 Marc Talló-Parra
Marc Talló-Parra Elena Muscolino
Elena Muscolino Juana Díez
Juana Díez
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Virol., 06 December 2022
Sec. Fundamental Virology
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.1073619
This article is part of the Research TopicPost-transcriptional regulation of viral protein expression and functionView all 5 articles
Viruses completely depend on the host translation machineries to express the viral proteins. Recent data reveal an unprecedented interaction of positive strand RNA ((+)RNA) viruses with the host tRNA epitranscriptome to favor viral protein expression via a specific reprogramming of codon optimality that ultimately favors decoding of the viral codons. We propose that this feature is shared by multiple RNA viruses and that the involved tRNA modifying enzymes represent promising novel targets for the development of broad-spectrum antivirals.
All steps of viral life cycles require factors from the host they infect. However, unlike replication and transcription where viral proteins direct these functions, viruses completely depend on the host translation machinery to express the viral proteins. This dependence is particularly apparent for positive strand RNA ((+)RNA) viruses. Since they do not carry their polymerases in the virions, upon release of their genomes into the cytoplasm, (+)RNA viruses first need to translate their genomes to express the viral replicases (1). To maximize viral protein expression, they display different strategies to take control of the host translation machinery. In turn, the host reprograms this machinery in an attempt to repress virus infection in an ever-evolving race to gain control of it (2). The translation process requires three major steps: initiation, in which the ribosomes are recruited to the initiation codon; elongation, in which the amino acids are brought to the ribosomes by transfer RNAs (tRNAs) and linked together to form a chain; and termination, in which the finished polypeptide is released. Most of our knowledge on the strategies used by (+)RNA viruses to efficiently translate their genomes relate to events affecting the translation initiation step. The involvement of the other steps of the translation process has been almost unexplored. We have recently uncovered an unprecedented interplay of (+)RNA viruses with the host tRNA epitranscriptome to favor translation elongation of the viral RNA genomes to maximize viral protein expression (3). These results place the tRNA epitranscriptome as a novel key regulatory point of (+)RNA virus infections. tRNAs are adapter molecules of the translation machinery that convert the transcriptome into the proteome. They carry specific amino acids to the ribosome according to complementary base pairing between positions 34, 35 and 36 of their anticodon and positions 3, 2 and 1, respectively, of the codon in mRNAs (4) (Figures 1, 2). tRNAs are heavily modified with a plethora of chemical modifications that comprise from simple addition or substitution of functional groups to complex structures whose biosynthesis involve multiple enzymes (4). This set of modifications is known as the tRNA epitranscriptome. From the around 200 modifications described to occur in RNA molecules, half of them are present in tRNAs (5). On average, tRNA molecules contain 13 modifications located throughout the tRNA molecule (Figure 1) (6). This represents around 1 modification every 5 residues. The high number of chemical modifications in tRNAs implies a major role in tRNA function. Most of the tRNA modifications are located at the anticodon loop. For example, in Homo sapiens 41 types of modifications are found in cytoplasmic tRNAs, of which 29 are located at the anticodon loop. In agreement with these variety and abundance, modifications at the anticodon loop have been shown to play fundamental roles in the regulation and accuracy of multiples steps of the translation elongation process, including decoding, aminoacylation and translocation (7). Modifications outside of the anticodon loop affect proper tRNA folding and stability (8).
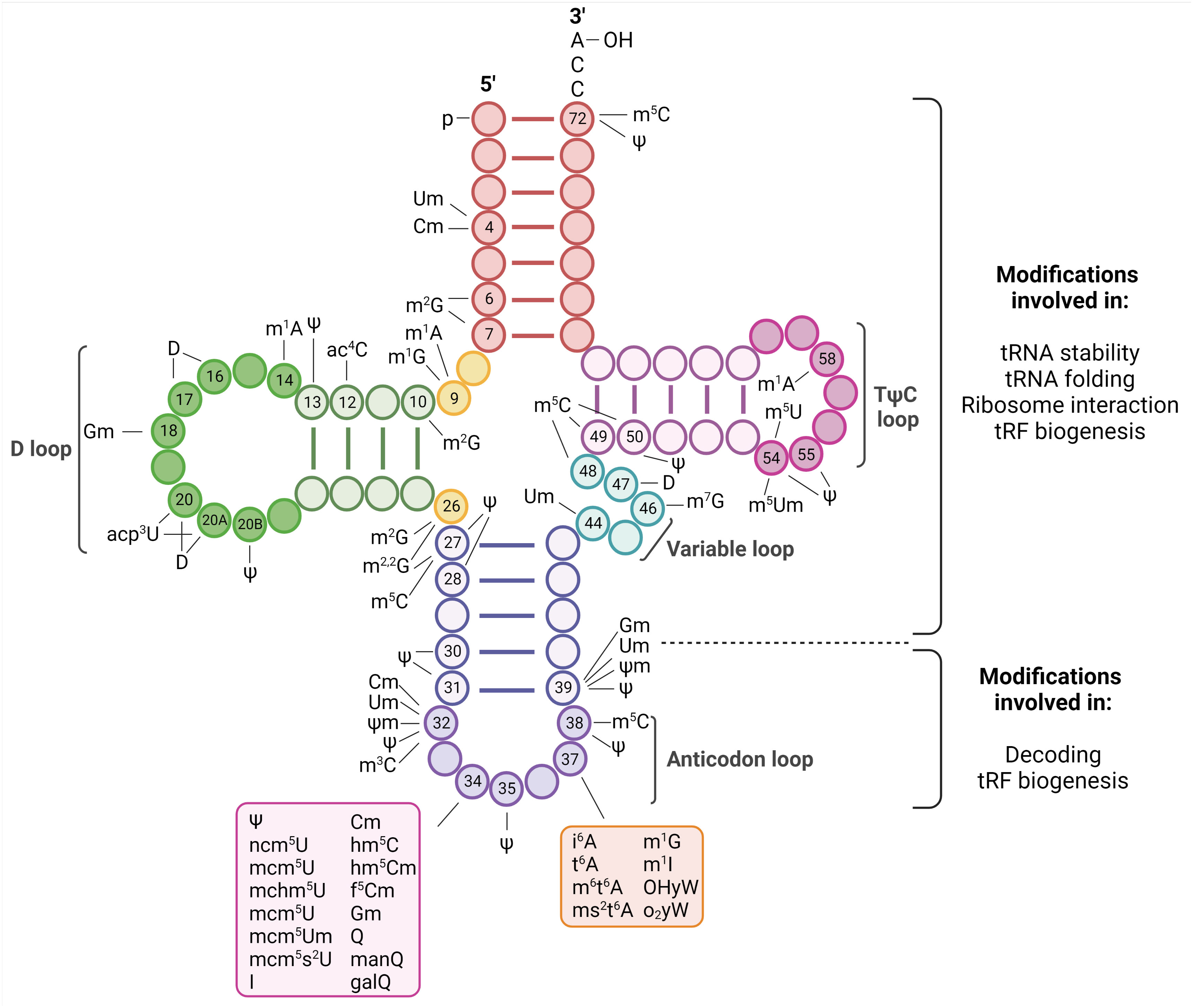
Figure 1 Representation of tRNA modifications. tRNA are 70-100 nucleoside in length and fold into a cloverleaf secondary structure. The structure is characterized by a 5’ phosphate group, a 3’ acceptor stem that contains the CCA terminal group required to attach the amino acid, the D-loop, the TΨC-loop (where Ψ is a pseudouridine), a Variable loop and the anticodon loop. Changes occurring in specific nucleotides of human cytoplasmic tRNAs are indicated [as reviewed in (4)]. These modifications have fundamental roles in tRNA biology and they have been shown to affect stability, folding, ribosome interaction, tRNA fragments (tRF) biogenesis, accurate aminoacylation, stabilizing codon-anticodon interaction, pairing and decoding. Created with BioRender.com.
The effect of tRNA modifications on decoding results mainly from two hot spots of modification located at position 37 and 34. Modifications at position 37 enhance translation fidelity preventing frame shift events (9). Modifications at position 34 of tRNA (wobble position) increase the flexibility of base pairing and contribute to the decoding capacity. The redundancy of the genetic code allows amino acids to be encoded by more than one codon. The frequency of these synonymous codons is universally biased and specific for each organism. For a given organism, the tRNA concentration is adapted to the codon usage. Thus, optimal codons speed up translation rate and rare codons slow it down because the cognate tRNAs are lowly abundant in comparison to those tRNAs used for optimal codons (10–12). Modifications at the wobble positions affect their base pairing with the third position of the codon ultimately expanding or restricting the decoding efficiency of certain synonymous codons. Well-studied examples include the mcm5 and mcm5s2 modifications at uridine 34 (U34) of cytoplasmic tRNAs. In the tRNA anticodon, position 35 and 36 follow the base pairing rules defined by Watson and Crick. However, when an unmodified U is located at position 34 (wobble U34), U recognizes both adenine (A) and guanine (G) in the third position of the codon. Modifications at this position might favor base pairing with A (A-ending codons) or G (G-ending codons). For example, the Glutamic acid (Glu) amino acid is encoded by two synonymous codons GAA and GAG. When the U34 position of the cognate tRNA is not modified, the tRNA decodes both GAG and GAA codons (Figure 2A). However, when U34 is mcm5/mcm5s2 modified, the tRNA favors decoding of the GAA codon (13, 14) (Figure 2A). The fact that U34 is modified in practically all organisms implies a strong evolutionary pressure and a major role in translation regulation (15). Indeed, it is now established that tRNA modifications are highly dynamic and their prevalence varies in response to levels of cellular metabolite and environmental stresses (4, 16). Moreover, global analyses have demonstrated a coordinated interplay between changes in the modifications of the wobble position, including U34, and selective translation of stress-dependent gene expression enriched in synonymous codons which decoding is favored by the induced modifications [reviewed in (16)]. Together, this indicates a coordinated interplay between the tRNA epitranscriptome and the codon bias to adapt translation during stress responses (17).
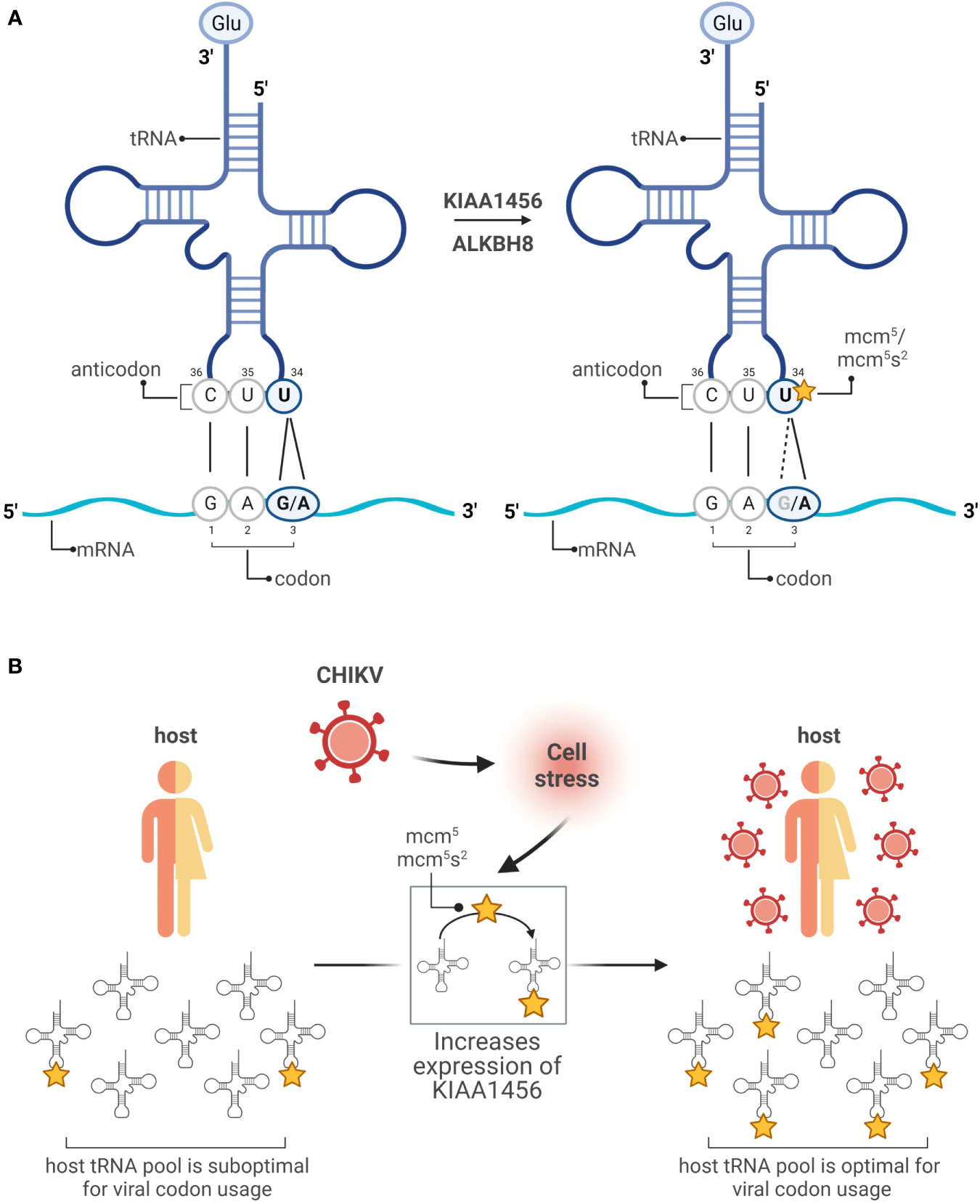
Figure 2 Modifications in the wobble uridine changes the reading. (A) In the anticodon of a tRNA coding for Glu (GAG/A), unmodified U34 base pairs with G or A in the third position of the mRNA codon (left). Following modifications promoted by KIAA1456 or ALKBH8 enzymes, U34 is favored to pair with an A (right). (B) Working model: the host tRNA pool is enriched toward host mRNAs containing optimal codons, while the CHIKV genome is enriched in suboptimal codons. CHIKV infection triggers a DNA damage response that promotes the expression of the tRNA-modifying enzyme KIAA1456. This ultimately results in a reprogramming of codon optimality that favors translation of suboptimal codons, enriched in both the host mRNAs encoding DNA damage response genes and the CHIKV RNAs. Created with BioRender.com.
The genomes of numerous and diverse (+)RNA viruses, including SARS-CoV-2, dengue (DENV), zika (ZIKV) or chikungunya (CHIKV) viruses, are enriched in rare codons. While highly expressed human mRNAs are enriched in G/C-ending codons, the genome of multiple (+)RNA viruses is enriched in A/U-ending codons (17). How (+)RNA genomes are able to express their proteins so efficiently in spite of their unfavorable codon usage? To address this fundamental question, we use CHIKV as a model system because its genome is translated to extremely high levels. Genome-wide transcriptome and translatome analyses together with fractionation studies showed that CHIKV infection dramatically alters the host mRNA translatome at the endoplasmic reticulum, the translation compartment where CHIKV efficiently translates, while few significant changes were observed at the cytosol, the other major cellular translational compartment (3). The set of translationally repressed host mRNAs were enriched for genes related to mitochondria, ribosomes and RNA translation. The set of translationally activated host mRNAs were enriched for genes related to DNA damage response, cell cycle and RNA transport. Importantly, these translationally activated mRNA, as the CHIKV RNA, displayed a poor codon usage, mostly enriched in GAA (Glu), AAA (Lys), CAA (Gln), AGA (Arg) and GGA (Gly) codons. In contrast the translationally repressed mRNAs displayed an optimal one enriched in the corresponding G-ending synonymous codons (3). This indicates a virus-induced reprogramming of codon optimality that favors translation of viral RNAs. Mechanistically, it involves CHIKV-induced translational activation of the KIAA1456 enzyme, a paralog of the ALKBH8 enzyme involved in the mcm5/mcm5s2 modifications (described above) that favor decoding of the GAA (Glu), AAA (Lys), CAA (Gln), AGA (Arg) and GGA (Gly) codons over the G-ending ones (Figure 2B) (18, 19). Interestingly, the KIAA1456 mRNA is itself enriched in these five codons, suggesting a positive feedback loop. Importantly, DENV, whose genome is enriched in these codons, also induces in the infected cell overexpression of the KIAA1456 enzyme and a corresponding increase in mcm5 tRNA modification levels while hepatitis C virus, whose genome is not enriched, does not (3). Notably, the use of KIAA1456-enzymes by viruses seems to be conserved beyond human viruses as in Escherichia coli the tRNALys U34 modification is required for the phage lambda to express gpG and gpGT, two viral replication proteins. However, in this case the tRNA modification modulates a specific ribosomal frameshift essential for proper expression of the gpG:gpGT radio instead of affecting global reprogramming of codon usage (20).
The DNA damage response is a regulatory network that detects DNA damage and mediates its repair (21). Interestingly, in yeast, alkylation stress that cause DNA damage result in an increase of (i) the expression of TRM9, the yeast homolog of human ALKBH8/KIAA1456, (ii) the levels of mcm5/mcm5s2 tRNA modification and (iii) the expression of genes related to DNA damage and cell cycle control, essential to survive alkylation stress. These genes are enriched in GAA and AGA codons. The similarities of these findings with the CHIKV observations strongly suggest that CHIKV has adapted its genome to the host translation environment under infection conditions. CHIKV infection triggers a DNA damage response that results in a reprogramming of codon optimality that ultimately would favor CHIKV RNA genomes enriched in the same codons as the DNA damage response genes. We propose that this mechanism is shared by other (+)RNA viruses beyond DENV and that the DNA damage response triggered by viral infection might have played a role in the shaping of the (+)RNA virus codon usage. This proposal is based on the next observations. First, as CHIKV and DENV, many other (+)RNA viruses are enriched in GAA, AAA, CAA, AGA and GGA codons (unpublished observation). Of note, as the genomes of some viruses outside the (+)RNA viral group are also enriched in these codons (unpublished observation), this mechanisms might be conserved across viral groups. Second, growing evidence indicates a complex interplay between RNA viruses and DNA damage responses. The activation and manipulation of the DNA-damage response by DNA viruses and retroviruses to optimize their replication processes is well-studied (22–24). This activation is triggered by the incoming viral DNA, the integration of retroviruses or in response to the aberrant DNA structures generated upon replication of DNA viruses among other factors. Although at present, only a few RNA viruses have been reported to interact with the DNA damage response, growing evidence indicates that RNA viruses also can manipulate this pathway to their benefit (22, 24, 25). Two examples include ZIKV and Porcine Reproductive and Respiratory Syndrome Virus (PRRSV). Two main components of the DNA-damage response regulatory network are the ataxia–telangiectasia mutated (ATM) and ATM Rad3-related (ATR) kinases (26). In ZIKV infection, the induction of the ATM signaling and corresponding S-phase arrest increases viral replication (27). Moreover, both ATM and ATR kinases have been described to be activated by PRRSV infection, co-localize with the viral replication complexes and promote replication (28). How (+)RNA viruses that replicate at the cytosol induce the DNA-damage network remains unclear. Putative mechanisms would include promoting inappropriate S phase entry or direct modification or components of the DNA damage response.
Novel emerging evidence highlights the roles of tRNA modifications in viral infections. Besides the described GAA, AAA, CAA, AGA and GGA codons CHIKV-induced translationally activated host mRNAs and CHIKV RNAs were enriched in other A/U ending codons whose recognition would depend on modifications at the U34 position still to be identified. Thus, it is much likely that besides KIAA1456, other tRNA modifying enzymes play major roles in CHIKV infection. We propose that this feature is shared by multiple RNA viruses and that the involved tRNA modifying enzymes represent promising novel targets for the development of broad- spectrum antivirals. Excitingly, recent work demonstrates that the host immune system also relies on tRNA modifications. A specific tRNA methylation (m1A58) selectively enhances protein translation in a codon-usage manner of a set of proteins that ultimately drive T cell proliferation (29). Together, these findings represent the starting study point of a previously unappreciated tRNA epitranscriptome-virus-immune system regulatory network that is awaiting to be deciphered and will provide novel opportunities for therapeutic interventions.
The tRNA epitranscriptome is a growing field whose complexity presents major challenges. First, to elucidate the tRNA expression patterns in the different tissues and cell types. The human genome contains around 400 genes, including isodecoders and isoacceptors (tRNA genes that share the same anticodon but have different body sequences and vice versa, respectively) that are not all transcribed in the same cell and whose expression is cell-specific. Although major advances have been made in the tRNA sequencing methods (30–32), to obtain and analyze a complete tRNA profile remains challenging in comparison with RNAseq. Second, to complete the mapping of tRNA modifications and the assignment of their roles. The diversity, dynamic nature and low abundance of some modifications are major challenges for their identification. Next-generation sequence (NGS), mass-spectrometry and nanopore-based detection are the main techniques towards the characterization of RNA modifications. However, the current methods have many limitations (31, 33). Moreover, only around 50% of tRNA modifying enzymes have been experimentally validated. This hampers the study of the function of many tRNA modifications. Thus, there is much fundamental biology of the tRNA epitranscriptome still to be learnt. Viruses are powerful tools to uncover cellular processes. Indeed, virus research has identified fundamental translation features such as capping, polyadenylation or splicing. We thus expect that the encounter of the virology and the tRNA epitranscriptome fields will help revealing new insights not only into viral infections and their control but also into tRNA biology.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Conceptualization and writing of the perspective: MT-P, EM, JD. All authors contributed to the article and approved the submitted version.
This work was supported by the Spanish Ministry of Economy, Industry and Competitiveness (AEI/MINECO/FEDER, UE; PID2019-106959RB-I00) and the “Unidad de Excelencia María de Maeztu” funded by the AEI (CEX2018-000792-M).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ahlquist P. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat Rev Microbiol (2006) 4(5):371–82. doi: 10.1038/nrmicro1389
2. Hoang HD, Neault S, Pelin A, Alain T. Emerging translation strategies during virus–host interaction. Wiley Interdiscip Rev RNA (2021) 12(1):1–19. doi: 10.1002/wrna.1619
3. Jungfleisch J, Böttcher R, Talló-Parra M, Pérez-Vilaró G, Merits A, Novoa EM, et al. CHIKV infection reprograms codon optimality to favor viral RNA translation by altering the tRNA epitranscriptome. Nat Commun (2022) 13(1):1–12. doi: 10.1038/s41467-022-31835-x
4. Suzuki T. The expanding world of tRNA modifications and their disease relevance. Nat Rev Mol Cell Biol (2021) 22(6):375–92. doi: 10.1038/s41580-021-00342-0
5. Krutyhołowa R, Zakrzewski K, Glatt S. Charging the code - tRNA modification complexes. Curr Opin Struct Biol (2019) 55:138–46. doi: 10.1016/j.sbi.2019.03.014
6. Pan T. Modifications and functional genomics of human transfer RNA. Cell Res (2018) 28(4):395–404. doi: 10.1038/s41422-018-0013-y
7. Lin H, Miyauchi K, Harada T, Okita R, Takeshita E, Komaki H, et al. CO2-sensitive tRNA modification associated with human mitochondrial disease. Nat Commun (2018) 9(1):1–17. doi: 10.1038/s41467-018-04250-4
8. Koh CS, Sarin LP. Transfer RNA modification and infection – implications for pathogenicity and host responses. Biochim Biophys Acta - Gene Regul Mech (2018) 1861(4):419–32. doi: 10.1016/j.bbagrm.2018.01.015
9. Björk GR, Wikström PM, Byström AS. Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Sci (80-) (1989) 244(4907):986–9. doi: 10.1126/science.2471265
10. Darnell AM, Subramaniam AR, O’Shea EK. Translational control through differential ribosome pausing during amino acid limitation in mammalian cells. Mol Cell (2018) 71(2):229–243.e11. doi: 10.1016/j.molcel.2018.06.041
11. Gardin J, Yeasmin R, Yurovsky A, Cai Y, Skiena S, Futcher B. Measurement of average decoding rates of the 61 sense codons in vivo. Elife (2014) 3:1–20. doi: 10.7554/eLife.03735
12. Peil L, Starosta AL, Lassak J, Atkinson GC, Virumäe K, Spitzer M, et al. Distinct XPPX sequence motifs induce ribosome stalling, which is rescued by the translation elongation factor EF-p. Proc Natl Acad Sci U S A (2013) 110(38):15265–70. doi: 10.1073/pnas.1310642110
13. Deng W, Babu IR, Su D, Yin S, Begley TJ, Dedon PC. Trm9-catalyzed tRNA modifications regulate global protein expression by codon-biased translation. PloS Genet (2015) 11(12):1–23. doi: 10.1371/journal.pgen.1005706
14. Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, et al. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell (2007) 28(5):860–70. doi: 10.1016/j.molcel.2007.09.021
15. Helm M, Alfonzo JD. Posttranscriptional RNA modifications: Playing metabolic games in a cell’s chemical legoland. Chem Biol (2014) 21(2):174–85. doi: 10.1016/j.chembiol.2013.10.015
16. Huber SM, Leonardi A, Dedon PC, Begley TJ. The versatile roles of the tRNA epitranscriptome during cellular responses to toxic exposures and environmental stress. Toxics (2019) 7(1):1–18. doi: 10.3390/toxics7010017
17. Chan C, Pham P, Dedon PC, Begley TJ. Lifestyle modifications: Coordinating the tRNA epitranscriptome with codon bias to adapt translation during stress responses. Genome Biol (2018) 19(1):1–11. doi: 10.1186/s13059-018-1611-1
18. De Crécy-Lagard V, Boccaletto P, Mangleburg CG, Sharma P, Lowe TM, Leidel SA, et al. Survey and summary: Matching tRNA modifications in humans to their known and predicted enzymes. Nucleic Acids Res (2019) 47(5):2143–59. doi: 10.1093/nar/gkz011
19. Yoshida M, Kataoka N, Miyauchi K, Ohe K, Iida K, Yoshida S, et al. Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia. Proc Natl Acad Sci U S A (2015) 112(9):2764–9. doi: 10.1073/pnas.1415525112
20. Maynard ND, MacKlin DN, Kirkegaard K, Covert MW. Competing pathways control host resistance to virus via tRNA modification and programmed ribosomal frameshifting. Mol Syst Biol (2012) 8(567):1–13. doi: 10.1038/msb.2011.101
21. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature (2009) 461(7267):1071–8. doi: 10.1038/nature08467
22. Lopez A, Nichols Doyle R, Sandoval C, Nisson K, Yang V, Fregoso OI. Viral modulation of the DNA damage response and innate immunity: Two sides of the same coin. J Mol Biol (2022) 434(6):167327. doi: 10.1016/j.jmb.2021.167327
23. Mertens ME, Knipe DM. Herpes simplex virus 1 manipulates host cell antiviral and proviral DNA damage responses. MBio (2021) 12(1):1–22. doi: 10.1128/mBio.03552-20
24. Luftig MA. Viruses and the DNA damage response: Activation and antagonism. Annu Rev Virol (2014) 1(1):605–25. doi: 10.1146/annurev-virology-031413-085548
25. Ryan EL, Hollingworth R, Grand RJ. Activation of the DNA damage response by RNA viruses. Biomolecules (2016) 6(1):2–24. doi: 10.3390/biom6010002
26. Lee JH, Paull TT. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat Rev Mol Cell Biol (2021) 22(12):796–814. doi: 10.1038/s41580-021-00394-2
27. Hammack C, Ogden SC, Madden JC, Medina A, Xu C, Phillips E. Zika virus infection induces DNA damage response in human. J Virol (2019) 93(20):1–20. doi: 10.1128/JVI.00638-19
28. Li P, Xu C, Zhang X, Cao C, Wang X, Cai G. Single-stranded RNA viruses activate and hijack host apical DNA damage response kinases for efficient viral replication. Genome Instab Dis (2022) 3(2):83–7. doi: 10.1007/s42764-022-00064-3
29. Liu Y, Zhou J, Li X, Zhang X, Shi J, Wang X, et al. tRNA-m1A modification promotes T cell expansion via efficient MYC protein synthesis. Nat Immunol (2022) 23(October):1433–44. doi: 10.1038/s41590-022-01301-3
30. Behrens A, Rodschinka G, Nedialkova DD. High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq. Mol Cell (2021) 81(8):1802–1815.e7. doi: 10.1016/j.molcel.2021.01.028
31. Zhang W, Foo M, Eren AM, Pan T. tRNA modification dynamics from individual organisms to metaepitranscriptomics of microbiomes. Mol Cell (2022) 82(5):891–906. doi: 10.1016/j.molcel.2021.12.007
32. Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, et al. Efficient and quantitative high-throughput tRNA sequencing. Nat Methods (2015) 12(9):835–7. doi: 10.1038/nmeth.3478
Keywords: tRNA, tRNA modifications, tRNA epitranscriptome, (+)RNA viruses, CHIKV, KIAA1456, viruses, codon usage
Citation: Talló-Parra M, Muscolino E and Díez J (2022) The host tRNA epitranscriptome: A new player in RNA virus infections. Front. Virol. 2:1073619. doi: 10.3389/fviro.2022.1073619
Received: 18 October 2022; Accepted: 22 November 2022;
Published: 06 December 2022.
Edited by:
Sonia Zuñiga, Spanish National Research Council (CSIC), SpainReviewed by:
Phuong Cao Thi Ngoc, Lund University, SwedenCopyright © 2022 Talló-Parra, Muscolino and Díez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juana Díez, anVhbmEuZGllekB1cGYuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.