
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Virol. , 04 January 2023
Sec. Viral Diversification and Evolution
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.1064882
 Maja Vukovikj1*
Maja Vukovikj1* Golubinka Boshevska1Elizabeta Janchevska1
Golubinka Boshevska1Elizabeta Janchevska1 Teodora Buzharova1
Teodora Buzharova1 Ardian Preshova1Milica Simova1Aneta Peshnacka1Dragan Kocinski2
Ardian Preshova1Milica Simova1Aneta Peshnacka1Dragan Kocinski2 Gordana Kuzmanovska2Shaban Memeti2Icko Gjorgoski3
Gordana Kuzmanovska2Shaban Memeti2Icko Gjorgoski3The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has a persistent negative impact on both the public health and the global economy. To comprehend the origin, transmission routes and discover the mutations that alter the virus’s transmissibility and pathogenicity, full-length SARS-CoV-2 genomes have to be molecularly characterized. Focusing on a two-year time frame (2020-2021), we provide an in-depth virologic and epidemiological overview of the SARS-CoV-2 pandemic in the Republic of North Macedonia by assessing the frequency and distribution of the circulating SARS-CoV-2 variants. Using genetic characterization and phylogenetic analysis we shed light on the molecular evolution of the virus as well as test for a possible connection between specific SARS-CoV-2 haplotypes and the severity of the clinical symptoms. Our results show that one fifth (21.51%) of the tested respiratory samples for SARS-CoV-2 were positive. A noticeable trend in the incidence and severity of the COVID-19 infections was observed in the 60+ age group between males and females. Of the total number of positive cases, the highest incidence of SARS-CoV-2 was noticed in 60+ males (4,170.4/100,000), with a statistically significant (0,0001) difference between the two sexes. Additionally, a 1.8x increase in male mortality and consequentially significantly higher number of death cases was observed compared to females of the same age group (0.001). A total of 327 samples were sequenced in the period March 2020 - August 2021, showing the temporal distribution of SARS-CoV-2 variants circulating in North Macedonia. The phylogenetic analysis showed that most of the viral genomes were closely related and clustered in four distinctive lineages, B.1, B.1.1.7, B.1.351 and B.1.617.2. A statistically significant difference was observed in the 2C_1 haplotype (p=0.0013), where 10.5% of the patients were hospitalized due to severe clinical condition. By employing genetic sequencing, coupled with epidemiological investigations, we investigated viral distribution patterns, identified emerging variants and detected vaccine breakthrough infections. The present work is the first molecular study giving a comprehensive overview of the genetic landscape of circulating SARS-CoV-2 viruses in North Macedonia in a period of two years.
A significant global public health issue emerged in 2019, now well-known as the coronavirus disease (COVID-19), caused by the SARS-CoV-2 coronavirus (1). The SARS-CoV-2 outbreak has caused an estimated 617 million cases of COVID-19 and 6,5 million deaths worldwide by September 19, 2022 (https://www.worldometers.info/coronavirus/).
The 29,903 nucleotides long, single-stranded RNA beta-coronavirus known as SARS-CoV-2 encodes both structural and non-structural proteins (2). In order to mediate membrane fusion and cell entrance, the spike (S) glycoprotein, is critical in recognizing the human host cell surface receptor angiotensin-converting enzyme-2 (ACE-2) (3, 4). The virus disrupts the host cell molecular functions as soon as it enters the cell, inducing interferon responses and ultimately apoptosis. Variants of concern (VOCs) caused by mutations in the S gene have the potential to improve “viral fitness” by enhancing ACE-2 receptor affinity, infectivity, viral replication, transmissibility, resistance to neutralizing antibodies, immunological escape, thus leading to increased illness severity and risk of reinfection (5).
To date, five variants of concern (VOCs) have been designated by the World Health Organization: Five VOCs have been recognized and include B.1.1.7 (Alpha), B.1.351 (Beta), P.1 (Gamma), B.1.617.2 (Delta), and B.1.1.529 (Omicron) (https://www.who.int/activities/tracking-SARS-CoV-2-variants). The Alpha variant was characterized by amino acid mutations D614G, N501Y and the H69-V70 deletion, Beta by three critical mutations in the receptor-binding domain (RBD) of the S protein - K417N, E484K and N501Y and Delta’s main changes were D614G, T478K and L452R (6). The Omicron variant contains 15 mutations in the RBD alone and over 30 mutations in the S protein, which may reduce the effectiveness of therapeutic antibodies and increase the ACE2 binding (7). Despite having the same origin, these strains differ in terms of pathogenesis, transmissibility, illness severity, vaccine efficacy and disease severity.
Therefore, molecular characterization of full-length SARS-CoV-2 genomes is essential to comprehend the virus’s origin, transmission routes and genome variations that affect the virus’s pathogenicity and transmissibility. For those involved in the fight against infectious disease, sequencing data present essential information. They help with the development of vaccines and antivirals, phylogenetic analysis, tracking the spread of viruses, monitoring the evolution of pathogens, developing other diagnostic tests and identifying any primary and intermediate zoonotic hosts (8).
The aims of our study were to give an in-depth virologic and epidemiological overview of the SARS-CoV-2 pandemics in the period January 2020 to December 2021 in North Macedonia, to assess the frequency and distribution of the circulating SARS-CoV-2 variants, to discern the molecular evolution of SARS-CoV-2 through genetic characterization and phylogenetic analysis and to discover, if any, connection between the SARS-CoV-2 haplotypes and the severity of the clinical symptoms. Additionally, we compared the sequencing results obtained with Illumina and Nanopore as second and third generation sequencing platforms.
A total number of 284,549 respiratory samples (nasal and throat swabs) were prospectively collected from January 2020 to December 2021 in the laboratory of virology at the Institute of Public Health. The analyzed samples include patients referred to the laboratory for routine testing from the regional Centers of Public Health, clinical samples of hospitalized patients in COVID-19 centres and private testing for travelling. The sampling was carried out in the first 1–7 days of the symptom’s onset for the symptomatic patients, while asymptomatic positive cases were detected usually among individuals getting tested for travel purposes or family members/contacts of confirmed cases. All samples were transported in virus transport medium (VTM) (COPAN Diagnostics, Inc., Murrieta, CA, United States), accompanied by patients’ anonymous forms, including age, gender, sampling date, place of residence and symptoms. The exclusion criteria include use of unsuitable transport media, prolonged transport time without suitable refrigeration or media spillage. From the total number of tested samples, 327 whole SARS-CoV-2 genomes were sequenced. The samples for sequencing were selected randomly, taking into consideration an equal geographical distribution of the samples. The selected samples with low cycle threshold values (≤30) were further subjected to sequencing.
Ethical approval was received from the Ethics Committee of the Medical Faculty, University “Ss. Cyril and Methodius”, North Macedonia (No. 03-4731/4).
Viral RNA extractions from the respiratory samples were performed using the RNeasy Mini Kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Shortly after the start of the pandemic, due to the high throughput we started using automatic extraction - KingFisher™ Flex Purification System (Thermo Fisher, Massachusetts, United States) and SaMag-96 (Sacace, Como, Italy) according to the manufacturer’s instructions. All SARS-CoV-2 samples selected for sequencing were re-isolated with the RNeasy Mini Kit, to obtain higher integrity and quality of the viral RNA. Viral RNA detections were carried out with the SARS-CoV-2 real-time RdRP gene duplex reverse transcription (RT)-polymerase chain reaction (PCR) developed and provided by Institute Pasteur, Paris, France (9). In addition, we made an in-house improvement of the assay by adding an internal RP control, which was validated according to the FIND recommendations (10). The second kit used for testing was TaqPath COVID-19 CE-IVD RT-PCR Kit (Thermo Fisher, Massachusetts, United States) that targets three genes (ORF1ab, N, and S), according to the manufacturer’s instructions. Due to the H69-V70 deletion in the Alpha variant, a failure to amplify the S gene occurred when using the TaqPath RT-PCR Kit. Samples were considered SARS-CoV-2 positive if the internal control was amplified and at least two SARS-CoV-2 gene targets were detected by the RT-PCR assays.
The ARTIC Network nCoV-2019 sequencing protocol was used for the multiplexed PCR amplicon approach (11). A reverse transcription reaction was carried out using LunaScript RT SuperMix (NEB, United States) coupled with non-specific primers such as random hexamers and anchored polyT, from previously diluted extract in accordance with Ct values. To amplify the whole genome, two pools (A and B) of the multiplex PCR primer set v1 were utilized (12). 2,5 µl of the synthesized cDNA was utilized as the template for each pool. Using 32 cycles for all samples, tiling 400 nt-amplicons with 20 base pair overlaps (excluding primers) were produced. With the use of the UltraII End Prep Reaction Module (NEB, United States), PCR amplicon pools were end-repaired and dA-tailed before the native barcodes were ligated with the NEBNext UltraII Ligation module (NEB, United States). After performing library clean-up with AMpure XP beads and short fragment buffer (SFB), the libraries were eluted in 15 μl of ONT’s elution buffer. The dsDNA HS Assay Kit was used to quantify the amplicons with the Qubit 2.0 fluorometer (Thermo Fisher Scientific, United States). Using the Natives Barcoding kits EXP-NBD104/EXP-NBD114 (ONT) and the Ligation Sequencing kit SQK-LSK109, the library preparation for the MinION sequencing was carried out in accordance with the manufacturer’s instructions and modifications as per Josh Quick et al. (13). The final library was sequenced on a FLO-MIN106 (R9.5) flow cell, while multiplexing twenty-four samples per run. The length of the ONT sequencing runs varied depending on the genome coverage in real-time, although they typically lasted for about 24 hours.
Real-time visualization of genome coverage and reference matching for each barcode were performed using the RAMPART tool (Read Assignment, Mapping, and Phylogenetic Analysis in Real Time), developed by the ARTIC network (14). To create fast5 and fastQ files, real-time basecalling was done using MinKNOW and its integrated Guppy high accuracy model (v3.5.2)(ONT). The minimum q-score a read must attain to pass qscore filtering was 7, roughly corresponding to a basecall accuracy of 85%. Following basecalling, we used EPI2ME and its associated tool WIMP for quick species identification as well as comprehensive QC metrics to gain an understanding in the performance of the run. Using ONT Guppy Barcoding Software v3.1.5 + 781ed575, the quality-checked reads were demultiplexed and trimmed for adapters. Using CLC Genomic Workbench version 20.0.4, mapping, alignment, consensus creation, and variant calling were completed (Qiagen, Hilden, Germany). We conducted data analysis in parallel using a variety of GALAXY tools (https://usegalaxy.eu). Using Minimap2 (v2.9), the readings were mapped against the SARS-CoV-2 reference (NC 045512) (15). SAMtools depth was used to obtain the coverage data, while ivar consensus (v 1.3.1) was used for generating a consensus sequence. For variant calling we used nanopolish (v0.13.2) (16). Data were manually examined using Tablet (v1.19) (17).
Hybrid capture-based targeted enrichment approach was performed using the Illumina DNA Prep with Enrichment kit. NEBNext® UltraTM II RNA First Strand Synthesis Module and NEBNext® UltraTM II Non-Directional RNA Second Strand kit New England Biolabs, MA, USA) were used to create double stranded cDNA from 5 µL (<10 ng) of RNA. According to manufacturer instructions, the generated cDNA was utilized as input for library preparation with the Illumina DNA Prep with Enrichment kit (18). Purified amplicons were used for the library preparation, in accordance with the manufacturer’s instructions. These instructions call for tagmentation, stop tagmentation, three washes, PCR with indexing, dual-sided size selection, and purification before individual libraries are pooled (18). Pre-enriched libraries were pooled by mass. A respiratory virus biotinylated adjacent oligoprobes panel, extended to include SARS-CoV-2 served as the basis for the enrichment process (19). Forty-eight samples were sequenced with the Illumina MiniSeq system using a MiniSeq High Output Reagent Kit (150-cycles). A detailed laboratory method can be found on protocols.io (20).
With the use of the Illumina Dynamic Read Analysis for GENomics (DRAGEN; v3.5.13; Illumina) Bio-IT Platform, sequencing data from the RVP capture method were processed. Using CLC Genomic Workbench version 20.0.4, mapping, alignment, consensus creation, and variant calling were completed (Qiagen, Hilden, Germany). Additionally, we used the GALAXY platform tools (https://usegalaxy.eu/) and reads were mapped using the Burrows-Wheeler Aligner MEM algorithm (BWA-MEM) (v0.7.7) against the SARS-CoV-2 reference (NC 045512). SAMtools depth was used to assess the genome coverage. We utilized ivar consensus (v1.3.1) to create the consensus sequence and ivar varaints to identify variants (v 1.3.1). Data were manually inspected using Tablet (v1.19) (17).
The basic quality metrics used for Illumina sequencing remained high and met the expected values on sequencing runs. For the six MiniSeq runs, the Q-score >30 was in range between 91 to 97.8%.
All statistical tests and visualizations were performed with R (v4.1.1), using ggplot2 (v3.3.5), dplyr (v1.0.7), and tidyr (v1.1.3), packages. Chi-square test and Fischer exact test were carried out with the “stats” package. Additionally, Microsoft Excel 2016 for Windows 10 and OpenEpi (http://www.openepi.com/Menu/OE_Menu.htm) were used for statistical data analyses. P-values < 0,05 were considered statistically significant.
ClustalW (21) was used to create nucleotide alignments and MEGA (Molecular Evolutionary Genetics Analysis v.11) was used to align codon positions (22). Using MEGA, a maximum likelihood phylogenetic tree with the General Time Reversible Model (GTR) was inferred, and the bootstrap analysis with 1,000 replications was used to determine the reliability of the tree’s topology. For better graphic representation of the phylogenetic trees Figtree v1.4.4 was used (23). The sequence data were deposited in the Global Initiative on Sharing Avian Influenza Data (GISAID) EpiCoV database (24).
A total of 284,549 respiratory samples were tested for SARS-CoV-2 in a two-year time frame, of which 61,206 (21.51%) tested positive. During the examined period, three major waves were observed caused by three different variants. Namely, the first wave spanned from week 41 to 52/2020 and the surge of cases was caused by the B.1 (20A and 20B) lineage. Week 46/2020 was marked by the highest number of laboratory confirmed cases throughout the observed period (n=2,544). The emergence of the Alpha variant drove the second wave, starting from week 7/2021 and reaching the highest number of positive cases in week 12 (n=2,187). The epidemiological situation in North Macedonia exhibited a trend of stabilization by week 18/2021, leading to a few months with very low number of positive detections. The third SARS-CoV-2 wave started as a result of the emergence of the Delta variant, with numbers growing rapidly from week 31/2021 (n=85) to week 33/2022 (n=775) (Figure 1). The predominant circulating variant in N. Macedonia was Delta until week 52/2021, before the newest Omicron variant took over.
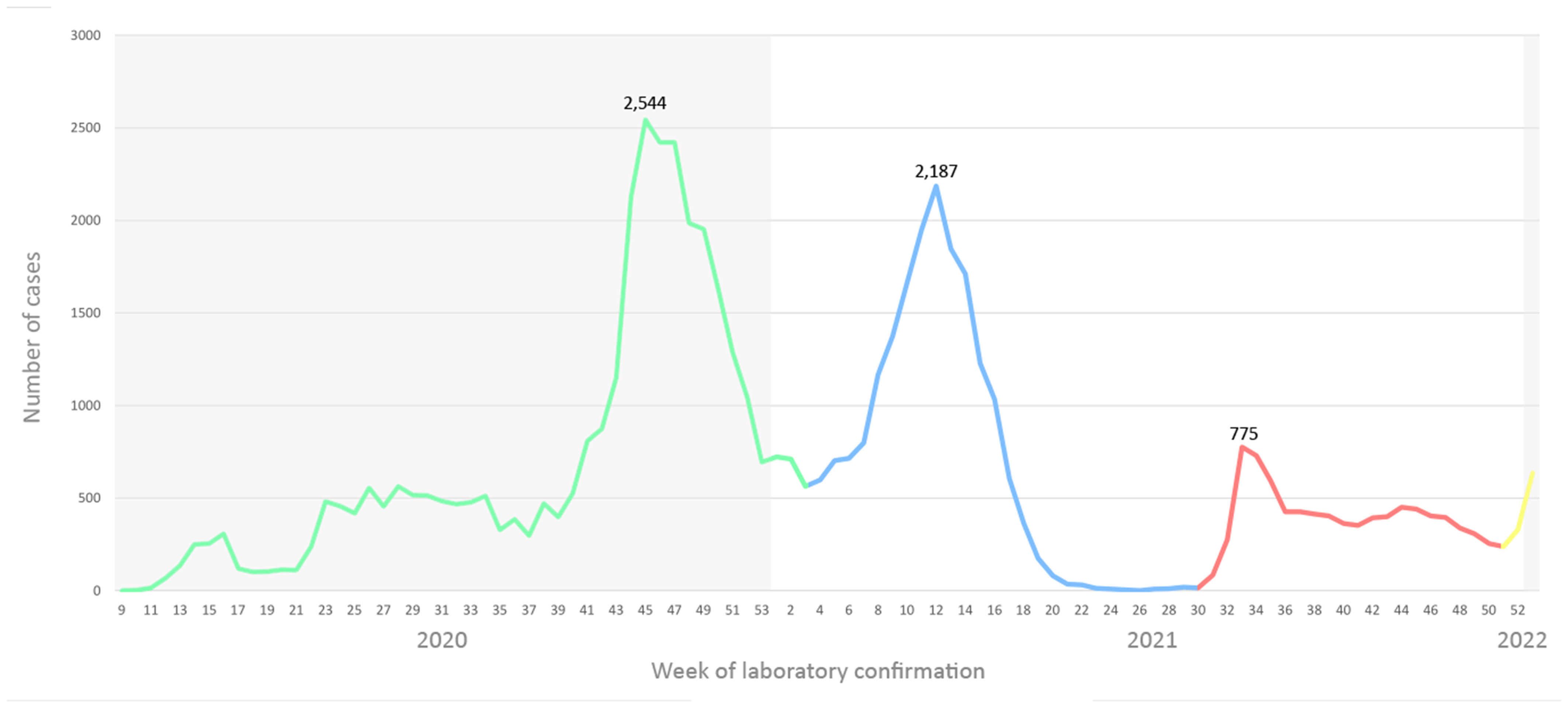
Figure 1 Number of SARS-CoV-2 cases on a weekly basis in a two-year span, color coded by variant. B.1 lineage designated with green, Alpha with blue, Delta with red and Omicron with yellow.
Additionally, we analyzed the SARS-CoV-2 positive cases by age group and sex, to check for any possible significant differences. Regarding the age groups, the highest number of SARS-CoV-2 positive cases was detected in the age group 60+ (n=16,276), whereas the highest specific morbidity of 4,101.9/100,000 was registered in the age group 50-59 (n=11,496) (Figure 2). High morbidity was observed in the age group 40-49 (3,688.4/100,000) and 30-39 (3,322.7/100,000), where 11,160 and 10,770 positive SARS-CoV-2 cases were detected, respectively. Further on, when analyzing the incidence of SARS-CoV-2 positive cases by age and sex, the highest specific morbidity among males was observed in the 60+ group (4,170.4/100,000), while the highest incidence among females was in the 50-59 age group (4,376.2/100,000) (Figure 3). The number of positive cases among males in the 60+ group was significantly higher compared to females (0.0001).
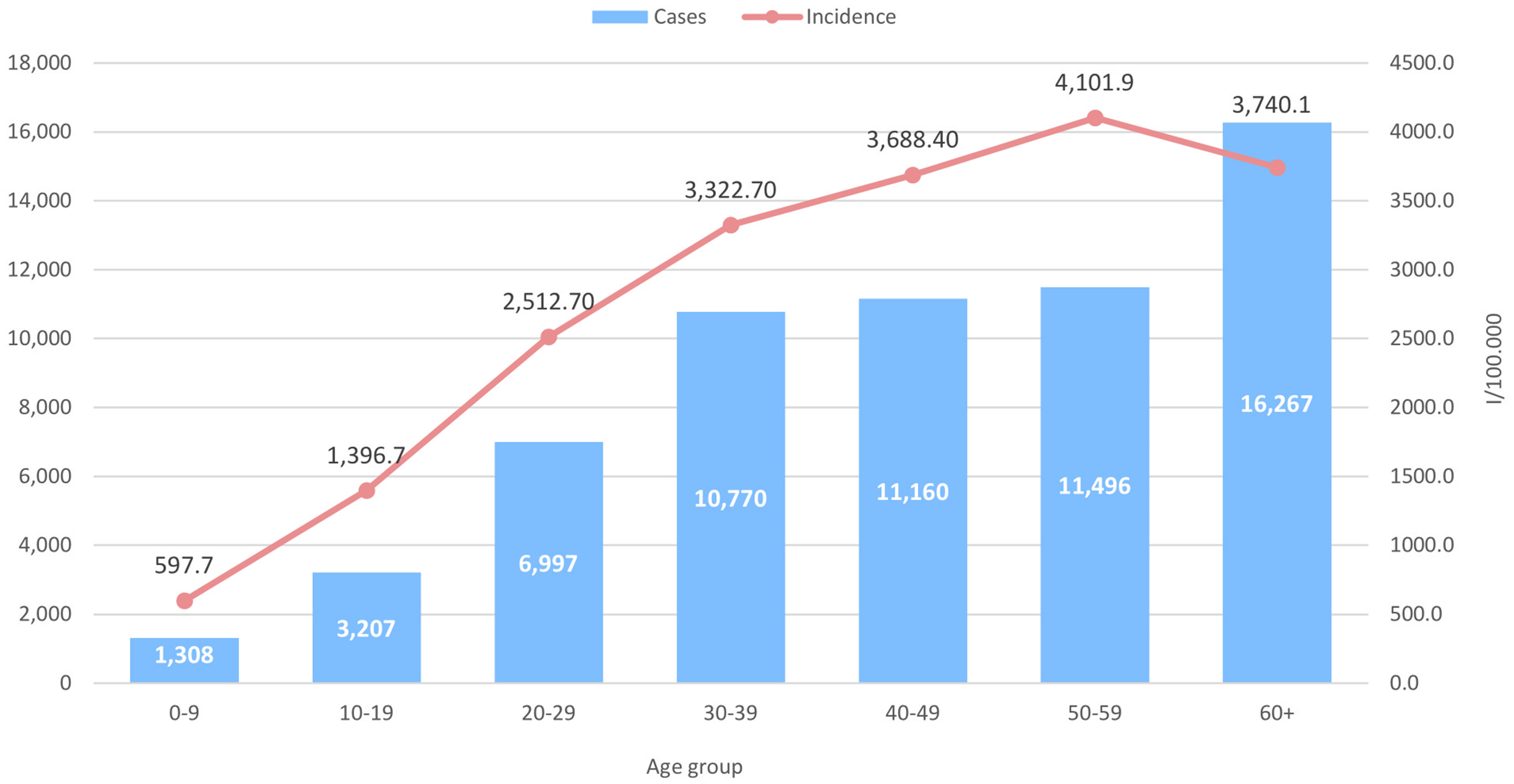
Figure 2 Number of SARS-CoV-2 cases in the period January 2020 – December 2021 and SARS-CoV-2 incidence by age groups.
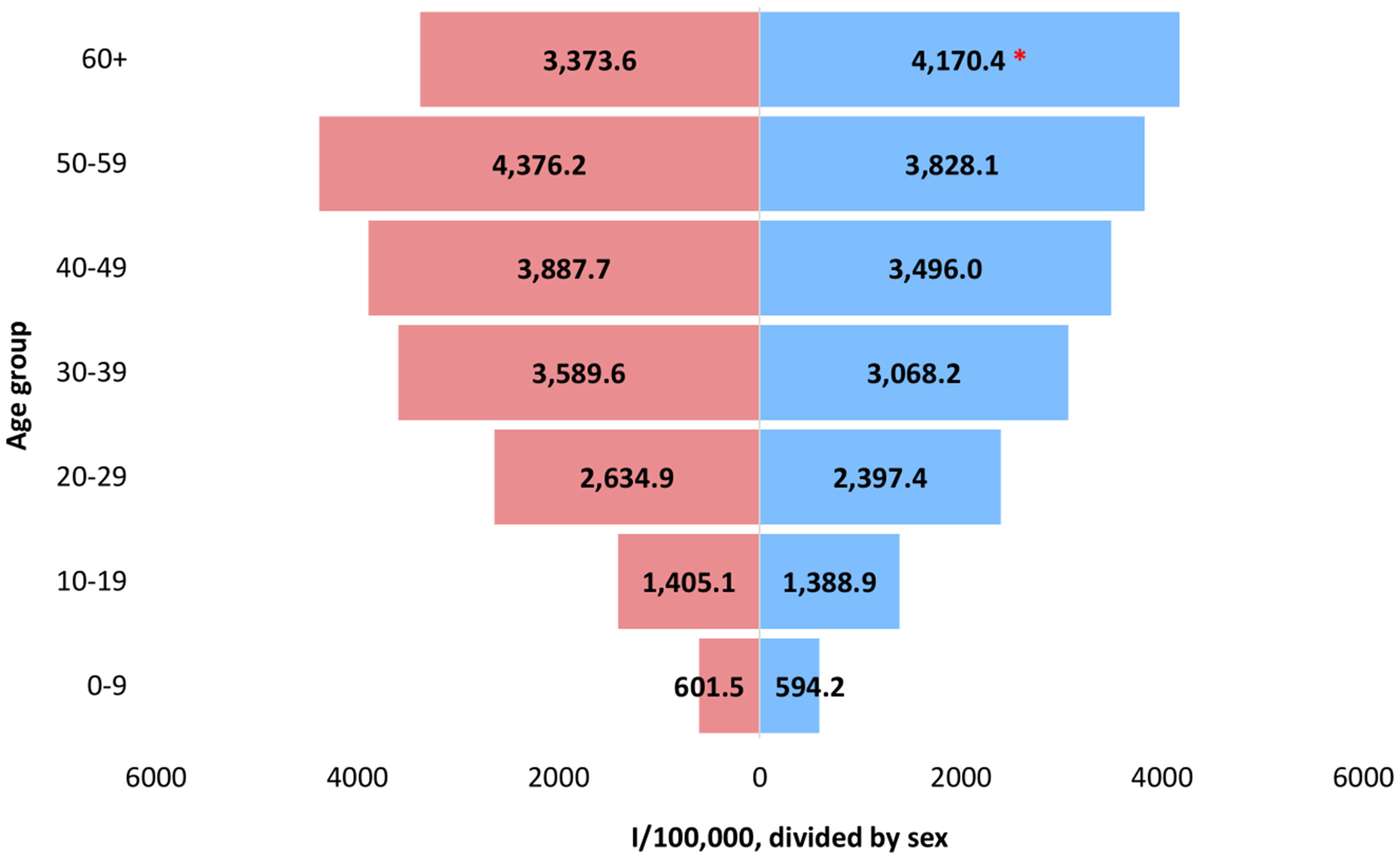
Figure 3 Incidence of SARS-CoV-2 positive cases in the period January 2020 – December 2021 by age group and sex. The statistically significant difference between males and females in the 60+ age group (0.0001) is marked with an asterisk (*). Males designated with blue, while females with red color, respectively.
Four distinctive peaks were observed in the number of death cases among SARS-CoV-2 positive patients, following the pattern of the emergence and introduction of new variants in the population (Figure 4). The highest number of death cases was recorded in week 12/2021 (n=81), during the Alpha wave.
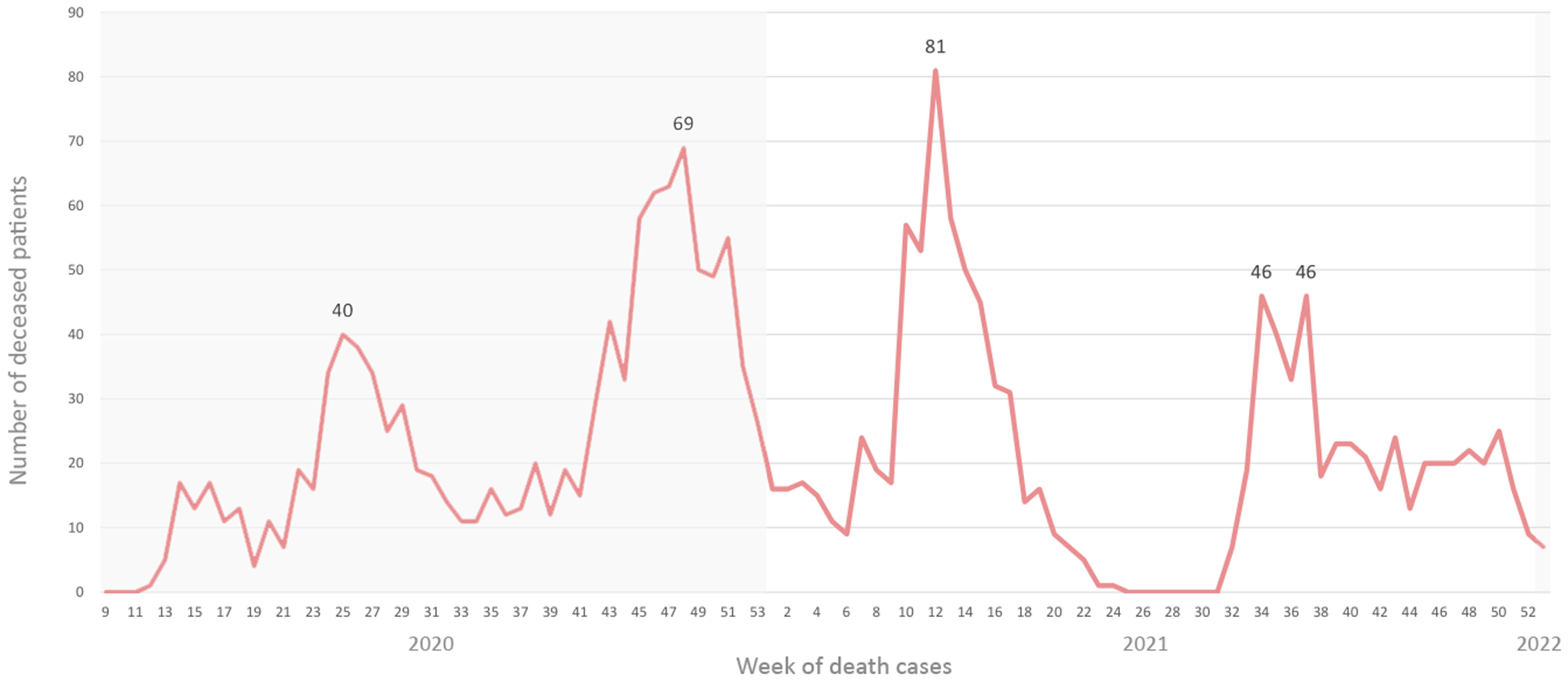
Figure 4 Number of death cases on a weekly basis in a two-year span.
We analyzed the death cases by age group and sex, to check for any possible significant differences. The incidence of deceased males and females was highest in the age group 60+ (5,213/100,000 and 294,7/100,000, respectively), however the mortality among males was 1.8 times higher compared to females in the 60+ group (Figure 5). Moreover, the number of death cases among males in the 60+ group was significantly higher compared to the female group (p=0.001).

Figure 5 Incidence of SARS-CoV-2 death cases in the period January 2020 – December 2021 by age and sex. The statistically significant difference in the number of death cases between males and females in the 60+ age group (p=0.001) was marked with asterisk (*). Males designated with blue, while females with red color, respectively.
A total of 327 samples were sequenced in the period March 2020 - August 2021. Most of the detected mutations (n=56) were located in the ORF1ab region, followed by the S protein (n=27) and the N protein (n=12) (Table 1). The ORF1ab region was one of the mutation hot spots with nsp12 or the RdRP protein harboring the most common mutation nsp12:P323L (98.78%) observed in almost all sequences. In the nsp6 protein, a triple deletion nsp6:F108del, nsp6:G107del, nsp6:S106del reached a frequency of 59.15%. Similarly, nsp3 harbored three mutations A890D, I1412T and T183I with frequency above 50% (54.88%. 50.91% and 57.01%, respectively). The D614G mutation in the S protein had the highest frequency (93.6%), followed by H69-V70del (64.63%), N501Y (60.98%), Y144del, S982A and D1118H (59.76%). Within the N protein, the R203K (69.82%) and the G204R (68.29%) were the most common mutations. All substitutions with frequency lower than 3% were not shown in the table.
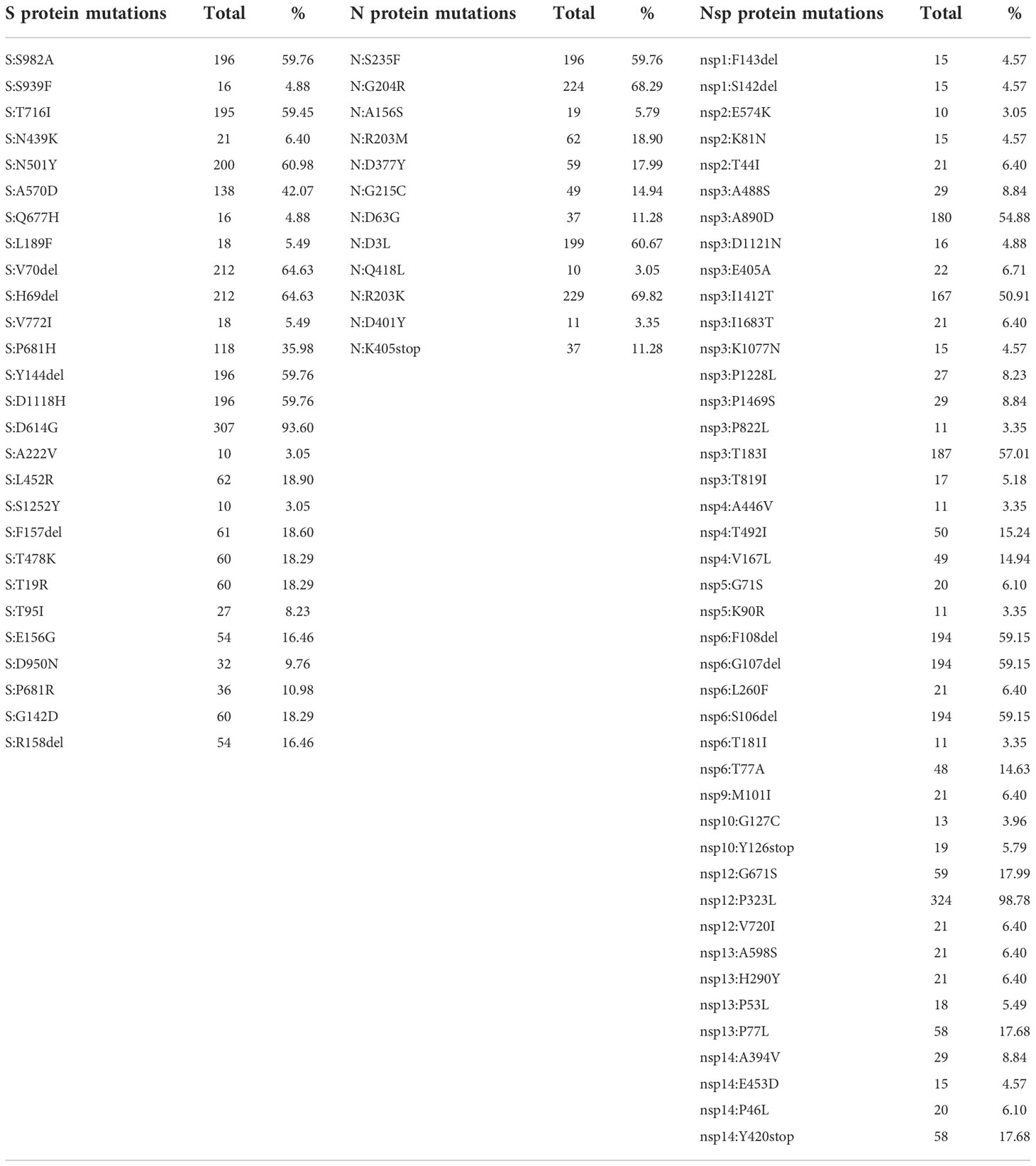
Table 1 A comprehensive overview of all detected mutations in the S, N and ORF1ab proteins.
All SARS-CoV-2 genomes obtained in this study (n=327) were selected for phylogenetic analysis, using the maximum likelihood method (Figure 6). Full-length virus genomes were annotated using the reference genome of hCoV-19/Wuhan/Hu-1/2019 (NC_045512.2). The viral phylogeny is based on the consensus sequence assembled of each sample and the branching indicates the evolutionary differences between samples. Based on the phylogenetic tree, the clustering of the sequences matched the previously established PANGO lineages: B.1, B.1.1.7, B.1.351 and B.1.617.2. The B.1 lineage was characterized by several distinctive branches, representing different clades. A large proportion of the sequenced viruses clustered together as they belonged to the B.1.1.7 lineage. The B.1.351 lineage was represented by only one detected case, clustering with viruses from Italy, Germany and Slovenia. The Delta lineage viruses clustered into several different clades, all being very closely related. Supplementary Table 1 contains all sequenced virus names for each lineage.
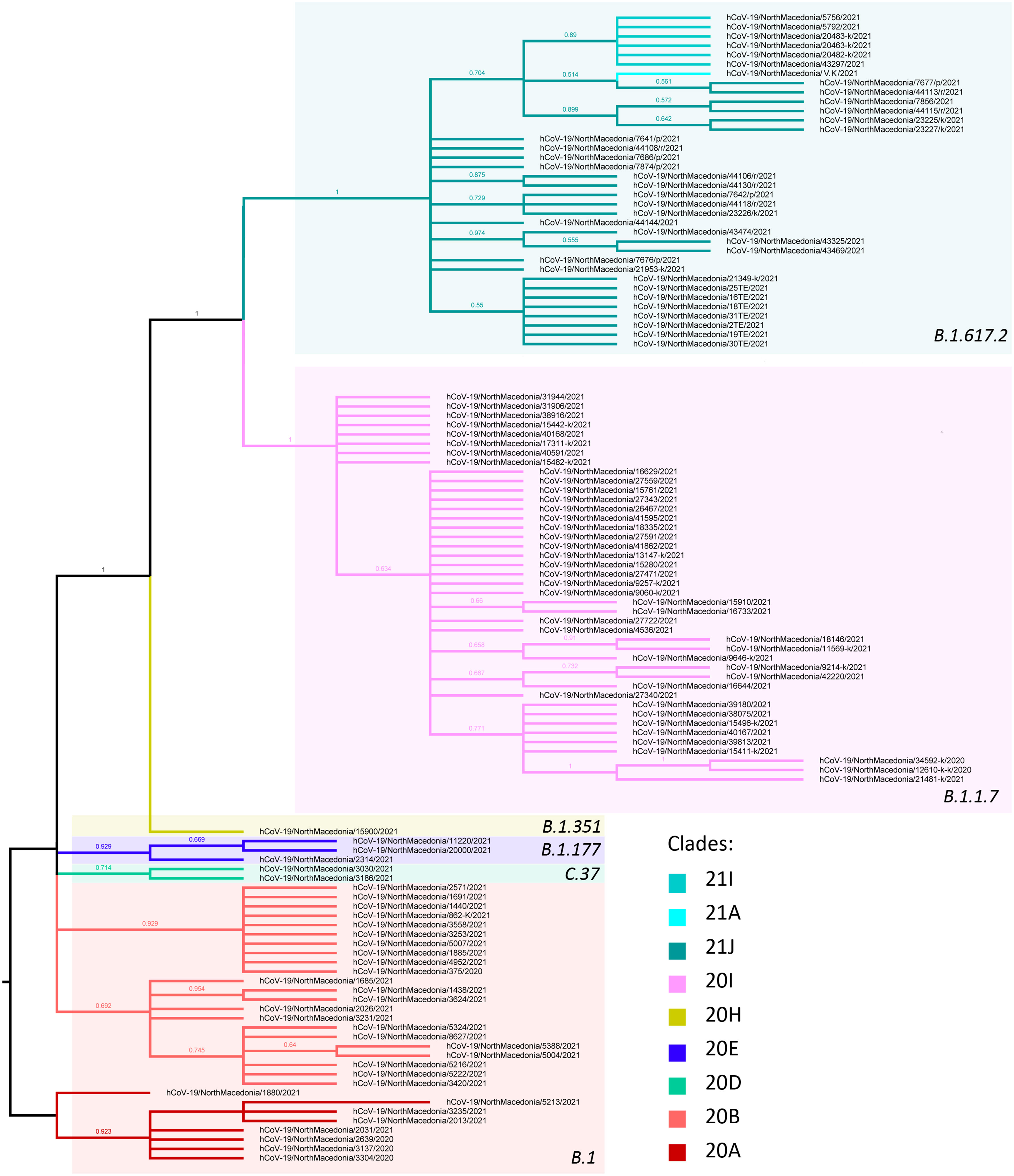
Figure 6 Phylogenetic tree of Macedonian sequenced strains of SARS-CoV-2. Lineages are separated into different color boxes, colors representing different clades are designated in figure legend.
In the period March 2020-January 2021 the B.1 line predominated in North Macedonia. Different B.1 clades circulated in the aforementioned period, with 20B being the dominant clade in March, August and September 2020 (100%). In April 2020, we noticed co-circulation of clades 20A and 20B (66.7% and 33.3%, respectively), however 20A was not detected again until January 2021 (38.6%). Clade 20D (Lambda) was observed only in January 2021, with a frequency of 4.5%. The viruses belonging to clade 20E were detected in three consecutive months January-March 2021 with low frequencies (2.3%, 2.2% and 2.1%, respectively). In January 2021 the first cases belonging to the B.1.1.7 lineage were confirmed with sequencing (11.4%), with a rapid increase in February (57.8%). The period March-June 2021 was predominated by the B.1.1.7 lineage with almost 100% frequency. In May 2021 we detected the first confirmed B.1.351 lineage (case, however this lineage did not continue its circulation within the population. In July 2021 we detected a co-circulation of 3 different Delta subclades, 21J (41.4%), 21I (8.6%) and 21A (1.4%), together with the Alpha variant (48.6%). In August 2021, it is clearly visible that Delta had completely taken over, with Delta 21J (87.5%) becoming the dominant variant (Figure 7). Figure 8 shows the geographical distribution of the SARS-CoV-2 variants in North Macedonia. The highest number of variants was detected in Skopje, as the one with the largest population and highest number of tested samples.
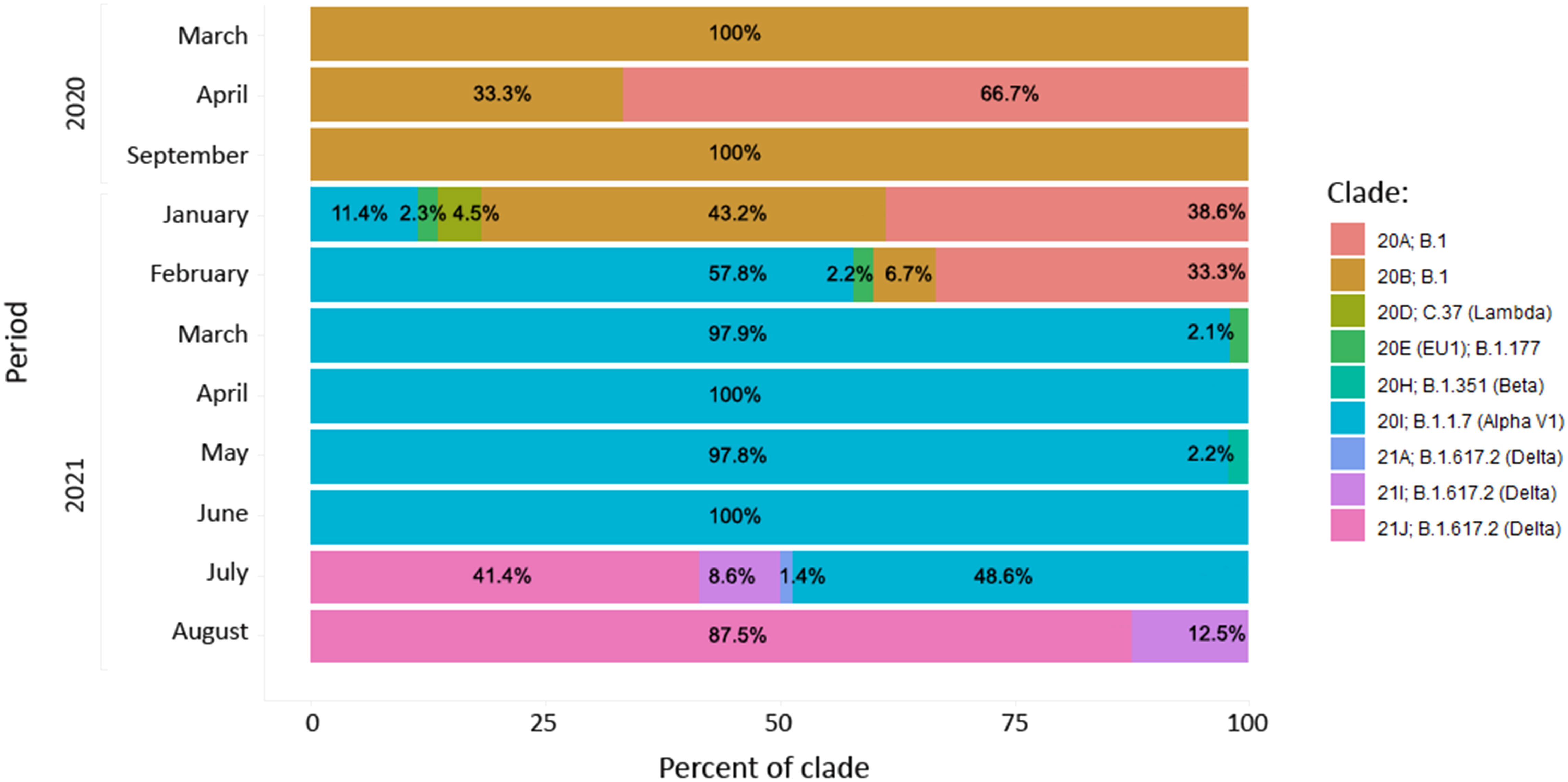
Figure 7 Distribution of SARS-CoV-2 clades in the period between March 2020 and August 2021.
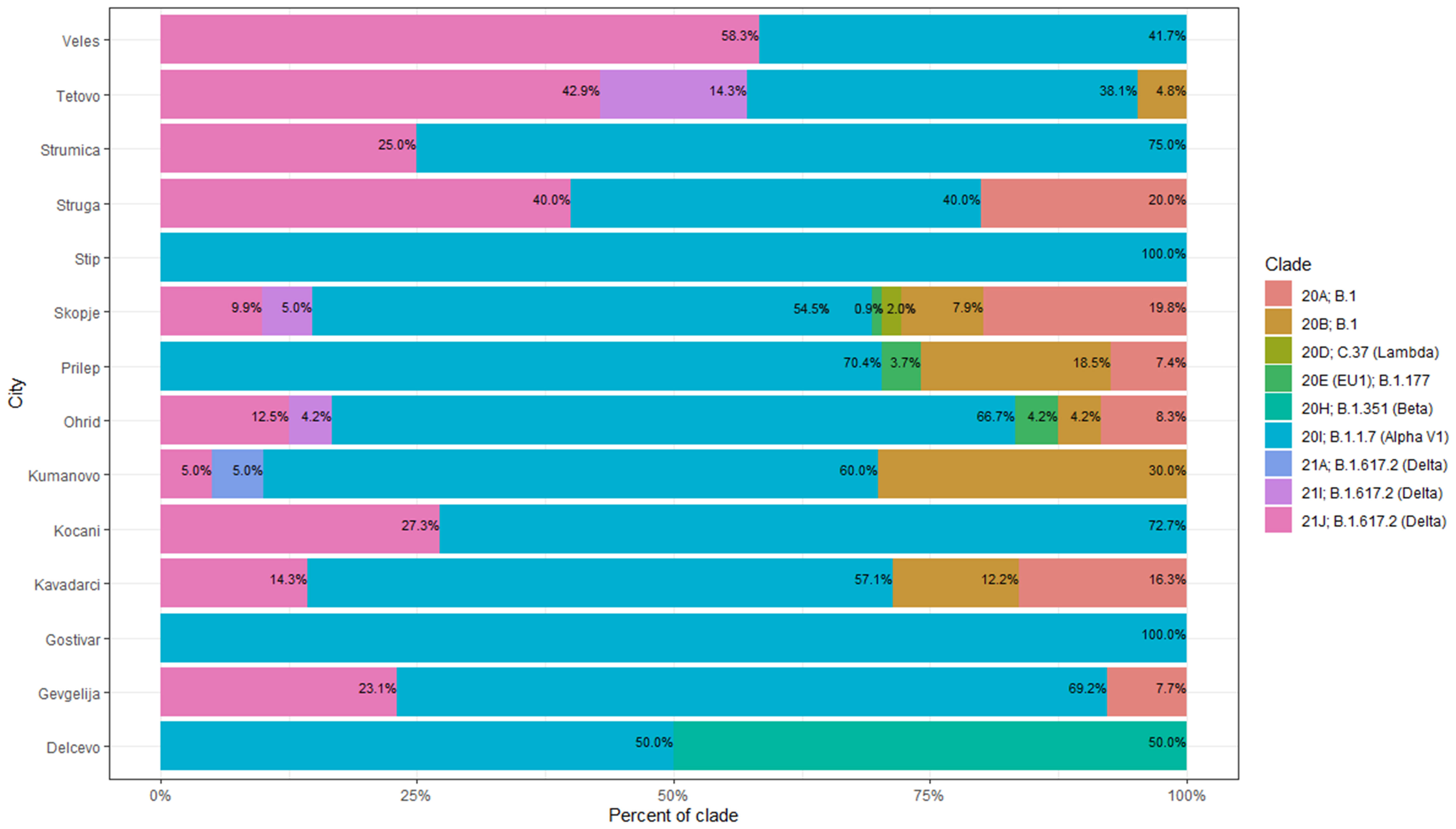
Figure 8 Geogrpahical distribution of SARS-CoV-2 clades in North Macedonia.
We went a step further and grouped the genomes into different haplotypes comprising the same unique combination of substitutions, within a particular lineage (Figure 9). The haplotypes comprised both spike and non-spike substitutions throughout the SARS-CoV-2 genome (Table 2). Haplotype 1 was characterized by the mutations N:K405*, nsp12:P323L, nsp12:M924R and nsp14:Y420stop and was the dominant haplotype circulating in 2020 (100%). In the period January-February 2021, haplotype 1A harboring the additional mutations N:R203K, N:G204R, nsp3:D1121N, nsp5:G71S, nsp12:P323L, S:D614G and S:Q677H reached a frequency of 36%. Haplotype 2 was characterized by several newly emerged mutations, especially in the S protein (S:L189F, S:N439K, S:D614G, S:V772I, S:S939F; S:H69- and S:V70-) with a frequency of 20.2%. Haplotype 2A, comprised all signature Alpha variant mutations (N:D3L, N:R203K, N:G204R, N:S235F, nsp3:T183I, nsp3:A890D, nsp3:I1412T, nsp12:P323L, NS8:Q27*, NS8:R52I, NS8:Y73C, S:N501Y, S:A570D, S:D614G, S:P681H, S:T716I, S:S982A, S:D1118H, nsp6:S106del, nsp6:G107del, nsp6:F108del, S:H69-V70del, S:Y144del) and it reaches its dominance in the period May-June 2021 with 73.5%. Haplotype 2A_1 harbored three additional mutations in the ORF1ab region when compared to haplotype 1 (nsp2:T44I, nsp3:E405A and nsp6:L260F) and reached a frequency of 12% in the period May-June 2021. Haplotype 2A_4 contained three different mutations, including one in the S protein (nsp12:N215Y, nsp14:H455Q, S:S221L), however it reached a frequency of only 3.6% in May-June 2021. Haplotype 2C, reached a frequency of 53.2% in July-August 2021, followed by haplotype 2C_1 (8.5%). The only characterized breakthrough infection belonged to haplotype 2C_2, as it was neither pure 21A Delta, nor it had all co-existing mutations of the 21I Delta clade. This haplotype harbored a signature set of 7 co-appearing mutations (nsp2:P129L, nsp3:H1274Y, nsp3:P822L, nsp4:A446V, nsp6:V149A, nsp15:H234Y and N:R385K).
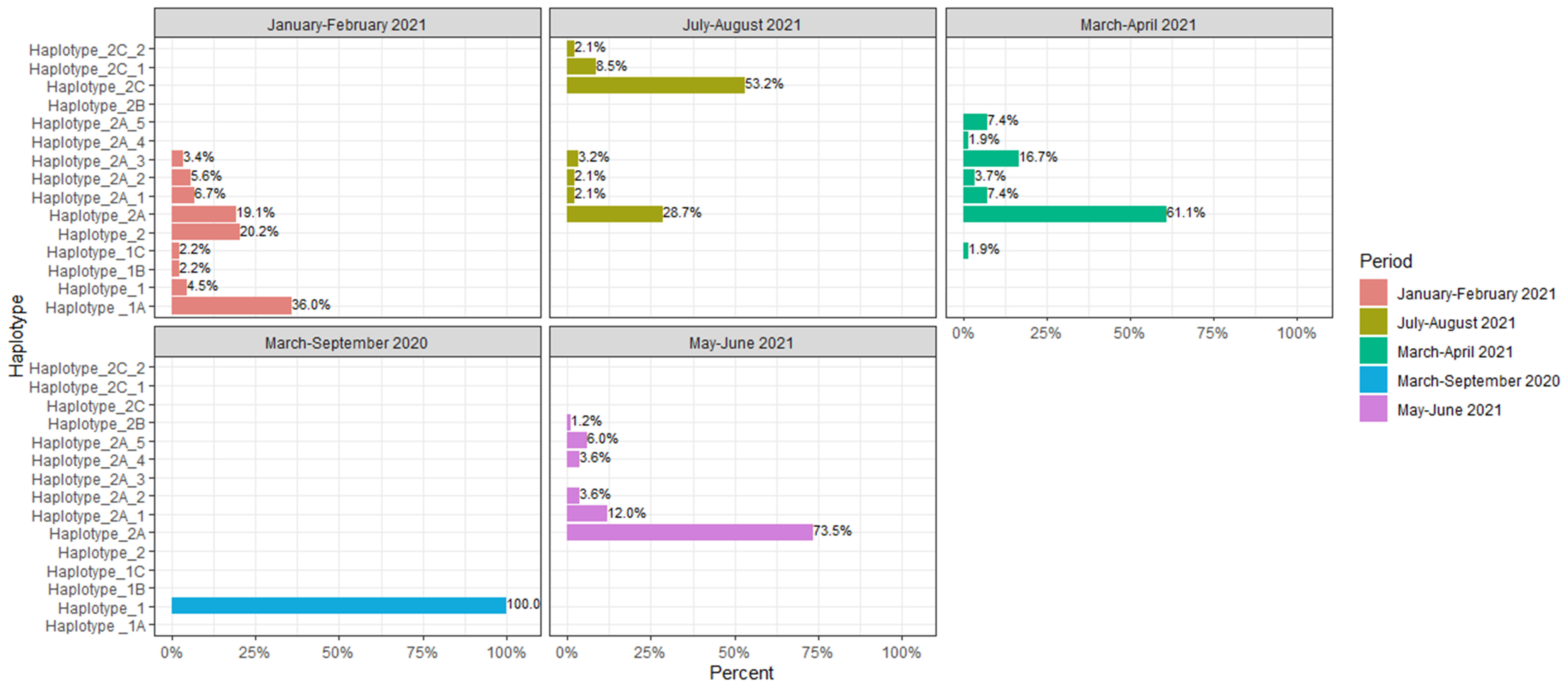
Figure 9 Schematic presentation of the distribution of haplotypes within different time periods of the pandemic.
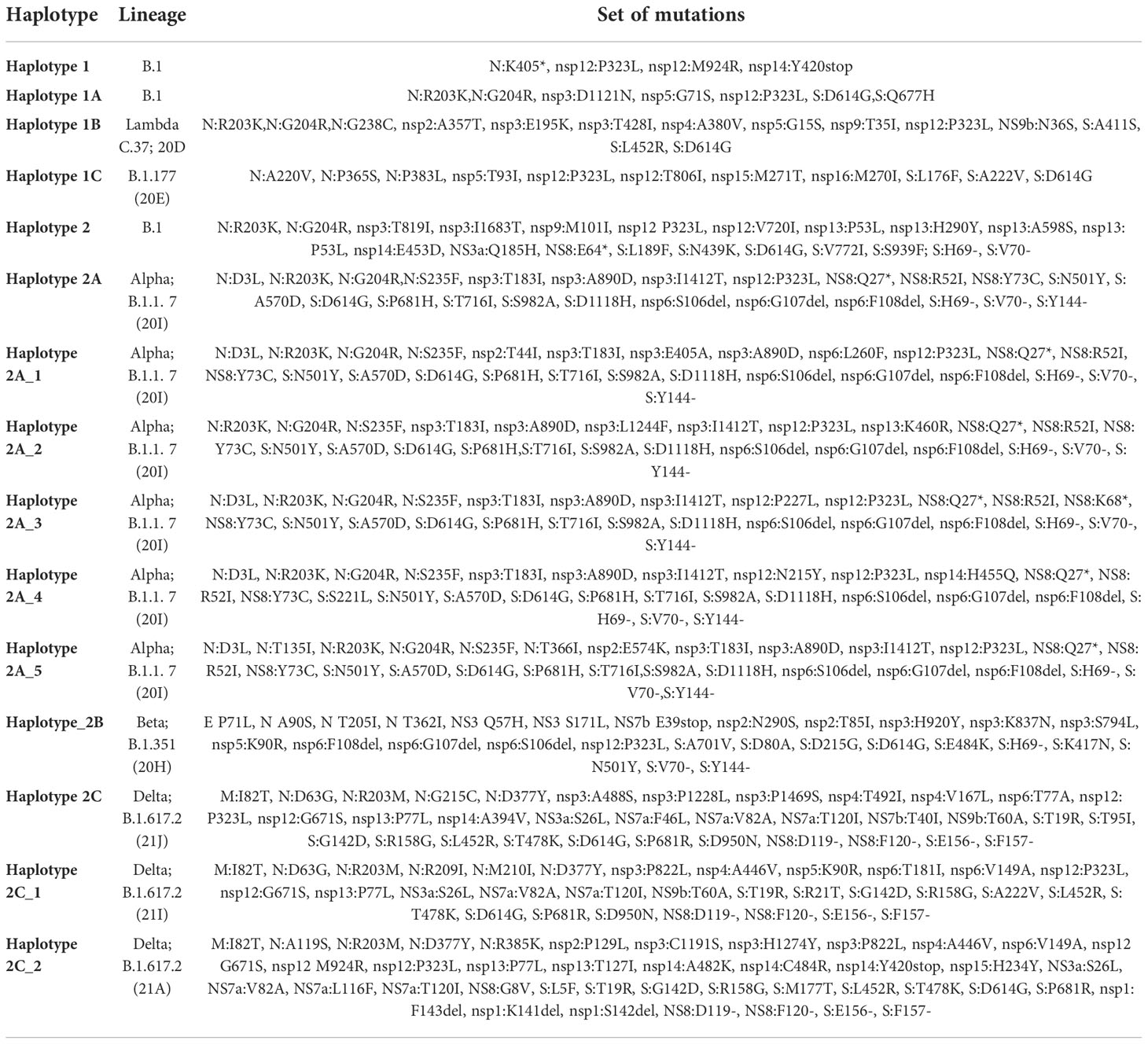
Table 2 Haplotypes and the corresponding mutations.
Additionally, by grouping the mutations into haplotypes, we attempted to correlate a signature set of both spike and non-spike constellation of mutations with the clinical manifestation of the disease (Figure 10). A statistically significant difference was observed in the haplotype 2C_1 group (p=0.0013), where 10.5% of the patients were hospitalized due to severe clinical condition. This haplotype was characterized by several distinctive mutations in the S, N and ORF1ab proteins. The epidemiological data showed that all haplotype 2C_1 cases were immediately hospitalized after receiving a confirmation for positive SARS-CoV-2 result, having remained in the hospital for at least three weeks. All had oxygen requirement, but none was put on mechanical ventilation. No comorbidities were reported. High percentage of hospitalized patients was observed in the haplotype 2C group (14%), but without statistical significance (p=0.2618). Additionally, almost half of the SARS-CoV-2 sequenced cases, collected from hospitalized patients belonged to haplotype 2A (49.1%), however when compared to the outpatient group with the same haplotype no significant difference were found (p= 0.3797).
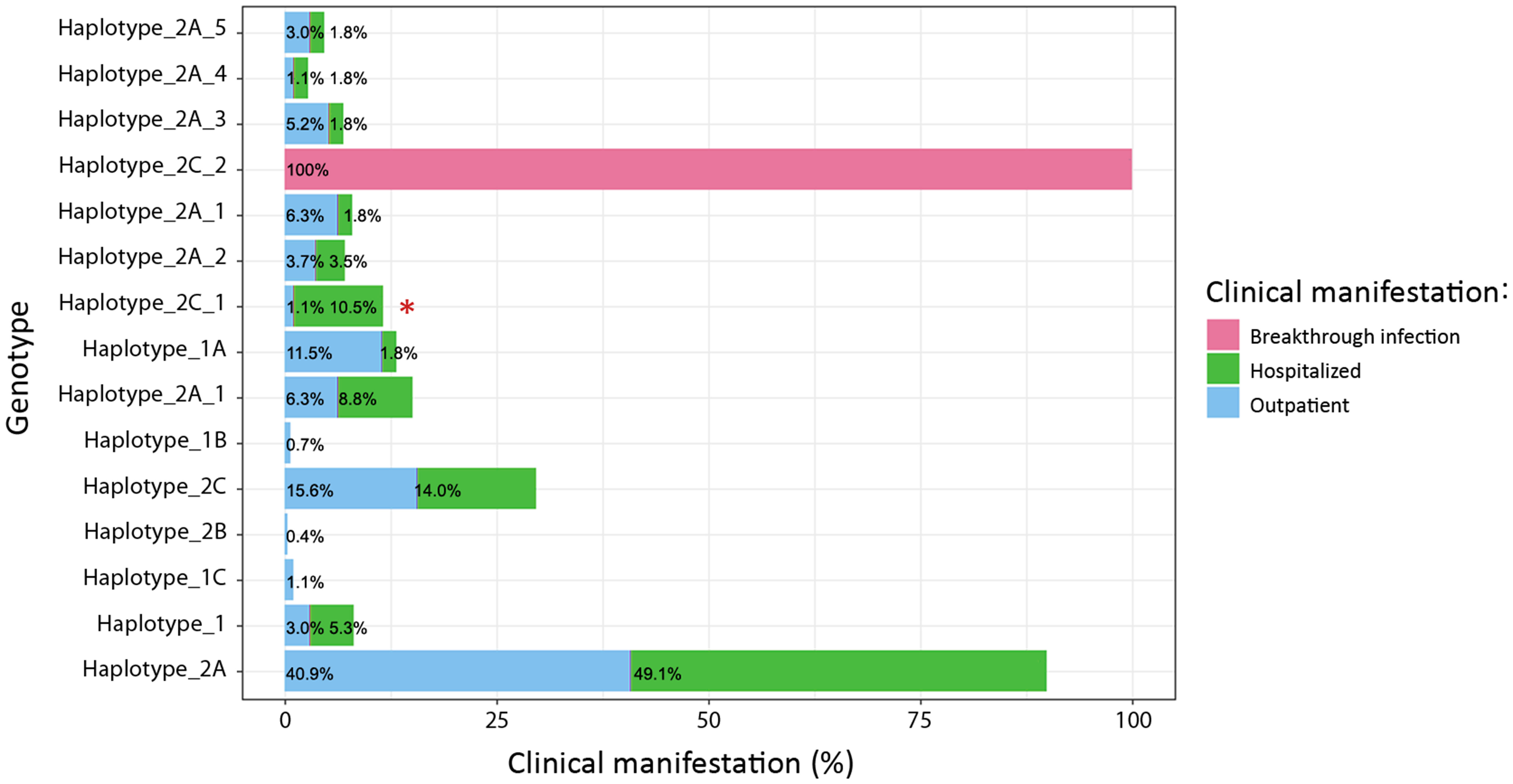
Figure 10 Distribuiton of haplotypes between hospitalized patients and outpatients. A statistically significant difference was observed in the haplotype 2C_1 group (p=0.0013) between hospitalized and outpatients and marked with asterisk (*).
The mean sequencing depths obtained with the Illumina platform vis-à-vis the Nanopore platform were 2450x coverage and 856X coverage, respectively. The average number of reads and sequence length for Illumina was 1,683.900 and 30-150bps, however the number of reads with Nanopore was notably lower (81,109), but as expected the length of the produced sequences was between 100-3000 bps. The passing filter was set to >85%, with coverage of the genome >10X. Twenty-one samples did not pass the sequencing metrics and all had a Ct value of above 30. The percentage of mapped reads was similar for both platforms 98.36% (Illumina) and 98.16% (Nanopore), however the PHRED score for the mapped reads differed (35 and 16, respectively). Additionally, the average error rate per base per read for Illumina was 0.005%, while the error rate for Nanopore was higher 0.12%. Despite the elevated error rates observed in the Nanopore sequencing reads, highly accurate consensus-level sequence determination was achieved above a minimum ~60x coverage depth.
Since its emergence in December 2019, the extremely contagious and dangerous SARS-CoV-2 virus has claimed millions of lives worldwide. The SARS-CoV-2 genome has been rapidly changing since the start of the pandemic, and there is evidence that these changes have an effect on the virus’s pathogenicity (25–28). Different variants of SARS-CoV-2 have emerged from geographic regions whose epidemiological conditions allowed for the stabilization of certain genetic combinations that had an impact on their fitness (25–28). In this study, we analyzed a two-year period of the greatest pandemic modern society has ever faced.
In the period January 2020 to December 2021, the epidemiological situation in North Macedonia was characterized by three major waves driven by the B.1, B.1.1.7 (Alpha) and B.1.617.2 (Delta) lineages, similar as around the world (29–31). Four distinctive peaks were observed in the number of deceased SARS-CoV-2 infected patients, following the pattern of the emergence and introduction of new variants in the population. The mortality among males was 1.8 times higher compared to females in the 60+ group and consequently the number of death cases among males in the same group was significantly higher compared to the females (p=0.001). To exclude the previous medical history of the patients as a factor influencing the co-relation between the sex of the patients and mortality, we assessed the percentage of comorbidities in the 60+ age group. Namely, in the 60+ group of deceased patients, 83.1% of the males had comorbidities (82.3% cardiovascular disease, 31.6% diabetes and 14.1% lung disease), whereas comorbidities were reported for 85.3% of the females (85.7% cardiovascular disease, 37.2% diabetes and 19.5% lung disease). Despite the fact that comorbidities significantly contribute to the development of more severe clinical conditions as well as death, their percentage was comparable in both groups, thus the significant difference in mortality between males and females is not due to their pre-existing medical conditions. Similar observations were made in different studies in Italy, USA and China (32–34), further supporting our finding that SARS-CoV-2 infected males are more at risk for worse outcomes and death. A meta-analysis revealed that age had a significant impact on the COVID-19 patient mortality, with an important cut-off point at age >50 and particularly >60 (35). These results are in line with the observation that older adult patients have a higher susceptibility to infection and more serious clinical manifestations due to physiological aging and particularly, a higher prevalence of comorbidities that contribute to the development of a more severe clinical condition (36).
SARS-CoV-2 has a modest pace of mutation in comparison to other widespread viruses like influenza (37). As a result, SARS-CoV-2 is less likely to undergo mutational changes, such as antigenic drift and antigenic shift, which change the virus’s genetic makeup and affects how infectious, transmissible and pathogenic is. However, over 3000 distinct point mutations have been found in virus isolates from around the world, indicating an increased frequency of alterations in the SARS-CoV-2 genome during the pandemic (38). Several SARS-CoV-2 genes, including S, N, and nsp12 (RdRP), have a wide mutational range (6). In our study most of the mutations were located in the ORF1ab gene region and the S protein. Soon after the start of the pandemic, ORF1ab became one of the mutational hot spots, particularly nsp2, nsp3, and nsp12 (39, 40). We observed similar results, with nsp3, nsp6 and nsp12 harboring mutations with the highest frequency in the ORF1ab region. Nsp12 coding the RdRp protein harbored the most common mutation nsp12:P323L (98.78%) observed in almost all sequences, similarly as in other studies (41). The spike protein has been documented to have more than 80 substitution mutations and deletions, with the most notable ones being L452R, followed by E484K, D614G, A222V, L18F, S477N, H69-V70del and N501Y, respectively (41). In North Macedonia the D614G mutation in the S protein had the highest frequency (93.6%), shown to lead to enhanced binding to the ACE2 receptor and increased replication in human bronchial and nasal airway epithelial cultures (42). As of early December 2021, this mutation is found in >99% of the 6 million viral genomes in the GISAID database (43). The RBD N501Y mutation increases the binding affinity to the ACE-2 receptor and transmissibility (6, 44) and in our study it reached a frequency of 60.98%.
The phylogenetic analysis showed that most of the viral genomes were closely related and clustered in four PANGO designated lineages, B.1, B.1.1.7, B.1.351 and B.1.617.2. The genetic distances between genomes were small, given the fact that the novel SARS-CoV-2 virus emerged quite recently and the timescale of evolution is very small. The sequencing analysis showed that in the period March 2020 - January 2021 the B.1 lineage predominated in North Macedonia, as everywhere in the world (30, 31). The B.1.1.7 variant was first discovered and transmitted in the United Kingdom (45) and it arrived in our country at the beginning of 2021. Its swift takeover and complete predominance in the following months due to its mutational landscape leading to 40-70% increase in transmissibility (46).
The specific key substitutions boost the affinity for the ACE2 receptor and have an effect on infection and transmission, specifically in the RBD (N501Y) and close to the furin cleavage site (P681H) (26). The Beta variant and B.1.1.7 both have few mutations in common as well as key amino acid changes K417N, E484K, N501Y, D614G and A701V in their spike proteins. Due to the presence of the E484K mutation in its spike protein, the Beta lineage has been reported to have greater transmissibility and immune evasion ability (47), however it was not successful in our country having detected only one case. One explanation could be that the Beta variant is not adequately represented due to the modest sequencing volumes. Another likely possibility could be the late introduction of this variant in the country, after Alpha had already established its dominance and Delta had started instituting its presence. The competing presence of the two variants and the higher transmissibility rate of Delta had most likely impeded the spread of Beta. When compared to Alpha, the significant spike mutations L452R, T478K, D614G and P681R in Delta are responsible for increasing its transmissibility and risk of hospitalization (48–50). The RBD and ACE-2 interaction and infectivity appear to be enhanced by the L452R mutation (51). Moreover, the RBD-ACE-2 complex is stabilized by the T478K mutation along with L452R, which increases the virus infectivity. Furthermore, increased transmissibility and viral load have been linked to the P681R mutation, which is present at the cleavage point between S1 and S2 (52).
A SARS-CoV-2 variant’s high transmissibility and infectivity are typically attributed to spike protein alterations, while the rapid accumulation of mutations across non-structural genes is causing the virus to continuously evolve and change in pathogenicity (53). By grouping the mutations into haplotypes, we attempted to find an association with a signature set of both spike and non-spike constellation substitutions with the clinical manifestation of the disease. During 2020 the dominant circulating haplotype was haplotype 1 harboring the mutations N:K405*, nsp12:P323L, nsp12:M924R and nsp14:Y420stop. Haplotype 1A comprised the substitutions N:R203K, N:G204R, nsp12:P323L and S:D614G, however it was first detected in January 2021. Strains carrying the combination of mutations S:D614G and nsp12:P323L have taken over on a global scale, although the individual presence of these two mutations did not produce successful lineages (54). In 2021, due to the emergence of many new substitutons and diversification of the mutational landscape of the SARS-CoV-2 virus, several different haplotypes co-circulated in the same period. High percentage of hospitalized patients was observed in the haplotype 2C but without statistical significance (p=0.2618). Given that it emerged relatively late in the pandemic, in June 2020, the mutation N:D63G is very intriguing. The nucleoprotein N, which is the most abundant protein and is responsible for packaging the viral genome, is most likely to have had an impact on infectious virus titers (43). The most striking and statistically significant difference was observed among the haplotype 2C_1 cases, where 10.5% of the patients were hospitalized due to severe clinical condition (p=0.0013). While other studies have found an association of B.1.617.2 with higher odds of oxygen requirement, ICU admission, or death (28), our study found strong correlation of a signature set of mutations (haplotype 2C_1) with severe clinical outcome. Almost 70% of the patients with haplotype 2C_1 were male, corroborating our finding that males are more prone to severe clinical conditions and death. All cases belonging to the haplotype 2C_1 were hospitalized for at least three weeks, due to the severity of their condition and all had oxygen requirement, but none was put on mechanical ventilation. Significant variations across the ORF1ab, N region and the S protein caused the divergence of this haplotype from the prototype 21A Delta, which might be a consequence of selection pressure due to growing vaccination rates (53). One of the signature mutations of the haplotype 2C_1, S:A222V, emerged first in 2020 in the B.1.177 lineage. The genetic context in which the S:A222V mutation arises appears to be essential for its viability and the epidemiological implications of the mutation may be influenced by epistatic contacts between mutations, with S: T478K and S:L452R, also possibly modifying the behavior of the open RBD (55). Thus we can consider S:A222V as an example of a mutation that has occurred more than once in the SARS-CoV-2 spike protein but has not been accompanied by an improved epidemiological fitness. Predicting the success of these novel mutations in the population might be possible by tracing their interactions with the genetic background in which they appear. The only characterized breakthrough infection belonged to haplotype 2C_2 and it harbored a signature set of 7 co-appearing mutations and the same set of mutations was found in a small number of samples from India and Europe (Denmark) (53). Even though we had only one breakthrough infection in our study, we suspect that the real number has probably been higher. The vaccination rate for the total population during the Delta wave in North Macedonia was 40.8% for one dose, 35.1% for two doses and 0.5% for booster vaccinated individuals. A large meta-analysis showed that the effectiveness of the COVID-19 vaccines against the Delta variant was 86% (RR = 0.14, 95% CI: 0.07–0.54) (56). Moreover, booster-vaccinated individuals demonstrated a significant reduction in infection rates compared with non-booster-vaccinated subjects (57). The loss of immunity caused by the poor booster vaccination coverage is a plausible reason for the breakthrough infections in North Macedonia, as it has been demonstrated that protection against the Delta variant waned over time in vaccinated persons and that an additional vaccine dose restored protection (58). Additionally, it was found that vaccination provides 78% protection against infection by the Delta variant, 90% protection against hospitalization and 91% protection against death (59).In this context, the high number of hospitalizations and severe clinical condition, especially in the 2C_1 haplotype, may be due to the overall low vaccination coverage in the country.
One of the main limitations of the study is not knowing which fraction of the patients referred for routine testing, due to aggravated clinical symptoms, were subsequently hospitalized. Therefore, by not having an accurate estimate of the ‘tested-to-subsequently hospitalized’ patients, our analysis is based solely on the samples received from clinics. Another drawback is the small number of sequenced samples, especially in the first ten months of the pandemic. The lack of funds and small sequencing volumes overall, led to under representation of certain periods and variants during the course of the pandemic. Additionally, systematic collection and analysis of SARS-CoV-2 breakthrough infections, might have given more clarity on the genetic landscape and mutation enrichment of such cases.
In the fight against the COVID-19 pandemic, sequencing was used to complement epidemiological investigations, study viral transmission, discover and identify emerging variants and comprehend vaccine breakthrough infection. Thanks to the widespread availability of NGS tools and the online sharing of genome information, mutations, and variants are routinely tracked and a large number of full-length genome information from various countries affected by the pandemic are available.
Any prolonged circulation of SARS-CoV-2 bears the risk of the virus evolving to increased fitness with sets of mutations that confer greater transmissibility, immune evasion, or severity than previous variants. In addition to the spike mutations, the leading spike-recombinant vaccines may also be affected by the co-occurring mutations within non-spike proteins. Therefore, monitoring the SARS-CoV-2 mutations in samples in real-time can reveal their function in immune evasion potential or neutralization efficacy (60).
The present work is the first molecular study giving a comprehensive overview of the genetic landscape of circulating SARS-CoV-2 viruses in North Macedonia in a period of two years. This study provides a through insight in the evolution and diversity of circulating SARS-CoV-2 viruses and their relationships with the clinical manifestation of the disease.
Virus names as deposited to GISAID and are used for the phylogenetic analysis (http://www.gisaid.org). Accession numbers are provided in Supplementary Table 1. Further inquiries should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics committee of the Medical Faculty, University “Ss. Cyril and Methodius”, North Macedonia. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
MV, GB, SM, and IG contributed to the conception and design of the study. GK and DK organized the database. DK and MV performed the statistical analysis. MV, TB, EJ, AP, MS, and APr carried out the laboratory analyses. MV wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.1064882/full#supplementary-material
1. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
2. Mercatelli D, Giorgi FM. Geographic and genomic distribution of SARS-CoV-2 mutations. Front Microbiol (2020) 11. doi: 10.3389/fmicb.2020.01800
3. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol (2022) 23(1):3–20. doi: 10.1038/s41580-021-00418-x
4. Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun (2020) 11(1):1620. doi: 10.1038/s41467-020-15562-9
5. Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell (2020) 181(5):1036–45.e9. doi: 10.1016/j.cell.2020.04.026
6. Focosi D, Maggi F. Neutralising antibody escape of SARS-CoV-2 spike protein: risk assessment for antibody-based covid-19 therapeutics and vaccines. Rev Med Virol (2021) 31(6):e2231. doi: 10.1002/rmv.2231
7. Shah M, Woo HG. Omicron: A heavily mutated SARS-CoV-2 variant exhibits stronger binding to ACE2 and potently escapes approved COVID-19 therapeutic antibodies. Front Immunol (2022) 12. doi: 10.3389/fimmu.2021.830527
8. Udugama B, Kadhiresan P, Kozlowski HN, Malekjahani A, Osborne M, Li VY, et al. Diagnosing COVID-19: The disease and tools for detection. ACS Nano (2020) 14(4):3822–35. doi: 10.1021/acsnano.0c02624
9. World Health Organization. Real-time RT-PCR assays for the detection of SARS-CoV-2. (Paris: Institute Pasteur) (2021). Available at: https://www.who.int/docs/default-source/coronaviruse/real-time-rt-pcr-assays-for-the-detection-of-sars-cov-2-institut-pasteur-paris.pdf.
10. Foundation for Innovative New Diagnostics (FIND). FIND In-house test development for molecular detection of SARS-COV-2. Geneva, Switzerland 20212020. Available at: https://www.finddx.org/wp-content/uploads/2020/09/COVID-19-Testing-in-the-Laboratory-Sept2020.pdf.
11. ARTIC network. ARTIC SARS-CoV-2. (2021). Available at: https://artic.network/ncov-2019.
12. Quick J. ARTIC nCoV-2019 sequencing protocol V.1. (2021). Available at: https://www.protocols.io/view/ncov-2019-sequencing-protocol-bp2l6n26rgqe/v1.
13. Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, et al. Multiplex PCR method for MinION and illumina sequencing of zika and other virus genomes directly from clinical samples. Nat Protoc (2017) 12(6):1261–76. doi: 10.1038/nprot.2017.066
14. Mapleson D, Drou N, Swarbreck D. RAMPART: a workflow management system for de novo genome assembly. Bioinformatics (2015) 31(11):1824–6. doi: 10.1093/bioinformatics/btv056
15. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics (2018) 34(18):3094–100. doi: 10.1093/bioinformatics/bty191
16. Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods (2015) 12(8):733–5. doi: 10.1038/nmeth.3444
17. Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, Cardle L, et al. Using tablet for visual exploration of second-generation sequencing data. Briefi Bioinf (2013) 14(2):193–202. doi: 10.1093/bib/bbs012
18. Illumina. Illumina DNA prep with enrichment. (2021). Available at: https://emea.support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/illumina_prep/illumina-dna-prep-with-enrichment-reference-guide-1000000048041-07.pdf.
19. O’Flaherty BM, Li Y, Tao Y, Paden CR, Queen K, Zhang J, et al. Comprehensive viral enrichment enables sensitive respiratory virus genomic identification and analysis by next generation sequencing. Genome Res (2018) 28(6):869–77. doi: 10.1101/gr.226316.117
20. Nguyen J HR, Lee T, Prystajecky N, Tyson J. Rapid, high throughput library preparation of SARS-CoV2 using illumina’s DNA prep library preparation kit V.3. (2022). Available at: https://www.protocols.io/view/rapid-high-throughput-library-preparation-of-sars-b3vgqn3w.
21. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and clustal X version 2.0. Bioinformatics (2007) 23(21):2947–8. doi: 10.1093/bioinformatics/btm404
22. Kumar S, Nei M, Dudley J, Tamura K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Briefings Bioinf (2008) 9(4):299–306. doi: 10.1093/bib/bbn017
24. Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance (2017) 22(13):30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494
25. Chadha J, Khullar L, Mittal N. Facing the wrath of enigmatic mutations: A review on the emergence of severe acute respiratory syndrome coronavirus 2 variants amid coronavirus disease-19 pandemic. Environ Microbiol (2022) 24(6):2615–29. doi: 10.1111/1462-2920.15687
26. Morris CP, Luo CH, Amadi A, Schwartz M, Gallagher N, Ray SC, et al. An update on severe acute respiratory syndrome coronavirus 2 diversity in the US national capital region: Evolution of novel and variants of concern. Clin Infect Dis (2022) 74(8):1419–28. doi: 10.1093/cid/ciab636
27. Hakim MS, Wibawa H, Trisnawati I, Supriyati E, Khair RE, Iskandar K, et al. Association between prognostic factors and the outcomes of patients infected with SARS-CoV-2 harboring multiple spike protein mutations. Sci Rep (2021) 11(1):1–24.
28. Ong SWX, Chiew CJ, Ang LW, Mak T-M, Cui L, Toh MPH, et al. Clinical and virological features of SARS-CoV-2 variants of concern: A retrospective cohort study comparing b. 1.1. 7 (Alpha), b. 1.315 (Beta) and b. 1.617. 2 (Delta). Clin Infect Dis (2022) 75(1):e1128–e1136. doi: 10.1093/cid/ciab721
29. Morawiec E, Miklasińska-Majdanik M, Bratosiewicz-Wąsik J, Wojtyczka RD, Swolana D, Stolarek I, et al. From alpha to delta–genetic epidemiology of SARS-CoV-2 (hCoV-19) in southern Poland. Pathogens (2022) 11(7):780.
30. Chan WS, Lam YM, Law JHY, Chan TL, Ma ESK, Tang BSF. Geographical prevalence of SARS-CoV-2 variants, august 2020 to July 2021. Sci Rep (2022) 12(1):4704.
31. Basheer A, Zahoor I. Genomic epidemiology of SARS-CoV-2 divulge B.1, B.1.36 and B.1.1.7 as the most dominant lineages in first, second, and third wave of SARS-CoV-2 infections in Pakistan. Microorganisms (2021) 9(12):2609. doi: 10.3390/microorganisms9122609
32. Jin J-M, Bai P, He W, Wu F, Liu X-F, Han D-M, et al. Gender differences in patients with COVID-19: Focus on severity and mortality. Front Public Health (2020) 8.
33. Quaresima V, Scarpazza C, Sottini A, Fiorini C, Signorini S, Delmonte OM, et al. Sex differences in a cohort of COVID-19 Italian patients hospitalized during the first and second pandemic waves. Biol Sex Diff (2021) 12(1):45.
34. Finelli L, Gupta V, Petigara T, Yu K, Bauer KA, Puzniak LA. Mortality among US patients hospitalized with SARS-CoV-2 infection in 2020. JAMA Netw Open (2021) 4(4):e216556. doi: 10.1001/jamanetworkopen.2021.6556
35. Bonanad C, García-Blas S, Tarazona-Santabalbina F, Sanchis J, Bertomeu-González V, Fácila L, et al. The effect of age on mortality in patients with COVID-19: A meta-analysis with 611,583 subjects. J Am Med Directors Assoc (2020) 21(7):915–8. doi: 10.1016/j.jamda.2020.05.045
36. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
37. AstraZeneca. The natural evolution of SARS-CoV-2: How science responds to these challenges. (2021). Available at: https://www.astrazeneca.com/what-science-can-do/topics/covid-19/the-natural-evolution-of-sars-cov-2.html.
38. Flores-Alanis A, Cruz-Rangel A, Rodríguez-Gómez F, González J, Torres-Guerrero CA, Delgado G, et al. Molecular epidemiology surveillance of SARS-CoV-2: Mutations and genetic diversity one year after emerging. Pathogens (2021) 10(2):184. doi: 10.3390/pathogens10020184
39. Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Trans Med (2020) 18(1):1–9. doi: 10.1186/s12967-020-02344-6
40. Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol (2020) 17(8):881–3. doi: 10.1038/s41423-020-0485-9
41. Sharif N, Alzahrani KJ, Ahmed SN, Khan A, Banjer HJ, Alzahrani FM, et al. Genomic surveillance, evolution and global transmission of SARS-CoV-2 during 2019–2022. PloS One (2022) 17(8):e0271074. doi: 10.1371/journal.pone.0271074
42. Zhou B, Thao TTN, Hoffmann D, Taddeo A, Ebert N, Labroussaa F, et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature (2021) 592(7852):122–7. doi: 10.1038/s41586-021-03361-1
43. Wassenaar TM, Wanchai V, Buzard G, Ussery DW. The first three waves of the covid-19 pandemic hint at a limited genetic repertoire for SARS-CoV-2. FEMS Microbiol Rev (2022) 46(3):fuac003. doi: 10.1093/femsre/fuac003
44. Graham MS, Sudre CH, May A, Antonelli M, Murray B, Varsavsky T, et al. Changes in symptomatology, reinfection, and transmissibility associated with the SARS-CoV-2 variant b. 1.1. 7: An ecological study. Lancet Public Health (2021) 6(5):e335–e45.
45. Public Health England. Investigation of novel SARS-CoV-2 variant: variant of concern 202012/01, technical briefing 3 (2020). Available at: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/950823/Variant_of_Concern_VOC_202012_01_Technical_Briefing_3_-_England.pdfpdf.
46. Washington NL, Gangavarapu K, Zeller M, Bolze A, Cirulli ET, Schiabor Barrett KM, et al. Emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the united states. Cell (2021) 184(10):2587–94.e7. doi: 10.1016/j.cell.2021.03.052
47. Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Detection of a SARS-CoV-2 variant of concern in south Africa. Nature (2021) 592(7854):438–43. doi: 10.1038/s41586-021-03402-9
48. Galloway SE, Paul P, MacCannell DR, Johansson MA, Brooks JT, MacNeil A, et al. Emergence of SARS-CoV-2 b. 1.1. 7 lineage–united states, december 29, 2020–january 12, 2021. Morbid Mort Wkly Rep (2021) 70(3):95.
49. Yang W, Shaman J. Epidemiological characteristics of three SARS-CoV-2 variants of concern and implications for future COVID-19 pandemic outcomes. medRxiv (2021) 2021:2021.05.19.21257476. doi: 10.1101/2021.05.19.21257476
50. Wang P, Casner RG, Nair MS, Wang M, Yu J, Cerutti G, et al. Increased resistance of SARS-CoV-2 variant p. 1 to antibody neutralization. Cell Host Microbe (2021) 29(5):747–51.e4. doi: 10.1016/j.chom.2021.04.007
51. Kirola L. Genetic emergence of b. 1.617. 2 in COVID-19. New Microbes New Infect (2021) 43:100929. doi: 10.1016/j.nmni.2021.100929
52. Cherian S, Potdar V, Jadhav S, Yadav P, Gupta N, Das M, et al. SARS-CoV-2 spike mutations, L452R, T478K, E484Q and P681R, in the second wave of COVID-19 in maharashtra, India. Microorganisms (2021) 9(7):1542. doi: 10.3390/microorganisms9071542
53. Banerjee A, Mazumder A, Roy J, Majumdar A, Chatterjee A, Biswas NK, et al. Evolution of delta variant by non-spike signature co-appearing mutations: Trailblazer of COVID-19 disease outcome. bioRxiv (2022) 2022:2022.04.05.487103. doi: 10.1101/2022.04.05.487103
54. Ilmjärv S, Abdul F, Acosta-Gutiérrez S, Estarellas C, Galdadas I, Casimir M, et al. Concurrent mutations in RNA-dependent RNA polymerase and spike protein emerged as the epidemiologically most successful SARS-CoV-2 variant. Sci Rep (2021) 11(1):13705.
55. Ginex T, Marco-Marín C, Wieczór M, Mata CP, Krieger J, Ruiz-Rodriguez P, et al. The structural role of SARS-CoV-2 genetic background in the emergence and success of spike mutations: The case of the spike A222V mutation. PloS Pathogens (2022) 18(7):e1010631.
56. Mahumud RA, Ali MA, Kundu S, Rahman MA, Kamara JK, Renzaho AMN. Effectiveness of COVID-19 vaccines against delta variant (B.1.617.2): A meta-analysis. Vaccines (2022) 10(2):277.
57. Zhu Y, Liu S, Zhang D. Effectiveness of COVID-19 vaccine booster shot compared with non-booster: A meta-analysis. Vaccines (2022) 10(9):1396. doi: 10.3390/vaccines10091396
58. Goldberg Y, Mandel M, Bar-On YM, Bodenheimer O, Freedman LS, Ash N, et al. Protection and waning of natural and hybrid immunity to SARS-CoV-2. N Engl J Med (2022) 386(23):2201–12. doi: 10.1056/NEJMoa2118946
59. Scobie HM, Johnson AG, Suthar AB, Severson R, Alden NB, Balter S, et al. Monitoring incidence of COVID-19 cases, hospitalizations, and deaths, by vaccination status–13 US jurisdictions, April 4–July 17, 2021. Morbid Mortal Wkly Rep (2021) 70(37):1284.
60. Grant R, Charmet T, Schaeffer L, Galmiche S, Madec Y, Von Platen C, et al. Impact of SARS-CoV-2 delta variant on incubation, transmission settings and vaccine effectiveness: Results from a nationwide case-control study in France. Lancet Reg Health-Europe (2022) 13:100278. doi: 10.1016/j.lanepe.2021.100278
Keywords: SARS-CoV-2, variants, pandemic, genetic characterization, molecular evolution, NGS
Citation: Vukovikj M, Boshevska G, Janchevska E, Buzharova T, Preshova A, Simova M, Peshnacka A, Kocinski D, Kuzmanovska G, Memeti S and Gjorgoski I (2023) In-depth genetic characterization of the SARS-CoV-2 pandemic in a two-year frame in North Macedonia using second and third generation sequencing technologies. Front. Virol. 2:1064882. doi: 10.3389/fviro.2022.1064882
Received: 08 October 2022; Accepted: 23 November 2022;
Published: 04 January 2023.
Edited by:
Manish Chandra Choudhary, Brigham and Women’s Hospital and Harvard Medical School, United StatesReviewed by:
Mohamed Husen Munshi, National Cancer Institute at Frederick (NIH), United StatesCopyright © 2023 Vukovikj, Boshevska, Janchevska, Buzharova, Preshova, Simova, Peshnacka, Kocinski, Kuzmanovska, Memeti and Gjorgoski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maja Vukovikj, a3V6bWFub3Zza2FtYWphMUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.