 Theresa A. Reno1†
Theresa A. Reno1† Lilas Tarnus2†
Lilas Tarnus2† Russell Tracy3
Russell Tracy3 Alan L. Landay4
Alan L. Landay4 Irini Sereti5
Irini Sereti5 Cristian Apetrei2,6
Cristian Apetrei2,6 Ivona Pandrea1,6*
Ivona Pandrea1,6*- 1Department of Pathology, School of Medicine, University of Pittsburgh, Pittsburgh, PA, United States
- 2Division of Infectious Diseases, Department of Medicine, School of Medicine, University of Pittsburgh, Pittsburgh, PA, United States
- 3Department of Pathology and Laboratory Medicine, Larner College of Medicine, University of Vermont, Burlington, VT, United States
- 4Department of Internal Medicine, Rush University Medical Center, Chicago, IL, United States
- 5Human Immunodeficiency Virus Pathogenesis Section, Laboratory of Immunoregulation, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, United States
- 6Department of Infectious Diseases and Immunology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA, United States
Chronic, systemic T-cell immune activation and inflammation (IA/INFL) have been reported to be associated with disease progression in persons with HIV (PWH) since the inception of the AIDS pandemic. IA/INFL persist in PWH on antiretroviral therapy (ART), despite complete viral suppression and increases their susceptibility to serious non-AIDS events (SNAEs). Increased IA/INFL also occur during pathogenic SIV infections of macaques, while natural hosts of SIVs that control chronic IA/INFL do not progress to AIDS, despite having persistent high viral replication and severe acute CD4+ T-cell loss. Moreover, natural hosts of SIVs do not present with SNAEs. Multiple mechanisms drive HIV-associated IA/INFL, including the virus itself, persistent gut dysfunction, coinfections (CMV, HCV, HBV), proinflammatory lipids, ART toxicity, comorbidities, and behavioral factors (diet, smoking, and alcohol). Other mechanisms could also significantly contribute to IA/INFL during HIV/SIV infection, notably, a hypercoagulable state, characterized by elevated coagulation biomarkers, including D-dimer and tissue factor, which can accurately identify patients at risk for thromboembolic events and death. Coagulation biomarkers strongly correlate with INFL and predict the risk of SNAE-induced end-organ damage. Meanwhile, the complement system is also involved in the pathogenesis of HIV comorbidities. Despite prolonged viral suppression, PWH on ART have high plasma levels of C3a. HIV/SIV infections also trigger neutrophil extracellular traps (NETs) formation that contribute to the elimination of viral particles and infected CD4+ T-cells. However, as SIV infection progresses, generation of NETs can become excessive, fueling IA/INFL, destruction of multiple immune cells subsets, and microthrombotic events, contributing to further tissue damages and SNAEs. Tackling residual IA/INFL has the potential to improve the clinical course of HIV infection. Therefore, therapeutics targeting new pathways that can fuel IA/INFL such as hypercoagulation, complement activation and excessive formation of NETs might be beneficial for PWH and should be considered and evaluated.
Introduction
Antiretroviral therapy (ART) has increased the life expectancy and reduced morbidity and mortality for persons with HIV (PWH), thus becoming one of the most successful interventions of the twentieth century, that turned a virtually 100% deadly condition into a chronic disease (1). Yet, over the years, it became apparent that, despite successful suppression of HIV replication, PWH still experience higher rates of comorbidities and have a decreased survival compared to age-matched subjects (2). Even in cases presenting with complete, prolonged viral suppression, such as elite controllers or compliant PWH on ART with undetectable viremia for decades, the levels of T-cell activation and systemic inflammation remain higher than in HIV-uninfected individuals (3–5). Residual immune activation and inflammation lead to an incomplete immune restoration and a steady decline in the proficiency and functions of the immune system, similar to immunosenescence, i.e., progressive deterioration of the immune system by natural aging. As a result, in the recent years, a new paradigm of HIV pathogenesis emerged, in which persistent T-cell immune activation and inflammation are the key drivers of HIV pathogenesis and response to ART (6). The observations that CD8+ T cells from individuals with chronic HIV infection express high levels of CD38 and HLA-DR, indicating persistent, chronic, T-cell activation, and that these T cell activation levels are inversely correlated with CD4+ T cell counts, and are highly predictive for disease progression to AIDS were reported in the early days of the AIDS pandemic (7, 8). A fundamental role of T-cell immune activation for disease progression has also been suggested by studies in persons living with HIV-2 (PWH-2) and elite controllers (individuals naturally controlling their HIV-1 infection, i.e., with undetectable plasma viral loads in the absence of ART). In HIV-2 infection, the decline of circulating CD4+ T cell counts is slower than in HIV-1 infection, and patients develop AIDS after a median duration of incubation of 14 years of untreated infection, compared to only 6 years for HIV-1 (9). The levels of T-cell immune activation are lower in PWH-2 than in PWH-1 (10, 11). While the magnitude of T-cell immune activation is generally correlated with plasma viral load (12, 13), some PWH-2 with undetectable viremia present with persistent chronic immune activation (14). This indicates that viral replication is not the only source of chronic immune activation. Similar findings have been reported in elite controllers. Despite controlling viral replication, the levels of immune activation in these patients are higher than in uninfected subjects, albeit lower than in untreated HIV-1 progressors (15). The decline of CD4+ T cells was correlated to the levels of CD4+ and CD8+ T cell activation (16) and individuals without increased immune activation do not experience a CD4+ T cell decline over time (17). As the absence of detectable viremia does not entirely avert the CD4+ T cell decline, nor the disease progression to AIDS, this strengthens the belief that dysfunctional, persistent immune activation is a key player in disease progression in humans.
Non-human primate (NHP) models also contributed to this paradigm of T-cell immune activation and inflammation driving the outcome of HIV infection. SIV impact on the NHPs depends on species and nature of the infection (18). As Asian macaques are not natural hosts of SIV, lentiviral infections are pathogenic in those species. Similar to humans with untreated HIV infection, SIV-infected macaques have a severely damaged gastrointestinal tract, generalized immune dysfunction, persistent high levels of systemic immune activation and inflammation, and eventually progress to AIDS (19). In the models of HIV/SIV disease progression, dysregulation of the cell cycle (20), infection/dysfunction and loss of regulatory T cells (Tregs)/Th17 cells (21–23), dendritic cells (24–26), and/or B cells (27, 28) are all indicative of failure to control T-cell activation/proliferation and systemic inflammation, and contribute to poor clinical outcomes. Conversely, SIV infections of African NHP species, including the African green monkeys (AGMs), sooty mangabeys (SM) and mandrills, that are natural hosts of SIV, generally do not progress to AIDS, despite high levels of viral replication and massive acute CD4+ T cell depletion (29–32). The lack of disease progression in natural hosts likely rely on their ability to control gut damage (33, 34), and chronic systemic T-cell activation and systemic inflammation (30, 35, 36). This is supported by the fact that, over a 20-year follow-up of SIV infections in natural hosts, only a handful of cases of AIDS have been observed, and, in every case, disease progression was associated with increased chronic T cell immune activation and inflammation (37–40). Moreover, when inflammation and T-cell immune activation were experimentally induced in AGMs, they presented changes in biological parameters that are usually associated with disease progression (increased plasma viral loads, loss of mucosal CD4+ T cells and microbial translocation) (41–43).
Altogether, comparative pathogenesis studies in NHP species with different pathogenic outcomes of SIV infection decisively contributed to the current paradigm in which chronic inflammation and immune activation are key drivers of immunopathogenesis, progression to AIDS, and the HIV-associated comorbidities called residual immune dysregulation syndrome (RIDS) (44).
Previous studies have implicated multiple contributing factors as sources of the immune dysregulation characteristic to acute HIV infection (Figure 1). HIV virions can directly activate lymphocytes and macrophages, triggering the secretion of inflammatory cytokines and chemokines, including type 1 IFNs, IP-10, TNF, IL-6, and IL-8 (45, 46). The production and release of these cytokines can be beneficial, as they contribute to suppression of viral replication (47–49). For example, IFN-1 expression causes a signaling cascade that leads to the production of restriction factors and thus establishes an antiviral milieu which inhibits viral replication (50). While the production of IFN-1 and other cytokines mediates the stimulation of innate and adaptive immune cell subsets that will fight the virus, the profound impact of these proinflammatory factors on the immune response can also be detrimental (51, 52), as demonstrated by experimental interventions in NHPs (53). IFN-1 also induces CD4+ T-cell loss by modulating the expression of TNF-related apoptosis-inducing ligand (TRAIL), which then binds to Death Receptor 5 (DR5) leading to apoptosis (53–56). T cell apoptosis during acute infection is also triggered by TNF binding to its cellular receptors (57, 58). Apoptosis of infected CD4+ T cells can initially curb viral propagation, yet uninfected T cells are not spared from destruction, which contributes to the massive T-cell loss observed during acute HIV infection. Consistent with observations in humans, in both non-pathogenic and pathogenic acute SIV infections, type 1 IFNs are highly upregulated through the activation of pDCs (59–62). The upregulation of IFN-1 leads to an increased expression of IFN-1-stimulated genes (ISG) that persists during chronic infection in pathogenic HIV/SIV infections but returns to baseline in natural hosts after a transient increase during acute infection (63, 64). TNFα enhancement by HIV proteins further fuels HIV replication in infected cells (65, 66). Meanwhile, the non-pathogenic SIV infection of AGMs causes induction of Forkhead Box P3 (FoxP3) (a transcription factor necessary for the development of Tregs), TGF-β, and IL-10, all of which contribute to the establishment of an anti-inflammatory environment (67). The maintenance of this anti-inflammatory milieu is enabled by the absence of persistent IFN expression (67). On the other hand, the pathogenic infection of macaques, associate a delayed IL-10 expression and only a slight increase in TGF-β levels, similarly to acute HIV infection (67). The late increase in IL-10 allows for inflammatory cytokines to go unchecked and may lead to the exhaustion of SIV/HIV-specific CD8+ T cells (68, 69).
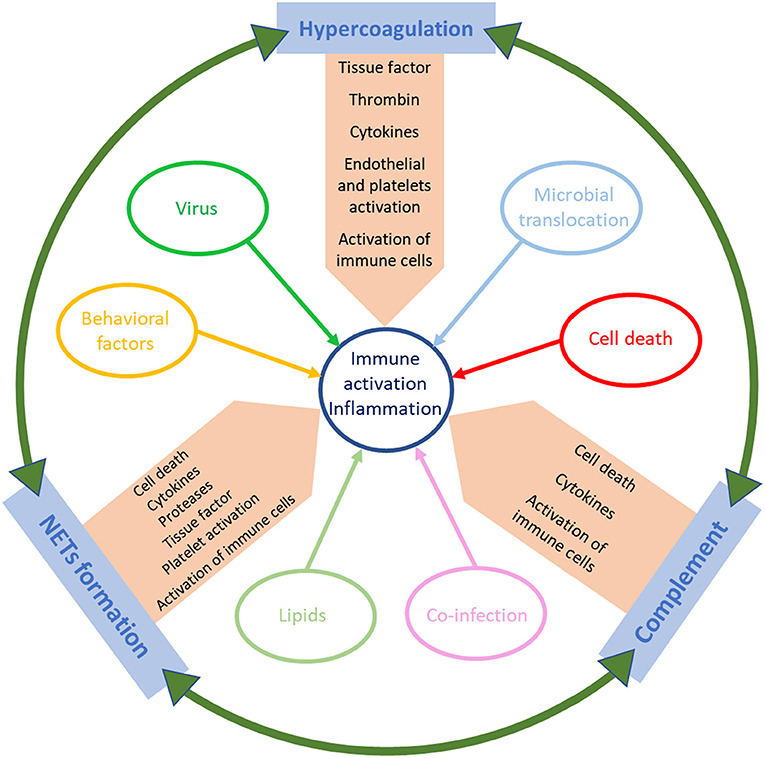
Figure 1. Schematic representation of the factors that trigger immune activation and inflammation in HIV/SIV infection.
As the virus begins to swiftly replicate, it disseminates to the lymphoid tissues throughout the body. One key location of the virus is the gut-associated lymphoid tissue (GALT). During acute infection, CCR5+ CD4+ memory T cells are depleted in the GI tract. Inflammatory cells, including neutrophils, monocytes, and dendritic cells, accumulate in the mucosa, while Th17 cells, the CD4+ T cells that regulate epithelial homeostasis, are lost, creating an environment prone to alterations of the mucosal integrity (70–72). The damaged epithelial mucosa allows microbial translocation from the gut lumen first into lamina propria and then into the systemic circulation (73). Natural hosts of SIVs do not exhibit microbial translocation because they have the ability to maintain the mucosal integrity by rapidly repairing the damages (33, 74) inflicted to their mucosal barrier by the SIV infection (34). In SIVsab-infected AGMs, administration of lipopolysaccharide (LPS) (41), to model microbial translocation seen in PWH, resulted in increased systemic immune activation and inflammation, leading to a rise in viremia. The same was true when gut damage was directly induced through administration of dextran sulfate sodium (DSS) in chronically infected AGMs (43). These experiments directly probed the role of the gut damage in inducing immune activation and inflammation. The loss of mucosal integrity and subsequent microbial translocation in humans and pathogenic NHP models trigger inflammation and T-cell immune activation by enhancing the activity of pattern recognition receptors which normally recognize molecular patterns associated to foreign pathogens (75). The activation of the innate immune system unleashes a multitude of immune cells that trigger the production of inflammatory cytokines, including IFN-α, IFN-β, IL-6, and TNF-α (76, 77). Another consequence of microbial translocation is an increase in circulating levels of sCD14, a marker of monocyte activation and LPS bioactivity, which has been shown to predict mortality, and of the scavenger receptor sCD163, the levels of which are associated with unstable non-calcified coronary plaques (18, 70, 78–80). Increased plasma levels of the translocated fungal polysaccharide (1 → 3)-β-D-Glucan (βDG) are also associated with immune activation (81). Microbial translocation is not the only way in which the gut microbiome contributes to the systemic immune activation, the composition of the gut microbiome can alter the degree of activation of monocytes and T cells (75). Furthermore, the composition of the translocated microbiome can impact the immune reconstitution in ART-treated PWH (82). Interventions aiming to introduce gut dysbiosis, e.g., through prolonged antibiotic administration, were not sufficient to lead to disease progression (83).
Meanwhile, microbiome alterations through antibiotics, such as cotrimoxazole and a combination of rifaximin and sulfasalazine, resulted in a transient reduction in systemic immune activation and inflammation (84, 85).
An additional contributor to the SNAEs may be the altered lipid profiles in PWH, which can also contribute to immune activation and inflammation (86). The SMART study established that varying sizes of HDL particles, but not other lipoproteins, were associated with an increased risk of cardiovascular disease (CVD) events. Meanwhile, the levels of the inflammatory and coagulation markers CRP, D-dimer, IL-6 and sCD14 could predict mortality from CVD in PWH (78, 87). Furthermore, the oxidation levels of HDL and LDL are modified in PWH, and HDLox levels positively correlate with the increases in IL-6 and sCD163, contributing to persistent immune activation for patients on ART (88) who are at an increased risk of serious non-AIDS events (SNAEs), including CVD (5). Administration of a diet rich in saturated fats and cholesterol to SIV-infected NHPs triggered immune activation and inflammation, thus confirming the importance of lipids as inductors of inflammation. In the NHPs fed with high fat diet, the increases in immune activation associated elevated levels of other cardiovascular biomarkers and a high incidence of cardiovascular lesions. Administration of the unhealthy diet also accelerated disease progression in the SIV-infected macaques and led to disease progression in one SIV-infected African green monkey (89).
Chronic coinfection with other viruses, notably human cytomegalovirus (hCMV), but also hepatitis B (HBV) and hepatitis C (HCV) viruses, can exacerbate HIV pathogenesis (90, 91). Exhaustion of virus-specific CD8+ T cell populations that regulate viral replication results in the reactivation of latent viruses and allows their active replication, boosting in turn systemic immune activation (92, 93). For example, coinfection with hCMV is associated with increased inflammatory and coagulation markers, including sCD14, IFN-γ-induced protein (IP-10), and D-dimer (94, 95). In PWH, chronic hCMV coinfection also exacerbates immunosenescence and the severity of CD4+ T cell depletion due to progressive thymic dysfunction (96). Individuals on ART still experience asymptomatic hCMV shedding, which triggers T-cell activation, proliferation, and exhaustion (97). Furthermore, an increased number of CD4+ T cells from the HIV/hCMV-coinfected subjects express the cellular senescence marker CD57, compared to individuals infected with either HIV or hCMV only (98). The increased pool of dysfunctional CD4+ and CD8+ T cells is associated with a loss of the effective control of hCMV and triggers a reversal of the CD4+/CD8+ ratio leading to an increased risk of morbidity and mortality (99, 100). This pathologic feature also occurs during the HIV/HCV coinfection, through the loss of CD4+ T cells and alterations of the Kupffer cells, the macrophage population resident in the liver, leading to elevated levels of circulating LPS and sCD14 (101–103). The loss of HCV-specific CD4+ T cells also results in an inadequate HCV control and reactivation (104, 105), which creates a positive feedback loop of viral replication fueling immune activation (102). Patients with HIV/HCV coinfection who received direct-acting antiviral agents for HCV eradication had significant decreases in the markers of immune activation and of increased HIV mortality: HIV DNA, sCD14, LPS, and D-dimers, indicating that coinfection suppression or eradication indeed help curb immune activation and inflammation (106).
As stated previously, ART is not able to completely reverse immune activation and inflammation, and low-level immune stimulation persists despite undetectable viremia, highlighting the importance of identifying and therapeutically targeting different mechanisms causing immune activation. In the following sections, we will focus on the role of additional factors that can fuel residual immune activation and inflammation under ART (Figure 1). We will thus discuss the relative contribution of hypercoagulation, complement pathway activation, and NET formation in driving systemic immune activation and inflammation in PWH and how can these pathways be therapeutically targeted to reduce SNAEs.
Hypercoagulation
The coagulation cascade is composed of a series of rapid and sequential enzymatic reactions leading to the generation of thrombin and eventually a fibrin clot (107). Among the positive and negative regulators of the coagulation cascade are immune activation and inflammation, the levels of which can trigger coagulation (108). The interplay between immune activation, inflammation and coagulation is critical for infection control, as the activation of coagulation can restrict pathogens movement, thus helping pathogen elimination (109). While this is beneficial for controlling infection, persistent inflammation can also trigger thrombosis and may result in blood vessel occlusion, causing pathologies, including peripheral thrombosis, pulmonary embolism, myocardial infarction, or stroke.
Contributing to the increased risk of thrombosis and CVD are deficiencies in proteins C and S, which hinder the inactivation of factors Va and VIIIa, and promote a procoagulant state (110, 111). Though immune activation and inflammation can initiate a procoagulant milieu, this interaction is now seen as a feedback loop where each can trigger the other (87, 112, 113) (Figure 2). This may be the reason for which the prevalence of CVDs and venous thromboembolism (VTE) is increased in untreated PWH and it is also not entirely abated by ART even when HIV replication is completely suppressed (114–116).
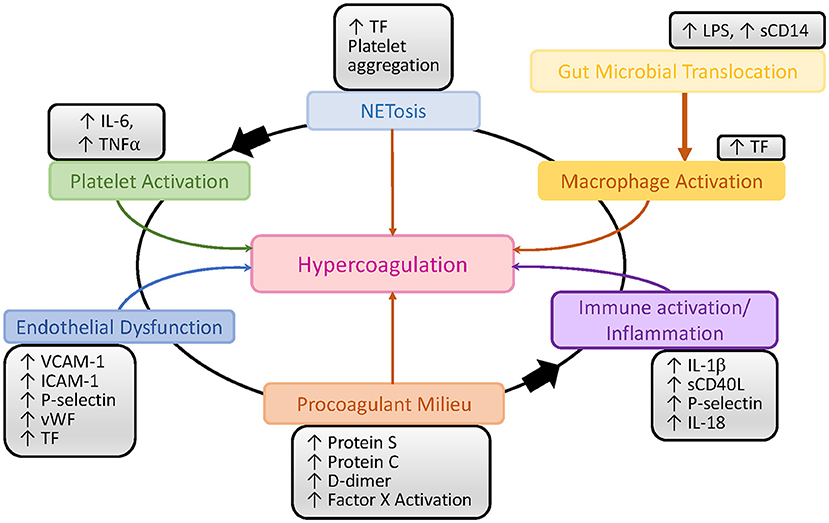
Figure 2. Schematic representation of the interactions between inflammation, immune activation and the coagulation pathways in HIV/SIV infection. LPS, lipopolysaccharide; TF, tissue factor; vWF, von Willebrand factor.
The altered coagulation associated with HIV infection was confirmed in NHPs (42), in which comparative studies of progressive and non-progressive SIV infections clearly demonstrated that the hypercoagulable state is specifically associated with the pathogenic, progressive SIV infections (42), providing strong validation of the SMART study data (87) and demonstrating the usefulness of animal models for the study of HIV-related comorbidities (117).
D-Dimer
One of the best biomarkers to assess the hypercoagulable state and the risk of death in the PWH and SIV-infected monkeys is D-dimer (42, 43, 87, 118–120), a byproduct of fibrin cleavage by plasmin. D-dimer plasma levels correlate with the total thrombolytic activity. The levels of D-dimer in the PWH included in the SMART cohort were 94% higher than in uninfected donors recruited in the MESA cohort (121). Two independent retrospective case control studies further confirmed that the increased levels of D-dimer in PWH were associated with CVD and VTE (122, 123). The elevation of D-dimer is not entirely correlated with plasma viral load, as virologically suppressed PWH still present increased levels of D-dimer (124). Further studies have confirmed that D-dimer levels were reduced but not normalized in ART-treated patients with undetectable viremia, and that higher levels of D-dimer in those patients were associated with an increased risk of SNAEs (87, 125, 126). Indeed, the frequency of myocardial infarction in PWH is significantly higher than in their non-infected peers (127). High D-dimer levels in both PWH and their uninfected counterparts have been shown to lead to immune activation and inflammation through the increase in cytokines, such as IL-6 (128, 129). Finally, high D-dimer levels are associated with immune reconstitution inflammatory syndrome (IRIS) and death, even after adjusting for other factors (130). The importance of D-dimer has also been shown in pathogenic SIV infections (42, 89, 117, 120), in which D-dimer increased early after infection (Figure 3) and correlated with immune activation and inflammation. Furthermore, in SIV-infected pigtailed macaques (PTM), the increases in D-dimer levels are associated with the presence of microthrombi in tissues (Figure 3). These microthrombi may promote hypoxia and tissue fibrosis (Figure 3) and thus significantly contribute to the lack of immune reconstitution and to the development of numerous comorbidities. This may provide a new mechanism responsible for the development of concomitant comorbidities and of end stage organ diseases in PWH. Conversely, non-progressive SIV infections of the natural hosts, in which D-dimer levels remain at the preinfection levels during chronic infection, do not develop CVD or thrombotic lesions (42, 131, 132).
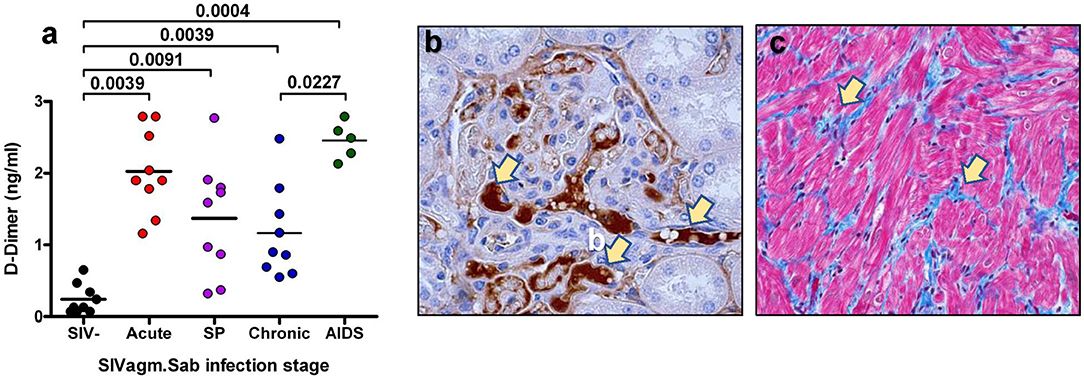
Figure 3. Coagulation disorders during SIV infection. (a) Changes in the levels of D-Dimer assessed at critical time points of SIVsab infection in pigtailed macaques. SP indicates set point. P-values were calculated with the paired t-test. (b) Thrombotic microangiopathy (TMA) during the pathogenic SIVsab infection of PTMs. Immunohistochemistry for fibrinogen (brown) in the kidney. (c) Fibrosis of the heart characterized by increased diffuse deposition of collagen (blue) replacing drop-out or lysed myocytes. Trichrome staining. The original magnification of all pictures was 400x. Reproduced with authorization from Pandrea et al. (42).
These data show the importance of D-dimer levels as a biomarker of morbidity and mortality in PWH, and underline the strong relationship between coagulation and inflammation.
Tissue Factor
TF initiates the extrinsic clotting pathway of the coagulation cascade (Figure 2), playing a pivotal role in thrombosis (133). TF is expressed on the surface of monocytes in PWH (134) and SIV-infected NHPs (135), and it is thought to be at least partly induced by the inflammation triggered by microbial translocation (134, 135) (Figure 2). Increased expression of TF on the surface of monocytes likely contributes to the atherosclerotic plaques that lead to CVD in PWH and to increased clot formation and thrombosis. These TF-expressing monocytes persist even when PWH are virologically suppressed with ART, and they can trigger both coagulation via the activation of factor X (135) and production of inflammatory cytokines, including interleukin-1β, IL-6, and TNFα. TF has also been found to be expressed on CD8+ and CD4+ T cells during HIV infection and its levels correlated with those of the T-cell immune activation marker CD38 (136). Similar findings have been observed in NHP models: in SIV-infected macaques, TF-expressing monocytes were crucial to the development of coagulopathy and its associated proinflammatory environment (135). Recently, we showed that in SIV-infected PTMs, TF is also expressed by neutrophils that release NETs and by the NETs themselves, predisposing these animals to pathological clotting (137) (Figure 4). Altogether these results place TF at the intersection between coagulation and inflammation, suggesting that TF inhibition may represent a valuable therapeutic strategy to reduce the risk of comorbidities in PWH (138).
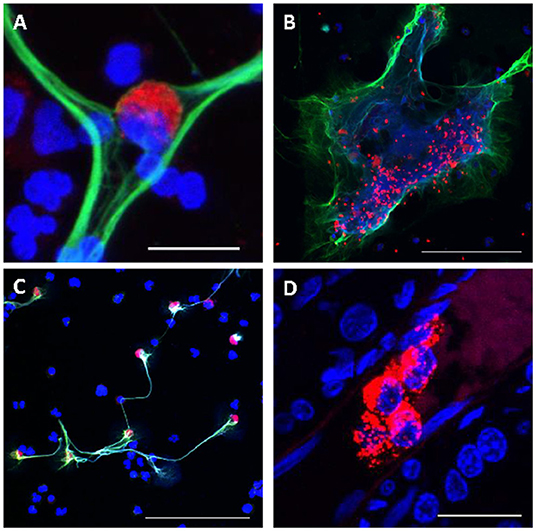
Figure 4. Consequences of Excessive NETosis in SIV-infected nonhuman primates. (A) Monocyte (red) trapped in a NET (green). (B) Trapping of aggregated platelets (red) in a NET (green). (C) Increased expression of TF (red) on polymononuclear neutrophils forming NETs (green), while others polymononuclear neutrophils do not express TF. (D) Occlusion of small blood vessels by NETs (red) in the kidney of a SIV-infected PTM. Nuclear staining was performed with DAPI (blue) for all figures. Scale bar lengths: 20 μm (A,D), 100 μm (B,C). NET, neutrophil extracellular trap; PTM, pig-tailed macaque; TF: Tissue factor. Reproduced with authorization from Sivanandham et al. (137).
Endothelial Dysfunction
Endothelium integrity is quintessential to hemostasis and its disruption leads to an inflammatory response that can promote VTEs, including platelet aggregation on the exposed subendothelial structures and clotting activation (139). HIV proteins can also cause endothelial dysfunction by reducing antioxidants through limiting nitric oxide bioavailability and increasing cell permeability (140). The association between HIV and endothelial dysfunction was reported early in the epidemic and has been confirmed in multiple studies which reported increases in VCAM-1, ICAM-1, P-selectin, sTF, and Von Willebrand factor in chronically HIV-infected individuals (140–142) (Figure 2). Studies have reported a short-term improvement of the endothelial dysfunction after initiation of ART (143, 144), while others reported that prolonged exposure to certain antiretrovirals has a detrimental effect on the integrity of the endothelium, leading to CVD and VTEs (145, 146).
Platelets
Platelets also play a significant role in HIV-induced hypercoagulation (Figures 2, 4). Platelet aggregation can limit pathogens access to the vascular system and they can also directly interact with pathogens and alter the inflammatory processes triggered by infection (147). Several studies have confirmed the interactions between platelets and HIV and have shown that these interactions lead to virus internalization and platelet activation which, in turn, can propagate the virus and lead to a thrombocytopenia (148, 149). Another plausible reason for the paradoxical thrombocytopenia associated with the SIV-induced hypercoagulable state is platelets being trapped and destroyed in NETs that are excessively released by neutrophils during SIV infection (137) (Figure 4). Platelet activation contributes to the inflammatory environment seen in PWH, which is characterized by increases in IL-1β, sCD40L, P-selectin, and IL-18 (150). Despite undetectable viremia, ART-treated PWH continue to present increased platelet activation and spreading (i.e., the process of platelet flattening at sites of vascular injury to increase their contact area) (151). Meanwhile, platelets from PWH also experience apoptosis and mitochondrial dysfunction (151), suggesting that monitoring platelet functions might be clinically relevant in PWH to assess their overall risk for VTE and CVD (152).
Targeting Hypercoagulation in PWH as an Adjunct Intervention to Control Immune Activation and Inflammation
Attempts have been made to reduce hypercoagulation in PWH. (i) The use of warfarin in PWH is limited by many drug-drug interactions that result in changes in warfarin metabolism, notably by inhibiting cytochrome P450 3A4, as well as the need to monitor INR levels through regular blood tests (153, 154). Therefore, its use has decreased sharply over the recent years in favor of direct oral anticoagulants (DOACs). (ii) Aspirin is one of the most widely used and well-studied anticoagulants that reduces platelet activity and aggregation. Aspirin also significantly reduced platelet activation and platelet hyperactivity in PWH (155). In addition, aspirin showed beneficial effects in reducing the levels of T-cell activation (CD38 and HLA-DR), monocyte activation (sCD14), platelet activation (P-selectin), and responsiveness of leukocytes to TLR agonist stimulation after only 1 week of treatment (156). Yet, no significant impact of aspirin on VTEs and inflammation has been reported in PWH (157, 158). Aspirin was reported to be less effective for the primary prevention of myocardial infarction in PWH compared to the normal population (159). It has also been shown that the risk of a recurrent acute coronary event was 6.5 times higher in ART-treated PWH than in matched, uninfected patients (160). These studies suggest that the current strategies for primary and secondary cardiovascular disease prevention in PWH should be tailored to this patient population, to include more potent anticoagulants, as well as other therapeutic targets, notably regulating lipids levels. Important insights could be obtained through the large, ongoing REPRIEVE clinical trial that evaluates the efficacy of statins for the primary prevention of cardiovascular events in PWH. This cohort confirmed the high incidence of coronary atherosclerosis in PWH on ART, the presence of plaques being associated with increased immune activation (161). (iii) Angiotensin receptor blockers have also been evaluated in PWH, to assess their potential benefits in reducing endothelial injury. After a 3-month prospective study, telmisartan administration led to an increase in endothelial progenitor cells, but had no effect on immune activation (162–164). (iv) Ixolaris, a TF inhibitor that blocks the extrinsic coagulation pathway at its origin, is a promising candidate for the treatment of hypercoagulation in PWH. In chronically SIV-infected pigtailed macaques, Ixolaris decreased D-dimer expression and TF activity, and reduced monocyte activation (135). Inflammation was not impacted in this study, except for IL-17, which was slightly reduced by Ixolaris administration. (v) Several studies assessed the benefits of DOAC administration in PWH. A longitudinal cohort study assessed DOAC safety among PWH who were undergoing ART (165) and concluded that there were similar effects compared to those taking warfarin, i.e., that these patients should be monitored as drug-drug interactions are likely, especially when DOACs are coadministered with protease inhibitors. This can lead to an increased risk of bleeding. Meanwhile, association with non-nucleoside reverse transcriptase inhibitors (NNRTIs), such as nevirapine and etravirine, can lead to reduced plasma DOAC levels and increase the risk of VTE (166). When dabigatran, a direct thrombin inhibitor, was administered to SIV-infected pigtailed macaques, it did not normalize coagulation parameters and resulted in a cardiovascular event in the study group. Edoxaban, a factor Xa inhibitor, significantly decreased the D-Dimer and TAT levels among PWH, but did not impact inflammation (167). Vorapaxar, a PAR-1 inhibitor did not impact HIV-related hypercoagulation, in PWH, as shown by the lack of effect on D-Dimer levels (168). In SIV-infected pigtailed macaques, Vorapaxar administration had only a modest impact on T-cell immune activation and did not normalize coagulation, nor did it impact disease progression.
In summary, the anticoagulant interventions performed so far resulted in some improvements in the levels of T-cell activation but did not normalize the inflammation biomarkers that are routinely measured in clinical trials to predict an improved clinical outcome.
Interventions aimed at blocking the coagulation cascade upstream seem to be more efficient than interventions targeting the extrinsic or common coagulation pathways. It can be hypothesized that, in HIV/SIV infections, once several coagulation factors have been activated and feedback loops are established between different pathways of the coagulation cascade, it is extremely difficult to counter the hypercoagulable state and its related inflammation.
HIV/SIV-Induced Complement Pathway Activation
The complement system is a key effector of the innate immune system, which is also involved in the adaptive immune responses. It is composed of over 30 proteins, interacting in three pathways: classical, alternative and lectin pathways, all converging toward the production of C3 convertases that cleave C3 into C3a and C3b (Figure 5). The terminal, lytic phase is common to all pathways and is initiated by the C5 convertase that severs C5 into C5a and C5b. The latter combines with C6, C7, C8, and C9 to form the membrane attack complex (MAC) (Figure 5).
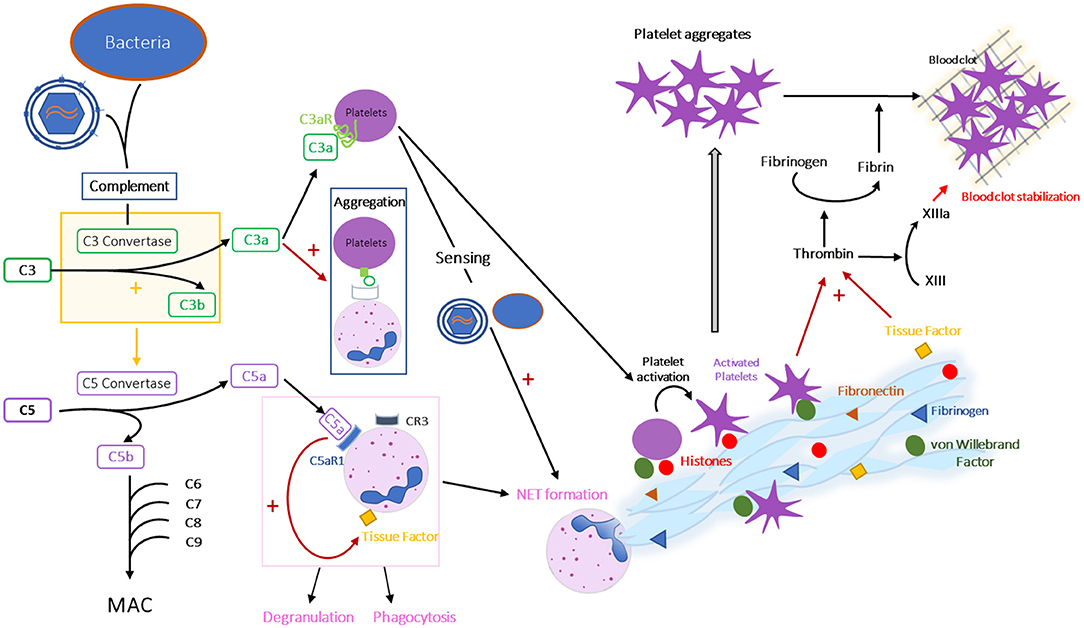
Figure 5. Interplay between complement, coagulation and formation of NETs. Schematic representation of the interactions between complement, coagulation and formation of NETs during HIV/SIV infection. CR, complement receptor; C3aR, C3a receptor; C5aR1, C5a receptor 1 or CD88.
Complement activation fights microorganisms through multiple effector functions: (i) MAC is inserted into the membrane of the pathogen or infected cell that has triggered complement activation, leading to the formation of pores in the membrane and causing osmotic lysis; (ii) C3b binds to microorganisms insuring their opsonization and phagocytosis; (iii) C3a, C4a, and C5a induce modifications in vascular permeability, facilitate and attract immune cell subsets that fight against the microorganisms and induce histamine release from mast cells (169).
Two types of regulators of complement activation (RCA) are described, soluble factors and membrane-bound regulators. The later play an important role in clearing immune complexes from circulation (e.g., CR1) and protecting host cells from the deleterious consequences of complement activation on self-cells (170). Those membrane-bound RCA include CD46, the membrane cofactor protein (MCP), a cofactor for complement factor I, which degrades C3b and C4b; CD55, the decay-accelerating factor (DAF), which accelerates the degradation of C3 and C5 convertases; and protectin (CD59), which limits C9 incorporation and thus MAC formation (170).
There are numerous triggers of the classic pathway, notably antibody-antigen complexes, apoptotic cells, C-reactive protein, beta-amyloid, and LPS (171, 172). The lectin pathway is activated by carbohydrates structures, such as oligosaccharides on bacterial membranes, as well as IgA or IgM (173, 174). The alternative pathway is activated by molecules expressed on non-self surfaces (microorganisms) or self surfaces (apoptotic and necrotic cells) (172). The alternative pathway is usually spontaneously activated at low levels, but can be further amplified after the activation of the classical pathway through interactions via C3b.
In HIV-1 infection, complement activation can be triggered through multiple mechanisms: (i) activation of classical pathway by C1q binding to anti-HIV antibodies (175) and C1s binding to gp41 (176); (ii) activation of lectin pathway through gp120 recognition by the mannose binding lectin (MBL) (177); and (iii) an amplification loop through the alternative pathway can also boost this complement activation. Furthermore, the microbial translocation observed during pathogenic HIV/SIV infection can lead to the activation of all complement pathways: (i) the lectin pathway via the recognition of Pathogen Associated Molecular Patterns (PAMPs), (ii) the alternative pathway via LPS, (iii) the classic pathway, which is triggered by the inflammation through LPS and the production of CRP, that can bind to C1q (178).
Through these multiple pathways, increased activation of the complement system occurs in PWH, as indicated by the increased levels of the complement cleavage products (179). Interestingly, complement activation increases with disease progression to AIDS, as well as in PWH developing IRIS (180–183).
While lower levels of the classic pathway activation were observed in PWH initiating ART, as emphasized by a modest increase in classic total complement activity and a 30% increase in intact C3 and C4 proteins after 12 weeks of ART, the levels of factor B (a factor involved in the formation of the alternate C3 convertase) are not completely restored on ART, suggesting that the alternative pathway is still activated (184). The residual complement activation in virologically-suppressed PWH was also observed in other studies reporting a 64% increase in C3a levels (185) and a 9% increase in soluble MAC (186), compared to uninfected subjects. Note that the levels of soluble MAC were particularly increased in virologically suppressed subjects with CD4+ T cell counts below 200/mm3 (186). Furthermore, while the levels of mannose-binding lectin increased in untreated PWH, they did not decrease over a 2-year period of ART (187). This residual complement activation may have multiple sources: (i) limited, but persistent viral replication in blood and/or lymphoid tissues leading to the activation of the classical and lectin pathways by gp41 and gp120 binding to C1s and MBL, respectively, as well as by anti-HIV antibodies triggering classic pathway, and (ii) residual microbial translocation causing an activation of all pathways (188).
Yet, despite this steady complement system activation, the complement-mediated HIV/SIV virolysis is limited (189). This high resistance of HIV and SIV to complement-mediated virolysis is due to the presence on viral membranes of membrane-bound RCA, such as CD46, CD55, and CD59, that are acquired during the budding from the cellular membrane (189–191). In addition to this viral escape mechanism, HIV and SIV can also hijack complement-induced opsonization. Immune cells recognize opsonized viruses through their FcR or complement receptors (CRs), and HIV and SIV can either infect these cells (192, 193) or use them as vehicles for transport to the germinal centers of lymphoid tissues (194). For example, follicular dendritic cells (FDC) exhibit CR1, CR2, and CR3, allowing them to bind opsonized viruses. In the follicles, viruses can either infect nearby T lymphocytes, or remain trapped by FDC, enabling affinity maturation of B cells. However, when trapped by FDC, viruses remain intact and infectious for months, constituting one of the most important viral reservoirs in the organism (195). Infection of the T CD4+ lymphocytes and viral trapping in the lymphoid tissues fuel immune activation in PWH and lead to histological alterations of the lymphoid tissues (196, 197). Intriguingly, while the pathogenic HIV and SIVmac are trapped in germinal centers of humans and macaques, respectively, this phenomenon is less frequent in non-pathogenic SIV infections of the natural African NHP hosts (32, 198–201), indicating that deposition of complement-opsonized viral particles in the lymphoid tissues might be critical for disease progression.
Complement may also impact HIV pathogenesis by fueling immune activation and inflammation. C3a is a proinflammatory factor that attracts macrophages, monocytes and mast cells, triggers histamine release by mast cells, induces the production of proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α by monocytes and macrophages, induces eosinophil degranulation, prompts platelet activation, and increases vascular permeability (202). C3a is also an immunomodulatory agent as it inhibits neutrophil mobilization (203). C5a is a potent inductor of inflammation after binding to its receptors, C5aR1 (CD88) or C5aR2. The inflammatory effects of C5a include, but are not limited to, recruitment of the immune effectors (macrophages, monocytes, dendritic cells, neutrophils, lymphocytes), activation of neutrophils, which result in enhanced production of the inflammatory cytokines IL-6 and TNF-α, increases in vascular permeability and tissue factor expression on endothelial cells, and suppression of Treg. Interestingly, CRP, IL-6, and TNF-α, whose expressions are increased during complement activation, have all been linked to disease progression and comorbidities in PWH (87, 126, 204–206). Thus, it is not surprising that complement activation itself has been linked to different comorbidities, notably neurological, kidney, and cardiovascular diseases, both in PWH and in the general population.
Complement and Neurological Comorbidities
HIV-associated neurocognitive disorders (HAND) have been described since the dawn of the AIDS pandemic (207). HAND aggregates wide clinical presentations ranging from HIV-related dementia, mostly characteristic to the pre-ART era, to milder neurocognitive (NC) impairments (208). With the advent of ART, incidence of HIV-related dementia has dramatically declined, but 30–50% of PWH still present NC disorders (209), with mild NC impairments now accounting for most NC diseases among PWH (210). There are multiple mechanisms through which HIV can induce neurocognitive disorders: neurotoxicity of viral proteins produced by infected cells (macrophages or glial cells, including astrocytes) (211), death of the productively-infected cells (212), brain damage (edema, weakening of the blood-brain barrier) induced by the immune and/or inflammatory responses (213), and ART neurotoxicity (214). These mechanisms are non-exclusive. Complement could also play a role in HIV neurotoxicity, as it leads to apoptosis and increased production of inflammatory cytokines. This is not unexpected as complement is involved in synapse pruning via C1q during brain development (215). Furthermore, complement might also play a role in some autoimmune neurological diseases, Alzheimer's disease, and multiple sclerosis (216).
In PWH that progress to AIDS, complement activation can be observed in the cerebrospinal fluid (CSF), with increased levels of C4 indicative of intrathecal synthesis (217). While most complement proteins are synthetized by hepatocytes, they can also be produced by other cells, notably astrocytes (218). Increased levels of C3 and C4 have been found in the CSF of PWH with NC dysfunction (219), and HIV-1 infection induces in vitro expression of C2 and C3 in infected-astrocytes (220, 221). This effect was observed in cell cultures treated with both whole virions and after incubation with only viral proteins gp41 or Nef (221, 222). HIV-1 also induces C3 expression in neurons (221, 222). In macaques with SIV encephalitis, high C3 and C1q expression on astrocytes and neurons was immunohistochemically detected, while the same cells were rarely positive in uninfected animals (223). Interestingly, C1q expression increased throughout the course of SIV infection and was correlated to CSF viral load (224), a marker of HIV-related dementia (225). Overexpression of C3 was confirmed in postmortem brain tissues from patients with HAND (226). Upregulation of the genes involved in the interferon responses and the complement pathway was higher in PWH with NC impairment and HIV encephalitis compared to those without NC impairment (227). Interestingly, a longitudinal study of PWH with NC impairment found that patients with improved neurocognition presented a down-regulation of the complement system, and vice versa (228). Another potential explanation for the neurological dysfunction in PWH is that neurons and astroglias of the PWH may be more susceptible to complement-mediated lysis, as the expression by neuronal and astroglial cell lines of CD59, one of the membrane-bound RCA, is down-regulated after incubation with gp41 peptides or inflammatory cytokines (229).
Despite a clear association between complement activation and NC impairment, the mechanism(s) through which complement activation impacts neurocognitive functions is not yet fully defined. As inhibitors of complement activation are developed for non-HIV related neurological diseases, this could pave the way for their evaluation in PWH presenting with HAND or with other NC disorders.
Complement and Renal Comorbidities
Increased incidence of kidney diseases was reported early in PWH (230). In addition to the HIV-associated nephropathy (HIVAN), other kidney diseases are described in PWH, notably HIV-associated immune complex kidney diseases (HIVICD) (231). HIVICD are glomerulopathies with glomerular deposition of immune complexes. Kidney involvement in PWH varies according to the location of the deposits of immune complexes. Positive staining of renal biopsies for IgG, IgM, IgA, C3, C4, and C1q, and hypocomplementemia is frequently reported (232). The high frequency of the immune complexes in the kidney may play a role in the complement activation and/or in tissue damage in PWH (231). Unlike HIVAN, HIVICD is not significantly impacted by ART in PWH (233).
Complement and Other Comorbidities
Recently, complement activation, particularly the increased C3 levels, has been associated with various metabolic and CVD in the general population. In a large cohort of patients followed over 6 years, patients with high C3 levels at inclusion had an increased risk of developing diabetes (234). Increased C3 levels were also associated with insulin resistance and non-alcoholic fatty liver disease (NAFLD), after adjusting for other variables (235, 236). The role of C3a in NAFLD and insulin resistance has also been demonstrated in animal studies, with C3aR−/− mice being resistant to diet-induced diabetes (237, 238). Prospective studies reported an association of increased C3 or C4 levels with higher risks of myocardial infarction (239, 240).
In HIV-uninfected subjects with advanced atherosclerosis, increased C5a was linked to an exacerbated CVD risk (241). This might be explained by the proinflammatory and procoagulant properties of C3a and C5a, notably platelet activation and TF induction on endothelial cells, respectively. An increase in C5 and factor H-related protein 5 levels in PWH on ART and higher levels of C5 in patients with non-AIDS comorbidities were also recently reported (242).
Higher C3 levels were associated with metabolic syndrome in PWH on ART (243), even after adjusting for confounding factors, but no data are available regarding association of complement proteins with NAFLD or CVD among PWH. Thus, the involvement of complement activation in driving the high frequency of CVD in PWH remains to be determined.
Targeting Complement Pathways
Currently, only four drugs targeting the complement system are approved by the FDA: two C1 esterase inhibitors and two C5 inhibitors (244). The C1 esterase inhibitors hinder C1s, C1r and mannan-binding lectin-associated serine protease (MASP), and they are used to treat patients with hereditary angioedema caused by a deficiency in C1 (SERPING1) inhibitor. The C5 inhibitors, eculizumab and ravulizumab, are monoclonal antibodies that bind to C5 and prevent its cleavage, and thus the formation of MAC. Eculizumab was developed for paroxysmal nocturnal hemoglobinuria, but its off-label use for kidney diseases of PWH has been reported (245). Numerous complement inhibitors are currently under development, including: a MASP-2 inhibitor (narsoplimab), C3 inhibitors (compstatin and its analogs, such as AMY-101, and pegcetacoplan), a C5aR1 antagonist (avacopan), an anti-C5aR1 monoclonal antibody (avdoralimab) and a factor B inhibitor (244). Their evaluation in PWH in clinical trials aiming to limit development of comorbidities may therefore be considered.
Net Formation
Generation of neutrophil extracellular traps (NETs) by polymorphonuclear neutrophils is a recently described mechanism of the innate immune response (246). Neutrophils are key effectors of the innate immune system, being traditionally considered to be involved in the defense against bacteria and fungi (246). More recently, they have been reported to be involved in the innate immune response against viruses (247). The principal mechanism of action of neutrophils is phagocytosis and killing of microorganisms through degranulation and production of reactive oxygen species. It was recently discovered that they can also control infection through generation of extracellular chromatin fibers called NETs (248). Neutrophils that release NETs develop a unique cellular morphology with decondensed nuclei, and ultimately lose their DNA (249). The extracellular chromatin fibers of the NETs are decorated with numerous granule proteins, such as lactoferrin, myeloperoxidase (MPO), histones, and neutrophil elastase (NE) (250). NETs can capture and kill bacteria, fungi, and viruses (246, 250, 251). In vitro generated NETs are long, thin stranded, web-like, extracellular fibers (246). NETs with a thicker morphology were identified in vivo in the gut, liver, skin, and lung in numerous diseases (249, 252, 253).
NETs are beneficial for the organism by eliminating microorganisms and preventing their spread, yet long-lasting or excessive NET formation can be deleterious, as they induce a proinflammatory environment, damage the tissues and can trigger immunothrombosis (254).
The involvement of NETs in the pathogenesis of HIV and SIV has only been studied recently. NETs can capture and eliminate HIV-1 in vitro (255, 256) and their production seems to be regulated by IL-10 (255). Formation of NETs by neutrophils isolated from SIV-infected macaques, upon stimulation with phorbol myristate acetate (PMA), is enhanced compared to neutrophils isolated from uninfected non-human primates (137).
Increased NET formation occurs early during the acute SIV infection, probably triggered directly by the high virus replication. Interestingly, NET formation by neutrophils is higher during chronic infection than during acute infection. This is not unexpected, as the triggers for NET formation diversify and increase in magnitude during later stages of infection. They include bacterial products translocated from the gut, and opportunistic pathogens, in addition to the retrovirus. NETosis is reduced by ART, but the number of NETs remains higher than in uninfected animals (137). Besides capturing virions and SIV/HIV infected cells, NETs can, especially when they are released in excess, also indiscriminately trap other immune cells (such as neutrophils, T, B cells, macrophages, etc.) and induce their apoptosis or lysis (Figure 4) (137). Thus, NETs might play a significant role in the destruction and/or lack of recovery of numerous immune cell subsets during SIV/HIV infection and thus provide a plausible explanation for a key unsolved aspect of HIV/SIV pathogenesis: the bystander loss of cells that are not directly infected with HIV/SIV. Besides the negative consequences related to the death of multiple immune cells, the overly stimulated neutrophils may represent a neglected source of inflammation in SIV-infected macaques and PWH. As neutropenia is often observed during SIV/HIV infection, the impact of neutrophils on SIV/HIV pathogenesis could be questioned but, similarly to CD4+ T cells, the issue might not rely on the absolute neutrophil counts, but rather on the activation status and the production of inflammatory cytokines by this immune cell subset. By trapping immune cells in the NETs, the neutrophils may contribute to their activation and death, both processes being associated with release of inflammatory cytokines or chemotaxis of other cells resulting in a fueling of inflammation. As such, normalizing NET formation may be critical to avoid tissue damages and comorbidities characteristic to HIV/SIV infection (117).
NETs can also trap and destroy platelets (Figure 4), contributing to platelet loss. Interestingly, while this observation provides a plausible explanation for the thrombocytopenia associated with SIV/HIV infection, it also links NETs to the coagulation issues seen in PWH and SIV-infected macaques (Figure 4) (137). It was recently showed that TF expression by NETs (Figure 4) and their ability to capture platelets potentiate each other to promote a proaggregative environment and platelet activation, leading to a hypercoagulable state. TF production by neutrophils may also play an important role in triggering systemic and tissue INFL, as does the TF expression by monocytes.
The contribution of NETs to the pathogenesis of HIV/SIV infection was recently supported by multiple findings in other infectious diseases. Thus, excessive formation of NETs is detected in severe influenza and SARS-CoV-2 infections (257, 258), where it is correlated with an higher mortality (259). Meanwhile, NETs also play a deleterious effect in cancer by promoting metastasis (260), and in autoimmune diseases and atherosclerosis by promoting macrophage-mediated inflammation (261). NETs can also facilitate venous thrombosis (262, 263), through expression of tissue factor (TF) (264, 265), platelet deposition, aggregation and activation (262), activation of factor XII (266), and cleavage of TF pathway inhibitor (267).
Targeting Net Formation
No drug inhibiting NET formation is currently FDA-approved. However, several clinical trials are ongoing, most of them using nebulized DNAse that degrades NETs. Other compounds are currently investigated for autoimmunity diseases, infectious diseases or cancers, either in humans or in animal models. This includes PAD-4 inhibitor (Cl-amidine), neutrophil elastase inhibitor (GW311616A), serine protease inhibitor (camostat mesylate), gasdermin D inhibitor (disulfiram) and C3 inhibitor (Cp40) (268–272). If any of these drugs is proven to be efficient in reducing NET formation in other pathologic conditions, it could be interesting to evaluate them in the context of HIV/SIV infection. However, preliminary studies using NHP models will be needed, both to confirm their efficacy on reducing the virus-driven NETosis and to collect pharmacokinetics data on those different compounds. Indeed, based on their administration route (per os, intravenous or subcutaneous), tissue distribution of those drugs could vary and modulate their efficacy in preventing NET formation.
Interactions Between Complement, Coagulation and NETs
It is now well-understood that complement, coagulation, and formation of NETs are intertwined mechanisms (Figures 1, 5). Indeed, complement protein C3a can activate platelets, by binding to its receptor C3aR present on platelets (273), and platelet-bound C3a can promote the platelet aggregation to neutrophils by binding to complement receptor 3 (CR3) expressed on the neutrophil membrane (274). Meanwhile, platelets are involved in the NET formation, as they can sense LPS through TLR4, which leads to their activation and adhesion to neutrophils, eventually inducing NETs (275). C5a is a neutrophil chemoattractant, leading to neutrophil recruitment and activation (276). C3a and C5a have also been linked to an induction of NETs, C3a through a mechanism remaining to be determined (277) and C5a by increasing TF expression and upregulating complement receptors on neutrophil surface (278, 279). Conversely, complement activation can also be triggered by activated neutrophils and their NETs, as complement proteins can deposit on their surface and form C3 convertases, leading to the production of new anaphylatoxins (280). Note that regulatory mechanisms can also be involved in these interactions. For example, factor H, a known complement inhibitor, can also bind to neutrophils and inhibit NET formation (281).
In addition to these interactions between complement proteins and NET formation, both complement and NETs interact with the coagulation cascade. Thus, both C3a and C5a participate in thrombus formation (282, 283). C3a is linked to platelet activation, while C5, after binding to its receptor C5aR1 (CD88), induces tissue factor expression on endothelial cells and neutrophils (284, 285). C5a is also thought to play a role in TF activation (282). Multiple effectors of coagulation are detected on the extracellular fibers of the NETs, including von Willebrand factor, fibrinogen, fibronectin (262) and TF (137). Besides, some proteins associated with NETs (neutrophil elastase and myeloperoxidase) can inhibit anticoagulant factors, such as thrombomodulin (286). When platelets adhere to NETs by binding to histones or to von Willebrand factor, they are activated and form platelet aggregates (137, 262) (Figure 3). Thrombin is then generated by platelets trapped in NETs after the activation of the coagulation cascade by the TF exposed by NETs and/or by NET-damaged endothelial cells (287), while the contact coagulation pathway can be initiated by the activation of factor XII by NETs and platelets (266). Transformation of fibrinogen by thrombin leads to fibrin deposition on the NETs, and to the formation of an immunothrombus (262). During infections, NETs and immunothrombosis prevent the spread of microorganisms throughout the body. As this process is constrained to microvessels, it prevents deleterious effects. However, in some cases, an uncontrolled immunothrombosis can lead to formation of pathological thrombi. It is therefore conceivable that such a scenario is present during HIV/SIV infection, especially in PWH off ART. Immunothrombosis in small blood vessels (Figure 4D) could drive the development of comorbidities and end-stage organ diseases.
Conclusion
Numerous mechanisms are involved in the persistence of low levels of inflammation and T-cell immune activation in ART-treated PWH. The intertwined roles of hypercoagulation, complement system and NET formation has recently emerged, highlighting different pathways that could be targeted to reduce the risk of SNAEs that remains elevated in ART-treated PWH.
Anticoagulant drugs have been available for decades, with DOACs recently expanding the panel of drugs, even though drug-drug interactions remain an issue for PWH. Meanwhile, drugs targeting the complement system and/or NET formation are either newly approved or still in research or industry pipelines. Interestingly, some drugs, such as C3 inhibitors, can inhibit two pathways (complement and NET formation) in vitro. Evaluating these compounds in NHP models, then in PWH if deemed promising, would be of great interest, as they might be able to thwart different mechanisms involved in the residual inflammation in ART-treated PWH. Studies gathering both clinical and biological data would be necessary, to observe if those drugs effectively reduced inflammation and immune activation, and if this translates into a reduction in the risk of SNAEs.
Author Contributions
TR, LT, RT, AL, IS, CA, and IP wrote, edited the manuscript, prepared, and selected the figures. All authors contributed to the article and approved the submitted version.
Funding
TR, LT, CA, and IP were supported by grants from the National Institutes of Health/ National Institute of Diabetes and Digestive and Kidney Diseases/National Heart, Lung and Blood Institute/National Institute of Allergy and Infectious Diseases: RO1 HL117715 (IP), R01 HL123096 (IP), R01 DK130481 (IP) R01 DK113919 (IP/CA), R01 DK119936 (CA), R01 AI119346 (CA), R01 DK131476 (CA), R01 HL154862 (MF/IP). The work of IS was supported by the intramural research program of NIAID/NIH. AL was supported by UM1-AI106701 AIDS Clinical Trials Group Immunology Support Laboratory. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Disclaimer
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Adam Kleinman for helpful discussion and critical reading of the manuscript.
References
1. Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet. (2013) 382:1525–33. doi: 10.1016/S0140-6736(13)61809-7
2. Guaraldi G, Orlando G, Zona S, Menozzi M, Carli F, Garlassi E, et al. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin Infect Dis. (2011) 53:1120–6. doi: 10.1093/cid/cir627
3. Ploquin MJ, Silvestri G, Müller-Trutwin M. Immune activation in HIV infection: what can the natural hosts of simian immunodeficiency virus teach us? Curr Opin HIV AIDS. (2016) 11:201–8. doi: 10.1097/COH.0000000000000238
4. Sereti I, Altfeld M. Immune activation and HIV: an enduring relationship. Curr Opin HIV AIDS. (2016) 11:129–30. doi: 10.1097/COH.0000000000000244
5. Zicari S, Sessa L, Cotugno N, Ruggiero A, Morrocchi E, Concato C, et al. Immune activation, inflammation, and non-AIDS co-morbidities in HIV-infected patients under long-term ART. Viruses. (2019) 11. doi: 10.3390/v11030200. [Epub ahead of print].
6. Douek DC, Roederer M, Koup RA. Emerging concepts in the immunopathogenesis of AIDS. Annu Rev Med. (2009) 60:471–84. doi: 10.1146/annurev.med.60.041807.123549
7. Lane HC, Masur H, Edgar LC, Whalen G, Rook AH, Fauci AS. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N Engl J Med. (1983) 309:453–8. doi: 10.1056/NEJM198308253090803
8. Giorgi JV, Fahey JL, Smith DC, Hultin LE, Cheng HL, Mitsuyasu RT, et al. Early effects of HIV on CD4 lymphocytes in vivo. J Immunol. (1987) 138:3725–30.
9. Esbjornsson J, Mansson F, Kvist A, da Silva ZJ, Andersson S, Fenyo EM, et al. Long-term follow-up of HIV-2-related AIDS and mortality in Guinea-Bissau: a prospective open cohort study. Lancet HIV. (2018) 6:e25–31. doi: 10.1016/S2352-3018(18)30254-6
10. Michel P, Balde AT, Roussilhon C, Aribot G, Sarthou JL, Gougeon ML. Reduced immune activation and T cell apoptosis in human immunodeficiency virus type 2 compared with type 1: correlation of T cell apoptosis with beta2 microglobulin concentration and disease evolution. J Infect Dis. (2000) 181:64–75. doi: 10.1086/315170
11. Hegedus A, Nyamweya S, Zhang Y, Govind S, Aspinall R, Mashanova A, et al. Protection versus pathology in aviremic and high viral load HIV-2 infection-the pivotal role of immune activation and T-cell kinetics. J Infect Dis. (2014) 210:752–61. doi: 10.1093/infdis/jiu165
12. Leligdowicz A, Feldmann J, Jaye A, Cotten M, Dong T, McMichael A, et al. Direct relationship between virus load and systemic immune activation in HIV-2 infection. J Infect Dis. (2010) 201:114–22. doi: 10.1086/648733
13. Sousa AE, Carneiro J, Meier-Schellersheim M, Grossman Z, Victorino RM. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J Immunol. (2002) 169:3400–6. doi: 10.4049/jimmunol.169.6.3400
14. Buggert M, Frederiksen J, Lund O, Betts MR, Biague A, Nielsen M, et al. Jansson, group SC. CD4+ T cells with an activated and exhausted phenotype distinguish immunodeficiency during aviremic HIV-2 infection. AIDS. (2016) 30:2415–26. doi: 10.1097/QAD.0000000000001223
15. Kamya P, Tsoukas CM, Boulet S, Routy JP, Thomas R, Cote P, et al. T cell Activation does not drive CD4 decline in longitudinally followed HIV-infected Elite Controllers. AIDS Res Ther. (2011) 8:20. doi: 10.1186/1742-6405-8-20
16. Hunt PW, Brenchley J, Sinclair E, McCune JM, Roland M, Page-Shafer K, et al. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis. (2008) 197:126–33. doi: 10.1086/524143
17. Bansal A, Sterrett S, Erdmann N, Westfall AO, Dionne-Odom J, Overton ET, et al. Normal T-cell activation in elite controllers with preserved CD4+ T-cell counts. AIDS. (2015) 29:2245–54. doi: 10.1097/QAD.0000000000000860
18. Lederer S, Favre D, Walters KA, Proll S, Kanwar B, Kasakow Z, et al. Transcriptional profiling in pathogenic and non-pathogenic SIV infections reveals significant distinctions in kinetics and tissue compartmentalization. PLoS Pathog. (2009) 5:e1000296. doi: 10.1371/journal.ppat.1000296
19. Evans DT, Silvestri G. Nonhuman primate models in AIDS research. Curr Opin HIV AIDS. (2013) 8:255–61. doi: 10.1097/COH.0b013e328361cee8
20. Bostik P, Dodd GL, Villinger F, Mayne AE, Ansari AA. Dysregulation of the polo-like kinase pathway in CD4+ T cells is characteristic of pathogenic simian immunodeficiency virus infection. J Virol. (2004) 78:1464–72. doi: 10.1128/JVI.78.3.1464-1472.2004
21. Cecchinato V, Trindade CJ, Laurence A, Heraud JM, Brenchley JM, Ferrari MG, et al. Altered balance between Th17 and Th1 cells at mucosal sites predicts AIDS progression in simian immunodeficiency virus-infected macaques. Mucosal Immunol. (2008) 1:279–88. doi: 10.1038/mi.2008.14
22. Favre D, Lederer S, Kanwar B, Ma ZM, Proll S, Kasakow Z, et al. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. (2009) 5:e1000295. doi: 10.1371/journal.ppat.1000295
23. Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood. (2008) 112:2826–35. doi: 10.1182/blood-2008-05-159301
24. Hu J, Gardner MB, Miller CJ. Simian immunodeficiency virus rapidly penetrates the cervicovaginal mucosa after intravaginal inoculation and infects intraepithelial dendritic cells. J Virol. (2000) 74:6087–95. doi: 10.1128/JVI.74.13.6087-6095.2000
25. Bruel T, Dupuy S, Demoulins T, Rogez-Kreuz C, Dutrieux J, Corneau A, et al. Plasmacytoid dendritic cell dynamics tune interferon-alfa production in SIV-infected cynomolgus macaques. PLoS Pathog. (2014) 10:e1003915. doi: 10.1371/journal.ppat.1003915
26. Wijewardana V, Kristoff J, Xu C, Ma D, Haret-Richter G, Stock JL, et al. Kinetics of myeloid dendritic cell trafficking and activation: impact on progressive, nonprogressive and controlled SIV infections. PLoS Pathog. (2013) 9:e1003600. doi: 10.1371/journal.ppat.1003600
27. Brocca-Cofano E, Kuhrt D, Siewe B, Xu C, Haret-Richter GS, Craigo J, et al. Pathogenic correlates of simian immunodeficiency virus-associated B cell dysfunction. J Virol. (2017) 91:e01051–17. doi: 10.1128/JVI.01051-17
28. Titanji K, Velu V, Chennareddi L, Vijay-Kumar M, Gewirtz AT, Freeman GJ, et al. Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIV-infected macaques. J Clin Investig. (2010) 120:3878–90. doi: 10.1172/JCI43271
29. Pandrea I, Ribeiro RM, Gautam R, Gaufin T, Pattison M, Barnes M, et al. Simian immunodeficiency virus SIVagm dynamics in African green monkeys. J Virol. (2008) 82:3713–24. doi: 10.1128/JVI.02402-07
30. Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. Natural SIV hosts: showing AIDS the door. Science. (2012) 335:1188–93. doi: 10.1126/science.1217550
31. Silvestri G, Paiardini M, Pandrea I, Lederman MM, Sodora DL. Understanding the benign nature of SIV infection in natural hosts. J Clin Invest. (2007) 117:3148–54. doi: 10.1172/JCI33034
32. VandeWoude S, Apetrei C. Going wild: lessons from naturally occurring t-lymphotropic lentiviruses. Clin Microbiol Rev. (2006) 19:728–62. doi: 10.1128/CMR.00009-06
33. Pandrea I, Sodora DL, Silvestri G, Apetrei C. Into the wild: simian immunodeficiency virus (SIV) infection in natural hosts. Trends Immunol. (2008) 29:419–28. doi: 10.1016/j.it.2008.05.004
34. Raehtz KD, Barrenäs F, Xu C, Busman-Sahay K, Valentine A, Law L, et al. African green monkeys avoid SIV disease progression by preventing intestinal dysfunction and maintaining mucosal barrier integrity. PLoS Pathog. (2020) 16:e1008333. doi: 10.1371/journal.ppat.1008333
35. Pandrea I, Apetrei C. Where the wild things are: pathogenesis of SIV infection in African nonhuman primate hosts. Curr HIV AIDS Rep. (2010) 7:28–36. doi: 10.1007/s11904-009-0034-8
36. Liovat AS, Jacquelin B, Ploquin MJ, Barré-Sinoussi F, Müller-Trutwin MC. African non human primates infected by SIV - why don't they get sick? Lessons from studies on the early phase of non-pathogenic SIV infection. Curr HIV Res. (2009) 7:39–50. doi: 10.2174/157016209787048546
37. Pandrea I, Onanga R, Rouquet P, Bourry O, Ngari P, Wickings EJ, et al. Chronic SIV infection ultimately causes immunodeficiency in African non-human primates. AIDS. (2001) 15:2461–2. doi: 10.1097/00002030-200112070-00019
38. Ling B, Apetrei C, Pandrea I, Veazey RS, Lackner AA, Gormus B, et al. Classic AIDS in a sooty mangabey after an 18-year natural infection. J Virol. (2004) 78:8902–8. doi: 10.1128/JVI.78.16.8902-8908.2004
39. Apetrei C, Gormus B, Pandrea I, Metzger M, ten Haaft P, Martin LN, et al. Direct inoculation of simian immunodeficiency virus from sooty mangabeys in black mangabeys (Lophocebus aterrimus): first evidence of AIDS in a heterologous African species and different pathologic outcomes of experimental infection. J Virol. (2004) 78:11506–18. doi: 10.1128/JVI.78.21.11506-11518.2004
40. Pandrea I, Silvestri G, Apetrei C. AIDS in african nonhuman primate hosts of SIVs: a new paradigm of SIV infection. Curr HIV Res. (2009) 7:57–72. doi: 10.2174/157016209787048456
41. Pandrea I, Gaufin T, Brenchley JM, Gautam R, Monjure C, Gautam A, et al. Cutting edge: experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. J Immunol. (2008) 181:6687–91. doi: 10.4049/jimmunol.181.10.6687
42. Pandrea I, Cornell E, Wilson C, Ribeiro RM, Ma D, Kristoff J, et al. Coagulation biomarkers predict disease progression in SIV-infected nonhuman primates. Blood. (2012) 120:1357–66. doi: 10.1182/blood-2012-03-414706
43. Hao XP, Lucero CM, Turkbey B, Bernardo ML, Morcock DR, Deleage C, et al. Experimental colitis in SIV-uninfected rhesus macaques recapitulates important features of pathogenic SIV infection. Nat Commun. (2015) 6:8020. doi: 10.1038/ncomms9020
44. Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW. Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol. (2013) 119:51–83. doi: 10.1016/B978-0-12-407707-2.00002-3
45. Sokoya T, Steel HC, Nieuwoudt M, Rossouw TM. HIV as a cause of immune activation and immunosenescence. Mediators Inflamm. (2017) 2017:6825493. doi: 10.1155/2017/6825493
46. Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. (2009) 83:3719–33. doi: 10.1128/JVI.01844-08
47. Vandergeeten C, Fromentin R, Chomont N. The role of cytokines in the establishment, persistence and eradication of the HIV reservoir. Cytokine Growth Factor Rev. (2012) 23:143–9. doi: 10.1016/j.cytogfr.2012.05.001
48. Reuter MA, Pombo C, Betts MR. Cytokine production and dysregulation in HIV pathogenesis: lessons for development of therapeutics and vaccines. Cytokine Growth Factor Rev. (2012) 23:181–91. doi: 10.1016/j.cytogfr.2012.05.005
49. Muema DM, Akilimali NA, Ndumnego OC, Rasehlo SS, Durgiah R, Ojwach DBA, et al. Association between the cytokine storm, immune cell dynamics, and viral replicative capacity in hyperacute HIV infection. BMC Med. (2020) 18:81. doi: 10.1186/s12916-020-01529-6
50. Gendelman HE, Baca LM, Turpin J, Kalter DC, Hansen B, Orenstein JM, et al. Regulation of HIV replication in infected monocytes by IFN-alpha. Mechanisms for viral restriction. J Immunol. (1990) 145:2669–76.
51. O'Brien M, Manches O, Sabado RL, Baranda SJ, Wang Y, Marie I, et al. Spatiotemporal trafficking of HIV in human plasmacytoid dendritic cells defines a persistently IFN-α-producing and partially matured phenotype. J Clin Invest. (2011) 121:1088–101. doi: 10.1172/JCI44960
52. Rajasuriar R, Khoury G, Kamarulzaman A, French MA, Cameron PU, Lewin SR. Persistent immune activation in chronic HIV infection: do any interventions work? AIDS. (2013) 27:1199–208. doi: 10.1097/QAD.0b013e32835ecb8b
53. Utay NS, Douek DC. Interferons and HIV infection: the good, the bad, the ugly. Pathog Immun. (2016) 1:107–16. doi: 10.20411/pai.v1i1.125
54. Nganou-Makamdop K, Billingsley JM, Yaffe Z, O'Connor G, Tharp GK, Ransier A, et al. Type I IFN signaling blockade by a PASylated antagonist during chronic SIV infection suppresses specific inflammatory pathways but does not alter T cell activation or virus replication. PLoS Pathog. (2018) 14:e1007246. doi: 10.1371/journal.ppat.1007246
55. Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. (2014) 511:601–5. doi: 10.1038/nature13554
56. Herbeuval JP, Boasso A, Grivel JC, Hardy AW, Anderson SA, Dolan MJ, et al. TNF-related apoptosis-inducing ligand (TRAIL) in HIV-1-infected patients and its in vitro production by antigen-presenting cells. Blood. (2005) 105:2458–64. doi: 10.1182/blood-2004-08-3058
57. Ye LL, Wei XS, Zhang M, Niu YR, Zhou Q. The significance of tumor necrosis factor receptor type II in CD8(+) regulatory T cells and CD8(+) effector T cells. Front Immunol. (2018) 9:583. doi: 10.3389/fimmu.2018.00583
58. Pasquereau S, Kumar A, Herbein G. Targeting TNF and TNF receptor pathway in HIV-1 infection: from immune activation to viral reservoirs. Viruses. (2017) 9:64. doi: 10.3390/v9040064
59. Jochems SP, Jacquelin B, Chauveau L, Huot N, Petitjean G, Lepelley A, et al. Plasmacytoid dendritic cell infection and sensing capacity during pathogenic and nonpathogenic simian immunodeficiency virus infection. J Virol. (2015) 89:6918–27. doi: 10.1128/JVI.00332-15
60. Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest. (2009) 119:3556–72. doi: 10.1172/JCI40115
61. Jacquelin B, Mayau V, Targat B, Liovat A-S, Kunkel D, Petitjean G, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Investig. (2009) 119:3544–55. doi: 10.1172/JCI40093
62. Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol. (2010) 84:7886–91. doi: 10.1128/JVI.02612-09
63. Bosinger SE, Sodora DL, Silvestri G. Generalized immune activation and innate immune responses in simian immunodeficiency virus infection. Curr Opin HIV AIDS. (2011) 6:411–8. doi: 10.1097/COH.0b013e3283499cf6
64. Echebli N, Tchitchek N, Dupuy S, Bruel T, Peireira Bittencourt Passaes C, Bosquet N, et al. Stage-specific IFN-induced and IFN gene expression reveal convergence of type I and type II IFN and highlight their role in both acute and chronic stage of pathogenic SIV infection. PLoS ONE. (2018) 13:e0190334. doi: 10.1371/journal.pone.0190334
65. Utay NS, Hunt PW. Role of immune activation in progression to AIDS. Curr Opin HIV AIDS. (2016) 11:131–7. doi: 10.1097/COH.0000000000000242
66. Appay V, Sauce D. Immune activation and inflammation in HIV-1 infection: causes and consequences. J Pathol. (2008) 214:231–41. doi: 10.1002/path.2276
67. Kornfeld C, Ploquin MJY, Pandrea I, Faye A, Onanga R, Apetrei C, et al. Antiinflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J Clin Investig. (2005) 115:1082–91. doi: 10.1172/JCI23006
68. Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. (2006) 12:1301–9. doi: 10.1038/nm1492
69. Freeman ML, Shive CL, Nguyen TP, Younes SA, Panigrahi S, Lederman MM. Cytokines and T-cell homeostasis in HIV infection. J Infect Dis. (2016) 214(Suppl. 2):S51–7. doi: 10.1093/infdis/jiw287
70. Marchetti G, Tincati C, Silvestri G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev. (2013) 26:2–18. doi: 10.1128/CMR.00050-12
71. Mudd JC, Brenchley JM. Gut mucosal barrier dysfunction, microbial dysbiosis, and their role in HIV-1 disease progression. J Infect Dis. (2016) 214(Suppl. 2):S58–66. doi: 10.1093/infdis/jiw258
72. Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol. (2012) 10:655–66. doi: 10.1038/nrmicro2848
73. Gordon SN, Cervasi B, Odorizzi P, Silverman R, Aberra F, Ginsberg G, et al. Disruption of intestinal CD4+ T cell homeostasis is a key marker of systemic CD4+ T cell activation in HIV-infected individuals. J Immunol. (2010) 185:5169–79. doi: 10.4049/jimmunol.1001801
74. Barrenas F, Raehtz K, Xu C, Law L, Green RR, Silvestri G, et al. Macrophage-associated wound healing contributes to African green monkey SIV pathogenesis control. Nat Commun. (2019) 10:5101. doi: 10.1038/s41467-019-13816-9
75. Neff CP, Krueger O, Xiong K, Arif S, Nusbacher N, Schneider JM, et al. Fecal microbiota composition drives immune activation in HIV-infected individuals. EBioMedicine. (2018) 30:192–202. doi: 10.1016/j.ebiom.2018.03.024
76. Vázquez-Castellanos JF, Serrano-Villar S, Latorre A, Artacho A, Ferrús ML, Madrid N, et al. Altered metabolism of gut microbiota contributes to chronic immune activation in HIV-infected individuals. Mucosal Immunol. (2015) 8:760–72. doi: 10.1038/mi.2014.107
77. Gootenberg DB, Paer JM, Luevano JM, Kwon DS. HIV-associated changes in the enteric microbial community: potential role in loss of homeostasis and development of systemic inflammation. Curr Opin Infect Dis. (2017) 30:31–43. doi: 10.1097/QCO.0000000000000341
78. Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis. (2011) 203:780–90. doi: 10.1093/infdis/jiq118
79. Burdo TH, Lentz MR, Autissier P, Krishnan A, Halpern E, Letendre S, et al. Soluble CD163 made by monocyte/macrophages is a novel marker of HIV activity in early and chronic infection prior to and after anti-retroviral therapy. J Infect Dis. (2011) 204:154–63. doi: 10.1093/infdis/jir214
80. Burdo TH, Lo J, Abbara S, Wei J, DeLelys ME, Preffer F, et al. Soluble CD163, a novel marker of activated macrophages, is elevated and associated with noncalcified coronary plaque in HIV-infected patients. J Infect Dis. (2011) 204:1227–36. doi: 10.1093/infdis/jir520
81. Mehraj V, Ramendra R, Isnard S, Dupuy FP, Ponte R, Chen J, et al. Circulating (1 → 3)-β-D-glucan is associated with immune activation during human immunodeficiency virus infection. Clin Infect Dis. (2020) 70:232–41. doi: 10.1093/cid/ciz212
82. Nganou-Makamdop K, Talla A, Sharma AA, Darko S, Ransier A, Laboune F, et al. Translocated microbiome composition determines immunological outcome in treated HIV infection. Cell. (2021) 184:3899–914 e16. doi: 10.1016/j.cell.2021.05.023
83. Ortiz AM, Flynn JK, DiNapoli SR, Vujkovic-Cvijin I, Starke CE, Lai SH, et al. Experimental microbial dysbiosis does not promote disease progression in SIV-infected macaques. Nat Med. (2018) 24:1313–6. doi: 10.1038/s41591-018-0132-5
84. Bourke CD, Gough EK, Pimundu G, Shonhai A, Berejena C, Terry L, et al. Cotrimoxazole reduces systemic inflammation in HIV infection by altering the gut microbiome and immune activation. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aav0537. [Epub ahead of print].
85. Pandrea I, Xu C, Stock JL, Frank DN, Ma D, Policicchio BB, et al. Antibiotic and antiinflammatory therapy transiently reduces inflammation and hypercoagulation in acutely SIV-infected pigtailed macaques. PLoS Pathog. (2016) 12:e1005384. doi: 10.1371/journal.ppat.1005384
86. Brites-Alves C, Luz E, Netto EM, Ferreira T, Diaz RS, Pedroso C, et al. Immune activation, proinflammatory cytokines, and conventional risks for cardiovascular disease in HIV patients: a case-control study in Bahia, Brazil. Front Immunol. (2018) 9:1469. doi: 10.3389/fimmu.2018.01469
87. Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. (2008) 5:e203. doi: 10.1371/journal.pmed.0050203
88. Kelesidis T, Jackson N, McComsey GA, Wang X, Elashoff D, Dube MP, et al. Oxidized lipoproteins are associated with markers of inflammation and immune activation in HIV-1 infection. AIDS. (2016) 30:2625–33. doi: 10.1097/QAD.0000000000001238
89. He T, Xu C, Krampe N, Dillon SM, Sette P, Falwell E, et al. High-fat diet exacerbates SIV pathogenesis and accelerates disease progression. J Clin Invest. (2019) 129:5474–88. doi: 10.1172/JCI121208
90. Paiardini M, Müller-Trutwin M. HIV-associated chronic immune activation. Immunol Rev. (2013) 254:78–101. doi: 10.1111/imr.12079
91. Hunt PW. HIV and inflammation: mechanisms and consequences. Curr HIV AIDS Rep. (2012) 9:139–47. doi: 10.1007/s11904-012-0118-8
92. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. (2006) 443:350–4. doi: 10.1038/nature05115
93. Sauce D, Larsen M, Fastenackels S, Pauchard M, Ait-Mohand H, Schneider L, et al. HIV disease progression despite suppression of viral replication is associated with exhaustion of lymphopoiesis. Blood. (2011) 117:5142–51. doi: 10.1182/blood-2011-01-331306
94. Freeman ML, Lederman MM, Gianella S. Partners in crime: the role of CMV in immune dysregulation and clinical outcome during HIV infection. Curr HIV AIDS Rep. (2016) 13:10–9. doi: 10.1007/s11904-016-0297-9
95. Lurain NS, Hanson BA, Hotton AL, Weber KM, Cohen MH, Landay AL. The association of human cytomegalovirus with biomarkers of inflammation and immune activation in HIV-1-infected women. AIDS Res Hum Retroviruses. (2016) 32:134–43. doi: 10.1089/aid.2015.0169
96. Christensen-Quick A, Vanpouille C, Lisco A, Gianella S. Cytomegalovirus and HIV persistence: pouring gas on the fire. AIDS Res Hum Retroviruses. (2017) 33:S23–30. doi: 10.1089/aid.2017.0145
97. Márquez M, Romero-Cores P, Montes-Oca M, Martín-Aspas A, Soto-Cárdenas M-J, Guerrero F, et al. Immune activation response in chronic HIV-infected patients: influence of Hepatitis C virus coinfection. PLoS ONE. (2015) 10:e0119568. doi: 10.1371/journal.pone.0119568
98. Chen B, Morris SR, Panigrahi S, Michaelson GM, Wyrick JM, Komissarov AA, et al. Cytomegalovirus coinfection is associated with increased vascular-homing CD57+ CD4 T cells in HIV infection. J Immunol. (2020) 204:2722–33. doi: 10.4049/jimmunol.1900734
99. Maidji E, Somsouk M, Rivera JM, Hunt PW, Stoddart CA. Replication of CMV in the gut of HIV-infected individuals and epithelial barrier dysfunction. PLoS Pathog. (2017) 13:e1006202. doi: 10.1371/journal.ppat.1006202
100. Freeman ML, Mudd JC, Shive CL, Younes SA, Panigrahi S, Sieg SF, et al. CD8 T-cell expansion and inflammation linked to CMV coinfection in ART-treated HIV infection. Clin Infect Dis. (2016) 62:392–6. doi: 10.1093/cid/civ840
101. Balagopal A, Ray SC, De Oca RM, Sutcliffe CG, Vivekanandan P, Higgins Y, et al. Kupffer cells are depleted with HIV immunodeficiency and partially recovered with antiretroviral immune reconstitution. AIDS. (2009) 23:2397–404. doi: 10.1097/QAD.0b013e3283324344
102. Operskalski EA, Kovacs A. HIV/HCV co-infection: pathogenesis, clinical complications, treatment, and new therapeutic technologies. Curr HIV AIDS Rep. (2011) 8:12–22. doi: 10.1007/s11904-010-0071-3
103. Zhang L, Bansal MB. Role of kupffer cells in driving hepatic inflammation and fibrosis in HIV infection. Front Immunol. (2020) 11:1086. doi: 10.3389/fimmu.2020.01086
104. Netski DM, Mosbruger T, Astemborski J, Mehta SH, Thomas DL, Cox AL. CD4+ T cell-dependent reduction in hepatitis C virus-specific humoral immune responses after HIV infection. J Infect Dis. (2007) 195:857–63. doi: 10.1086/511826
105. Kim AY, Schulze zur Wiesch J, Kuntzen T, Timm J, Kaufmann DE, Duncan JE, et al. Impaired hepatitis C virus-specific T cell responses and recurrent hepatitis C virus in HIV coinfection. PLoS Med. (2006) 3:e492. doi: 10.1371/journal.pmed.0030492
106. López-Cortés LF, Trujillo-Rodríguez M, Báez-Palomo A, Benmarzouk-Hidalgo OJ, Dominguez-Molina B, Milanés-Guisado Y, et al. Eradication of hepatitis C virus (HCV) reduces immune activation, microbial translocation, and the HIV DNA level in HIV/HCV-coinfected patients. J Infect Dis. (2018) 218:624–32. doi: 10.1093/infdis/jiy136
107. Delvaeye M, Conway EM. Coagulation and innate immune responses: can we view them separately? Blood. (2009) 114:2367–74. doi: 10.1182/blood-2009-05-199208
108. Schulz C, Engelmann B, Massberg S. Crossroads of coagulation and innate immunity: the case of deep vein thrombosis. J Thromb Haemostasis. (2013) 11(Suppl. 1):233–41. doi: 10.1111/jth.12261
109. Antoniak S. The coagulation system in host defense. Res Prac Thromb Haemostasis. (2018) 2:549–57. doi: 10.1002/rth2.12109
110. Bissuel F, Berruyer M, Causse X, Dechavanne M, Trepo C. Acquired protein S deficiency: correlation with advanced disease in HIV-1-infected patients. J Acquired Immune Deficiency Syndr. (1992) 5:484–9. doi: 10.1097/00126334-199205000-00009
111. Sorice M, Griggi T, Arcieri P, Circella A, d'Agostino F, Ranieri M, et al. Protein S and HIV infection. The role of anticardiolipin and anti-protein S antibodies. Thromb Res. (1994) 73:165–75. doi: 10.1016/0049-3848(94)90095-7
112. Levi M, van der Poll T, Büller HR. Bidirectional relation between inflammation and coagulation. Circulation. (2004) 109:2698–704. doi: 10.1161/01.CIR.0000131660.51520.9A
113. Levi M, van der Poll T. Two-way interactions between inflammation and coagulation. Trends Cardiovasc Med. (2005) 15:254–9. doi: 10.1016/j.tcm.2005.07.004
114. Feffer SE, Fox RL, Orsen MM, Harjai KJ, Glatt AE. Thrombotic tendencies and correlation with clinical status in patients infected with HIV. Southern Med J. (1995) 88:1126–30. doi: 10.1097/00007611-199511000-00008
115. Palella FJ Jr, Phair JP. Cardiovascular disease in HIV infection. Curr Opin HIV AIDS. (2011) 6:266–71. doi: 10.1097/COH.0b013e328347876c
116. Agrati C, Mazzotta V, Pinnetti C, Biava G, Bibas M. Venous thromboembolism in people living with HIV infection (PWH). Transl Res. (2020) 227:89–99. doi: 10.1016/j.trsl.2020.07.007
117. Pandrea I, Landay A, Wilson C, Stock J, Tracy R, Apetrei C. Using the pathogenic and nonpathogenic nonhuman primate model for studying non-AIDS comorbidities. Curr HIV AIDS Rep. (2015) 12:54–67. doi: 10.1007/s11904-014-0245-5
118. Borges AH, O'Connor JL, Phillips AN, Baker JV, Vjecha MJ, Losso MH, et al. Factors associated with D-dimer levels in HIV-infected individuals. PLoS ONE. (2014) 9:e90978. doi: 10.1371/journal.pone.0090978
119. Wilson EMP, Singh A, Hullsiek KH, Gibson D, Henry WK, Lichtenstein K, et al. Study to understand the natural history of, monocyte-activation phenotypes are associated with biomarkers of inflammation and coagulation in chronic HIV infection. J Infect Dis. (2014) 210:1396–406. doi: 10.1093/infdis/jiu275
120. Kristoff J, Haret-Richter G, Ma D, Ribeiro RM, Xu C, Cornell E, et al. Early microbial translocation blockade reduces SIV-mediated inflammation and viral replication. J Clin Invest. (2014) 124:2802–6. doi: 10.1172/JCI75090
121. Neuhaus J, Jacobs DR Jr, Baker JV, Calmy A, Duprez D, La Rosa A, et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J Infect Dis. (2010) 201:1788–95. doi: 10.1086/652749
122. Musselwhite LW, Sheikh V, Norton TD, Rupert A, Porter BO, Penzak SR, et al. Markers of endothelial dysfunction, coagulation and tissue fibrosis independently predict venous thromboembolism in HIV. AIDS. (2011) 25:787–95. doi: 10.1097/QAD.0b013e3283453fcb
123. Ford ES, Greenwald JH, Richterman AG, Rupert A, Dutcher L, Badralmaa Y, et al. Traditional risk factors and D-dimer predict incident cardiovascular disease events in chronic HIV infection. AIDS. (2010) 24:1509–17. doi: 10.1097/QAD.0b013e32833ad914
124. Freiberg MS, Bebu I, Tracy R, So-Armah K, Okulicz J, Ganesan A, et al. D-dimer levels before HIV seroconversion remain elevated even after viral suppression and are associated with an increased risk of non-AIDS events. PLoS ONE. (2016) 11:e0152588. doi: 10.1371/journal.pone.0152588
125. Grund B, Baker JV, Deeks SG, Wolfson J, Wentworth D, Cozzi-Lepri A, et al. Relevance of interleukin-6 and D-dimer for serious non-AIDS morbidity and death among HIV-positive adults on suppressive antiretroviral therapy. PLoS ONE. (2016) 11:e0155100. doi: 10.1371/journal.pone.0155100
126. Boulware DR, Hullsiek KH, Puronen CE, Rupert A, Baker JV, French MA, et al. Higher levels of CRP, D-dimer, IL-6, and hyaluronic acid before initiation of antiretroviral therapy (ART) are associated with increased risk of AIDS or death. J Infect Dis. (2011) 203:1637–46. doi: 10.1093/infdis/jir134
127. Freiberg MS, Chang CC, Kuller LH, Skanderson M, Lowy E, Kraemer KL, et al. HIV infection and the risk of acute myocardial infarction. JAMA Internal Med. (2013) 173:614–22. doi: 10.1001/jamainternmed.2013.3728
128. Cohen HJ, Harris T, Pieper CF. Coagulation and activation of inflammatory pathways in the development of functional decline and mortality in the elderly. Am J Med. (2003) 114:180–7. doi: 10.1016/S0002-9343(02)01484-5
129. Baker J, Quick H, Hullsiek KH, Tracy R, Duprez D, Henry K, et al. Interleukin-6 and d-dimer levels are associated with vascular dysfunction in patients with untreated HIV infection. HIV Med. (2010) 11:608–9. doi: 10.1111/j.1468-1293.2010.00835.x
130. Sereti I, Sheikh V, Shaffer D, Phanuphak N, Gabriel E, Wang J, et al. Prospective international study of incidence and predictors of immune reconstitution inflammatory syndrome and death in people living with human immunodeficiency virus and severe lymphopenia. Clin Infect Dis. (2020) 71:652–60. doi: 10.1093/cid/ciz877
131. Ma D, Jasinska A, Kristoff J, Grobler JP, Turner T, Jung Y, et al. International Vervet Research, SIVagm infection in wild African green monkeys from South Africa: epidemiology, natural history, evolutionary considerations. PLoS Pathog. (2013) 9:e1003011. doi: 10.1371/journal.ppat.1003011
132. Ma D, Jasinska AJ, Feyertag F, Wijewardana V, Kristoff J, He T, et al. International Vervet Research, Factors associated with siman immunodeficiency virus transmission in a natural African nonhuman primate host in the wild. J Virol. (2014) 88:5687–705. doi: 10.1128/JVI.03606-13
133. Mackman N Role Role of tissue factor in hemostasis thrombosis vascular development. Arterioscl Thromb Vasc Biol. (2004) 24:1015–22. doi: 10.1161/01.ATV.0000130465.23430.74
134. Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. (2010) 115:161–7. doi: 10.1182/blood-2009-03-210179
135. Schechter ME, Andrade BB, He T, Richter GH, Tosh KW, Policicchio BB, et al. Inflammatory monocytes expressing tissue factor drive SIV and HIV coagulopathy. Sci Transl Med. (2017) 9. doi: 10.1126/scitranslmed.aam5441. [Epub ahead of print].
136. Teer E, Essop MF. HIV and cardiovascular disease: role of immunometabolic perturbations. Physiology. (2018) 33:74–82. doi: 10.1152/physiol.00028.2017
137. Sivanandham R, Brocca-Cofano E, Krampe N, Falwell E, Venkatraman SMK, Ribeiro RM, et al. Neutrophil extracellular trap production contributes to pathogenesis in SIV-infected nonhuman primates. J Clin Investig. (2018) 128:5178–83. doi: 10.1172/JCI99420
138. He T, Falwell E, Brocca-Cofano E, Pandrea I. Modeling aging in HIV infection in nonhuman primates to address an emerging challenge of the post-ART era. Curr Opin Virol. (2017) 25:66–75. doi: 10.1016/j.coviro.2017.07.012
139. Yau JW, Teoh H, Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord. (2015) 15:130. doi: 10.1186/s12872-015-0124-z
140. Anand AR, Rachel G, Parthasarathy D. HIV proteins and endothelial dysfunction: implications in cardiovascular disease. Front Cardiovasc Med. (2018) 5:185. doi: 10.3389/fcvm.2018.00185
141. Nordøy I, Aukrust P, Müller F, Frøland SS. Abnormal levels of circulating adhesion molecules in HIV-1 infection with characteristic alterations in opportunistic infections. Clin Immunol Immunopathol. (1996) 81:16–21. doi: 10.1006/clin.1996.0151
142. Graham SM, Chen J, Le J, Ling M, Chung DW, Liles WC, et al. Von Willebrand factor adhesive activity and ADAMTS13 protease activity in HIV-1-infected men. Int J Med Sci. (2019) 16:276–84. doi: 10.7150/ijms.28110
143. Wolf K, Tsakiris DA, Weber R, Erb P, Battegay M. Antiretroviral therapy reduces markers of endothelial and coagulation activation in patients infected with human immunodeficiency virus type 1. J Infect Dis. (2002) 185:456–62. doi: 10.1086/338572
144. Torriani FJ, Komarow L, Parker RA, Cotter BR, Currier JS, Dubé MP, et al. Endothelial function in human immunodeficiency virus-infected antiretroviral-naive subjects before and after starting potent antiretroviral therapy: the ACTG (AIDS Clinical Trials Group) Study 5152s. J Am Coll Cardiol. (2008) 52:569–76. doi: 10.1016/j.jacc.2008.04.049
145. Zhong DS, Lu XH, Conklin BS, Lin PH, Lumsden AB, Yao Q, et al. HIV protease inhibitor ritonavir induces cytotoxicity of human endothelial cells. Arterioscl Thromb Vasc Biol. (2002) 22:1560–6. doi: 10.1161/01.ATV.0000034707.40046.02
146. Hsue PY, Hunt PW, Wu Y, Schnell A, Ho JE, Hatano H, et al. Association of abacavir and impaired endothelial function in treated and suppressed HIV-infected patients. AIDS. (2009) 23:2021–7. doi: 10.1097/QAD.0b013e32832e7140
147. Zucker-Franklin D, Seremetis S, Zheng ZY. Internalization of human immunodeficiency virus type I and other retroviruses by megakaryocytes and platelets. Blood. (1990) 75:1920–3. doi: 10.1182/blood.V75.10.1920.bloodjournal75101920
148. Youssefian T, Drouin A, Massé JM, Guichard J, Cramer EM. Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood. (2002) 99:4021–9. doi: 10.1182/blood-2001-12-0191
149. Scaradavou A. HIV-related thrombocytopenia. Blood Rev. (2002) 16:73–6. doi: 10.1054/blre.2001.0188
150. Ahmad R, Iannello A, Samarani S, Morisset R, Toma E, Grosley M, et al. Contribution of platelet activation to plasma IL-18 concentrations in HIV-infected AIDS patients. AIDS. (2006) 20:1907–9. doi: 10.1097/01.aids.0000244217.46445.45
151. Mesquita EC, Hottz ED, Amancio RT, Carneiro AB, Palhinha L, Coelho LE, et al. Persistent platelet activation and apoptosis in virologically suppressed HIV-infected individuals. Sci Rep. (2018) 8:14999. doi: 10.1038/s41598-018-33403-0
152. Nkambule BB, Mxinwa V, Mkandla Z, Mutize T, Mokgalaboni K, Nyambuya TM, et al. Platelet activation in adult HIV-infected patients on antiretroviral therapy: a systematic review and meta-analysis. BMC Med. (2020) 18:357. doi: 10.1186/s12916-020-01801-9
153. Anderson AM, Chane T, Patel M, Chen S, Xue W, Easley KA. Warfarin therapy in the HIV medical home model: low rates of therapeutic anticoagulation despite adherence and differences in dosing based on specific antiretrovirals. AIDS Patient Care STDs. (2012) 26:454–62. doi: 10.1089/apc.2012.0068
154. Liedtke MD, Rathbun RC. Drug interactions with antiretrovirals and warfarin. Expert Opin Drug Safety. (2010) 9:215–23. doi: 10.1517/14740330903493458
155. Falcinelli E, Francisci D, Schiaroli E, Minuz P, Orsini S, Malincarne L, et al. Effect of aspirin treatment on abacavir-associated platelet hyperreactivity in HIV-infected patients. Int J Cardiol. (2018) 263:118–24. doi: 10.1016/j.ijcard.2018.04.052
156. O'Brien M, Montenont E, Hu L, Nardi MA, Valdes V, Merolla M, et al. Aspirin attenuates platelet activation and immune activation in HIV-1-infected subjects on antiretroviral therapy: a pilot study. J Acquired Immune Deficiency Syndr. (2013) 63:280–8. doi: 10.1097/QAI.0b013e31828a292c
157. Maggi P, De Socio GV, Cicalini S, D'Abbraccio M, Dettorre G, Di Biagio A, et al. Statins and aspirin in the prevention of cardiovascular disease among HIV-positive patients between controversies and unmet needs: review of the literature and suggestions for a friendly use. AIDS Res Ther. (2019) 16:11. doi: 10.1186/s12981-019-0226-2
158. O'Brien MP, Hunt PW, Kitch DW, Klingman K, Stein JH, Funderburg NT, et al. A randomized placebo controlled trial of aspirin effects on immune activation in chronically human immunodeficiency virus-infected adults on virologically suppressive antiretroviral therapy. Open Forum Infect Dis. (2017) 4:ofw278. doi: 10.1093/ofid/ofw278
159. Suchindran S, Regan S, Meigs JB, Grinspoon SK, Triant VA. Abstract 030: aspirin use and myocardial infarction in HIV versus Non-HIV patients. Circulation. (2013) 127:A030. doi: 10.1161/circ.127.suppl_12.A030
160. Boccara F, Mary-Krause M, Teiger E, Lang S, Lim P, Wahbi K, et al. Prognosis of Acute Coronary Syndrome in, Acute coronary syndrome in human immunodeficiency virus-infected patients: characteristics and 1 year prognosis. Eur Heart J. (2011) 32:41–50. doi: 10.1093/eurheartj/ehq372
161. Hoffmann U, Lu MT, Foldyna B, Zanni MV, Karady J, Taron J, et al. Assessment of coronary artery disease with computed tomography angiography and inflammatory and immune activation biomarkers among adults with HIV eligible for primary cardiovascular prevention. JAMA Netw Open. (2021) 4:e2114923. doi: 10.1001/jamanetworkopen.2021.14923
162. Lake JE, Seang S, Kelesidis T, Liao DH, Hodis HN, Stein JH, et al. Telmisartan to reduce cardiovascular risk in older HIV-infected adults: a pilot study. HIV Clin Trials. (2015) 16:197–206. doi: 10.1179/1945577115Y.0000000006
163. Lake JE, Seang S, Kelesidis T, Currier JS, Yang OO. Telmisartan increases vascular reparative capacity in older HIV-infected adults: a pilot study. HIV Clin Trials. (2016) 17:225–32. doi: 10.1080/15284336.2016.1234222
164. Pelliccia F, Pasceri V, Cianfrocca C, Vitale C, Speciale G, Gaudio C, et al. Angiotensin II receptor antagonism with telmisartan increases number of endothelial progenitor cells in normotensive patients with coronary artery disease: a randomized, double-blind, placebo-controlled study. Atherosclerosis. (2010) 210:510–5. doi: 10.1016/j.atherosclerosis.2009.12.005
165. George JM, Kuriakose SS, Monroe A, Hou Q, Byrne M, Pau AK, et al. District of Columbia cohort executive, utilization of direct oral anticoagulants in people living with human immunodeficiency virus: observational data from the district of Columbia cohort. Clin Infect Dis. (2020) 71:e604–13. doi: 10.1093/cid/ciaa284
166. Bates D, Dalton B, Gilmour J, Kapler J. Venous thromboembolism due to suspected interaction between rivaroxaban and nevirapine. Can J Hosp Pharm. (2013) 66:125–9. doi: 10.4212/cjhp.v66i2.1235
167. Baker JV, Wolfson J, Peterson T, Mooberry M, Gissel M, Mystakelis H, et al. Factor Xa inhibition reduces coagulation activity but not inflammation among people with HIV: a randomized clinical trial. Open Forum Infect Dis. (2020) 7. doi: 10.1093/ofid/ofaa026. [Epub ahead of print].
168. Group AS. Vorapaxar for HIV-associated inflammation and coagulopathy (ADVICE): a randomised, double-blind, placebo-controlled trial. Lancet HIV. (2018) 5:e553–9. doi: 10.1016/S2352-3018(18)30214-5
169. Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol. (2009) 46:2753–66. doi: 10.1016/j.molimm.2009.04.027
170. Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev. (2016) 274:152–71. doi: 10.1111/imr.12475
171. Kishore U, Gupta SK, Perdikoulis MV, Kojouharova MS, Urban BC, Reid KB. Modular organization of the carboxyl-terminal, globular head region of human C1q A, B, C chains. J Immunol. (2003) 171:812–20. doi: 10.4049/jimmunol.171.2.812
172. Loos M, Bitter-Suermann D, Dierich M. Interaction of the first (C1), the second (C2) and the fourth (C4) component of complement with different preparations of bacterial lipopolysaccharides and with lipid A. J Immunol. (1974) 112:935–40.
173. Neth O, Jack DL, Dodds AW, Holzel H, Klein NJ, Turner MW. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect Immun. (2000) 68:688–93. doi: 10.1128/IAI.68.2.688-693.2000
174. Roos A, Bouwman LH, van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol. (2001) 167:2861–8. doi: 10.4049/jimmunol.167.5.2861
175. Spear GT, Takefman DM, Sullivan BL, Landay AL, Zolla-Pazner S. Complement activation by human monoclonal antibodies to human immunodeficiency virus. J Virol. (1993) 67:53–9. doi: 10.1128/jvi.67.1.53-59.1993
176. Ebenbichler CF, Thielens NM, Vornhagen R, Marschang P, Arlaud GJ, Dierich MP. Human immunodeficiency virus type 1 activates the classical pathway of complement by direct C1 binding through specific sites in the transmembrane glycoprotein gp41. J Exp Med. (1991) 174:1417–24. doi: 10.1084/jem.174.6.1417
177. Haurum JS, Thiel S, Jones IM, Fischer PB, Laursen SB, Jensenius JC. Complement activation upon binding of mannan-binding protein to HIV envelope glycoproteins. AIDS. (1993) 7:1307–13. doi: 10.1097/00002030-199310000-00002
178. McGrath FD, Brouwer MC, Arlaud GJ, Daha MR, Hack CE, Roos A. Evidence that complement protein C1q interacts with C-reactive protein through its globular head region. J Immunol. (2006) 176:2950–7. doi: 10.4049/jimmunol.176.5.2950
179. Huson MA, Wouters D, van Mierlo G, Grobusch MP, Zeerleder SS, van der Poll T. HIV coinfection enhances complement activation during sepsis. J Infect Dis. (2015) 212:474–83. doi: 10.1093/infdis/jiv074
180. Perricone R, Fontana L, de Carolis C, Carini C, Sirianni MC, Aiuti F. Evidence for activation of complement in patients with AIDS related complex (ARC) and/or lymphoadenopathy syndrome (LAS). Clin Exp Immunol. (1987) 70:500–7.
181. Carini C, Perricone R, Fratazzi C, Fontana L, De Carolis C, D'Amelio R, et al. Complement activation is associated with the presence of specific human immunodeficiency virus (HIV)-anti-HIV immune complexes in patients with acquired immunodeficiency syndrome-related complex or lymphoadenopathy syndrome. Scand J Immunol. (1989) 30:347–53. doi: 10.1111/j.1365-3083.1989.tb01220.x
182. Lage SL, Wong CS, Amaral EP, Sturdevant D, Hsu DC, Rupert A, et al. Classical complement and inflammasome activation converge in CD14highCD16- monocytes in HIV associated TB-immune reconstitution inflammatory syndrome. PLoS Pathog. (2021) 17:e1009435. doi: 10.1371/journal.ppat.1009435
183. Tran HT, Van den Bergh R, Loembe MM, Worodria W, Mayanja-Kizza H, Colebunders R, et al. Modulation of the complement system in monocytes contributes to tuberculosis-associated immune reconstitution inflammatory syndrome. AIDS. (2013) 27:1725–34. doi: 10.1097/QAD.0b013e328361648b
184. Spear GT, Olinger G, Sullivan BL, Landay AL, Kessler H, Connick E, et al. Alteration of complement protein levels after antiretroviral therapy in HIV-infected persons. AIDS Res Hum Retroviruses. (1999) 15:1713–5.
185. Rossheim AEB, Cunningham TD, Hair PS, Shah T, Cunnion KM, Troy SB. Effects of well-controlled HIV infection on complement activation and function. J Acquired Immune Deficiency Syndr. (2016) 73:20–6. doi: 10.1097/QAI.0000000000001079
186. Schein TN, Blackburn TE, Heath SL, Barnum SR. Plasma levels of soluble membrane attack complex are elevated despite viral suppression in HIV patients with poor immune reconstitution. Clin Exp Immunol. (2019) 198:359–66. doi: 10.1111/cei.13366
187. Heggelund L, Mollnes TE, Ueland T, Christophersen B, Aukrust P, Froland SS. Mannose-binding lectin in HIV infection: relation to disease progression and highly active antiretroviral therapy. J Acquired Immune Deficiency Syndr. (2003) 32:354–61. doi: 10.1097/00126334-200304010-00002
188. Yu Q, Yu R, Qin X. The good and evil of complement activation in HIV-1 infection. Cell Mol Immunol. (2010) 7:334–40. doi: 10.1038/cmi.2010.8
189. Saifuddin M, Parker CJ, Peeples ME, Gorny MK, Zolla-Pazner S, Ghassemi M, et al. Role of virion-associated glycosylphosphatidylinositol-linked proteins CD55 and CD59 in complement resistance of cell line-derived and primary isolates of HIV-1. J Exp Med. (1995) 182:501–9. doi: 10.1084/jem.182.2.501
190. Schmitz J, Zimmer JP, Kluxen B, Aries S, Bogel M, Gigli I, et al. Antibody-dependent complement-mediated cytotoxicity in sera from patients with HIV-1 infection is controlled by CD55 and CD59. J Clin Invest. (1995) 96:1520–6. doi: 10.1172/JCI118190
191. Montefiori DC, Cornell RJ, Zhou JY, Zhou JT, Hirsch VM, Johnson PR. Complement control proteins, CD46, CD55, and CD59, as common surface constituents of human and simian immunodeficiency viruses and possible targets for vaccine protection. Virology. (1994) 205:82–92. doi: 10.1006/viro.1994.1622
192. Boyer V, Desgranges C, Trabaud MA, Fischer E, Kazatchkine MD. Complement mediates human immunodeficiency virus type 1 infection of a human T cell line in a CD4- and antibody-independent fashion. J Exp Med. (1991) 173:1151–8. doi: 10.1084/jem.173.5.1151
193. Robinson WE Jr, Montefiori DC, Mitchell WM. Antibody-dependent enhancement of human immunodeficiency virus type 1 infection. Lancet. (1988) 1:790–4. doi: 10.1016/S0140-6736(88)91657-1
194. Ho J, Moir S, Kulik L, Malaspina A, Donoghue ET, Miller NJ, et al. Role for CD21 in the establishment of an extracellular HIV reservoir in lymphoid tissues. J Immunol. (2007) 178:6968–74. doi: 10.4049/jimmunol.178.11.6968
195. Smith BA, Gartner S, Liu Y, Perelson AS, Stilianakis NI, Keele BF, et al. Persistence of infectious HIV on follicular dendritic cells. J Immunol. (2001) 166:690–6. doi: 10.4049/jimmunol.166.1.690
196. Banki Z, Kacani L, Rusert P, Pruenster M, Wilflingseder D, Falkensammer B, et al. Complement dependent trapping of infectious HIV in human lymphoid tissues. AIDS. (2005) 19:481–6. doi: 10.1097/01.aids.0000162336.20439.8d
197. Q. Le Hingrat, Sereti I, Landay AL, Pandrea I, Apetrei C. The hitchhiker guide to CD4+ T-cell depletion in lentiviral infection. A critical review of the dynamics of the CD4+ T cells in SIV and HIV infection. Front Immunol. (2021) 12:695674. doi: 10.3389/fimmu.2021.695674
198. Kacani L, Stoiber H, Speth C, Banki Z, Tenner-Racz K, Racz P, et al. Complement-dependent control of viral dynamics in pathogenesis of human immunodeficiency virus and simian immunodeficiency virus infection. Mol Immunol. (2001) 38:241–7. doi: 10.1016/S0161-5890(01)00046-3
199. Beer B, Scherer J, zur Megede J, Norley S, Baier M, Kurth R. Lack of dichotomy between virus load of peripheral blood and lymph nodes during long-term simian immunodeficiency virus infection of African green monkeys. Virology. (1996) 219:367–75. doi: 10.1006/viro.1996.0262
200. Huot N, Bosinger SE, Paiardini M, Reeves RK, Muller-Trutwin M. Lymph node cellular and viral dynamics in natural hosts and impact for HIV cure strategies. Front Immunol. (2018) 9:780. doi: 10.3389/fimmu.2018.00780
201. Apetrei C, Robertson DL, Marx PA. The history of SIVs and AIDS: epidemiology, phylogeny and biology of isolates from naturally SIV infected non-human primates (NHP) in Africa. Front Biosci. (2004) 9:225–54. doi: 10.2741/1154
202. Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J Immunol. (2015) 194:3542–8. doi: 10.4049/jimmunol.1403068
203. Wu MC, Brennan FH, Lynch JP, Mantovani S, Phipps S, Wetsel RA, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci USA. (2013) 110:9439–44. doi: 10.1073/pnas.1218815110
204. Lau B, Sharrett AR, Kingsley LA, Post W, Palella FJ, Visscher B, et al. C-reactive protein is a marker for human immunodeficiency virus disease progression. Arch Intern Med. (2006) 166:64–70. doi: 10.1001/archinte.166.1.64
205. Rodger AJ, Fox Z, Lundgren JD, Kuller LH, Boesecke C, Gey D, et al. Activation and coagulation biomarkers are independent predictors of the development of opportunistic disease in patients with HIV infection. J Infect Dis. (2009) 200:973–83. doi: 10.1086/605447
206. Vaidya SA, Korner C, Sirignano MN, Amero M, Bazner S, Rychert J, et al. Tumor necrosis factor alpha is associated with viral control and early disease progression in patients with HIV type 1 infection. J Infect Dis. (2014) 210:1042–6. doi: 10.1093/infdis/jiu206
207. Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Ann Neurol. (1986) 19:517–24. doi: 10.1002/ana.410190602
208. Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. (2007) 69:1789–99. doi: 10.1212/01.WNL.0000287431.88658.8b
209. Heaton RK, Clifford DB, Franklin DR Jr, Woods SP, Ake C, Vaida F, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER study. Neurology. (2010) 75:2087–96. doi: 10.1212/WNL.0b013e318200d727
210. Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder–pathogenesis and prospects for treatment. Nat Rev Neurol. (2016) 12:234–48. doi: 10.1038/nrneurol.2016.27
211. Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. (1994) 367:188–93. doi: 10.1038/367188a0
212. Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci USA. (1986) 83:7089–93. doi: 10.1073/pnas.83.18.7089
213. Abassi M, Morawski BM, Nakigozi G, Nakasujja N, Kong X, Meya DB, et al. Cerebrospinal fluid biomarkers and HIV-associated neurocognitive disorders in HIV-infected individuals in Rakai, Uganda. J Neurovirol. (2017) 23:369–75. doi: 10.1007/s13365-016-0505-9
214. Ciccarelli N, Fabbiani M, Di Giambenedetto S, Fanti I, Baldonero E, Bracciale L, et al. Efavirenz associated with cognitive disorders in otherwise asymptomatic HIV-infected patients. Neurology. (2011) 76:1403–9. doi: 10.1212/WNL.0b013e31821670fb
215. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
216. Dalakas MC, Alexopoulos H, Spaeth PJ. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat Rev Neurol. (2020) 16:601–17. doi: 10.1038/s41582-020-0400-0
217. Reboul J, Schuller E, Pialoux G, Rey MA, Lebon P, Allinquant B, et al. Immunoglobulins and complement components in 37 patients infected by HIV-1 virus: comparison of general (systemic) and intrathecal immunity. J Neurol Sci. (1989) 89:243–52. doi: 10.1016/0022-510X(89)90026-9
218. Levi-Strauss M, Mallat M. Primary cultures of murine astrocytes produce C3 and factor B, two components of the alternative pathway of complement activation. J Immunol. (1987) 139:2361–6.
219. Jongen PJ, Doesburg WH, Ibrahim-Stappers JL, Lemmens WA, Hommes OR, Lamers KJ. Cerebrospinal fluid C3 and C4 indexes in immunological disorders of the central nervous system. Acta Neurol Scand. (2000) 101:116–21. doi: 10.1034/j.1600-0404.2000.101002116.x
220. Speth C, Stockl G, Mohsenipour I, Wurzner R, Stoiber H, Lass-Florl C, et al. Human immunodeficiency virus type 1 induces expression of complement factors in human astrocytes. J Virol. (2001) 75:2604–15. doi: 10.1128/JVI.75.6.2604-2516.2001
221. Bruder C, Hagleitner M, Darlington G, Mohsenipour I, Wurzner R, Hollmuller I, et al. HIV-1 induces complement factor C3 synthesis in astrocytes and neurons by modulation of promoter activity. Mol Immunol. (2004) 40:949–61. doi: 10.1016/j.molimm.2003.10.016
222. Speth C, Schabetsberger T, Mohsenipour I, Stockl G, Wurzner R, Stoiber H, et al. Mechanism of human immunodeficiency virus-induced complement expression in astrocytes and neurons. J Virol. (2002) 76:3179–88. doi: 10.1128/JVI.76.7.3179-3188.2002
223. Speth C, Williams K, Hagleitner M, Westmoreland S, Rambach G, Mohsenipour I, et al. Complement synthesis and activation in the brain of SIV-infected monkeys. J Neuroimmunol. (2004) 151:45–54. doi: 10.1016/j.jneuroim.2004.02.013
224. Depboylu C, Schafer MK, Schwaeble WJ, Reinhart TA, Maeda H, Mitsuya H, et al. Increase of C1q biosynthesis in brain microglia and macrophages during lentivirus infection in the rhesus macaque is sensitive to antiretroviral treatment with 6-chloro-2',3'-dideoxyguanosine. Neurobiol Dis. (2005) 20:12–26. doi: 10.1016/j.nbd.2005.01.030
225. McArthur JC, McClernon DR, Cronin MF, Nance-Sproson TE, Saah AJ, St Clair M, et al. Relationship between human immunodeficiency virus-associated dementia and viral load in cerebrospinal fluid and brain. Ann Neurol. (1997) 42:689–98. doi: 10.1002/ana.410420504
226. Nitkiewicz J, Borjabad A, Morgello S, Murray J, Chao W, Emdad L, et al. HIV induces expression of complement component C3 in astrocytes by NF-kappaB-dependent activation of interleukin-6 synthesis. J Neuroinflamm. (2017) 14:23. doi: 10.1186/s12974-017-0794-9
227. Gelman BB, Chen T, Lisinicchia JG, Soukup VM, Carmical JR, Starkey JM, et al. National Neuro, The National NeuroAIDS Tissue Consortium brain gene array: two types of HIV-associated neurocognitive impairment. PLoS ONE. (2012) 7:e46178. doi: 10.1371/journal.pone.0046178
228. Ubaida-Mohien C, Lamberty B, Dickens AM, Mielke MM, Marcotte T, Sacktor N, et al. Modifications in acute phase and complement systems predict shifts in cognitive status of HIV-infected patients. AIDS. (2017) 31:1365–78. doi: 10.1097/QAD.0000000000001503
229. Chong YH, Lee MJ. Expression of complement inhibitor protein CD59 in human neuronal and glial cell lines treated with HIV-1 gp41 peptides. J Neurovirol. (2000) 6:51–60. doi: 10.3109/13550280009006382
230. Rao TK, Filippone EJ, Nicastri AD, Landesman SH, Frank E, Chen CK, et al. Associated focal and segmental glomerulosclerosis in the acquired immunodeficiency syndrome. N Engl J Med. (1984) 310:669–73. doi: 10.1056/NEJM198403153101101
231. Nobakht E, Cohen SD, Rosenberg AZ, Kimmel PL. HIV-associated immune complex kidney disease. Nat Rev Nephrol. (2016) 12:291–300. doi: 10.1038/nrneph.2015.216
232. Kimmel PL, Phillips TM, Ferreira-Centeno A, Farkas-Szallasi T, Abraham AA, Garrett CT. HIV-associated immune-mediated renal disease. Kidney Int. (1993) 44:1327–40. doi: 10.1038/ki.1993.386
233. Foy MC, Estrella MM, Lucas GM, Tahir F, Fine DM, Moore RD, et al. Comparison of risk factors and outcomes in HIV immune complex kidney disease and HIV-associated nephropathy. Clin J Am Soc Nephrol. (2013) 8:1524–32. doi: 10.2215/CJN.10991012
234. Engstrom G, Hedblad B, Eriksson KF, Janzon L, Lindgarde F. Complement C3 is a risk factor for the development of diabetes: a population-based cohort study. Diabetes. (2005) 54:570–5. doi: 10.2337/diabetes.54.2.570
235. Muscari A, Antonelli S, Bianchi G, Cavrini G, Dapporto S, Ligabue A, et al. Pianoro Study, Serum C3 is a stronger inflammatory marker of insulin resistance than C-reactive protein, leukocyte count, and erythrocyte sedimentation rate: comparison study in an elderly population. Diabetes Care. (2007) 30:2362–8. doi: 10.2337/dc07-0637
236. Jia Q, Li C, Xia Y, Zhang Q, Wu H, Du H, et al. Association between complement C3 and prevalence of fatty liver disease in an adult population: a cross-sectional study from the Tianjin Chronic Low-Grade Systemic Inflammation and Health (TCLSIHealth) cohort study. PLoS ONE. (2015) 10:e0122026. doi: 10.1371/journal.pone.0122026
237. Mamane Y, Chung Chan C, Lavallee G, Morin N, Xu LJ, Huang J, et al. The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes. (2009) 58:2006–17. doi: 10.2337/db09-0323
238. Pritchard MT, McMullen MR, Stavitsky AB, Cohen JI, Lin F, Edward Medof M, et al. Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology. (2007) 132:1117–26. doi: 10.1053/j.gastro.2007.01.053
239. Muscari A, Bozzoli C, Puddu GM, Sangiorgi Z, Dormi A, Rovinetti C, et al. Association of serum C3 levels with the risk of myocardial infarction. Am J Med. (1995) 98:357–64. doi: 10.1016/S0002-9343(99)80314-3
240. Szeplaki G, Prohaszka Z, Duba J, Rugonfalvi-Kiss S, Karadi I, Kokai M, et al. Association of high serum concentration of the third component of complement (C3) with pre-existing severe coronary artery disease and new vascular events in women. Atherosclerosis. (2004) 177:383–9. doi: 10.1016/j.atherosclerosis.2004.07.022
241. Speidl WS, Exner M, Amighi J, Kastl SP, Zorn G, Maurer G, et al. Complement component C5a predicts future cardiovascular events in patients with advanced atherosclerosis. Eur Heart J. (2005) 26:2294–9. doi: 10.1093/eurheartj/ehi339
242. Vujkovic-Cvijin I, Sortino O, Verheij E, Wit FW, Kootstra NA, Sellers B, et al. The complement pathway is activated in people with HIV and is associated with non-AIDS comorbidities. J Infect Dis. (2021) 224:1405–09. doi: 10.1093/infdis/jiab096
243. Bryant AK, Fazeli PL, Letendre SL, Ellis RJ, Potter M, Burdo TH, et al. Complement component 3 is associated with metabolic comorbidities in older HIV-positive adults. AIDS Res Hum Retroviruses. (2016) 32:271–8. doi: 10.1089/aid.2015.0179
244. Mastellos DC, Ricklin D, Lambris JD. Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov. (2019) 18:707–29. doi: 10.1038/s41573-019-0031-6
245. Freist M, Garrouste C, Szlavik N, Coppo P, Lautrette A, Heng AE. Efficacy of eculizumab in an adult patient with HIV-associated hemolytic uremic syndrome: a case report. Medicine. (2017) 96:e9358. doi: 10.1097/MD.0000000000009358
246. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
247. Galani IE, Andreakos E. Neutrophils in viral infections: current concepts and caveats. J Leukoc Biol. (2015) 98:557–64. doi: 10.1189/jlb.4VMR1114-555R
248. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
249. Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. (2012) 18:1386. doi: 10.1038/nm.2847
250. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
251. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. (2011) 179:199–210. doi: 10.1016/j.ajpath.2011.03.013
252. Savchenko AS, Inoue A, Ohashi R, Jiang S, Hasegawa G, Tanaka T, et al. Long pentraxin 3 (PTX3) expression and release by neutrophils in vitro and in ulcerative colitis. Pathology international. (2011) 61:290–7. doi: 10.1111/j.1440-1827.2011.02651.x
253. de Buhr N, von Kockritz-Blickwede M. How neutrophil extracellular traps become visible. J Immunol Res. (2016) 2016:4604713. doi: 10.1155/2016/4604713
254. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
255. Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. (2012) 12:109–16. doi: 10.1016/j.chom.2012.05.015
256. Barr FD, Ochsenbauer C, Wira CR, Rodriguez-Garcia M. Neutrophil extracellular traps prevent HIV infection in the female genital tract. Mucosal Immunol. (2018) 11:1420–8. doi: 10.1038/s41385-018-0045-0
257. Zhu L, Liu L, Zhang Y, Pu L, Liu J, Li X, et al. High level of neutrophil extracellular traps correlates with poor prognosis of severe influenza A infection. J Infect Dis. (2018) 217:428–37. doi: 10.1093/infdis/jix475
258. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.138999. [Epub ahead of print].
259. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. (2020) 136:1169–79. doi: 10.1182/blood.2020007008
260. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. (2020) 583:133–8. doi: 10.1038/s41586-020-2394-6
261. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. (2015) 349:316–20. doi: 10.1126/science.aaa8064
262. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
263. Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA. (2013) 110:8674–9. doi: 10.1073/pnas.1301059110
264. Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS ONE. (2012) 7:e45427. doi: 10.1371/journal.pone.0045427
265. Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J. (2015) 36:1405–14. doi: 10.1093/eurheartj/ehv007
266. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
267. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. (2010) 16:887–96. doi: 10.1038/nm.2184
268. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. (2020) 217. doi: 10.1084/jem.20201129. [Epub ahead of print].
269. Knight JS, Subramanian V, O'Dell AA, Yalavarthi S, Zhao W, Smith CK, et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis. (2015) 74:2199–206. doi: 10.1136/annrheumdis-2014-205365
270. Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. (2017) 6. doi: 10.7554/eLife.24437. [Epub ahead of print].
271. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. (2018) 3. doi: 10.1126/sciimmunol.aar6689. [Epub ahead of print].
272. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Investig. (2020) 130:6151–7. doi: 10.1172/JCI141374
273. Polley MJ, Nachman RL. Human platelet activation by C3a and C3a des-arg. J Exp Med. (1983) 158:603–15. doi: 10.1084/jem.158.2.603
274. Hamad OA, Mitroulis I, Fromell K, Kozarcanin H, Chavakis T, Ricklin D, et al. Contact activation of C3 enables tethering between activated platelets and polymorphonuclear leukocytes via CD11b/CD18. Thromb Haemost. (2015) 114:1207–17. doi: 10.1160/TH15-02-0162
275. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
276. Fernandez HN, Henson PM, Otani A, Hugli TE. Chemotactic response to human C3a and C5a anaphylatoxins. I. Evaluation of C3a and C5a leukotaxis in vitro and under stimulated in vivo conditions. J Immunol. (1978) 120:109–15.
277. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun. (2016) 7:11037. doi: 10.1038/ncomms11037
278. Huang YM, Wang H, Wang C, Chen M, Zhao MH. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol. (2015) 67:2780–90. doi: 10.1002/art.39239
279. Mollnes TE, Brekke OL, Fung M, Fure H, Christiansen D, Bergseth G, et al. Essential role of the C5a receptor in E. coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood. (2002) 100:1869–77. doi: 10.1182/blood.V100.5.1869.h81702001869_1869_1877
280. Yuen J, Pluthero FG, Douda DN, Riedl M, Cherry A, Ulanova M, et al. NETosing neutrophils activate complement both on their own NETs and bacteria via alternative and non-alternative pathways. Front Immunol. (2016) 7:137. doi: 10.3389/fimmu.2016.00137
281. Schneider AE, Sandor N, Karpati E, Jozsi M. Complement factor H modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol Immunol. (2016) 72:37–48. doi: 10.1016/j.molimm.2016.02.011
282. Subramaniam S, Jurk K, Hobohm L, Jackel S, Saffarzadeh M, Schwierczek K, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. (2017) 129:2291–302. doi: 10.1182/blood-2016-11-749879
283. Sauter RJ, Sauter M, Reis ES, Emschermann FN, Nording H, Ebenhoch S, et al. Functional relevance of the anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation. (2018) 138:1720–35. doi: 10.1161/CIRCULATIONAHA.118.034600
284. Ikeda K, Nagasawa K, Horiuchi T, Tsuru T, Nishizaka H, Niho Y. C5a induces tissue factor activity on endothelial cells. Thromb Haemost. (1997) 77:394–8. doi: 10.1055/s-0038-1655974
285. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. (2006) 177:4794–802. doi: 10.4049/jimmunol.177.7.4794
286. Glaser CB, Morser J, Clarke JH, Blasko E, McLean K, Kuhn I, et al. Oxidation of a specific methionine in thrombomodulin by activated neutrophil products blocks cofactor activity. A potential rapid mechanism for modulation of coagulation. J Clin Invest. (1992) 90:2565–73. doi: 10.1172/JCI116151
Keywords: HIV, SIV, coagulation, complement, neutrophil extracellular traps
Citation: Reno TA, Tarnus L, Tracy R, Landay AL, Sereti I, Apetrei C and Pandrea I (2022) The Youngbloods. Get Together. Hypercoagulation, Complement, and NET Formation in HIV/SIV Pathogenesis. Front. Virol. 1:795373. doi: 10.3389/fviro.2021.795373
Received: 15 October 2021; Accepted: 21 December 2021;
Published: 18 January 2022.
Edited by:
Alessandra Bandera, University of Milan, ItalyReviewed by:
Rupert Kaul, University of Toronto, CanadaWilliam Anderson Paxton, University of Liverpool, United Kingdom
Copyright © 2022 Reno, Tarnus, Tracy, Landay, Sereti, Apetrei and Pandrea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivona Pandrea, cGFuZHJlYUBwaXR0LmVkdQ==
†These authors have contributed equally to this work