73L_alt1, 73L_alt2 and 73L_alt3) (Figure 5A and Table 2). All new primers were successfully validated by PCR done in isolation from other primers present in the large primer pools (Figure 5B). We chose to include all three primers to ensure proper amplification of the ROI in case a specific primer would be less efficient than others. Next, to confirm that the newly designed primers increased the coverage of the ROI, we added them to the original pool of ARTIC v3 primers. We chose four different samples comprising three different lineages (B.1.617.2, AY.4, and AY.12). Two different concentrations were used to test the new primers: 1) The primers were added in the same concentration as the other ARTIC primers (i.e., 0.5 μl of 100 mM primer solution), or as 2) twice the standard primer concentrations to favor their use during PCR amplification. First, we observed an increase in coverage for both B.1.617.2 samples (Samples 1 and 2) when using the modified pool of primers (Figure 6). For Sample 3 (AY.4), we observed a clear enhancement of the coverage in the ROI, especially with the higher concentration of primers (Figure 6). Surprisingly, for Sample 4 (AY.12) the region between nucleotide 21,716 and 21,989 did not display any reads at all. These results indicate that the modifications of the primers may allow obtaining complete coverage of Delta variants of lineages B.1.617.2 and AY.4 but not in AY.12, at least for the samples being tested. Differences in Ct values could not explain this unexpected result as both AY.12 samples had Ct values that were in range with the rest of the samples (data not shown). Additionally, both AY.12 and AY.4 have similar mutations in the ROI (Table 1). Thus, we suspect that competing primers might cause this lack of coverage.
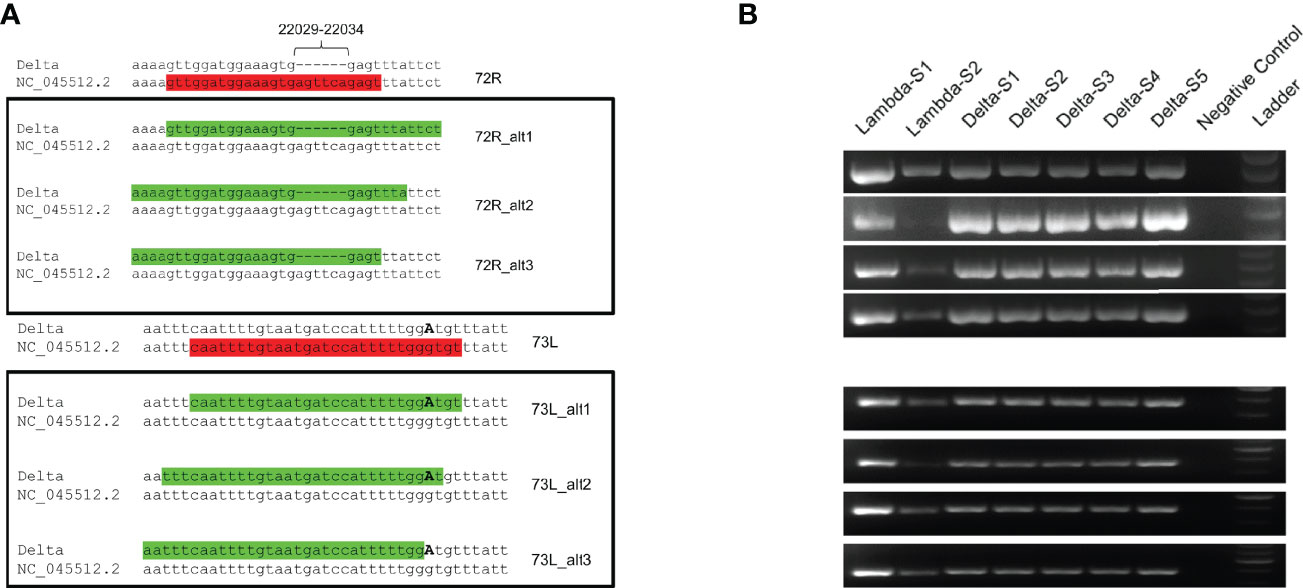
Figure 5 Sequence analysis of proposed primers. (A) Alternative primers for 72R (72R_alt1, 72R_alt2, 72R_alt3) and for 73L (73R_alt1, 73R_alt2, 73R_alt3) primers were generated with Delta variant sequences as template and aligned back to the NC_045512.2 reference sequence. The original primers are indicated in red font, and modified primers in green font, highlighting the presence of missing bases and substitution (G21987A). (B) The alternative primers were validated by PCR under the same conditions as the ones used in the ARTIC v3 protocol.
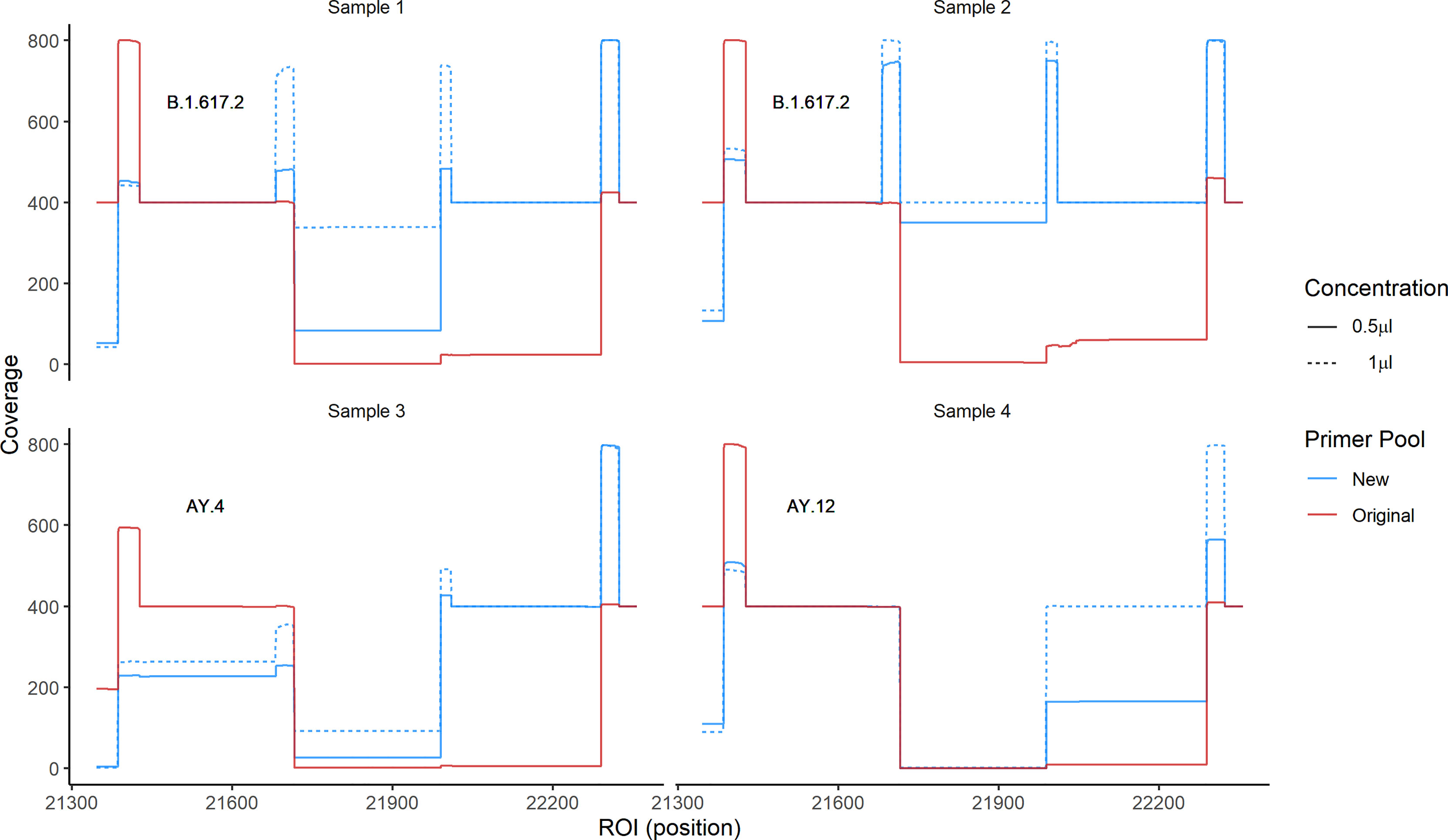
Figure 6 Coverage of the ROI using primer pools containing Delta-specific primers. We sequenced four samples of three different lineages using two pools of primers: 1) The original ARTIC v3 and 2) the ARTIC v3 pools where we added all newly designed primers in two concentrations, with concentration 1 corresponding to the standard primer concentration, and concentration 2 to twice that amount. The coverage using the new pool of primers (blue) and the original pool (red) are shown.
In the context of routine genomic sequencing in our laboratory, we confirmed the presence of a large under-sequenced region in the S gene of the SARS-CoV-2 genome, and that the issue originated from mismatches of ARTIC v3 primers 72R and 73L to the sequences of the then predominant Delta variants. We revealed the presence of this USR in the ROI of genome sequences originating from most laboratories worldwide, which started early with the first appearance of Delta variants. Although others have released updated primers to circumvent such issues (12, 19), the USR continued to appear to large extent in the reported genome sequences globally. Finally, we provided a series of alternative primers for primers 72R and 73L, particularly addressing users of the ARTIC v3 protocol and targeting circulating Delta variants in Switzerland in Sommer 2021. These primers were tested on a reduced dataset of circulating delta variant samples in Switzerland, and they could be added to the large pools of the ARTIC v3 primers to ensure better amplification and sequencing of the ROI for most, but not all tested cases (75% success rate). There is for sure an inherent difficulty to establish new, universal primer sequences given that the large amount of low-covered sequences may hide unknown sequence diversity in the ROI.
To our knowledge, a systematic analysis of the presence of this USR has not been performed elsewhere. Nonetheless, we acknowledge the existence of previous reports updating the primer 72R to address observed mutations in the target genomes (19, 20). Although we could identify and provide a solution for this ROI, our data analysis at the global scale indicated that many USR might also be present in other regions of the genomes of SARS-CoV-2 (Figure 3). Therefore, it is of major importance to regularly control for the presence of USRs and determine how geographically and temporally consistent they become, as they may indicate the apparition of new viral lineages. Although it may be easier to track the laboratory protocols used for SARS-CoV-2 genomic sequencing for countries reporting smaller data volumes, it may be less so for countries providing large amounts of data from many sequencing facilities. Currently, little information is available in the sequence metadata regarding some of the key procedures involved in generating SARS-CoV-2 genome sequences, such as reverse transcription conditions, choice of primer sets, PCR amplification conditions, while more focus is rather given to sequencing technologies. We encourage genome data submitters to provide complete wet laboratory information in the future to allow for higher methodological comparability and for identifying protocols that would need improvement.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: European Nucleotide Archive, PRJEB50374.
LB and AR wrote the first draft of the manuscript. AR supervised the project. LB, CG, SN, and AR analyzed the data bioinformatically. FS-R, SL, and PB provided epidemiological data. FS-R, SL, and PB provided samples. RD provided ethical documentation. SG, MM, CB, and PB performed the experiments. All authors edited and provided valuable feedback which helped to shape the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank the diagnostic department of the Institute for Infectious Diseases, University of Bern for collecting and storing the samples. This article has appeared online in a preprint on the website medRxiv.
1. Walls AC, Park YJ, Tortorici MA, Wall A, Mcguire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell (2020) 181:281–92.e286. doi: 10.1016/j.cell.2020.02.058
2. V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat Rev Microbiol (2021) 19:155–70. doi: 10.1038/s41579-020-00468-6
3. Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics (2018) 34:4121–3. doi: 10.1093/bioinformatics/bty407
4. Rambaut A, Holmes EC, O'toole A, Hill V, Mccrone JT, Ruis C, et al. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat Microbiol (2020) 5:1403–7. doi: 10.1038/s41564-020-0770-5
5. Alm E, Broberg EK, Connor T, Hodcroft EB, Komissarov AB, Maurer-Stroh S, et al. Geographical and Temporal Distribution of SARS-CoV-2 Clades in the WHO European Region, January to June 2020. Euro Surveill (2020) 25(32):2001410. doi: 10.2807/1560-7917.ES.2020.25.32.2001410
6. Alizon S, Haim-Boukobza S, Foulongne V, Verdurme L, Trombert-Paolantoni S, Lecorche E, et al. Rapid Spread of the SARS-CoV-2 Delta Variant in Some French Regions, June 2021. Euro Surveill (2021) 26(28):2100573. doi: 10.2807/1560-7917.ES.2021.26.28.2100573
7. Lopez Bernal J, Andrews N, Gower C, Gallagher E, Simmons R, Thelwall S, et al. Effectiveness of Covid-19 Vaccines Against the B.1.617.2 (Delta) Variant. N Engl J Med (2021) 385:585–94. doi: 10.1056/NEJMoa2108891
8. Alaa Abdel Latif JLM, Manar A, Ginger T, Cano M, Haag E, Zhou J, et al. Outbreak.Info, in: Delta Variant Report (2021). Available at: https://outbreak.info/situation-reports/delta (Accessed 16 February 2022).
9. Charre C, Ginevra C, Sabatier M, Regue H, Destras G, Brun S, et al. Evaluation of NGS-Based Approaches for SARS-CoV-2 Whole Genome Characterisation. Virus Evol (2020) 6:veaa075. doi: 10.1093/ve/veaa075
10. Nasir JA, Kozak RA, Aftanas P, Raphenya AR, Smith KM, Maguire F, et al. A Comparison of Whole Genome Sequencing of SARS-CoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture. Viruses (2020) 12(8):895. doi: 10.3390/v12080895
11. Plitnick J, Griesemer S, Lasek-Nesselquist E, Singh N, Lamson DM, St George K. Whole-Genome Sequencing of SARS-CoV-2: Assessment of the Ion Torrent AmpliSeq Panel and Comparison With the Illumina MiSeq ARTIC Protocol. J Clin Microbiol (2021) 59(12):e0064921. doi: 10.1128/JCM.00649-21
12. Itokawa K, Sekizuka T, Hashino M, Tanaka R, Kuroda M. Disentangling Primer Interactions Improves SARS-CoV-2 Genome Sequencing by Multiplex Tiling PCR. PloS One (2020) 15:e0239403. doi: 10.1371/journal.pone.0239403
13. Shu Y, Mccauley J. GISAID: Global Initiative on Sharing All Influenza Data - From Vision to Reality. Euro Surveill (2017) 22(13):30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494
14. Shen W, Le S, Li Y, Hu F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PloS One (2016) 11:e0163962. doi: 10.1371/journal.pone.0163962
15. Pagès H, Aboyoun P, Gentleman R, Debroy S. Biostrings: Efficient Manipulation of Biological Strings (2021). Available at: https://rdrr.io/bioc/Biostrings/.
16. Gradel C, Terrazos Miani MA, Barbani MT, Leib SL, Suter-Riniker F, Ramette A. Rapid and Cost-Efficient Enterovirus Genotyping From Clinical Samples Using Flongle Flow Cells. Genes (Basel) (2019) 10(9):659. doi: 10.3390/genes10090659
17. Neuenschwander SM, Terrazos Miani MA, Amlang H, Perroulaz C, Bittel P, Casanova C, et al. A Sample-To-Report Solution for Taxonomic Identification of Cultured Bacteria in the Clinical Setting Based on Nanopore Sequencing. J Clin Microbiol (2020) 58(6):e00060–20. doi: 10.1128/JCM.00060-20
18. Li H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation From Sequencing Data. Bioinformatics (2011) 27:2987–93. doi: 10.1093/bioinformatics/btr509
19. Davis JJ, Long SW, Christensen PA, Olsen RJ, Olson R, Shukla M, et al. Analysis of the ARTIC Version 3 and Version 4 SARS-CoV-2 Primers and Their Impact on the Detection of the G142D Amino Acid Substitution in the Spike Protein. Microbiol Spectr (2021) 9:e0180321. doi: 10.1128/Spectrum.01803-21
Keywords: Betacoronaviruses, whole-genome sequencing, next generation sequencing, genomics, SARS-CoV-2
Citation: Borcard L, Gempeler S, Terrazos Miani MA, Baumann C, Grädel C, Dijkman R, Suter-Riniker F, Leib SL, Bittel P, Neuenschwander S and Ramette A (2022) Investigating the Extent of Primer Dropout in SARS-CoV-2 Genome Sequences During the Early Circulation of Delta Variants. Front.Virol. 2:840952. doi: 10.3389/fviro.2022.840952
Received: 21 December 2021; Accepted: 16 March 2022;
Published: 13 April 2022.
Edited by:
Manuela Sironi, Eugenio Medea (IRCCS), ItalyReviewed by:
Richard Johnathan Orton, University of Glasgow, United KingdomCopyright © 2022 Borcard, Gempeler, Terrazos Miani, Baumann, Grädel, Dijkman, Suter-Riniker, Leib, Bittel, Neuenschwander and Ramette. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alban Ramette, YWxiYW4ucmFtZXR0ZUBpZmlrLnVuaWJlLmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.