 Kei Miyakawa
Kei Miyakawa Mayuko Nishi1
Mayuko Nishi1 Sundararaj Stanleyraj Jeremiah
Sundararaj Stanleyraj Jeremiah Yuko Morikawa
Yuko Morikawa Akihide Ryo
Akihide Ryo- 1Department of Microbiology, School of Medicine, Yokohama City University, Kanagawa, Japan
- 2Graduate School of Infection Control Sciences, Kitasato University, Tokyo, Japan
The host innate immune response is the first line of defense against human immunodeficiency virus (HIV) infection. The type I interferon (IFN) response is a robust anti-viral response that induces the transcription of several IFN-stimulated genes (ISGs). However, the effects of ISGs, particularly on the HIV-1 Gag protein, remain largely unknown. Hence, we screened ISG-encoded proteins by bioluminescence resonance energy transfer to identify the crucial host effectors that suppressed Gag function. Consequently, we identified the transmembrane protein MAL as a Gag-interacting ISG product. In fact, ectopic expression of MAL substantially inhibited the production of HIV-1 particles, leading to the translocation, accumulation, and eventual lysosomal degradation of Gag in the host endosomal compartments. Owing to the conserved N-terminal region of MAL, which specifically interacts with HIV-1 Gag, this particular antiviral function of MAL targeting Gag is also conserved among orthologs of various animal species. Notably, the antiviral activity of MAL was partially antagonized by the viral accessory protein Nef, as it interfered with the interaction between MAL and Gag. Therefore, this study reveals a previously unidentified antiviral function of MAL and its viral counteraction. It also sheds new light on therapeutic strategies against HIV-1 infection based on the intrinsic antiviral immunity of host cells.
Introduction
Human immunodeficiency virus type 1 (HIV-1) is the causative agent of acquired immunodeficiency syndrome (AIDS). Its genome comprises nine genes: three structural genes (gag, pol, and env), two regulatory genes (rev and tat), and four accessory genes (nef, vpu, vif, and vpr) (1). The Gag structural protein is synthesized as a precursor polyprotein (Pr55Gag) consisting of four major domains: matrix (MA), capsid (CA), nucleocapsid (NC), and p6, and two spacer sequences: SP1 and SP2. During late HIV-1 replication, the Gag protein is transported to the plasma membrane (PM) where it is assembled and multimerized for viral budding (2, 3). The MA domain of the Gag polyprotein helps transport it to the PM by interacting with anionic membrane lipids, such as phosphatidylinositol 4,5-bisphosphate (PIP2) (4, 5).
In the infected cells, HIV-1 targets various host factors to promote the production of progeny virions. On the other hand, host cells express antiviral factors that inhibit key steps of viral replication (6). Interferons (IFNs) are antiviral signaling proteins that are secreted by an infected cell during viral infection. They induce the expression of an array of antiviral proteins in both the infected and the surrounding non-infected cells. Furthermore, IFNs are classified into three categories: types I, II, and III. Notably, type I and type III IFNs are involved in establishing an antiviral tissue environment (7). These IFNs inhibit HIV replication at various stages of the viral life cycle by promoting the expression of IFN-stimulated gene (ISG) products. For example, the ISG-encoded proteins APOBEC3G and BST2/tetherin abrogate HIV replication by targeting reverse transcription of the HIV genome and the release of the virus, respectively (8). However, HIV has evolved and acquired accessory proteins, such as Vif and Vpu, to counter the function of the antiviral ISG products (9, 10). Indeed, these ISG products are specifically targeted for degradation by the host ubiquitin-proteasomal pathway or lysosome pathway, sustaining viral replication.
Previously studies have shown that several host proteins induced by type I and type III IFNs suppress the production of viral particles (11, 12). However, which ISG product targets the translocation of Pr55Gag to the PM and its multimerization for the production and assembly of viral particles during HIV-1 biogenesis is not fully clarified. We therefore screened for intracellular protein-protein interactions by generating a library of ISGs using NanoLuciferase (NanoLuc)-based bioluminescence resonance energy transfer (NanoBRET). In this study, we utilized the NanoBRET technology to identify putative host proteins that bind to Gag and potently restrict HIV replication. Consequently, we identified the tetraspanin membrane protein MAL as a novel ISG product that targets Gag and exhibits anti-HIV activities. Additionally, we found that the HIV accessory protein Nef antagonizes the antiviral function of MAL, sustaining viral replication.
Materials And Methods
Cells and Plasmids
We cultured HEK293 cells (ATCC, #CRL-1573) in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS). Additionally, we cultured PC3 (ATCC, #CRL-1435) and CEM cells in RPMI supplemented with 10% FBS. The human codon-optimized HIV-1NL4-3 Gag and Nef, and non-human MAL cDNAs were synthesized and subcloned into pEGFP- or pcDNA-based vectors. Mutants were generated by PCR-based mutagenesis procedures. The HIV-1 molecular clone pNL4-3, pNL4-3/Gag-EGFP, pNL4-3/Fyn10WT and pNL4-3/Fyn10ΔMA have been described previously (13, 14). We obtained the pNL4-3 plasmid defective in nef from the NIH HIV reagent program (catalogue number ARP-12755). Expression vectors encoding HaloTag-conjugated ISG-encoded proteins were collected by Kazusa Genome Technologies (Chiba, Japan) and purchased from Promega. Plasmids encoding GFP-tagged LC3 and RhoB were obtained from Addgene (catalogue number #11546) and Takara-Bio (catalogue number #632490), respectively. NCBI accession numbers of MAL genes are: Homo sapiens (AB529205), Macaca mulatta (XM_015112833), Mus musculus (NM_010762), Gallus gallus (NM_001199391), and Danio rerio (NM_001017686).
NanoBRET
We transfected HEK293 cells in 96-well white plates with vectors encoding the HaloTag-fused ISG-encoded protein (100 ng) and NanoLuc-fused Gag or Nef (1 ng). The NanoBRET activity was measured at 48 h post-transfection using the NanoBRET Nano-Glo Detection System (Promega). First, cells were co-transfected with C-terminal NanoLuciferase (NL)-conjugated Gag and N-terminal HaloTag (HT)-fused ISG expression vectors. Subsequently, the HT-618 ligand and furimazine substrate were added. If two proteins were within 200 nm of each other, NanoBRET signals were detected. The NanoBRET signals of the interaction between Gag-NL and Gag-HT was used as a positive control.
HIV-1 Production Assay
HEK293 or PC3 cells seeded in 12-well plates were co-transfected with pNL4-3 and either an ISG-encoded protein expression vector or an empty vector using Effectene (Qiagen) for HEK293 cells or Lipofectamine 3000 (Thermo) for PC3 cells. In experiments with siRNA, cells were transfected with 20 pmol of siRNA using Lipofectamine RNAiMAX (Thermo) one day prior to transfection with pNL4-3. Two days post-transfection, cell lysates and supernatants were harvested and subjected to immunoblotting. In some experiments, 10 µM MG132 (Merck #474790) or 100 µM chloroquine (Cayman Chemical #14194) were added 16 h before cells were harvested. The p24 antigen levels in the supernatants were measured using an ELISA kit (Zepto Metrix). siRNAs were purchased from Qiagen (catalogue number #SI03650318 for the negative control and #SI00627214 and #SI00627228 for MAL).
HIV-1 Multicycle Assay
Replication-competent HIV-1 stocks were produced by transient transfection of HEK293 cells with the pNL4-3 or pNL4-3ΔNef plasmids. Culture supernatants containing HIV-1were collected 48 h after transfection and filtered through a 0.45 µm Millex-HV filter (Merck).
CEM cells (106 cells) were transfected with 50 pmol of siRNAs using a 4D-Nucleofector (Lonza) with electroporation program CL-120, following the manufacturer’s instructions. At 24 h post-transfection, the CEM cells were infected with 25 ng of the HIV-1 p24 antigen. The cells were then centrifuged for 90 min at 800 x g with 5 µg/ml polybrene, and subsequently washed three times with PBS to remove the added viruses. Thereafter, we periodically collected the nascent virions that were produced by the infected cells and measured the p24 levels as described in section 2.3.
Immunoprecipitation
HEK293 cells seeded in six-well plates were transfected with Gag-FLAG-encoding vector and the HT-tagged protein expression plasmid with or without the Nef expression plasmid (500 ng each). At 48 h post-transfection, the cells were lysed with an HBST buffer (10 mM HEPES pH 7.4, 150 mM NaCl, and 0.5% Triton-X-100) containing a protease inhibitor cocktail (Merck #11697498001). The cell lysates were immunoprecipitated using an anti-FLAG EZview Red Affinity Gel (Merck #F2426) for 16 h, and the bound proteins were analyzed by immunoblotting as follows. Samples dissolved in SDS loading buffer were loaded and electrophoresed using 10% polyacrylamide gels, and the separated proteins were blotted onto PVDF membranes (Merck). Subsequently, the membranes were probed with primary antibodies and horseradish peroxidase-conjugated secondary antibodies (Cytiva). The detected proteins were visualized using a FluorChem imaging system (Alpha Innotech) or LuminoGraph imaging system (Atto). Protein bands were analyzed using ImageJ software (NIH). Primary antibodies used in this study included the following: HaloTag (Promega #G9211), FLAG (Merck #F3165), GFP (MBL #598), vinculin (Merck #V9131), p24 (NIH HIV reagent program #ARP-3537), and Nef (Thermo #MA1-71501).
Immunofluorescence
For the immunofluorescence assay, HepG2 cells were seeded onto glass cover slips one day before transfection. At 24 or 48 h post-transfection, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. Thereafter, the cells were stained with anti-HaloTag (Promega #G9281) and Alexa Fluor594-conjugated secondary antibody (Thermo #A48284).
Results
Identification of Gag-Binding Proteins That Inhibit HIV-1 Production
We prepared a human cDNA library comprising 723 ISG products based on the Interferome database (http://www.interferome.org/). While preparing this library, we only selected the ISG products whose expression was induced more than 1.5-fold following treatment with type I or III IFNs (15). Subsequently, to identify the ISG products that interacted with Gag, we performed a high-throughput screening of the library by co-expressing the C-terminus of the NanoLuc-conjugated HIV-1 Gag protein with each of the 723 ISG products in HEK293 cells. Moreover, we analyzed the protein–protein interactions by NanoBRET. In the first screening, we selected 62 ISGs as potential candidates whose products bind to the Gag protein (Figure 1A). Since Gag is the major structural protein required for viral particle formation, we investigated whether these candidates hindered viral particle production. To this end, we performed a second screening wherein we co-transfected HEK293 cells with the candidate ISG products and pNL4-3 and estimated the Gag p24 antigen levels in the culture supernatant by ELISA. We found that five ISG-encoded proteins (MAL, ZNF36L2, ZNF36, TCIRG1, and BCMP1) significantly inhibited the production of HIV-1 particles without causing apparent cytotoxicity (Figure 1B). Indeed, they inhibited viral particle production more prominently than the known antiviral factor IFITM2, which also targets Gag (16). Moreover, a single round of infection using VSVg-pseudotyped reporter viruses revealed that these proteins did not interfere with the early stages of the HIV life cycle (from cell entry to reporter gene expression; Supplementary Figure S1). Therefore, we selected these five candidate proteins for further in-depth functional analyses.
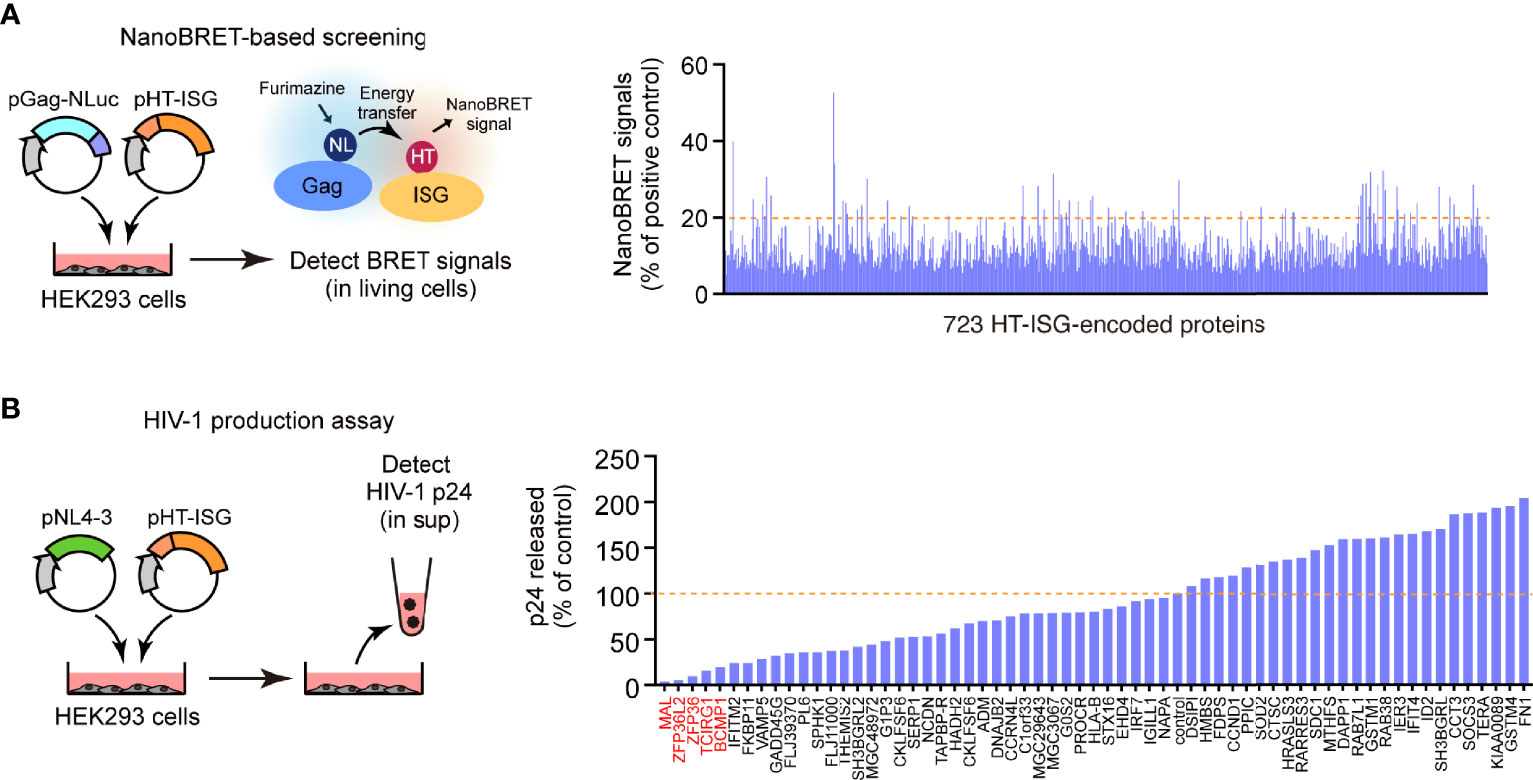
Figure 1 Identification of Gag-binding proteins that inhibit HIV-1 production. (A) NanoBRET-based screening identified 723 Gag-interacting proteins encoded by interferon-stimulated genes (ISGs) in HEK293 cells. (B) HIV particle production assay. HEK293 cells were co-transfected with the HIV molecular clone pNL4-3 and ISG-encoded protein expression vectors. Two days post-transfection, viral p24 antigen levels in the culture supernatants were measured by ELISA.
MAL Inhibits Gag Protein Assembly at the Plasma Membrane
Our immunoprecipitation analysis validated the interactions between the candidate proteins and Gag, as all the candidate proteins co-immunoprecipitated with Gag (Figure 2A and Supplementary Figure S2A). We also found that these proteins decreased viral particle production in a dose-dependent manner; this decrease was accompanied by a massive reduction in intracellular Gag levels (Figure 2B). Since, the Gag protein accumulates, assembles, and multimerizes on the PM prior to HIV-1 particle formation, we examined its localization upon overexpressing the five candidate proteins with Gag-GFP. Notably, Gag was localized on the PM in cells that overexpressed ZNF36, TCIRG1, BCMP1, and ZNF36L2 (Supplementary Figure S2B). However, Gag was mostly located in the cytoplasmic puncta and hardly localized to the PM in cells overexpressing MAL (Figure 3A and Supplementary Figure S3). Since MAL primarily localizes to the PM and endosomes (17), we hypothesized that Gag accumulated in the endosomal puncta in the MAL-overexpressing cells. Consistent with this hypothesis, we discovered that Gag was co-localized with the endosomal marker RhoB, but not with the autophagosomal marker LC3 (Figure 3B). Furthermore, we found that chloroquine (a lysosome inhibitor) and not MG132 (a proteasome inhibitor) inhibited MAL-mediated Gag degradation (Figure 3C). Thus, we deduced that MAL hindered the translocation of Gag to the PM by sequestering it to endosomal compartments, eventually leading to its lysosomal degradation.
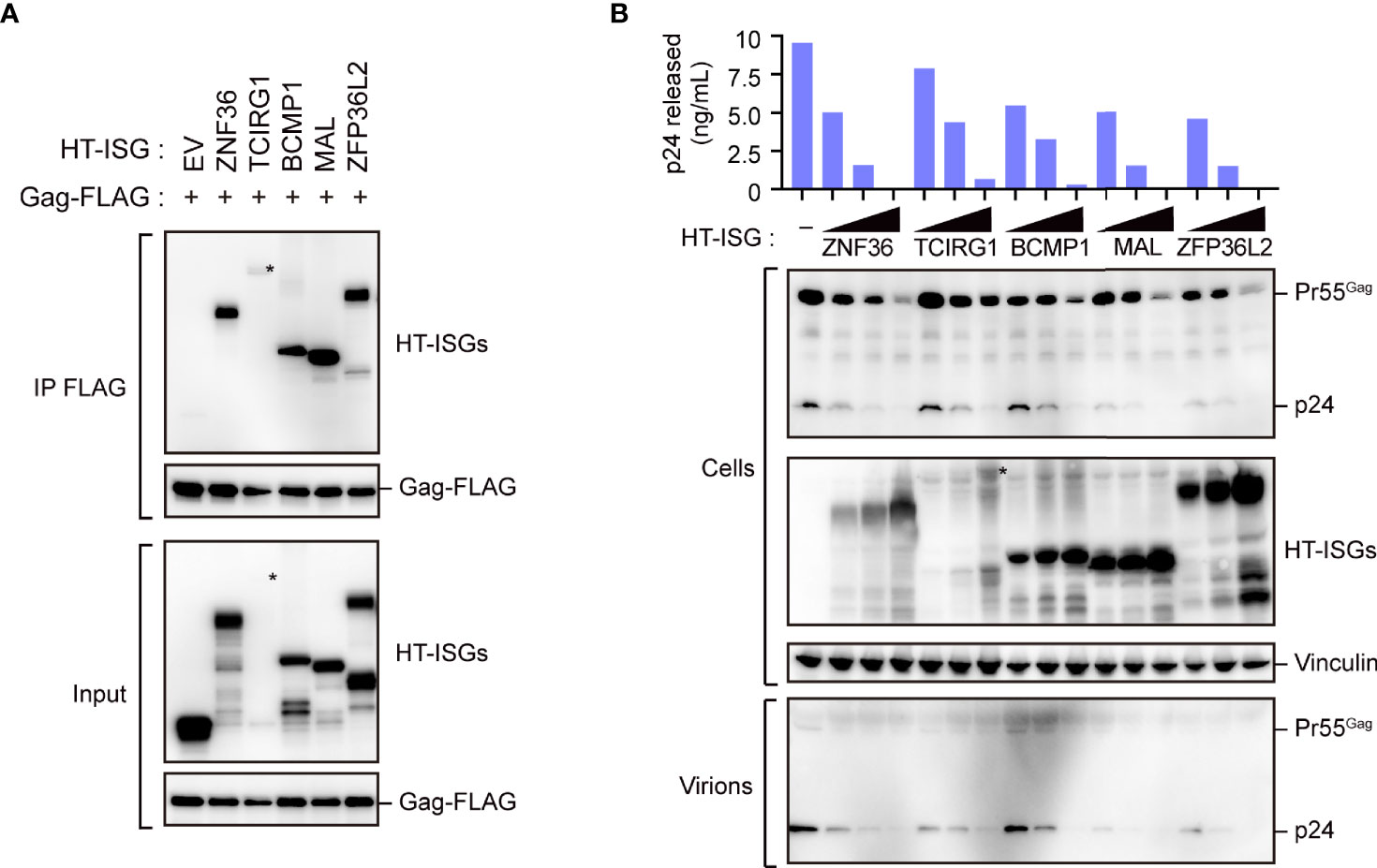
Figure 2 Characterization of Gag-binding proteins. (A) Immunoprecipitation of HEK293 cells with Gag-FLAG and indicated HaloTag (HT)- interferon-stimulated gene (ISG)-encoded protein expression vectors. Cell lysates were precipitated with anti-FLAG, followed by immunoblotting. Asterisks on the blot indicate TCIRG1 expression. Longer exposure blot of input sample is shown in Supplementary Figure S2A. (B) HIV particle production assay. HEK293 cells were co-transfected with the HIV molecular clone pNL4-3 and the indicated ISG-encoded protein expression vectors. Two days post-transfection, cell lysates (Cells) and the culture supernatants (Virions) were subjected to immunoblotting. Viral p24 antigen in the culture supernatants was measured by ELISA.
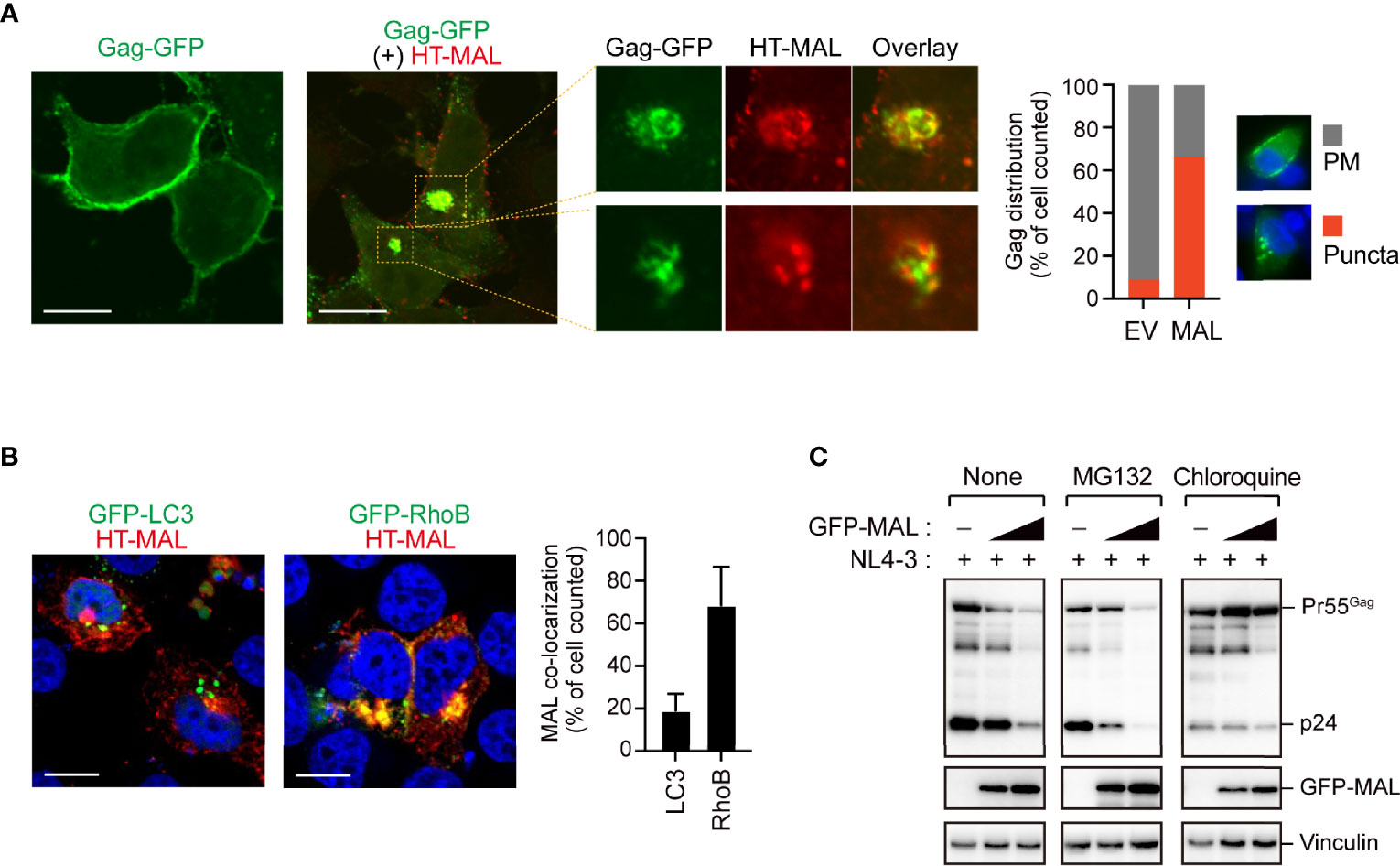
Figure 3 MAL inhibits translocation of Gag to the plasma membrane. (A) Confocal microscopy images of HepG2 cells expressing pNL4-3/Gag-EGFP and HaloTag (HT)-conjugated MAL proteins. The third image in this panel is the expanded view. Note that other viral proteins except Pol were expressed in this experiment. Scale bar, 10µm. Low magnification images, including other cells, are shown in Supplementary Figure S3. The graph on the right shows the percentage of cells in which Gag-GFP localized at the PM or in cytoplasmic puncta (n > 20 cells). (B) Confocal micrographs showing HepG2 cells expressing HT-MAL and either GFP-LC3 (autophagosomal marker) or GFP-Rho3 (endosomal marker). Nuclei were stained with DAPI. Scale bar, 10µm. The graph on the right shows the percentage of cells in which MAL localized to LC3- or RhoB-positive structures (n > 20 cells). (C) Lysosomal inhibitor blocks MAL-mediated Gag degradation. HEK293 cells were co-transfected with pNL4-3 and the MAL expression vectors. Two days post-transfection, cell lysates were subjected to immunoblotting. The compounds indicated in the figure were added 16 h before the cells were harvested.
Antiviral Effect of Endogenous MAL Against HIV-1
We next examined the effect of endogenous expression of MAL on HIV-1 production in PC3 cells, which are known to functionally express MAL (18). We treated the cells with two different MAL-targeting siRNAs, which had 90% knockdown efficiencies, and then transfected them with pNL4-3 (Figure 4A and Supplementary Figure S4). Compared with the control cell supernatants, the MAL-knockdown cell supernatants had a 3- to 4-fold increase in p24 levels, reflecting increased HIV-1 production (Figure 4B). To further investigate the function of MAL in multi-round HIV-1 infection, we transduced CEM cells with MAL-targeting siRNA and infected them with replication-competent HIV-1. Subsequent calculation of p24 levels revealed that viral infection was significantly enhanced in MAL-silenced CEM cells compared to those in control cells (Figures 4C, D). We confirmed an incremental increase in cellular Pr55Gag in cells with MAL knockdown (Figures 4B, D). Together, these results indicate that endogenous MAL negatively regulates HIV-1 replication by degrading Pr55Gag.
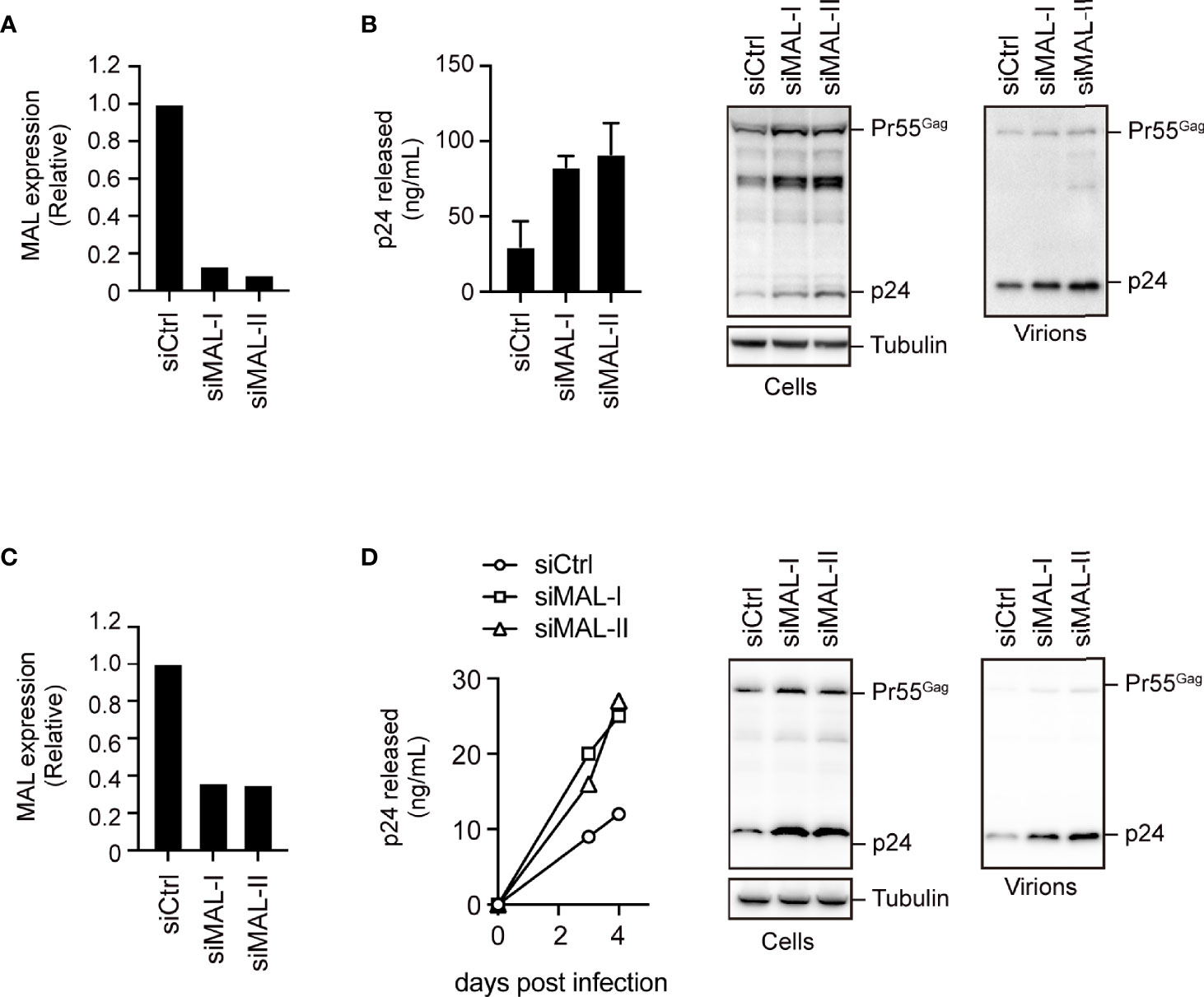
Figure 4 Endogenous MAL expression inhibits HIV-1 production. (A, B) PC3 cells transduced with control-(Ctrl) or MAL-targeting siRNAs (siMAL-I and siMAL-II) were transfected with pNL4-3. MAL knockdown was confirmed by RT-PCR (A). Following transfection, cell lysates (Cells) and the culture supernatants (Virions) were subjected to immunoblotting and p24 levels were measured by ELISA (B). (C, D) CEM T cells transduced with indicated siRNAs were infected with HIV-1. MAL knockdown was confirmed by RT-PCR (C). The supernatants were harvested at 3 and 4 days post-infection and p24 levels were measured by ELISA. At 4 days post-infection, cell lysates and the culture supernatants (virions) were subjected to immunoblotting (D).
The Gag MA Domain Is a Key Target of MAL-Mediated Inhibition
Since the MA and NC domains of the Gag protein are specifically involved in localizing Gag to the PM (3), we next mapped the MAL-binding sites on Pr55Gag using several domain mutants. Our immunoprecipitation analysis revealed that the polyprotein mutant lacking the MA domain, but not the NC domain, exhibited reduced binding to MAL (Figure 5A). This suggested that the MA domain is responsible for the interaction of Gag with MAL. Since MAL has four transmembrane domains, we generated MAL mutants lacking either the N-terminus or the C-terminus for further immunoprecipitation analysis. Consequently, we observed that Gag could interact with the MAL mutant that lacked the C-terminal region (Figure 5B). Consistently, the MA domain of Gag interacted with the N-terminal half of MAL, indicating that Gag binds to the N-terminal domain of MAL. To investigate whether this interaction is a prerequisite for the anti-HIV activity of MAL, we examined its inhibitory effect on Gag mutants lacking the MA domain. To this end, we co-transfected cells with GFP-MAL and pNL4-3 encoding either wild-type Gag or MA-deleted Gag with the Fyn peptide (Fyn10WT or Fyn10ΔMA), which can associate with the PM (5). Subsequent immunoblotting analysis revealed that the amount of MA-deleted Gag was not reduced in the cells, whereas that of the wild-type Gag was prominently reduced under identical conditions (Figure 5C). These results suggested that MAL inhibits Gag via the MA domain. We further found that both Gag mutants, which lack PM-targeting activity due to the absence of a myristoyl group (G2A) or highly basic domain (6A2T), had low binding activity to MAL (Supplementary Figures S5A, B). Together, these results suggest that the PM targeting of Gag is necessary for MAL interaction.
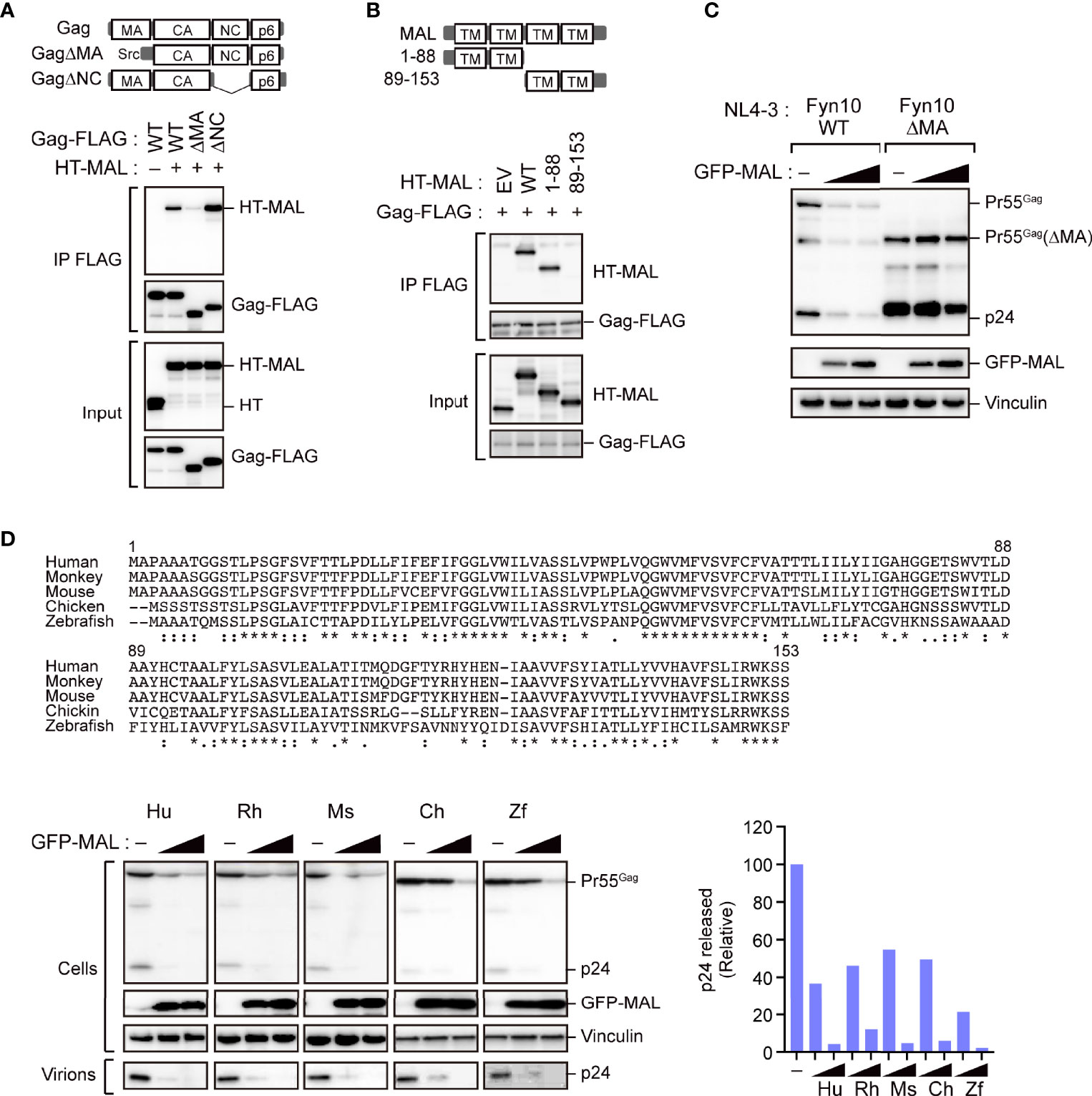
Figure 5 The matrix domain of Gag is a major target for MAL-mediated inhibition. (A) MAL binds to the MA domain of Gag. Immunoprecipitation assays of HEK293 cells expressing the specified Gag-FLAG mutants and HaloTag (HT)-MAL. Cell lysates were precipitated with anti-FLAG antibodies, followed by immunoblotting. WT, wild-type; Δ, gene deletion. (B) N-terminus of MAL binds to Gag. Immunoprecipitation assays of HEK293 cells expressing the indicated HT-MAL mutants and Gag-FLAG. Cell lysates were precipitated with anti-FLAG antibodies, followed by immunoblotting. (C) MA-deleted Gag is resistant to MAL. HEK293 cells were co-transfected with pNL4-3/Fyn(10)full MA or pNL4-3/Fyn(10)ΔMA and GFP-MAL. Two days post-transfection, cell lysates were subjected to immunoblotting. (D) Antiviral activity of MAL is conserved across animal species. Sequence alignment of MAL across the specified animal species (top). The symbols below the alignment show the conserved residues (*), conservative substitutions (:) and semi-conservative substitutions (.). HEK293 cells were co-transfected with the HIV molecular clone pNL4-3 and the specified MAL expression vectors. Two days post-transfection, cell lysates (Cells) and the culture supernatants (Virions) were subjected to immunoblotting (bottom left). p24 antigen levels in the culture supernatants were also measured by ELISA (bottom right).
Since the N-terminus of MAL is conserved among animal species (Figure 5D), we analyzed whether the inhibitory function of MAL is also conserved across the orthologs of other animal species, including Macaca mulatta (rhesus monkey), Mus musculus (mouse), Gallus gallus (chicken), and Danio rerio (zebrafish). Our results revealed that all orthologs could reduce intracellular Gag levels (Figure 5D), suggesting that the antiviral function of MAL has been conserved during evolution.
Nef Partially Overcomes the Antiviral Activity of MAL
As our above results suggested that HIV-1 can replicate to an extent even in MAL-expressing T cells (Figure 4D), we hypothesized that HIV-1 can possibly counteract the antiviral activity of endogenous MAL. A previous study reported that the HIV-1 accessory protein Nef induces MAL-dependent massive secretion of exosomal markers, inducing the efficient fusion of the endosomes with the PM (19). Thus, we examined whether Nef could regulate the antiviral effect of MAL using wild-type HIV-1 and HIV-1 lacking the Nef, Vpu, and Env proteins, which might bind to the host membrane proteins. Our results demonstrated that the antiviral activity of MAL was enhanced in HIV-1 lacking Nef, but not in HIV-1 lacking Vpu and Env (Figure 6A). In MAL-expressing cells, the levels of Pr55Gag in the ΔNef virus were reduced compared to that of the WT virus (Figure 6B). In PC3 cells, the production of ΔNef virus was lower than that of the WT virus, but this reduction was not observed in MAL-depleted PC3 cells (Figure 6C). Consistent with this, intracellular Pr55Gag levels of ΔNef virus were recovered by MAL knockdown (Figure 6D). Moreover, multi-round HIV-1 infection analysis revealed that reduced ΔNef virus replication was also recovered by MAL knockdown (Figure 6E). Together, these results suggest that Nef may rescue MAL-mediated Gag degradation, promoting HIV production.
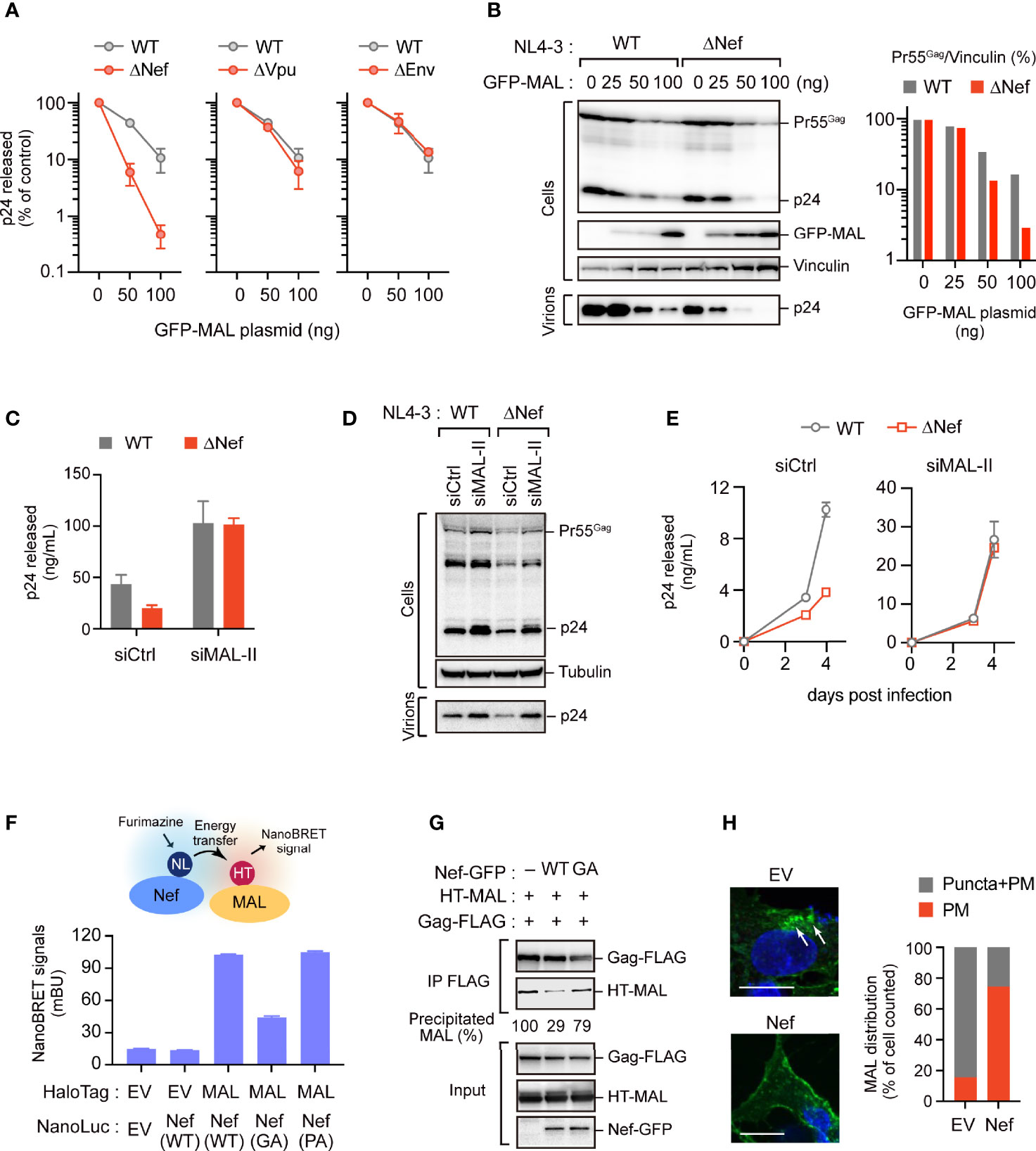
Figure 6 Nef partially overcomes the antiviral activity of MAL. (A, B) HEK293 cells were transfected with the HIV-1 molecular clone pNL4-3 lacking nef, vpu, or env, and the GFP-MAL expression vector. Two days post-transfection, p24 levels in the supernatants were assessed by ELISA (A). Cell lysates were subjected to immunoblotting (B). Bar graph shows the relative Pr55Gag levels normalized to vinculin levels as determined by densitometry. WT, wild-type; Δ, gene deletion. (C, D) PC3 cells transduced with control-(Ctrl) or MAL-targeting siRNA (siMAL-II) were transfected with pNL4-3 or pNL4-3ΔNef. Cell lysates (Cells) and supernatants (Virions) were subjected to immunoblotting and p24 levels were measured by ELISA. (E) CEM T cells transduced with Ctrl or siMAL-II were infected with HIV-1 or Nef-deficient HIV-1. The supernatants were harvested at 3 and 4 days post-infection and p24 levels were measured by ELISA. (F) Nef associates with MAL. HEK293 cells were co-transfected with C-terminal NanoLuciferase (NL)-conjugated Nef and HaloTag (HT)-MAL expression vectors. Two days post-transfection, NanoBRET signals from cells were measured. WT, wild-type; GA, G2A mutation; PA, P72,75A mutation. (G) Nef inhibits the Gag-MAL interaction. Immunoprecipitation assays of HEK293 cells expressing Gag-FLAG and HT-MAL in the presence or absence of Nef. Cell lysates were precipitated with anti-FLAG antibodies, followed by immunoblotting analysis. Precipitated MAL levels (relative) were also shown, determined by densitometry. (H) Nef inhibits the internalization of MAL. Confocal microscopy images of HepG2 cells expressing GFP-MAL and Nef-FLAG. Note that no other viral proteins were expressed in this experiment. Nuclei were stained with DAPI. Arrows indicate internalized MAL. Scale bar, 10µm. Low magnification images, including other cells, are shown in Supplementary Figure S6. The graph on the right shows the percentage of cells in which GFP-MAL localized at the PM or in cytoplasmic puncta (n > 20 cells).
In fact, we detected a proximal interaction between Nef and MAL using NanoBRET (Figure 6F), suggesting that they are in close proximation in living cells. Interestingly, Nef mutants devoid of their membrane-targeting ability (G2A mutants) exhibited reduced NanoBRET signals. In contrast, Nef mutants lacking the PxxP motifs, which are important for binding of Nef to Src kinases (20), could still bind to MAL (Figure 6F). These results suggested that Nef was adjacent to MAL at the host PM. Notably, our immunoprecipitation analysis revealed that Nef interfered with the binding between MAL and Gag (Figure 6G), implying that Nef impedes the antiviral function of MAL against Gag. The immunofluorescence analysis also showed that in the absence of Nef, MAL was present in both the PM and intracellular endosomes. However, in the presence of Nef, MAL was primarily localized to the PM without localizing to the endosomes (Figure 6H and Supplementary Figure S6). These results suggest that Nef attenuates the antiviral activity of MAL by interfering with its localization to the PM and the endosomes, inhibiting accumulation of Gag in the endosomal compartments.
Discussion
In this study, we identified several novel Gag-binding ISG-products that negatively regulate HIV-1 particle production. We revealed that MAL has an anti-HIV function, as it sequesters Gag into the endosomal components. We also found that the viral accessory protein Nef antagonizes the antiviral function of MAL by interfering with the binding between MAL and Gag (Figure 7).
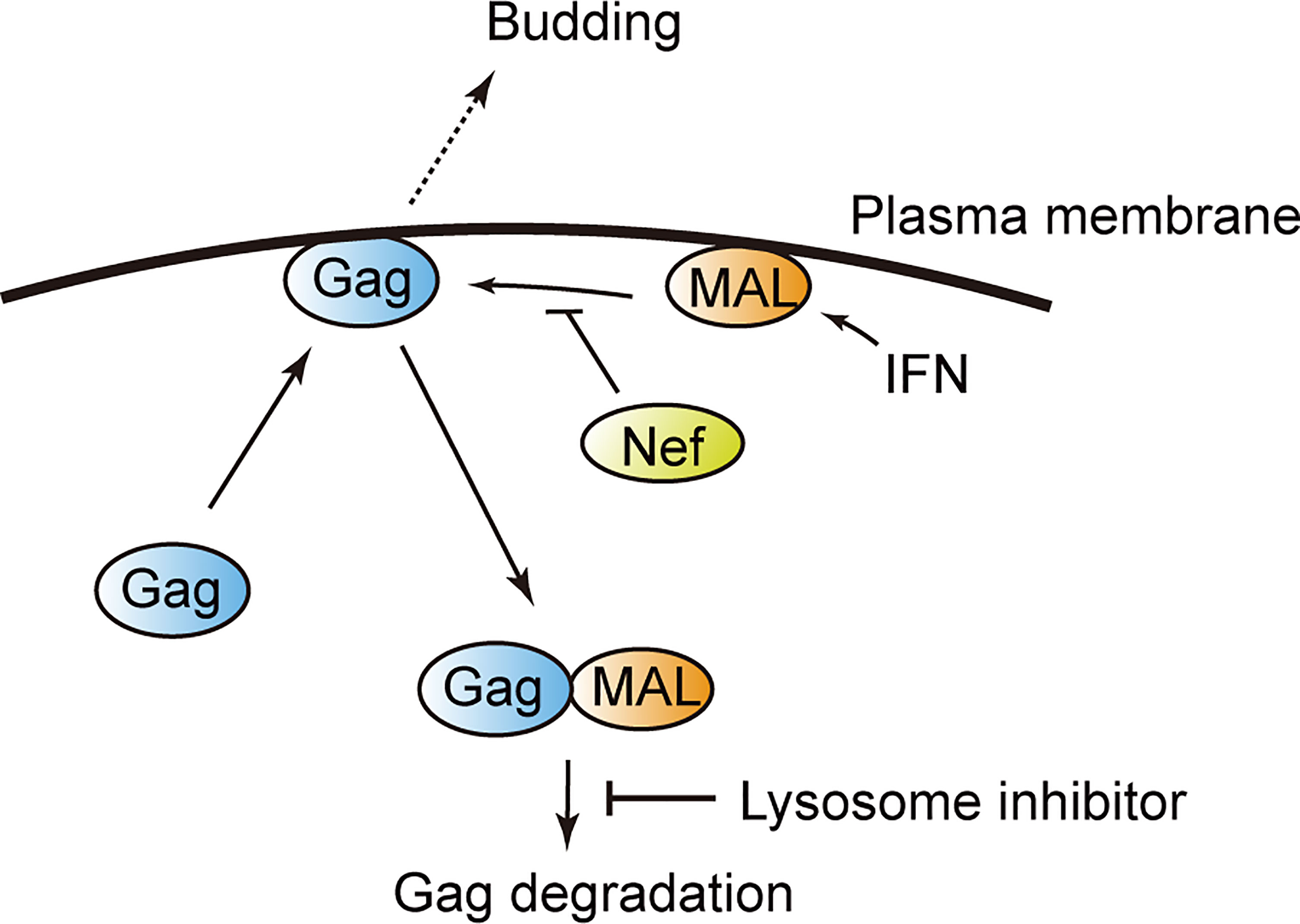
Figure 7 Model illustrating MAL-induced Gag degradation. MAL targets the matrix domain of the HIV-1 Gag protein and induces its degradation via the endolysosomal pathway. The viral protein Nef inhibits this interaction, overcoming the antiviral activity of MAL.
MAL is a 17 kDa membrane protein consisting of four transmembrane domains. It was first identified in human T lymphocytes (21). Moreover, it localizes to lipid raft microdomains of the PM, endosomes, and trans-Golgi networks (17), and is expressed in human T cells, epithelial cells, and myelin-forming cells (22). In T cells, MAL is involved in transporting proteins to the cell membrane, the secretion of exosomes or multivesicular bodies (MVBs), and in organizing immune synapses (23–25). Although the role of MAL in HIV-1 replication has not been reported to date, a previous study did report its involvement in the viral propagation of HSV-1 (26). Here, we demonstrate that MAL also targets the HIV-1 protein Gag and interferes with its transport to the PM, where HIV-1 particles are formed.
Although previous studies have shown that translocation of Gag to the PM and sorting of endosomes is crucial for efficient viral production, the precise molecular mechanisms underlying the intracellular dynamics of Gag remain unclear. The Gag protein is transported to the PM via N-terminal myristoylation and the basic patch on the MA domain (27, 28). However, the role of endosomes or MVBs in active transportation has not been well characterized. Indeed, the MA domain contains a myristoyl group that enables the stable binding of Gag to PIP2 on the PM and is involved in its stable localization to the PM (4, 5, 29). This localization is necessary and sufficient to trigger viral assembly. Our results revealed that MAL promotes the localization of Gag to intracellular endosomal vesicles rather than the PM via interacting with the MA domain; however, the detailed molecular mechanism underlying this phenomenon remains elusive.
We also found that Nef antagonizes the antiviral function and interaction of MAL with Gag. Previous studies have demonstrated that Nef stabilizes the localization of Gag to the PM; nevertheless, the underlying mechanism is as yet unknown (30, 31). In this study, we found that Nef possibly stabilizes the localization of Gag to the PM by antagonizing the antiviral function of endogenous MAL. Consistently, we also observed that despite endogenous MAL exhibiting antiviral activity, a low level of virus particles was still produced. These results suggest that the stoichiometric relationship between Nef and MAL can determine the efficiency of viral replication.
Multiple studies have reported the functional interactions between HIV-1 Nef and MAL. Indeed, Nef promotes the secretion of exosomes and microvesicles, which is mediated by MAL (32–34). Consistently, the activity of Nef is inhibited upon knockdown of MAL (19). In our current study, we revealed an additional function of MAL in inhibiting HIV-1 particle production via sequestrating Gag to endosomal compartments. It is possible that Nef directs the function of MAL for exosome secretion rather than HIV-1 inhibition, sustaining HIV-1 particle production. This interesting hypothesis needs to be addressed in the future.
Our immunoprecipitation data reveal that Nef physically associates with MAL, blocking its binding to Gag. However, other mechanism(s) of its action on Gag need to be further explored. For example, Nef is known to alter the lipid content and protein properties of the PM (35, 36). Likewise, it is possible that Nef also alters the content, properties, and structure of MAL in the PM.
Owing to our analysis of the MAL orthologs in different species, we verified the antiviral effects of MAL derived from zebrafish; the MAL amino acid sequence of zebrafish has approximately 50% homology to that of human MAL. Thus, we deduced that a conserved region of these sequences is possibly crucial for HIV-1 inhibition. In this regard, we discovered that the N-terminus of the MAL protein is conserved among different animal species and is important for its binding to Gag. Nevertheless, the functional significance of the N-terminus of MAL binding to the Gag MA domain and sequestering Gag to the endosomal compartments is still undetermined. Although we found that the membrane binding activity of the MA domain is required for the interaction between Gag and MAL, it is interesting that the anti-retroviral function of MAL is conserved across multiple animal species.
In summary, our results demonstrate that MAL is an anti-HIV factor that targets Gag. It also facilitates the internalization and degradation of Gag-expressing viral particles. However, its antiviral effect is antagonized by the viral protein Nef. Thus, our findings shed new light on the molecular mechanisms underlying the HIV-1 degradation pathway in the innate immune system. Importantly, MAL and other molecules of this pathway are potential novel therapeutic targets for AIDS and related disorders.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author Contributions
KM designed and performed the research, analyzed the data, and wrote the manuscript. MN and SJ performed the research and analyzed the data. YM contributed reagents and analyzed the data. AR directed the research, analyzed the data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by JSPS KAKENHI Grants (JP19K07594 and JP20H03498) and AMED Grants (JP20fk0410020 and JP21fk0410039).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Dr. Akira Ono for providing NL4-3/Fyn10WT and pNL4-3/Fyn10ΔMA, and Kazufusa Takahashi and Kenji Yoshihara for their technical assistance. The following reagents were obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: Anti- HIV-1 p24 Monoclonal (183-H12-5C), ARP-3537, contributed by Dr. Bruce Chesebro and Kathy Wehrly; and HIV-1 NL4-3 ΔNef Infectious Molecular Clone, ARP-12755, contributed by Dr. Olivier Schwartz.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.836125/full#supplementary-material
Abbreviations
HIV-1, Human immunodeficiency virus 1; IFN, Interferon; ISG, Interferon-stimulated gene; AIDS, Acquired immunodeficiency syndrome; MA, Matrix; MVBs, Multivesicular bodies; PM, Plasma membrane; NanoLuc, NanoLuciferase; NanoBRET, NanoLuciferase-based bioluminescence resonance energy transfer.
References
2. Adamson CS, Freed EO. Human Immunodeficiency Virus Type 1 Assembly, Release, and Maturation. Adv Pharmacol (2007) 55:347–87. doi: 10.1016/S1054-3589(07)55010-6
3. Freed EO. HIV-1 Assembly, Release and Maturation. Nat Rev Microbiol (2015) 13:484–96. doi: 10.1038/nrmicro3490
4. Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) Bisphosphate Regulates HIV-1 Gag Targeting to the Plasma Membrane. Proc Natl Acad Sci USA (2004) 101:14889–94. doi: 10.1073/pnas.0405596101
5. Chukkapalli V, Ono A. Molecular Determinants That Regulate Plasma Membrane Association of HIV-1 Gag. J Mol Biol (2011) 410:512–24. doi: 10.1016/j.jmb.2011.04.015
7. Lazear HM, Schoggins JW, Diamond MS. Shared and Distinct Functions of Type I and Type III Interferons. Immunity (2019) 50:907–23. doi: 10.1016/j.immuni.2019.03.025
8. Colomer-Lluch M, Ruiz A, Moris A, Prado JG. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front Immunol (2018) 9:2876. doi: 10.3389/fimmu.2018.02876
9. Strebel K. HIV Accessory Proteins Versus Host Restriction Factors. Curr Opin Virol (2013) 3:692–9. doi: 10.1016/j.coviro.2013.08.004
10. Malim MH, Bieniasz PD. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb Perspect Med (2012) 2:a006940. doi: 10.1101/cshperspect.a006940
11. Barr SD, Smiley JR, Bushman FD. The Interferon Response Inhibits HIV Particle Production by Induction of TRIM22. PLoS Pathog (2008) 4:e1000007. doi: 10.1371/journal.ppat.1000007
12. Wilson SJ, Schoggins JW, Zang T, Kutluay SB, Jouvenet N, Alim MA, et al. Inhibition of HIV-1 Particle Assembly by 2',3'-Cyclic-Nucleotide 3'-Phosphodiesterase. Cell Host Microbe (2012) 12:585–97. doi: 10.1016/j.chom.2012.08.012
13. Miyakawa K, Nishi M, Matsunaga S, Okayama A, Anraku M, Kudoh A, et al. The Tumour Suppressor APC Promotes HIV-1 Assembly via Interaction With Gag Precursor Protein. Nat Commun (2017) 8:14259. doi: 10.1038/ncomms14259
14. Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. Interaction Between the Human Immunodeficiency Virus Type 1 Gag Matrix Domain and Phosphatidylinositol-(4,5)-Bisphosphate Is Essential for Efficient Gag Membrane Binding. J Virol (2008) 82:2405–17. doi: 10.1128/JVI.01614-07
15. Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, et al. Interferome V2.0: An Updated Database of Annotated Interferon-Regulated Genes. Nucleic Acids Res (2013) 41:D1040–6. doi: 10.1093/nar/gks1215
16. Tartour K, Appourchaux R, Gaillard J, Nguyen XN, Durand S, Turpin J, et al. IFITM Proteins Are Incorporated Onto HIV-1 Virion Particles and Negatively Imprint Their Infectivity. Retrovirology (2014) 11:103. doi: 10.1186/s12977-014-0103-y
17. Puertollano R, Alonso MA. MAL, an Integral Element of the Apical Sorting Machinery, Is an Itinerant Protein That Cycles Between the Trans-Golgi Network and the Plasma Membrane. Mol Biol Cell (1999) 10:3435–47. doi: 10.1091/mbc.10.10.3435
18. Llorente A, de Marco MC, Alonso MA. Caveolin-1 and MAL Are Located on Prostasomes Secreted by the Prostate Cancer PC-3 Cell Line. J Cell Sci (2004) 117:5343–51. doi: 10.1242/jcs.01420
19. Ventimiglia LN, Fernandez-Martin L, Martinez-Alonso E, Anton OM, Guerra M, Martinez-Menarguez JA, et al. Cutting Edge: Regulation of Exosome Secretion by the Integral MAL Protein in T Cells. J Immunol (2015) 195:810–4. doi: 10.4049/jimmunol.1500891
20. Saksela K, Cheng G, Baltimore D. Proline-Rich (PxxP) Motifs in HIV-1 Nef Bind to SH3 Domains of a Subset of Src Kinases and Are Required for the Enhanced Growth of Nef+ Viruses But Not for Down-Regulation of CD4. EMBO J (1995) 14:484–91. doi: 10.1002/j.1460-2075.1995.tb07024.x
21. Millan J, Alonso MA. MAL, a Novel Integral Membrane Protein of Human T Lymphocytes, Associates With Glycosylphosphatidylinositol-Anchored Proteins and Src-Like Tyrosine Kinases. Eur J Immunol (1998) 28:3675–84. doi: 10.1002/(SICI)1521-4141(199811)28:11<3675::AID-IMMU3675>3.0.CO;2-5
22. Copie-Bergman C, Plonquet A, Alonso MA, Boulland ML, Marquet J, Divine M, et al. MAL Expression in Lymphoid Cells: Further Evidence for MAL as a Distinct Molecular Marker of Primary Mediastinal Large B-Cell Lymphomas. Mod Pathol (2002) 15:1172–80. doi: 10.1097/01.MP.0000032534.81894.B3
23. Anton O, Batista A, Millan J, Andres-Delgado L, Puertollano R, Correas I, et al. An Essential Role for the MAL Protein in Targeting Lck to the Plasma Membrane of Human T Lymphocytes. J Exp Med (2008) 205:3201–13. doi: 10.1084/jem.20080552
24. Anton OM, Andres-Delgado L, Reglero-Real N, Batista A, Alonso MA. MAL Protein Controls Protein Sorting at the Supramolecular Activation Cluster of Human T Lymphocytes. J Immunol (2011) 186:6345–56. doi: 10.4049/jimmunol.1003771
25. Ventimiglia LN, Alonso MA. Biogenesis and Function of T Cell-Derived Exosomes. Front Cell Dev Biol (2016) 4:84. doi: 10.3389/fcell.2016.00084
26. Lopez-Guerrero JA, de la Nuez C, Praena B, Sanchez-Leon E, Krummenacher C, Bello-Morales R. Herpes Simplex Virus 1 Spread in Oligodendrocytic Cells Is Highly Dependent on MAL Proteolipid. J Virol (2020) 94:e01739–19. doi: 10.1128/JVI.01739-19
27. Vlach J, Saad JS. Trio Engagement via Plasma Membrane Phospholipids and the Myristoyl Moiety Governs HIV-1 Matrix Binding to Bilayers. Proc Natl Acad Sci USA (2013) 110:3525–30. doi: 10.1073/pnas.1216655110
28. Olety B, Ono A. Roles Played by Acidic Lipids in HIV-1 Gag Membrane Binding. Virus Res (2014) 193:108–15. doi: 10.1016/j.virusres.2014.06.015
29. Monde K, Chukkapalli V, Ono A. Assembly and Replication of HIV-1 in T Cells With Low Levels of Phosphatidylinositol-(4,5)-Bisphosphate. J Virol (2011) 85:3584–95. doi: 10.1128/JVI.02266-10
30. Malbec M, Sourisseau M, Guivel-Benhassine F, Porrot F, Blanchet F, Schwartz O, et al. HIV-1 Nef Promotes the Localization of Gag to the Cell Membrane and Facilitates Viral Cell-to-Cell Transfer. Retrovirology (2013) 10:80. doi: 10.1186/1742-4690-10-80
31. Collette Y, Olive D. The Primate Lentivirus-Encoded Nef Protein can Regulate Several Steps of the Viral Replication Cycle. Virology (1999) 265:173–7. doi: 10.1006/viro.1999.0053
32. Campbell TD, Khan M, Huang MB, Bond VC, Powell MD. HIV-1 Nef Protein Is Secreted Into Vesicles That Can Fuse With Target Cells and Virions. Ethn Dis (2008) 18:S2–14-9.
33. Muratori C, Cavallin LE, Kratzel K, Tinari A, De Milito A, Fais S, et al. Massive Secretion by T Cells Is Caused by HIV Nef in Infected Cells and by Nef Transfer to Bystander Cells. Cell Host Microbe (2009) 6:218–30. doi: 10.1016/j.chom.2009.06.009
34. Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y, et al. HIV Nef is Secreted in Exosomes and Triggers Apoptosis in Bystander CD4+ T Cells. Traffic (2010) 11:110–22. doi: 10.1111/j.1600-0854.2009.01006.x
35. Bregnard C, Zamborlini A, Leduc M, Chafey P, Camoin L, Saib A, et al. Comparative Proteomic Analysis of HIV-1 Particles Reveals a Role for Ezrin and EHD4 in the Nef-Dependent Increase of Virus Infectivity. J Virol (2013) 87:3729–40. doi: 10.1128/JVI.02477-12
Keywords: HIV-1, interferon-stimulated genes, Gag, antiviral immunity, innate immunity
Citation: Miyakawa K, Nishi M, Jeremiah SS, Morikawa Y and Ryo A (2022) MAL Inhibits the Production of HIV-1 Particles by Sequestering Gag to Intracellular Endosomal Compartments. Front.Virol. 2:836125. doi: 10.3389/fviro.2022.836125
Received: 15 December 2021; Accepted: 07 March 2022;
Published: 23 March 2022.
Edited by:
Akio Adachi, Kansai Medical University, JapanCopyright © 2022 Miyakawa, Nishi, Jeremiah, Morikawa and Ryo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kei Miyakawa, a2VpbUB5b2tvaGFtYS1jdS5hYy5qcA==; Akihide Ryo, YXJ5b0B5b2tvaGFtYS1jdS5hYy5qcA==