 Manfred Weidmann
Manfred Weidmann Elena Graf
Elena Graf Daniel Lichterfeld3
Daniel Lichterfeld3 Ahmed Abd El Wahed
Ahmed Abd El Wahed Michaël Bekaert
Michaël Bekaert
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol., 25 March 2022
Sec. Bioinformatic and Predictive Virology
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.835707
An unprecedented use of high-throughput sequencing for routine monitoring of SARS-CoV-2 viruses in patient samples has created a dataset of over 6 million SARS-CoV-2 genomes. To monitor genomes, deposited in the GISAID database, and to track the continuous sequence evolution of molecular assay oligonucleotide target sequences. A simple pipeline tool for non-experts was developed to mine this database for nucleotide changes in oligonucleotides and tested with the long oligonucleotides of a Recombinase polymerase amplification (RPA) assay targeting the RNA-dependent RNA polymerase (RdRP) gene of the SARS-CoV-2. Results indicate the emergence of a single nucleotide change in the reverse oligonucleotide from 0.03 to 26.23% (January to May 2021) in Alpha variant genomes, which however reduced to 17.64% by September after which the Alpha variant was completely displaced by the Delta variant. For all other variants, no relevant nucleotide changes were observed. The oligonucleotide screening pipeline allows efficient screening of nucleotide changes in oligonucleotides of all sizes in minutes.
The use of high-throughput sequencing platforms for monitoring SARS-CoV-2 genomes from patient samples has yielded an unprecedented 5,556,541 SARS-CoV-2 genome sequence entries in the GISAID database (1) by end of November 2021 (https://www.gisaid.org; accessed 2021-11-29). This huge number eclipses the collection of 45,888 complete HIV-1 genomes gathered during a 30-year period and indeed the total of 4,719,307 of all virus sequences in NCBI.
Designing and monitoring changes in oligonucleotides to keep molecular tests up to date necessitates rapid and scalable technologies to cope with the size of this input. The data load renders, traditional multiple sequence alignments (MAFT) unfeasible; generic alignment search tools, such as BLAST, are insufficient to screen such a database because their core algorithms allow automatic error correction, which is inadequate in the case of monitoring for sequence aberrations. In contrast, K-mer search techniques, in which K-mers are short sequences of K characters, are commonly used to scan vast databases for frequent patterns. This has been used to guide primer selection, where a K-mer is a PCR oligonucleotide of a specific length; it can even, in some cases, allow for the detection of degenerate oligonucleotides (2–4).
In order to monitor the RdRp gene target regions for the oligonucleotides of a SARS-CoV-2 RT-RPA (5) we sought to design an easy-to-use tool to search the GISAID database for nucleotide changes in the target regions using the capabilities of K-mer. Continuous monitoring of mutations was used for a temporal analysis of the rise of emerging nucleotide changes in individual oligonucleotides.
All sequences of the SARS-CoV-2 variants B.1.1.17 (Alpha; United Kingdom), B.1.351 (Beta; South Africa), P.1 (Gamma; Brazil), B.1.617.2 (Delta, India), B.1.526 (Iota; NYC, USA), B.1.427/B.1.429 (Epsilon; USA, California), B.1.525 (Eta; Nigeria), C.37 (Lambda, Peru), B.1.621 (Mu, Colombia), B.1.1.529 (Omicron, South Africa) were retrieved from the GISAID database.
We devised a rapid and scalable method for searching the SARS-CoV-2 genome for oligonucleotide and used it to search the target sequences of an RdRP-RT-RPA amplicon, RdRP oligonucleotides, RdRP Probe, and complete amplicon (Supplementary Table S1; Supplementary Figure S1A).
To do so, we used K-mer of the exact size of the oligonucleotides to mine with great speed, through the genome using KAT v2.4.2 mode “sect” (6). Kat calculates the K-mer coverage across every genome and reports the primer-coverage/integrity. Partial genome sequences or sequence with substantial number of Ns, or gaps were excluded and marked as “fail.” Genome sequences reported with imperfect oligonucleotide presence (partial coverage) were then fully aligned against the oligonucleotides using Smith-Waterman local alignment (7) to accurately identify sequence variations. A python script, oligomutk, handles the pipeline and the reporting. Using the Docker platform, all processing can be done locally or on a cloud service. The outputs were organized hierarchically into the categories fail/pass, forward primer, reverse primer, amplicon. The script sortseq automatically sorts oligomutk output.
To allow temporal analysis, we downloaded the complete FASTA file, and the metadata file provided from GISAID and used the additional script typetempsort to sort the sequences according to type and month of collection. Graphs were generated using PRISM v9.1.
All scripts are freely available at https://github.com/aicuramedical/covidmutants. A readme file with guidance is provided.
In an early trial, a total of 505,666 whole genome sequences were retrieved for the five main SARS-CoV-2 variants. The number of individual genomes ranged from 202 to 167,069 per month (Supplementary Table S2). The oligomutk script provides a result table in a tabulation separated values (TSV) format which was sorted for the categories fail/pass, forward primer, reverse primer, probe. Failed sequences were analysed for degenerations and degenerated sequences were counted.
To monitor for emergence of individual mutations, the oligomutk outputs were analysed, and the percentages of mutations detected in all RPA oligonucleotides were plotted (Supplementary Figure S1B). The percentage of mutated sequences for Beta, Gamma, and Eta variants averaged at 0.38% (ranging from 0 to 1.82%; Supplementary Figure S1C). One nucleotide mutation in the forward primer emerged in 7.00% of Alpha variants in January, increasing to 23.36% percent in April (Supplementary Figure S1D). In contrast, we also observed that 19.50% of the Epsilon variants (USA, California) had altered sequences in January, which appeared to indicate a major deletion in the 5' region of the downstream primer of the RPA amplicon. However, a closer analysis of the sequences by MAFT showed sequencing/assembly errors. The number of discarded/erroneous sequences from January to April for this variant averaged at 6.56% (range 2.56–9.46%) and decreased to 2.71% by April, while the percentage of discarded sequences across all other variants averaged at 1.21% (range 0.48–2.07%) indicating most likely a general problem with sequencing quality for the sequencing performed for the Epsilon variant.
Since from April 2021, it was increasingly impossible to download monthly packages through the GISAID search function, we downloaded and analysed curated FASTA files available from GISAID for variants Alpha, Beta, Delta, Gamma and Iota at the beginning of September 2021, while continuing to manually download selected emerging variants Eta, Lambda, Kappa, and Mu. In total, 956,910 sequences were screened.
In general, the amount of mutated sequences across variants ranged from 0.12–0.66% but was determined at 13.30% for the Alpha variant, indicating that since April the emerging mutation in the forward primer sequence had not developed into a dominant sequence among the Alpha virus sequences.
In order to better manage the growing dataset of SARS-CoV-2 sequences on GISAID, all sequences downloaded from GISAID at the end of November were split into monthly packages for each lineage using the additional script typetempsort and analysed as before using oligomutk in combination with sortseq.
For the 3,679,450 sequences of the 10-month analysis discarded sequences ranged at a mean of 1.26 ± 0.59% (range 0.27–3.46%) across all variants while mutated sequences ranged at a lower level at a mean of 0.51 ± 0.48% (range 0.08–2.64%), respectively, with a significant difference as confirmed by unpaired t-test (P-value < 0.0001; Supplementary Figure S3).
The updated analysis confirmed that the emerging nucleotide change in the target sequence of the forward primer among the Alpha variant sequences soared from 0.03% in January to 26.23% by May but subsided to 17.64% by August, after which it disappeared entirely.
The sequence data submitted to GISAID until December 13th 2021 are dominated by sequences from the United Kingdom and the United States contributing in total 55.45% (24.12 and 31.33%, respectively) of the total of 6,022,997 SARS-CoV-2 sequences while the remaining sequences are submitted by 212 other entities. This constitutes a submission bias leading to an analysis bias, and it is necessary to rethink on how to extract a representative subset of SARS-CoV-2 sequences for analysis in futures studies. The percentage of mutated and discarded sequences across variants described here might be a starting point.
The mean percentage of discarded sequences due to sequencing errors at 1.26% was significantly higher (+0.75%) than the observed mean of mutated sequences in the RdRp oligonucleotide binding regions, indicating that frequencies > 1.26% need monitoring. Despite this bias and ongoing sequencing errors, the RdRp target gene sequence of the RPA, analysed as a whole, remained stable (Table 1), with a low-level occurrence of point mutations (Supplementary Tables S3, S4) which, however, did not develop into dominant strains. Since in general sequences discarded by oligomutk range at 0.91–2.11% (Figure 1; Supplementary Tables S3, S4) the surge of discarded sequences in the Epsilon variant from January to February to almost 30% and their subsequent rapid decline rapidly indicated a sequencing quality issue rather than the emergence of a deletion of half the reverse primer (Figure 1).
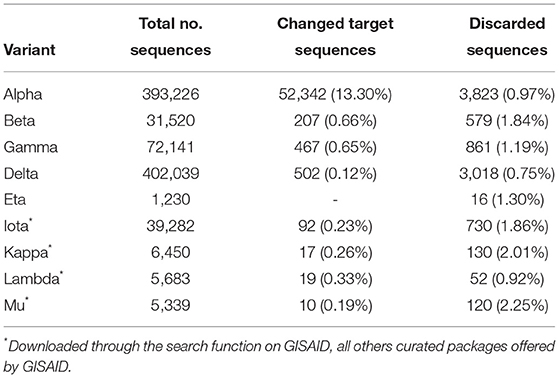
Table 1. Analysis of sequences downloaded from GISAID on 2021-09-06.
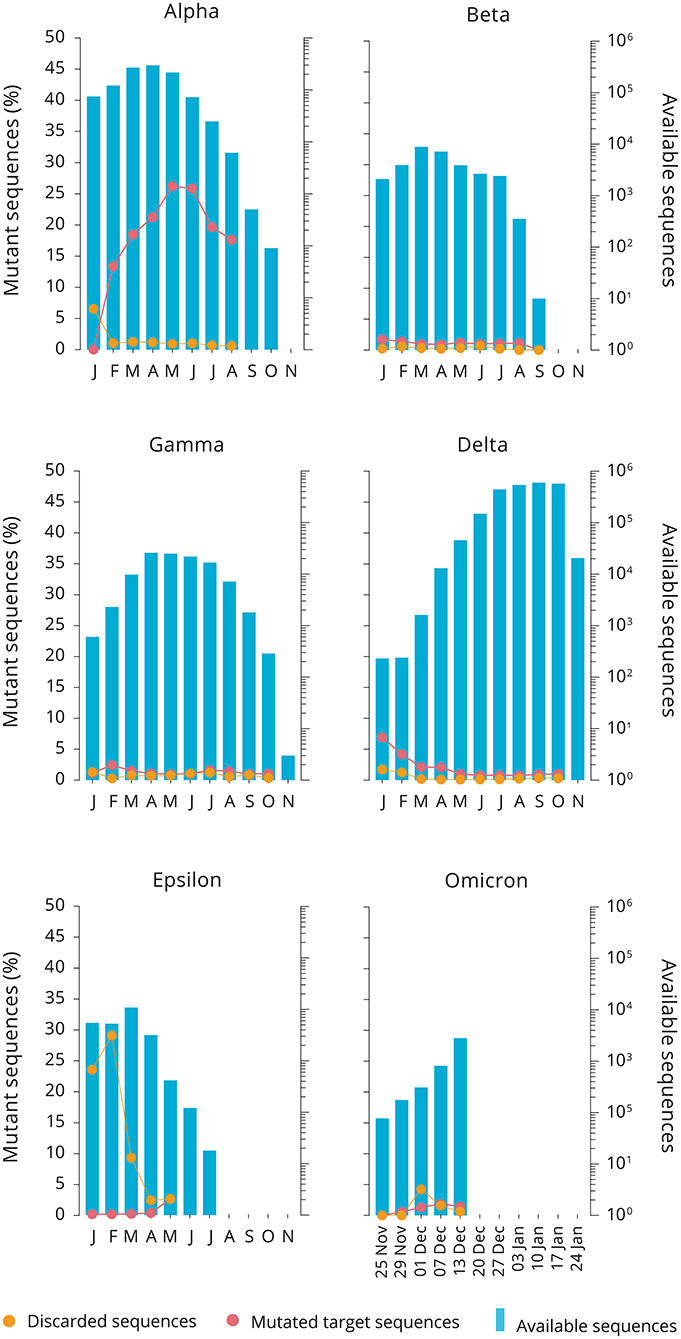
Figure 1. Monthly analysis of SARS-CoV-2 variant genomes available from GISAID. The total number of sequences per month depicted by the blue bars (scale on the right-hand side of each panel). Mutated target sequences are red dots, while discarded sequences poor quality are shown as orange dots. The red line in the Alpha variant depicts the emerging mutant sequence for the forward primer (....................c...........).
Only one nucleotide mutation emerged in the Alpha variant sequences, which slowly accumulated in the first quarter of the year (Figure 1). The occurrence of the mutation in the forward primer target sequence already observed in the first trial was corroborated by the second and third trial but dropped from a maximum of 26.23% in May to 17.64% by August, indicating that the variant with this particular nucleotide change was not able to expand and persist any further. It became obsolete, when Alpha variants were replaced by the emerging Delta variant, which since has dominated the pandemic. It is assumed that the Delta variant emerged and displaced the Alpha variant so quickly because it inherently produces higher viral loads in patients, thus rendering it more transmissible (8, 9). The depletion of the Alpha variant we observed may have been caught up in this displacement from the Delta variant and therefore subsided before it could generate a larger foothold in the Alpha variant population. It has been shown that a single mutation at position 14,408 in the RdRp gene was associated with a higher number of point mutations in the SARS-CoV genome overall (10). The mutation observed here at position 15,233 is a silent mutation and therefore can not be connected to inherent RdRp property changes or Alpha variant fitness.
The pipeline altogether is easy to use and was shown to handle sequence packages in excess of 600,000 sequences at a time (Supplementary Table S3) and can be used for closely monitoring new emerging variants such as currently the Omicron variant (Figure 1).
RPA amplicons are generically robust toward degenerations (11) and have been shown to tolerate up to nine nucleotide changes across all three oligonucleotides and robustness for detecting a wide range of genotypes of a virus (12, 13). Single nucleotide variations, even if they can occur at almost every position of an oligonucleotide (Supplementary Table S5) are therefore not considered to be of concern for the efficiency of the RPA amplicon. However, it indicates that RdRp mutations in general may occur more frequently than expected and need monitoring.
RPA oligonucleotides are longer than real-time PCR primers and therefore can not be entered into an oligonucleotide mutation scanner tool offered by GISAID with nucleotide length restrictions for PCR primers, while the approach presented here is able to easily scan with the complete amplicon target (128-mer).
This scalable algorithm is a desirable alternative to monitor SARS-CoV-2 genomes submitted in the GISAID database for molecular assay designers using oligonucleotides longer than the average oligonucleotides used in PCR design.
The tool enables rapid post-design sequence monitoring to assess emergent nucleotide mutations to adjust the design if necessary.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
MW, AA, and MB: study conception, design, drafting, and editing of the manuscript. MW: data acquisition. EG, DL, and MB: script and pipeline development. MW and AA: data analysis. All authors contributed to the article and approved the submitted version.
MW and EG work for Midge Medical GmbH, Berlin, which is developing a SARS-CoV-2 test based on RPA.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.835707/full#supplementary-material
1. Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall. (2017) 1:33–46. doi: 10.1002/gch2.1018
2. Lemmon GH, Gardner SN. Predicting the sensitivity and specificity of published real-time PCR assays. Ann Clin Microbiol Antimicrob. (2008) 7:18. doi: 10.1186/1476-0711-7-18
3. Hysom DA, Naraghi-Arani P, Elsheikh M, Carrillo AC, Williams PL, Gardner SN. Skip the alignment: degenerate, multiplex primer and probe design using K-mer matching instead of alignments. PLoS ONE. (2012) 7:e34560. doi: 10.1371/journal.pone.0034560
4. Kalendar R, Khassenov B, Ramankulov Y, Samuilova O, Ivanov KI. FastPCR: an in silico tool for fast primer and probe design and advanced sequence analysis. Genomics. (2017) 109:312–19. doi: 10.1016/j.ygeno.2017.05.005
5. El Wahed AA, Patel P, Maier M, Pietsch C, Rüster D, Böhlken-Fascher S, et al. Suitcase lab for rapid detection of SARS-CoV-2 based on recombinase polymerase amplification assay. Anal Chem. (2021) 93:2627–34. doi: 10.1021/acs.analchem.0c04779
6. Mapleson D, Garcia Accinelli G, Kettleborough G, Wright J, Clavijo BJ. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics. (2017) 33:574–6. doi: 10.1093/bioinformatics/btw663
7. Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. (1981) 147:195–7. doi: 10.1016/0022-2836(81)90087-5
8. Earnest R, Uddin R, Matluk N, Renzette N, Siddle KJ, Loreth C, et al. Comparative transmissibility of SARS-CoV-2 variants Delta and Alpha in New England, USA. medRxiv [Preprint]. (2021). doi: 10.1101/2021.10.06.21264641
9. Luo CH, Morris CP, Sachithanandham J, Amadi A, Gaston D, Li M, et al. Infection with the SARS-CoV-2 Delta variant is associated with higher infectious virus loads compared to the Alpha variant in both unvaccinated and vaccinated individuals. medRxiv [Preprint]. (2021). doi: 10.1101/2021.08.15.21262077
10. Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. (2020) 18:179. doi: 10.1186/s12967-020-02344-6
11. Boyle DS, Lehman DA, Lillis L, Peterson D, Singhal M, Armes N, et al. Rapid detection of HIV-1 proviral DNA for early infant diagnosis using recombinase polymerase amplification. mBio. (2013) 4:e00135-13. doi: 10.1128/mBio.00135-13
12. Daher RK, Stewart G, Boissinot M, Boudreau DK, Bergeron MG. Influence of sequence mismatches on the specificity of recombinase polymerase amplification technology. Mol Cell Probes. (2015) 29:116–21. doi: 10.1016/j.mcp.2014.11.005
Keywords: recombinase polymerase amplification, sequence mutation screening, GISAID, SARS-CoV-2 genomes, screening of oligonucleotides, mutations in oligonucleotides
Citation: Weidmann M, Graf E, Lichterfeld D, Abd El Wahed A and Bekaert M (2022) Efficient Screening of Long Oligonucleotides Against Hundred Thousands of SARS-CoV-2 Genome Sequences. Front. Virol. 2:835707. doi: 10.3389/fviro.2022.835707
Received: 20 December 2022; Accepted: 28 February 2022;
Published: 25 March 2022.
Edited by:
Brian Wigdahl, Drexel University, United StatesReviewed by:
Fuqing Wu, The University of Texas Health Science Center at Houston, United StatesCopyright © 2022 Weidmann, Graf, Lichterfeld, Abd El Wahed and Bekaert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michaël Bekaert, bWljaGFlbC5iZWthZXJ0QHN0aXIuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.