
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol. , 28 February 2022
Sec. Emerging and Reemerging Viruses
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.814114
This article is part of the Research Topic Emerging and Re-emerging Viral Diseases View all 37 articles
 Marianna Scrima1,2†
Marianna Scrima1,2† Alessia Maria Cossu1,2,3†
Alessia Maria Cossu1,2,3† Egildo Luca D'Andrea1,4
Egildo Luca D'Andrea1,4 Marco Bocchetti2,3Ylenia Abruzzese1Clara Iannarone1Cinzia Miarelli1Piera Grisolia2Federica Melisi2,3
Marco Bocchetti2,3Ylenia Abruzzese1Clara Iannarone1Cinzia Miarelli1Piera Grisolia2Federica Melisi2,3 Lucia Genua5,6Felice Di Perna7Paolo Maggi8,9
Lucia Genua5,6Felice Di Perna7Paolo Maggi8,9 Giovanbattista Capasso10
Giovanbattista Capasso10 Teresa Maria Rosaria Noviello11,12
Teresa Maria Rosaria Noviello11,12 Michele Ceccarelli11,12Alessandra Fucci1,4
Michele Ceccarelli11,12Alessandra Fucci1,4 Michele Caraglia1,2,3*
Michele Caraglia1,2,3*Coronavirus disease 2019 (COVID-19) emerged in December 2019 when the first case was reported in Wuhan, China, and turned into a pandemic. Whole-genome sequencing (WGS) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) plays a crucial role in understanding the disease. For this reason, we performed WGS of 101 SARS-CoV-2 strains obtained from individuals from two districts of Campania (Italy) from January to May 2021. The phylogenetic analysis of sequence data identified five types of clades including 10 different Pango lineages: 20A (Lineages B.1.258.17, B.1.258.14, and B.1.160) (n = 10; 9.9%), 20B (Lineages B.1.1.351 and B.1.374) (n = 5; 4.9%), 20E (EU1) (Lineages B.1.177.53, B.1.177.75, and B.1.177) (n = 5; 4.9%), 20I (Alpha.V1) (B.1.1.7) (n = 60; 59.4%), and 20J (Gamma.V3) (Lineage P.1.1) (n = 21; 20.7%). In the early time of the epidemic (January and February 2021), B.1.1.7 lineage was in 62% of samples only in Benevento district, while this lineage appears in Avellino later in 64% of samples from March to May. The occurrence of P.1.1 lineage spreading from March to the end of the study was recorded in all districts with the same frequencies of ~21%. The highest genomic distance was observed in Lineage P.1.1. Moreover, we identified 219 “known” missense mutations with different frequencies (114 in ORF1a/1b; 12 in ORF3a; 29 in S; 5 in M; 29 in N; and 5 each in ORF7a and ORF8). This report suggests the quickly spreading in Campania of new variants of SARS-CoV-2 and the strict surveillance of the occurrence of genetic variants of SARS-CoV-2.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in late 2019 (1). SARS-CoV-2 is a highly transmissible and pathogenic coronavirus and caused an acute respiratory disease named “coronavirus disease 2019” (COVID-19). As a novel betacoronavirus (species name: Severe acute respiratory syndrome-related coronavirus; family: Coronaviridae) (2), SARS-CoV-2 shares 79% genome sequence identity with SARS-CoV. SARS-CoV-2 has a single-stranded, positive-sense RNA (+RNA) genome of ~29.9 kb directly working as an mRNA, to initiate viral genome replication and transcription. The SARS-CoV-2 genome ORF1a and ORF1b regions encode two polyproteins, which are cleaved by two viral proteases into 16 nonstructural proteins (nsp1–16) (3). Moreover, they also encode four structural domains (S, spike; E, envelope; M, membrane; and N, nucleocapsid) and seven putative ORFs encoding accessory proteins interspersed between the structural genes (ORF3a, 6, 7a, 7b, 8, 9b, and 10) (4, 5). These domains are the main protagonists of genomic variants that alter the amino acid composition of any of these ORFs. More heavily mutated SARS-CoV-2 lineages emerged during the time and due to the rapid spreading of the virus with frequent inter-individual passages. The occurrence of these hypermutated viruses resulted in the change of infective characteristics of the pathogen with the consequent increase of the transmission abilities. The most relevant mutations are often located in the spike encoding region, with the S protein primarily involved in the entry of the virus in the host cells and in the recognition by neutralizing antibodies (6, 7). In July 2020, Rambaut et al. (8) described a possible nomenclature for the lineage of SARS-CoV-2 to define specific viral genomes with common nucleotide sequences that spread around the world during the pandemia. This is the reason that prompted to implement to use an algorithm named Phylogenetic Assignment of Named Global Outbreak LINeages (Pangolin). Pango lineages are particularly useful for investigations at national or regional scales and define a relevant phylogenetic cluster. Pangolin nomenclature was flanked by a new classification introduced by the WHO that identified different variants of SARS-CoV-2 that have been documented during the current pandemic, some of which are identified as variants of concern (VOCs) with influence on public health (9). Based on the epidemiological update of June 2021 by the WHO, four different SARS-CoV-2 VOCs have been identified since the start of the pandemic: i) Alpha (B.1.1.7), first VOC described in the United Kingdom in late December 2020; ii) Beta (B.1.351), first identified in South Africa in December 2020; iii) Gamma (P.1), first identified in Brazil in early January 2021; and iv) Delta (B.1.617.2), first identified in India in December 2020. All four identified VOCs show mutations in the receptor-binding domain (RBD) and the N-terminal domain (NTD) (7, 10). In addition to VOCs, there are variants of interest (VOIs) defined as variants with specific genetic modifications. VOI can enhance transmissibility or virulence and reduce neutralization by antibodies. In this light, despite the extraordinary speed of vaccine development against COVID-19 and continued mass vaccination efforts across the world, the emergence of these new variant strains of SARS-CoV-2 compromises the significant progress made so far in hindering the spread of SARS-CoV-2. For these reasons, in this study, we examined epidemiological and viral genetic data to recreate the pattern of SARS-CoV-2 diffusion in two districts of Campania (Avellino and Benevento). Whole-genome sequencing was performed for 101 SARS-CoV-2 strains obtained from individuals from two districts in a time span of 5 months from January to May 2021. Confirmed COVID-19 cases in Italy from January 2021 to May 2021 are 4,216,003; in Campania, there are 419,269 distributed in all districts. We were interested in Benevento (12, 373 cases) and Avellino (20, 006 cases) districts (https://coronalevel.com/Italy/Campania/), aiming, in conclusion, to trace local transmission and the insurgence of different SARS-CoV-2 variant strains in specific temporal and geographical contexts.
Nasopharyngeal swabs samples were collected in viral transport media (UTM®) from individuals in two Campania districts from January to May 2021: 54 from Benevento and 47 from Avellino.
Avellino and Benevento are neighboring districts in Campania, and they are 44 km away. As a consequence, the area of the two districts is limited, and the number of viral genomes sequenced should be appropriate to evaluate the diffusion of the variants. All specimens with suspected COVID-19 infection were confirmed to be SARS-CoV-2 positive by real-time PCR (qRT-PCR). A total of 101 successful whole-genome sequences from the following cases were analyzed.
Ethical review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements because the samples were collected as part of routine care. The participants provided their written informed consent to participate in this study.
Viral nucleic acid was isolated from 200 μl of nasopharyngeal swab using MagMAX™ Viral/Pathogen Nucleic Acid Isolation Kit (A42352, Thermo Fisher Scientific, Waltham, MA, USA) in high-throughput automated mode on KingFisher™ Purification System (Thermo Fisher Scientific, Waltham, MA, USA). The extracted RNA was directly used for amplification using TaqPath™ COVID-19 CE-IVD RT-PCR Kit (48102, Thermo Fisher Scientific, Waltham, MA, USA) to determine viral RNA copy number quantification, according to the manufacturer's protocol. Samples were considered positive when the cycle threshold (Ct) values were <37 for at least two out of three SARS-CoV-2 target genes. The analyzed viral targets were “S” gene, “N” gene, and “ORF1ab” gene of SARS-CoV-2. Negative and positive controls were run simultaneously with samples in order to ascertain the positivity of the expression of the viral genes. We used certified commercial kits in order to assess the presence of viral RNA.
Ion AmpliSeq™ SARS-CoV-2 Research Panel library (Thermo Fisher Scientific, Waltham, MA, USA) was prepared from samples containing 3,000 to 320,000 copies of viral RNA with Ct mean value of the three genes (S, N, and ORF1ab) equal to 16. The Ion AmpliSeq SARS-CoV-2 Research Panel is composed of 2 different primer pools to amplify 237 amplicons across the SARS-CoV-2 genome. The average of generated sequences is 200 bp (125–275 bp in length). The whole-genome sequencing was performed by using Ion PI Hi-Q Sequencing 200 Kit–Chef Kit (Thermo Fisher Scientific, Waltham, MA, USA) on the Ion Proton Sequencer and Ion S5 System (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer's protocol. The median depth of coverage for all the sequences is 13,780 (range 2,000–46,779).
Sequences are uploaded on Gisaid and Sequence Read Archive (SRA) database: ID of Gisaid uploading sequencing are summarized in Supplementary Table 3, and Accession of SRA data PRJNA791285 is accessible at the following link: https://www.ncbi.nlm.nih.gov/sra/PRJNA791285.
Torrent Server–Torrent Suite (version 5.12.2) was used to perform the alignment of the samples, and using the COVID19AnnotateSnpEff plug-in, we performed a variant annotation.
The default parameters were used to remove low-quality and short reads: BaseCaller trimming quality cutoff of 15, barcode-filter-minreads of 10, phasing-residual filter = 2.0, and number unfiltered of 1,000. These trimmed reads were mapped to the SARS-CoV-2 reference sequence (Accession: NC_045512), and a consensus sequence was generated thereafter using the Iterative Refinement Meta-Assembler (IRMAreport v1.3.0.2) (10), which produced a consensus sequence for each sample using a >50% cutoff for calling single-nucleotide polymorphisms. We used a default configuration module for IRMA with the same parameters, as follows: median read Q score filter of 30; minimum read length of 150 bases; frequency threshold for insertion and deletion of 0.25 and 0.6, respectively; minimum number of read patterns and read count to continue to attempt assembly per gene segment, both equal to 15; and the Smith–Waterman mismatch penalty of 5 and gap open penalty of 10.
The IRMA report plug-in was used to generate the FASTA sequences containing the SARS-CoV-2 genome. These FASTA sequences were further processed for genome annotation and strain classification. Variant Caller v5.12.0.4 and COVID19AnnotateSnpEff were used to detect variants and annotate variants, respectively.
Nextclade tool was used to check sequencing errors and to assess the quality of the assembled sequences. Particularly, assembled sequences containing a high divergence and a high volume of missing data have been removed from further analysis (11). Further analysis and the manual control of nucleic acid and amino acid changes were performed using web-based CoVsurver tool (GISAID-CoVsurver mutations App https://www.gisaid.org/epiflu-applications/covsurver-mutations-app/). Lolliplot of spike protein mutations was generated using a lollipops generator (12).
Multiple sequence alignment and genetic distance compared with reference sequence were performed using Clustal Omega Program (13). The complete reference genome sequence of SARS-CoV-2 was retrieved from the National Center for Biotechnology Information (NCBI) GenBank database (accession number NC_045512.2). Population data for Campania were retrieved from the Italian National Institute of Statistics (http://dati.istat.it/). Data visualization was performed using customized scripts based on the ggplot2 package (https://ggplot2.tidyverse.org) in the statistical environment R.
The phylogenetic analysis of the 101 SARS-CoV-2 consensus sequences obtained was assigned according to Phylogenetic Assignment of Named Global Outbreak (PANGO) (https://pangolin.cog-uk.io/) (8). Pangolin proposed dynamic nomenclature for SARS-CoV-2 lineages to assist genomic evolution. Clades were assigned using the Nextclade sequence analysis webapp https://clades.nextstrain.org/ and GISAID platform (11).
The present study aims to the genomic characterization of 101 SARS-CoV-2 whole-genome sequences in two districts of Campania (Italy), geographically and temporally distributed from January to May 2021, as reported in Figures 1A,B. In the early time of the epidemic (January and February 2021), B.1.1.7 lineage was in 62% of samples (16/26) only in Benevento district, while this lineage appears in Avellino later in 64% of samples (30/47) from March to May. The occurrence of P.1.1 lineage spreading from March to the end of the study was recorded in all districts with the same frequencies of ~21%.
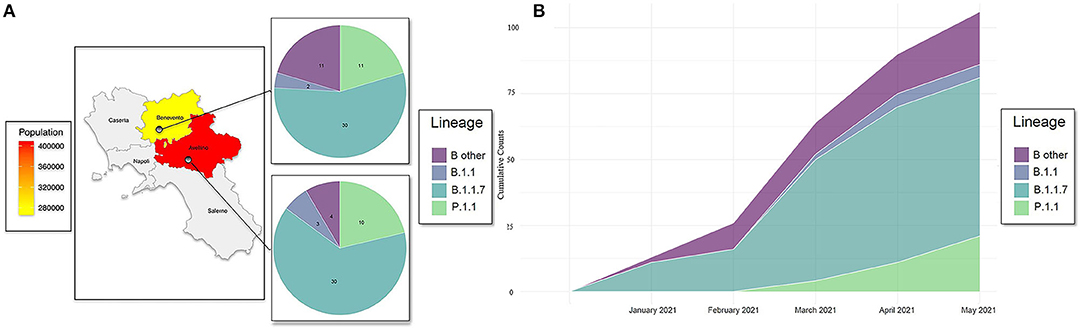
Figure 1. (A) Geographic distribution of COVID-19 cases, lineages detected, and population density among two provinces of Campania: Avellino and Benevento. (B) Distribution of SARS-CoV-2 genomes against collection date and lineages. Cumulative counts of the 101 SARS-CoV-2 genomes spreading in districts of Benevento and Avellino against collection date. Lineage “B 1.1” refers to haplotype B: B.1.1.351 and B.1.1.374. “B other” refers to B.1.177.53, B.1.177.75, B.1.177, B.1.258.14, B.1.258.17, and B.1.160.
Three available nomenclature systems were used to detect distinct variants, clades, and lineages—PANGO, GISAID, and NEXTSTRAIN—flanked by a new nomenclature system used by the WHO (10). In accordance with PANGO nomenclature, all sequences belonged to lineages B and P. Further lineage analysis resulted in the assignment of samples to 10 different circulating lineages; the most common were B.1.1.7 (59.4%) and P.1.1 (20%) (Table 1). In accordance with GISAID, samples were classified into different clades grouped into superclade G and clades GH, GV, GR, and GRY. The most dominant clade was GRY (50%). The third nomenclature system used was NEXTSTRAIN with its NEXTCLADE web-based application. This system revealed that our population can be grouped into five clades as organized in a phylogenetic tree as shown in Figure 2. The majority of sequences from our study were assigned to the 20I and 20J clades distributed as follows: 20A (Lineages B.1.258.17, B.1.258.14, and B.1.160) (n = 10; 9.9%), 20B (Lineages B.1.1.351 and B.1.374) (n = 5; 4.9%), 20E (EU1) (Lineages B.1.177.53, B.1.177.75, and B.1.177.77) (n = 5; 4.9%), 20I (Alpha.V1) (B.1.1.7) (n = 60; 59.4%), and 20J (Gamma.V3) (Lineage P.1.1) (n = 21; 20.7%) summarized in Table 1.
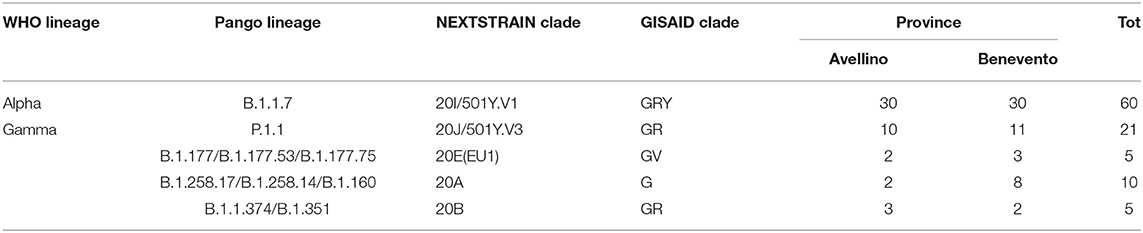
Table 1. Distribution of SARS-CoV-2 samples according to Pango lineage, clade, and WHO lineage.
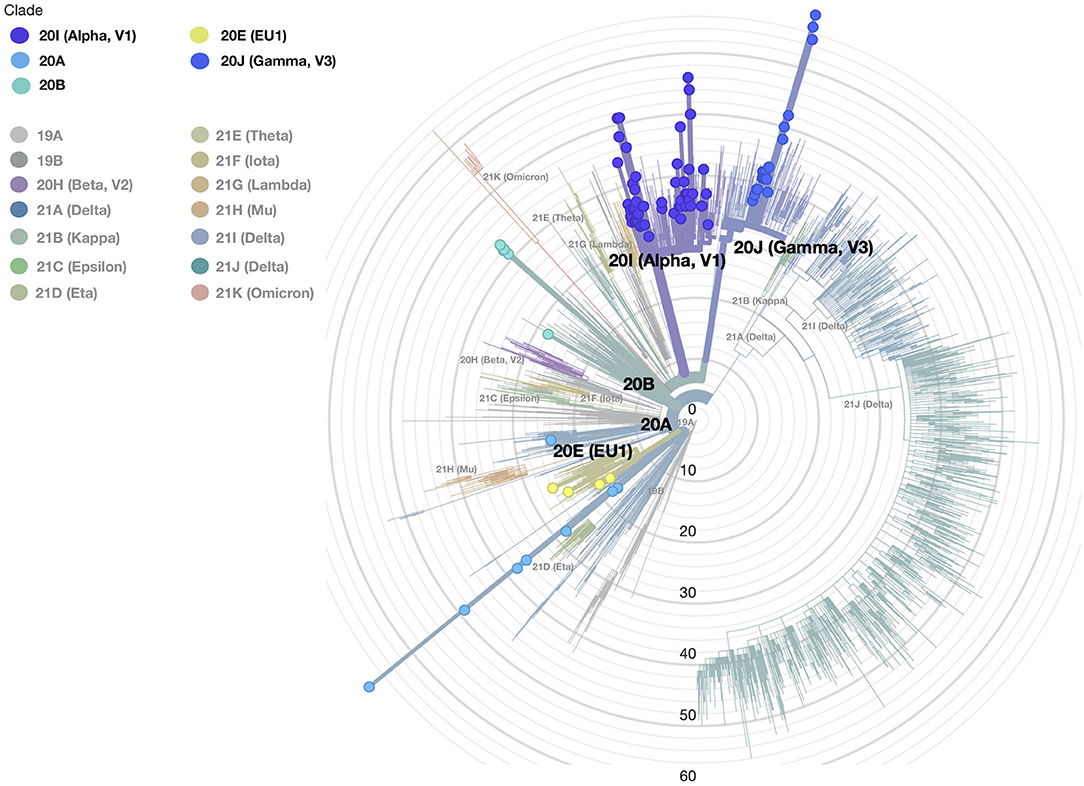
Figure 2. The phylogenetic tree of all SARS-CoV-2 samples was made with the Nextclade and globally represents position and clade clustering (Nextrain nomenclature). Our different clades are defined by colors (legend of colors is on the top left); the small circles show our samples, and the gray concentric circles represent the scale of the tree defined as the difference in mutation profiles between each sample and the reference sequence.
The genetic distance compared with the Wuhan SARS-CoV-2 reference genome (NC_045512.2) was lower in B.1.1 (B.1.1.351 and B.1.1.374) lineage than B.1.1.7 lineage (7.7 × 10−4 [95% CI 6.9 × 10−4; 8.5 × 10−4] vs. 14.5 × 10−4 [95% CI 13 × 10−4 to 15.8 × 10−4]) and other B lineages (B.1.177.53, B.1.177.75, B.1.177, B.1.258.14, B.1.258.17, and B.1.160) (10 × 10−4 [95% CI 6.4 × 10−4; 1.3 × 10−4]). The highest genomic distance was observed in lineage P.1.1 (17.9 × 10−4; [95% CI 14.2 × 10−4; 21.5 × 10−4]) (Figure 3). Concordant with the increase of lineage B.1.1.7 over time and passage to lineage P.1.1, the genetic pairwise distance indicated that the SARS-CoV-2 sequences evolved progressively during the time.
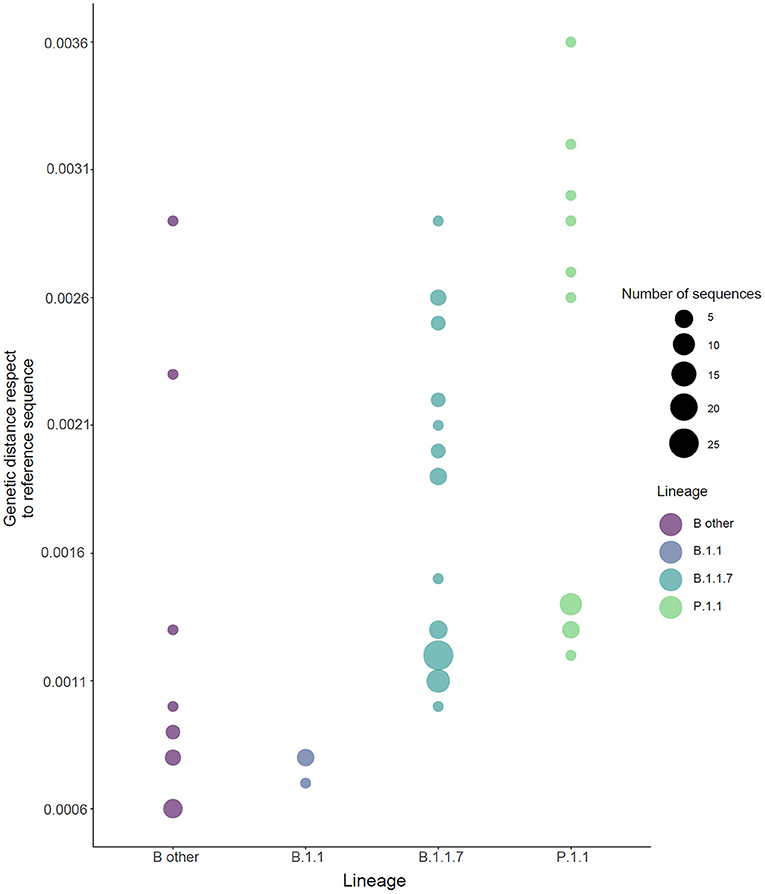
Figure 3. Genetic distance for the SARS-CoV-2 genome lineages. Sequences are colored by Pango lineage as shown in the legend. Reference sequence: NC_045512.2. Lineage “B.1.1” refers to haplotype B: B.1.1.351 and B.1.1.374. “B other” refers to B.1.177.53, B.1.177.75, B.1.177, B.1.258.14, B.1.258.17, and B.1.160. The size of the circle is related to the number of samples.
Compared with the reference Wuhan SARS-CoV-2 sequence, the whole-genome sequences of 101 individuals displayed 219 amino acid substitutions of different viral genes (114 in ORF1a/1b, 49 in S, 12 in ORF3a, 5 in M, 29 in N, and five each in ORF7a and 8) summarized in Supplementary Table 1. We focused our attention only on the non-synonymous “known” mutations as defined by GISAID CovSurver; among these mutations, 32 are on spike protein in two or more samples: 7 with high frequency (100%−60%) (D614G, N501Y, P681H, S982A, D1118H, A570D, and T716I-Lineage B.1.1.7) (14), 10 with an intermediate frequency (20%) (T20N, L18F, P26S, T1027I, V1176F, K417T, H655Y, D138Y, R190S, and E484K-Lineage P.1) (15), and 15 with low frequency in all lineages (L54F, R78K, T95I, Y144F, L189F, A222V, N439K, S477N, V772I, Y789N, P812S, S813N, H1101Y, T1117I, and Q1201K) distributed in all four different spike protein domains (S2, S1-N terminal, RBD, and S1-C terminal) as indicated in Figure 4. Referred only to spike protein molecular alteration, we identified 62 “unique” non-synonymous mutations summarized in Supplementary Table 2. The “unique” mutations were not reported previously and will be of interest to study.
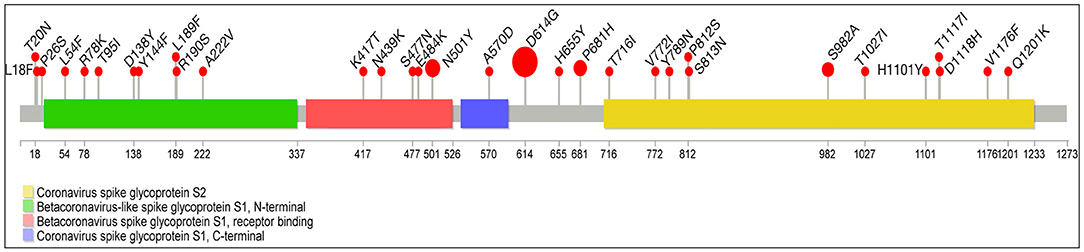
Figure 4. Lolliplot graph with mapping missense mutations in spike protein. Lollipop plot summarizes the frequency of SARS-CoV-2 mutations with prevalence ≥2% in our study cohort (n = 101). A SARS-CoV-2 genome map with base-pair positions is shown at the bottom. Mutation frequencies and details are indicated on the red bubbles. Spike protein domains are indicated by four different colors: green, Betacoronavirus-like spike glycoprotein S1, N-terminal; red, Betacoronavirus-like spike glycoprotein S1, receptor binding; violet, coronavirus-like spike glycoprotein S1, C-terminal; yellow, coronavirus-like spike glycoprotein S2. The size of the circle is related to the number of samples with a specific mutation.
SARS-CoV-2 is a single-strand RNA virus, and like all viruses, it accumulates mutations over time. However, the frequency of mutation and the consequences for transmission and disease in the host population depend on both the mutation rate (determined by the viral replication characteristics) and the impacts of mutation on the individual hosts. These factors characterize the viral variants' onset during the epidemic. Among the viruses, RNA viruses are more prone than DNA viruses to mutate (16). However, SARS-CoV-2, like related coronaviruses, possesses a proof-reading domain within its gene sequence (ExoN), which decreases the probability of mutations to occur, compared with well-known RNA viruses such as influenza, HIV, and hepatitis C viruses. The rapid diffusion and spreading of the SARS-CoV-2 infection and disease have caused a very high number of active cases of 229,921,289 and has pushed the virus to mutate to give rise to more infectious variants. Several viral lineages have probable impacts (VOI) or proven impacts (VOC) on human health. It is expected that new mutations and variants will emerge in the near future as the SARS-CoV-2 pandemic continues to persist despite the use of mass vaccination strategies in several countries (but, unluckily, those are not being equally applied worldwide). In this frame, it should be useful to predict the course of SARS-CoV-2 evolution, but the latter is complicated by the broad potential host range and known transmissibility to animals (17). In the present study, we evaluated the complete viral genome sequence in the SARS-CoV-2 from 101 infected individuals in the districts of Avellino and Benevento, in the south of Italy. We identified five types of clades that include ten different Pango lineages (1): 20A (Lineages B.1.258.17, B.1.258.14, and B.1.160) (n = 10; 9.9%), 20B (Lineages B.1.1.351 and B.1.374) (n = 5; 4.9%), 20E (EU1) (Lineages B.1.177.53, B.1.177.75, and B.1.177) (n = 5; 4.9%), 20I (Alpha.V1) (B.1.1.7) (n = 60; 59.4%), and 20J (Gamma.V3) (Lineage P.1.1) (n = 21; 20.7%). The two higher-frequency lineages are as follows: Lineage B.1.1.7, which appeared for the first time of the study (January) with total frequencies (from January to May) of 64 % (30/47) in Avellino and 56% (30/54) in Benevento, and P1.1 lineage spreading from March with total frequencies of 22% (10/47) in Avellino and 39% (11/28) in Benevento.
The results suggest that lineage B.1.1.7 had a higher transmission rate; in fact, in 2 months, it spread in two provinces very quickly, and the genomic surveillance gave us the chance to track genomic virus change. The highest genomic distance, which reflects a higher level of mutations compared with the reference sequence, was observed in lineage P.1.1, and the genetic pairwise distance indicated that the SARS-CoV-2 sequences evolved progressively over time and faster than other lineages.
The variable mutation rate detected in the different analyzed lineages of SARS-CoV2, compared with the backbone shows high genetic variability of SARS-CoV2 adapting to overcome the initial immune protection. The latter finding is in accordance with the emergence of new mutations in the population during the transmission of the virus together with the entrance of new variants in the population from outside. Interestingly, in our series of samples, all the SARS-CoV-2 genomes analyzed showed the presence of mutations, with the most representative being the D614G mutation, which is the symbol of all variants promoting viral spread by increasing the number of open spike protomers in the homotrimeric receptor complex (7). It is noteworthy that, only considering the “known” nonsynonymous mutations, 219 amino acid substitutions were affecting different viral genes (114 in ORF1a/1b, 49 in S, 12 in ORF3a, 5 in M, 29 in N, and five each in ORF7a and 8).
All “known” mutations of each analyzed lineage of SARS-CoV-2 in this study are on publicly accessible web application CoV-GLUE (http://cov-glue.cvr.gla.ac.uk). It is not clear if any of these mutations can affect the transmission and evolution of SARS-CoV-2 infection, and it should be studied in-depth in the future.
In conclusion, our data show the SARS-CoV-2 variants occurrence in the first months of 2021 in Benevento and Avellino districts (south of Italy). This was paralleled by a progressive increase of the P.1.1 variant and was also characterized by the appearance of several other mutations in the viral genome also affecting genes different from S. The impact of these mutations, some of which are new, is not presently predictable and needs to be additionally investigated. Our results suggest the mutational rate of SARS-CoV-2 is unexpectedly higher probably due to the high rate of transmission and to the frequent inter-human passages of the virus. However, other factors cannot be presently excluded. Our results suggest that strict surveillance of the occurrence of genetic variants of SARS-CoV-2 should be performed in order to predict its diffusion and monitor its morbidity and sensitivity to vaccination strategies.
Sequences are uploaded on Gisaid and SRA database: ID of Gisaid uploading sequencing are summarized in Supplementary Table 3 and Accession of SRA data PRJNA791285 is accessible at the following link: https://www.ncbi.nlm.nih.gov/sra/PRJNA791285.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study.
MS and AC contributed to conception and design of the study and performed experimental processes. ED'A, MB, YA, CI, CM, PG, and FM performed experimental processes. LG, FP, PM, and GC performed a supervision of work. TN and MCe performed bioinformatic analysis. AF wrote sections of the manuscript. MCa contributed to conception and design of the study. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.814114/full#supplementary-material
1. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 579:265–9. doi: 10.1038/s41586-020-2008-3
2. Coronaviridae Study Group of the International Committee on Taxonomy of V. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. (2020) 5:536–44. doi: 10.1038/s41564-020-0695-z
3. Yadav R, Chaudhary JK, Jain N, Chaudhary PK, Khanra S, Dhamija P, et al. Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells. (2021) 10:821. doi: 10.3390/cells10040821
4. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. (2020) 583:459–68. doi: 10.1038/s41586-020-2286-9
5. Singh J, Pandit P, McArthur AG, Banerjee A, Mossman K. Evolutionary trajectory of SARS-CoV-2 and emerging variants. Virol J. (2021) 18:166. doi: 10.1186/s12985-021-01633-w
6. Almubaid Z, Al-Mubaid H. Analysis and comparison of genetic variants and mutations of the novel coronavirus SARS-CoV-2. Gene Rep. (2021) 23:101064. doi: 10.1016/j.genrep.2021.101064
7. Winger A, Caspari T. The spike of concern-the novel variants of SARS-CoV-2. Viruses. (2021) 13:1002. doi: 10.3390/v13061002
8. Rambaut A, Holmes EC, O'Toole A, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. (2020) 5:1403–7. doi: 10.1038/s41564-020-0770-5
9. Aleem A, Akbar Samad AB, Slenker AK. Emerging Variants of SARS-CoV-2 And Novel Therapeutics Against Coronavirus (COVID-19). Treasure Island, FL: StatPearls (2021).
10. Otto SP, Day T, Arino J, Colijn C, Dushoff J, Li M, et al. The origins and potential future of SARS-CoV-2 variants of concern in the evolving COVID-19 pandemic. Curr Biol. (2021) 31:R918–R29. doi: 10.1016/j.cub.2021.06.049
11. Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. (2018) 34:4121–3. doi: 10.1093/bioinformatics/bty407
12. Jay JJ, Brouwer C. Lollipops in the clinic: information dense mutation plots for precision medicine. PLoS ONE. (2016) 11:e0160519. doi: 10.1371/journal.pone.0160519
13. Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. (2019) 47:W636–41. doi: 10.1093/nar/gkz268
14. Fiorentini S, Messali S, Zani A, Caccuri F, Giovanetti M, Ciccozzi M, et al. First detection of SARS-CoV-2 spike protein N501 mutation in Italy in August, 2020. Lancet Infect Dis. (2021) 21:e147. doi: 10.1016/S1473-3099(21)00007-4
15. Faria NR, Mellan TA, Whittaker C, Claro IM, Candido DDS, Mishra S, et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in manaus, Brazil. Science. (2021) 372:815–21. doi: 10.1126/science.abh2644
16. Holmes EC, Grenfell BT. Discovering the phylodynamics of RNA viruses. PLoS Comput Biol. (2009) 5:e1000505. doi: 10.1371/journal.pcbi.1000505
Keywords: SARS-CoV-2, genomic characterization, mutation, COVID-19, whole-genome sequencing (WGS)
Citation: Scrima M, Cossu AM, D'Andrea EL, Bocchetti M, Abruzzese Y, Iannarone C, Miarelli C, Grisolia P, Melisi F, Genua L, Di Perna F, Maggi P, Capasso G, Noviello TMR, Ceccarelli M, Fucci A and Caraglia M (2022) Genomic Characterization of the Emerging SARS-CoV-2 Lineage in Two Districts of Campania (Italy) Using Next-Generation Sequencing. Front. Virol. 2:814114. doi: 10.3389/fviro.2022.814114
Received: 12 November 2021; Accepted: 05 January 2022;
Published: 28 February 2022.
Edited by:
Shuofeng Yuan, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Richard Johnathan Orton, University of Glasgow, United KingdomCopyright © 2022 Scrima, Cossu, D'Andrea, Bocchetti, Abruzzese, Iannarone, Miarelli, Grisolia, Melisi, Genua, Di Perna, Maggi, Capasso, Noviello, Ceccarelli, Fucci and Caraglia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michele Caraglia, bWljaGVsZS5jYXJhZ2xpYUB1bmljYW1wYW5pYS5pdA==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.