 Robert M. Zink
Robert M. Zink Kenedi Holck2
Kenedi Holck2- 1School of Natural Resources, University of Nebraska-Lincoln, Lincoln, NE, United States
- 2School of Biological Sciences, University of Nebraska-Lincoln, Lincoln, NE, United States
- 3Nebraska State Museum, University of Nebraska-Lincoln, Lincoln, NE, United States
The hunt for the immediate non-human host of SARS-CoV-2 has centered on bats of the genus Rhinolophus. We explored the phylogenetic predictions of two models of viral transmission, the Spillover Model and the Circulation Model and suggest that the Spillover Model can be eliminated. The Circulation Model suggests that viral transmission occurs among susceptible hosts irrespective of their phylogenetic relationships. Susceptibility could be mediated by the ACE2 gene (important for viral docking) and we constructed a phylogeny of this gene for 159 mammal species, finding a phylogenetic pattern consistent with established mammalian relationships. The tree indicates that viral transfer occurs over large evolutionary distances. Although lacking consensus, some studies identify a virus from a particular R. affinis individual (RaTG13) as being most closely related phylogenetically to human SARS-CoV-2. However, other R. affinis harbor viruses that are relatively unrelated to human viruses, and viruses found in this species exhibit sequence differences of up to 20%, suggesting multiple transfers over time. There is little correspondence between viral and host (bat) species limits or phylogenetic relationships. An ACE2 phylogeny for Rhinolophus followed species limits, unlike the pattern in the viral phylogeny indicating that phylogenetic similarity of ACE2 is not a predictor of viral transmission at the bat species level. The Circulation Model could be modified to apply to any individual of any species of Rhinolophus; more individuals and species must be examined.
Introduction
Numerous publications have employed phylogenetic analyses of viral sequences and the mammalian host species in which they were found to identify the most likely source of the SARS CoV-2 virus that led to the COVID-19 pandemic. Most studies suggest either bats of the genus Rhinolophus or pangolins as the source of the virus that invaded humans [e.g., (1–5)], although some suggest it is premature to rule out a laboratory origin (6, 7). Wenzel (8) pointed out that many phylogenetic studies of routes of viral transmission were inadequate, being based on small samples or flawed analytical methods [e.g., (9); Raza et al. 2021]. Machado et al. (10) used a comprehensive phylogenetic analysis to confirm that viruses carried by bats are more closely related to viruses currently circulating in humans than to those that currently occurring in pangolins, a result consistent with most other studies (4, 5, 11–13).
Few studies have considered the phylogenetic predictions for models of routes of viral transmission. The implicit assumption in most studies is that whatever bat species harbors a virus whose sequence is most closely related phylogenetically to those circulating in humans is the likely source of the human COVID-19 pandemic. However, there is no consensus on which species of Rhinolophus harbors a virus most closely related to SARS-CoV-2 in humans. In the Spike protein gene tree of Frutos et al. (4), the viral sequence from Rhinolophus affinis (intermediate horseshoe bat) was closest to those recovered from eight humans. In the RdRp gene tree (4), the closest sequence to humans came from a virus found in Rhinolophus malayanus (Malayan horseshoe bat), with the next taxon being R. affinis (same individual for both proteins). However, there was insignificant (54%) bootstrap support for the grouping of R. malayanus with humans in their RdRp gene tree. Of course, bootstrap values provide a measure character agreement and not necessarily phylogenetic certainty. In Wacharapluesadee et al. (11) R. affinis was closest to humans in a RdRp gene tree. Comparing three recombinant segments, Temman et al. (12) produced trees that show R. affinis as the sister to humans, a cluster of R. marshalli, R. pusillus, and R. malayanus as sister to humans, and a mixture of Rhinolophus as sister to humans. In Zhou et al. (13) the sister clade to human SARS-CoV-2 included R. malayanus, R. pusillus, R. affinis and R. shameli. Latinne et al. (5) show a sister-group relationship between R. malayanus and human SARS-CoV-2. Thus, there is relatively little phylogenetic consistency across studies in terms of the species of Rhinolophus that harbors the virus lineage most closely related to that found in humans.
The inconsistent phylogenetic results could be a function of limited sampling of individuals per species and genes. For example, the four pangolins (Manis javanica) used by Frutos et al. (4) were apparently collected in 2017 and the spike protein sequences exhibit 14 variable sites (out of 4,718 bp) of which 13 are singletons. For the RdRp gene, the four pangolins exhibited no base substitutions (out of 385 bp). It is possible that these four pangolins were a family group or close relatives and might therefore be pseudo replicates. In fact, Zhou et al. (13) discovered two non-sister clades of viral sequences in pangolins, neither of which were sister to viral sequences found in humans. Thus, results for pangolins are influenced by number of individuals used in phylogenetic analyses, although it appears pangolins can be ruled out as the source for human SARS-CoV-2. Sampling concerns also exist for viral sequences derived from humans. In the 385 bp for RdRp there were no variable sites, and for the spike protein there were 0, 1, or 2 pairwise substitutions out of the 4,718 bp (4). These samples provide insufficient diversity for phylogenetic analysis.
The Hunt for the Donor of SARS-CoV-2 to Humans
Most studies in which R. affinis was shown as sister to humans used a sequence from the same individual bat, RaTG13 (Genbank MN996532), which was collected on July 24, 2013. Many studies have used the same publicly available sequences, often collected before the pandemic, and given the rapid rate of viral evolution the age of the specimens could be a concern. Sampling other individual R. affinis has shown that their viral sequences do not form a clade (4, 13) and viral lineages from this host exhibit considerable diversity; the spike proteins from R. affinis average 21% sequence divergence (n = 7), although two of the pairwise differences are 1.1% and 4.1%. There is as much sequence divergence between viruses from different individuals of R. affinis as there is between viruses from different bat species, which explains why several individuals of R. affinis and R. sinicus (Chinese rufus horseshoe bat) are in well supported clades separated on the tree from other conspecifics (13). The existence of closely and distantly related viral strains in R. affinis suggests recent as well as older viral transmission events, assuming that sequence divergence reflects time since colonization of a new host. The genetic distances from viral sequences (spike protein) recovered from R. affinis to human viruses is slightly greater (ca. 25%) than within-affinis comparisons, suggesting novel mutations during the colonization of humans (from whatever source). However, one cannot claim that R. affinis is “the” species that produced the viral strain that jumped to humans, because other members of the same species harbor viruses that are not most closely related to those circulating in humans. Rhinolophus includes at least 75 species (14) split between Africa and Southeast Asia, whereas no study included more than about 13 species (13). Machado et al. (10) suggested that viral transmission might be common throughout the Orthocoronavirinae, providing a phylogenetically testable hypothesis for future studies. Thus, many bat species (with multiple individuals from multiple localities) remain unsampled, and thus the actual distribution of SARS-CoV-2 is unclear.
Other species harbor SARS-CoV-2. Chandler et al. (15) reported that variants of SARS-CoV-2 occurring in white-tailed deer (Odocoileus virginianus) are the same as those found in humans from the local area (Iowa, USA), suggesting recent transfer without as yet subsequent mutations (or reverse transfer from deer to humans). Deer mice can be experimentally infected with SARS-CoV-2 (16). Mink also harbor the virus (17, 18). Hence, the search for species that could become reservoirs for the virus should be phylogenetically diverse.
Viral Lineages, Nomenclature, and Transmission Models
Frutos et al. (19) considered the mode of viral transmission and provided a discussion of the virus species concept stating “A virus is not spreading based on species and species barriers but simply based on its ability to recognize a receptor and circumvent the host immune defenses. This occurs regardless of the “species” status given by humans using classification criteria. There is no distinction between “animal hosts,” “human hosts,” hence there is no such thing as the crossing of the species barrier.” This caricature is puzzling given that many papers used a phylogeny of viral sequences to determine which host species harbored viruses that were most closely related to viruses in humans and therefore crossed the species barrier. However, it is fair to say that there has not been a rigorous comparison of host and viral phylogenies. Bat systematists classify species of bats by congruence of morphological, molecular, and behavioral evidence. Defining a species of virus, however, is more elusive, and usually terms such as “SARS-CoV-2 variants” are used (to circumvent calling each unique viral sequence a species), for which one can reconstruct evolutionary relationships. Frutos et al. (2021) statement “hence there is no such thing as the crossing of a species barrier” might apply to viral lineages but the virus has crossed species barriers in the host (bat) phylogeny [(20, 21); but see (22)].
Frutos et al. (19, 23) consider two (of perhaps many) conceptual models of viral transmission, the Spillover Model and the Circulation Model. Frutos et al. (19) suggest that the Spillover Model (Figure 1A) should be replaced by the Circulation Model (Figure 1B). The Spillover Model “postulates that an animal reservoir must be at the origin of zoonosis” (19). In theory, a disease would become prevalent within a reservoir species and “spillover” into another host, which might be facilitated by an intermediate species (24, 25). Several publications [e.g., Raza et al. (26)] have suggested bats as the reservoir, pangolins as the intermediate, and humans as the final host. Frutos et al. (19) note many problems with this hypothesis [e.g., (5, 11, 12, 12)]. Viral sequences originated from smuggled pangolins confiscated by Chinese customs before the pandemic. There were no related reports of epizootic disease in pangolins or other animals in China or elsewhere; in addition, the SARS-CoV-2 related viruses were only reported in smuggled Malayan pangolin and Rhinolophus bats (19), which might not be germane to the question of human invasion. Frutos et al. (19) concluded that the Spillover Model is unsupported.
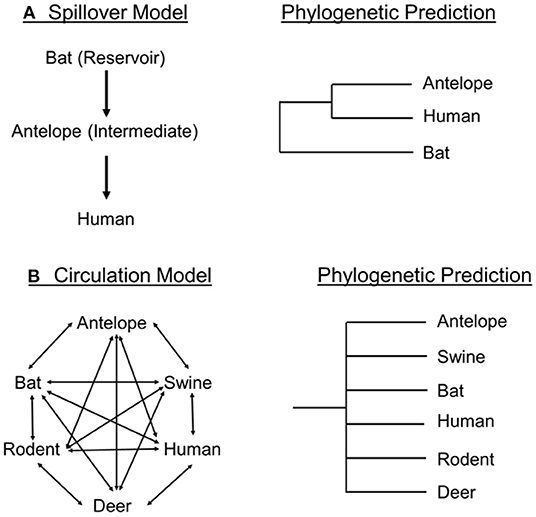
Figure 1. Spillover Model (A) and Circulation model (B) as presented by Frutos et al. (19) and their phylogenetic predictions for viral transmission.
Frutos et al. (19, 23) proposed the Circulation Model (Figure 1B) to account for the evolution, transmission, and achievement of epidemic status of viruses in human populations. The model predicts that viruses circulate through local populations on the basis of receptor-recognition compatibility of local hosts, irrespective of species barriers. That is, host compatibility and proximity create a “metapopulation” (19) of viruses within hosts of co-existing species. For instance, a Sarbecovirus population might be composed of multiple viral quasispecies within local populations of bats, rodents, humans, and boar. The virus is continually passed to and from compatible hosts in the area, irrespective of taxonomic boundaries, and each quasi-species experiences different host-specific selective pressures.
The Circulation Model proposes that a human epidemic is precipitated by intra-human evolution of a quasi-species “individual” that adapts this viral genotype to greater transmissibility within humans. According to Frutos et al. (19), this is termed the “Stochastic” Phase, or Latency Phase. At this point the activity of the host organism creates the conditions for epidemic transmission, termed the “amplification loop” (19). This “loop” is facilitated by activities such as gatherings, celebrations, and markets where infected individuals are in close physical proximity. Once an outbreak threshold is reached an epidemic results and viral transmission attains a polynomial rate. This aligns with work showing that population density is positively correlated with the spread of COVID-19 infections (27).
Phylogenetic Predictions of Two Possible Transmission Models
If phylogenetic hypotheses are to be used to make inferences about pathways of viral transmission, it is useful to determine if the Spillover and Circulation models make mutually exclusive phylogenetic predictions. In the Spillover model ((19); Figure 1A), one would expect a sister-group relationship between the intermediate species and the final host species. A spillover process could occur contemporaneously in multiple host and viral lineages and provide a more complicated phylogenetic pattern in which we would observe independent instances of human viral sequences dispersed on the phylogeny each sister to a different or same Rhinolophus bat species. However, a consistent pattern in the phylogenetic trees involving SARS-CoV-2 [e.g., (4, 13)] is the absence of multiple examples of viral and human sister-group relationships, instead, the viral sequences from the sampled humans cluster together, with some species or group of species of Rhinolophus as its sister taxon. Thus, the Spillover Model as envisioned by Frutos et al. (19) fails to meet phylogenetic predictions for single or multiple spillover events, which agrees with these author's conclusion.
According to the Circulation Model (Figure 1B), viruses need a host with a susceptible ACE2 genotype (12) and spread among species occurs irrespective of host phylogenetic boundaries, but should reflect pathways of compatibility. Their Circulation Model included bats, pigs, humans, antelope, deer, and rodents (and omitted pangolins), a broad sampling of mammalian taxa, unlike the phylogeny in Frutos et al. (4), which depicts humans, bats and pangolins. The Circulation Model (Figure 1B) predicts an evolutionary tree with no phylogenetic structure. In other words, the viral phylogeny would not reflect the host phylogenetic relationships, but as formulated, does not in dictate whether viral transmission would be coincident with host species limits.
ACE2 and the Circulation Model
The ACE2 gene is involved in viral docking and is thought to play a role in viral transmission of SARS-CoV-2 among species (12, 28–30). To explain the predicted phylogenetic outcome, the Circulation Model would have to assume that compatible ACE2 genotypes are phylogenetically randomly distributed among species. We examined this assumption by constructing a phylogenetic tree for the ACE2 gene for 159 mammal species (aligned amino acid sequences downloaded from Genbank; https://www.ncbi.nlm.nih.gov/gene/59272/ortholog/?scope = 40,674), finding that the gene tree is consistent with mammalian phylogeny (Supplementary Figure 1)–closely related mammal species have phylogenetically closely related ACE2 sequences irrespective of their viral lineages. This tree has implications for several aspects of the relationship between ACE2 and the occurrence of SARS-CoV-2. Palmer et al. (31) stated that the ACE2 of deer and humans “share a high degree of similarity” and suggest this could account for the recent transmission of SARS-CoV-2 from humans to deer (32). Fenollar et al. (17) stated “The mink's receptor for SARS-CoV-2 is very similar to that of humans.” However, Palmer et al. (31) and Fenollar et al. (17) confuse similarity with phylogenetic relationships, and the ACE2 genes from humans, mink, and deer are far apart phylogenetically (Supplementary Figure 1). In addition, bats, pangolins and humans are also phylogenetically distant, meaning that bat-human viral transmission occurs over relatively great evolutionary distances [as do transmissible spongiform encephalopathies; (33)]. This observation could be considered consistent with a Circulation Model, in that it appears to support phylogenetically random viral transmission among species albeit at higher taxonomic levels.
To explore further the relationship between viral phylogeny and the bat ACE2 gene tree, we constructed a phylogenetic hypothesis for species of Rhinolophus (Figure 2); most available sequences come from two species (R. affinis, R. sinicus) often implicated as candidates for transmission to humans (5). The ACE2 gene tree strongly reflects bat species limits. Individuals identified as R. affinis formed a clade (23 individuals; 19 distinct sequences; average number of base pair and amino acid differences 3.9, 1.2) as did those identified as R. sinicus (26 individuals; 26 distinct sequences; average number of base pair and amino acid differences 13.1, 7.2). If viral transmission is mediated by ACE2, viral lineages from species such as R. affinis and R. sinicus ought to form clades (given the low ACE2 intraspecific diversity in bats), which is not the case (13). That is, most closely related bats, even from the same species, do not harbor most closely related viruses, although closely related bats possess closely related ACE2 genotypes. It is difficult to reconcile ACE2 compatibility with viral phylogeny under the Circulation Model at the species level in bats.
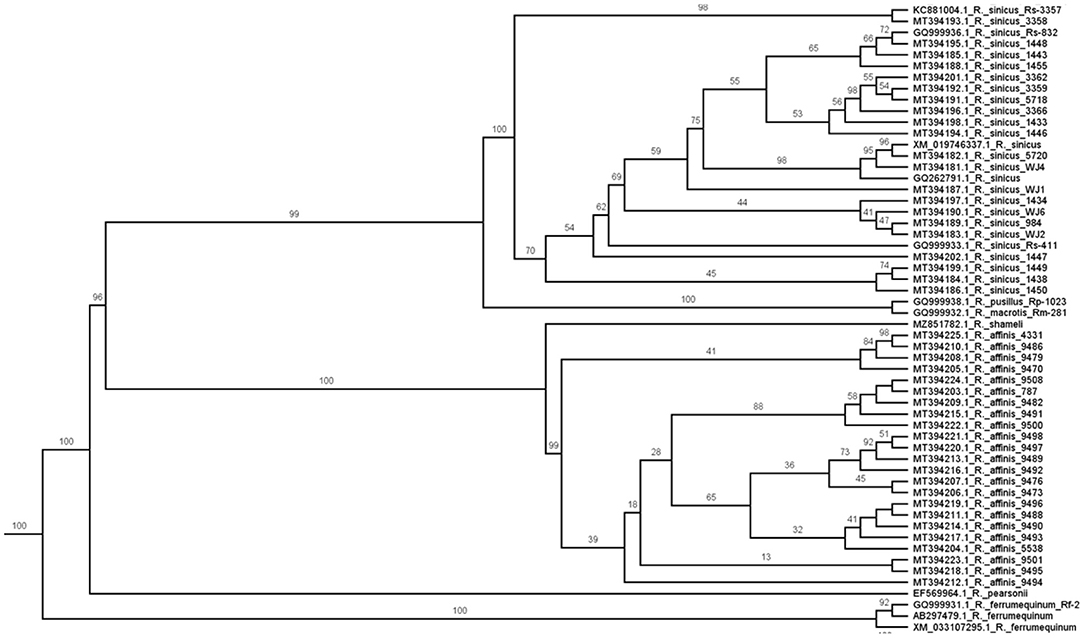
Figure 2. Maximum likelihood phylogeny of the ACE2 gene for bats of the genus Rhinolophus [see also (34)]. Numbers on branches are bootstrap values. Note that all 26 specimens of R. sinicus and all 23 specimens of R. affinis are reciprocally monophyletic. If ACE2 relationships indicate pathways for viral transmission, one would expect the viral phylogeny to be similarly structured but it is not (13).
One might ask why all R. affinis do not share the same viral sequence given their similar ACE2 genotypes? Perhaps other R. affinis are susceptible to SARS-CoV-2 but simply not exposed to the same viral lineage as that found in RaTG13. If all viruses need are hosts with compatible ACE2 genotypes, and the latter are phylogenetically structured, the mismatch between viral phylogeny and bat species limits suggests viral transmission is not phylogenetically random among species, a condition of the Circulation Model.
A valid test of the Circulation Model would be to sample vertebrate host populations from phylogenetically unrelated sympatric lineages and compare their circulating viruses and phylogenetic similarities in ACE2 to verify phylogenetic predictions (Figure 1B). For instance, we should (hypothetically) observe viral sister-linages in pigs and rodents, bats and deer, humans and antelope, and be able to identify what in their ACE2 genotype makes them compatible. To date, most studies use three major taxa, bats, humans and pangolins, which precludes a valid phylogenetic test of the Circulation Model. At present, it would be necessary to reformulate the Circulation Model to a taxonomic level below species, namely individuals, for it to have explanatory power. Latinne et al. (5) suggest that cross-species transfer is common in the genus Rhinolophus, but it goes beyond species of bats to seemingly random transmission among groups of individuals within the same species.
Conclusions and Recommendations
If a phylogenetic hypothesis is to provide a test of competing models of viral transmission, several factors merit consideration. A large number of individuals per species and species per genus from phylogenetically diverse lineages are needed to evaluate routes of viral transmission, but as of yet this has not accomplished in any single study. Phylogenetic analyses should meet stringent criteria (10). Published phylogenetic hypotheses of viral sequences do not support a Spillover Model, and only support a Circulation Model if it is modified to function at the level of individual bats, irrespective of species limits, in the genus Rhinolophus (5). That is, viral transmission does not track bat species boundaries or phylogenetic relationships, and hence, any particular species of bat is not the unit of transmission. Although transmission occurs among compatible hosts, the role of ACE2 in viral transmission is not phylogenetically straightforward, as bat species are reciprocally monophyletic on the ACE2 gene tree, whereas their viral lineages are not.
To facilitate assessment of ACE2 role in viral transmission, researchers should collect and preserve bat specimens from which they obtain virus samples and deposit them (study skins and tissue samples) in natural history collections. Guo et al. (30) stated “Bats were released after anal swabs [sic] sampling.” This practice precludes definitive identification of the bat by museum specialists, and without voucher specimens (tissue, study skin) one cannot compare ACE2 sequences or morphological characteristics from the same individual bat for which the viral genome was sequenced (35, 36). Given the many unanswered issues, we advocate the One Health approach to understanding the relationship between “people, animals, plants and their shared environment” (https://www.cdc.gov/onehealth/basics/index.html).
Author Contributions
RZ analyzed data and wrote the manuscript. KH and GM wrote sections on models of viral transmission, approved, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank R. Frutos, H. Vázquez-Miranda, and John Wenzel for discussion.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.832466/full#supplementary-material
Supplementary Figure 1. Maximum likelihood phylogeny produced by MegaX (37) from 159 aligned amino acids at ACE2 rooted with platypus (Ornithorhynchus anatinus). White-tailed deer (Odocoileus virginianus texanus), short-tailed weasel (Mustela erminea), pangolin (Manus javanica) are denoted with an arrow, human (Homo sapiens) is denoted with a large arrow, and the vertical bar corresponds to a monophyletic group of bats. Note that all major taxonomic groups (bats, rodents, primates) form distinct clades and all representatives from genera are clustered together. The percentage of 100 trees in which the associated taxa clustered together is shown next to the branches. The heuristic search employed the JTT model. A discrete Gamma distribution was used to model evolutionary rate differences among sites [4 categories (+G, parameter = 0.9136)]. The rate variation model allowed for some sites to be evolutionarily invariable [(+I), 18.37% sites]. All positions with < 95% site coverage were eliminated, i.e., fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option). A total of 796 amino acid positions were in the final dataset.
References
1. Forster P, Forster L, Renfrew C, Forster M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Nat Acad Sci. (2020) 117:9241–3. doi: 10.1073/pnas.2004999117
2. Mavian C, Pond SK, Marini S, Magalis BR, Vandamme A-M, Dellicou S, et al. Sampling bias and incorrect rooting make phylogenetic network tracing of SARS-COV-2 infections unreliable. Proc Nat Acad Sci. (2020) 117:12522–3. doi: 10.1073/pnas.2007295117
3. Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol. (2020) 30:1346–51. doi: 10.1016/j.cub.2020.03.022
4. Frutos R, Serra-Cobo J, Chen T, Devaux CA. COVID-19: time to exonerate the pangolin from the transmission of SARS-CoV-2 to humans. Infect Genet Evol. (2020) 84:104493. doi: 10.1016/j.meegid.2020.104493
5. Latinne A, Hu B, Olival KJ, Zhu G, Zhang L, Li H, et al. Origin and cross-species transmission of bat coronaviruses in China. Nat Commun. (2020) 11:1–15. doi: 10.1038/s41467-020-17687-3
6. Segreto R, Deigin Y. The genetic structure of SARS-CoV-2 does not rule out a laboratory origin: SARS-COV-2 chimeric structure and furin cleavage site might be the result of genetic manipulation. BioEssays. (2021) 43:2000240. doi: 10.1002/bies.202000240
7. Sallard E, Halloy J, Casane D, Decroly E, van Helden J. Tracing the origins of SARS-COV-2 in coronavirus phylogenies: a review. Environ Chem Lett. (2020) 2011:1–17. doi: 10.1007/s10311-020-01151-1
8. Wenzel J. Origins of SARS-CoV-1 and SARS-CoV-2 are often poorly explored in leading publications. Cladistics. (2020) 36:374–9. doi: 10.1111/cla.12425
9. Choo SW, Zhou J, Tian X, Zhang S, Qiang S, O'Brien SJ, et al. Are pangolins scapegoats of the COVID-19 outbreak-CoV transmission and pathology evidence? Conserv Lett. (2020) 13:e12754. doi: 10.1111/conl.12754
10. Machado DJ, Scott R, Guirales S, Janies DA. Fundamental evolution of all Orthocoronavirinae including three deadly lineages descendent from Chiroptera-hosted coronaviruses: SARS-CoV, MERS-CoV and SARS-CoV-2. Cladistics. (2021) 37:461–88. doi: 10.1111/cla.12454
11. Wacharapluesadee S, Tan CW, Maneeorn P, Duengkae P, Zhu F, Joyjinda Y, et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat Commun. (2021) 12:1–9. doi: 10.1038/s41467-021-21240-1
12. Temmam S, Vongphayloth K, Salazar EB, Munier S, Bonomi M, Régnault B, et al. Coronaviruses with a SARS-CoV-2-like receptor-binding domain allowing ACE2-mediated entry into human cells isolated from bats of Indochinese peninsula. (2021). doi: 10.21203/rs.3.rs-871965/v1
13. Zhou H, Ji J, Chen X, Bi Y, Li J, Wang Q, et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell. (2021) 184:4380–91. doi: 10.1016/j.cell.2021.06.008
14. Simmons NB. Order Chiroptera. mammal species of the world: a taxonomic and geographic reference. (2005) 1:312–529.
15. Chandler JC, Bevins SN, Ellis JW, Linder TJ, Tell RM, Jenkins-Moore M, et al. SARS-CoV-2 exposure in wild white-tailed deer (Odocoileus virginianus). Proc Natl Acad Sci USA. (2021) 118:e2114828118. doi: 10.1073/pnas.2114828118
16. Fagre A, Lewis J, Eckley M, Zhan S, Rocha SM, Sexton NR, et al. SARS-CoV-2 infection, neuropathogenesis and transmission among deer mice: Implications for spillback to New World rodents. PLoS ONE pathogens. (2021) 17:e1009585.
17. Fenollar F, Mediannikov O, Maurin M, Devaux C, Colson P, Levasseur A, et al. Mink, SARS-CoV-2, and the human-animal interface. Front Microbiol. (2021) 12:745. doi: 10.3389/fmicb.2021.663815
18. Devaux CA, Pinault L, Delerce J, Raoult D, Levasseur A, Frutos R. Spread of mink SARS-CoV-2 variants in humans: a model of sarbecovirus interspecies evolution. Front Microbiol. (2021) 12:675528. doi: 10.3389/fmicb.2021.675528
19. Frutos R, Gaotte L, Devaux CA. Understanding the origin of COVID-19 requires to change the paradigm on zoonotic emergence from the spillover model to the viral circulation model. Infect Genet Evol. (2021) 95:104812. doi: 10.1016/j.meegid.2021.104812
20. Stoffberg S, Jacobs DS, Mackie IJ, Matthee CA. Molecular phylogenetics and historical biogeography of Rhinolophus bats. Mol Phylogenet Evol. (2010) 54:1–9. doi: 10.1016/j.ympev.2009.09.021
21. Agnarsson I, Zambrana-Torrelio CM, Flores-Saldana NP, May-Collado LJ. A time-calibrated species-level phylogeny of bats (Chiroptera, Mammalia). PLoS Curr. (2011) 3:RRN1212. doi: 10.1371/currents.RRN1212
22. Liang J, Zhu C, Zhang L. Cospeciation of coronavirus and paramyxovirus with their bat hosts in the same geographical areas. BMC Ecol Evol. (2021) 21:1–11. doi: 10.1186/s12862-021-01878-7
23. Frutos R, Pliez O, Gavotte L, Devaux CA. There is no “origin” to SARS-CoV-2. Environ Res. (2021) 112173. doi: 10.1016/j.envres.2021.112173
24. Tazerji SS, Duarte PM, Rahimi P, Shahabinejad F, Dhakal S, et al. Transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) to animals: an updated review. J Transl Med. (2020) 18:1–11. doi: 10.3390/ani11010016
25. Bonilla-Aldana DK, Rodriguez-Morales AJ. A brief introduction to coronavirus disease 2019 (COVID-19) and the roles of zoonotic spillover. in Human Viruses: Diseases, Treatments and Vaccines. Springer: Cham (2021). p. 565–70. doi: 10.1007/978-3-030-71165-8_26
26. Raza S, Navid MT, Zahir W, Khan MN, Awais M, Yaqub T, et al. Analysis of the spike proteins suggest pangolin as an intermediate host of COVID-19 (SARS-CoV-2). Int J Agric Biol. (2021) 25:639–44.
28. Qiu Y, Zhao YB, Wang Q, Li JY, Zhou ZJ, Liao CH, et al. Predicting the angiotensin converting enzyme 2 (ACE2) utilizing capability as the receptor of SARS-CoV-2. Microbes Infect. (2020) 22:221–5.
29. Lopes LR, de Mattos Cardillo G, Paiva PB. Molecular evolution and phylogenetic analysis of SARS-CoV-2 and hosts ACE2 protein suggest Malayan pangolin as intermediary host. Braz J Microbiol. (2020) 51:1593–9. doi: 10.1007/s42770-020-00321-1
30. Guo H, Hu B, Si HR, Zhu Y, Zhang W, Li B, et al. Identification of a novel lineage bat SARS-related coronaviruses that use bat ACE2 receptor. Emerg Microbes Infect. (2021) 10:1507–14. doi: 10.1080/22221751.2021.1956373
31. Palmer MV, Martins M, Falkenberg S, Buckley A, Caserta LC, Mitchell PK, et al. Susceptibility of white-tailed deer (Odocoileus virginianus) to SARS-CoV-2. J virol. (2021) 95:e00083–21. doi: 10.1128/JVI.00083-21
32. Kuchipudi SV, Surendran-Nair M, Ruden RM, Yon M, Nissly RH, Nelli RK, et al. Multiple spillovers and onward transmission of SARS-CoV-2 in free-living and captive white-tailed deer. bioRxiv. (2021). doi: 10.1101/2021.10.31.466677
33. Zink RM. Genetic and evolutionary considerations of the chronic wasting disease–human species barrier. Infect Genet Evol. (2020) 84:104484. doi: 10.1016/j.meegid.2020.104484
34. Mao X, Tsagkogeorga G, Thong VD, Rossiter SJ. Resolving evolutionary relationships among six closely related taxa of the horseshoe bats (Rhinolophus) with targeted resequencing data. Mol Phylogenet Evol. (2019) 139:106551. doi: 10.1016/j.ympev.2019.106551
35. Colella JP, Agwanda BR, Anwarali Khan FA, Bates J, Carrion Bonilla CA, de la Sancha NU, et al. Build international biorepository capacity. Science. (2020) 370:773–4. doi: 10.1126/science.abe4813
36. Colella JP, Bates J, Burneo SF, Camacho MA, Carrion Bonilla C, Constable I, et al. Leveraging natural history biorepositories as a global, decentralized, pathogen surveillance network. PLoS Pathog. (2021) 17:e1009583. doi: 10.1371/journal.ppat.1009583
Keywords: phylogenetics, Spillover Model, species barriers, viral transmission, Circulation model
Citation: Zink RM, Holck K and Morgan GL (2022) Phylogenetic Tests of Models of Viral Transmission. Front. Virol. 2:832466. doi: 10.3389/fviro.2022.832466
Received: 09 December 2021; Accepted: 05 January 2022;
Published: 09 February 2022.
Edited by:
Mark Zwart, Netherlands Institute of Ecology (NIOO-KNAW), NetherlandsReviewed by:
Denis Jacob Machado, University of North Carolina at Charlotte, United StatesCopyright © 2022 Zink, Holck and Morgan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert M. Zink, cnppbmsyQHVubC5lZHU=