 Yusuke Higuchi
Yusuke Higuchi Jun-ichirou Yasunaga
Jun-ichirou Yasunaga Masao Matsuoka
Masao Matsuoka
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Virol., 03 January 2022
Sec. Virus and Host Immunity
Volume 1 - 2021 | https://doi.org/10.3389/fviro.2021.792659
This article is part of the Research TopicHost Immune Responses to Retroviral InfectionsView all 5 articles
Human T-cell leukemia virus type 1 (HTLV-1) is the causative agent of adult T-cell leukemia-lymphoma (ATL) and inflammatory diseases including HTLV-1-associated myelopathy (HAM). A remarkable feature of HTLV-1 is that this virus transmits primarily through cell-to-cell contact. HTLV-1 increases the number of infected cells in vivo to ensure its survival and transmission. Therefore, survival of HTLV-1-infected cells in vivo is very critical for transmission under the host immune surveillance. HTLV-1 possesses multiple strategies to evade host immune responses. Among viral genes, Tax and HTLV-1 bZIP factor (HBZ) play crucial roles in the proliferation of infected cells and the subsequent development of ATL. Although Tax strongly activates the NF-kB pathway, the immunogenicity of Tax is very high; it is a major target of cytotoxic T lymphocytes. Therefore, the virus minimizes Tax production, expressing it only intermittently in vivo. On the other hand, the immunogenicity of HBZ is low, and its expression is maintained in all ATL cases. HBZ transforms the immunophenotype of infected cells into regulatory T cell-like (CD4+ CD25+ CCR4+ TIGIT+ Foxp3+), and promotes the production of immunosuppressive cytokines. Furthermore, HBZ mRNA not only encodes the protein but also functions itself like long non-coding RNA. As a result, Tax and HBZ enable long-term escape from host immunity, persistent infection, and proliferation of infected cells. Here, we review the viral strategies to counteract to host immune surveillance system.
Human T-cell leukemia virus type 1 (HTLV-1) causes the neoplastic disease, adult T-cell leukemia-lymphoma (ATL), and various inflammatory diseases including HTLV-1 associated myelopathy (HAM) and uveitis (HU) (1). A part of HTLV-1 carriers (~5% in Japan) is estimated to develop ATL after a long latent period (2). HTLV-1 is derived from simian T-cell leukemia virus type 1 (STLV-1) (3). Interspecies transmission from monkeys to humans is estimated to have occurred ~ 50,000–20,000 years ago (4). Thus, this virus has survived for a long time in monkeys and humans. Since this virus causes persistent infection in the host, it must have strategies to survive in vivo and to enable its transmission to new hosts. To achieve these ends, the virus modulates the character of infected cells to make them resistant to host immune responses and advantageous for viral transmission. This article reviews these viral strategies, which are closely linked to the pathogenesis of HTLV-1.
An important attribute of HTLV-1 is that this virus transmits primarily through cell-to-cell contact (5). Cell-free virions have very poor infectivity even in in vitro culture (6). To facilitate viral transmission, HTLV-1 increases the number of infected T cells in vivo by causing them to proliferate (7). HTLV-1 transmits via three main routes: (1) mother-to-infant transmission through breast feeding, (2) sexual transmission, primarily male-to-female, and (3) blood transfusion and needle sharing. For transmission via breast-feeding and sexual contact, HTLV-1 infected cells must migrate into semen and breast milk. Viral genes must enable infected cells to have such attributes. T cells in the breast milk and semen have effector/memory phenotype (8). In HBZ transgenic (HBZ-Tg) mice, HBZ expressing T cells show effector/memory T-cell phenotype (9), indicating that HBZ coverts expressing T cells to effector/memory phenotype. Thus, the immunophenotype of infected cells is determined by HBZ (10). After entering into new host, Tax is essential for de novo infection (8).
The HTLV-1 provirus encodes structural genes (gag, pol, and env), regulatory genes (tax and rex) and accessory genes [p12, p13, p30 and HTLV-1 bZIP factor (HBZ)] (7). The HBZ gene is encoded in the minus strand of the provirus, and expressed as anti-sense transcripts (11), whereas all other viral genes are transcribed from the plus strand. Sense and anti-sense transcription of viral genes in vivo are differentially regulated and have different functions (Figure 1). Transcription of the sense strand genes depends on Tax. Tax trans-activates plus-strand transcription of HTLV-1 through Tax-responsive elements in long terminal repeat (LTR). Sense-strand genes encode Gag, Pol, and Env, which are essential for the formation of viral particles. Thus, sense-strand transcription is necessary for de novo infection. Tax mediated activation of sense strand genes also increases Rex expression, which inhibits splicing of viral genes, resulting in suppressed Tax expression. In contrast, the anti-sense transcript, HBZ, is not needed for de novo infection, but is critical for clonal proliferation of infected cells in vivo (12). Thus, Tax and HBZ have different roles in the life cycle of this virus.
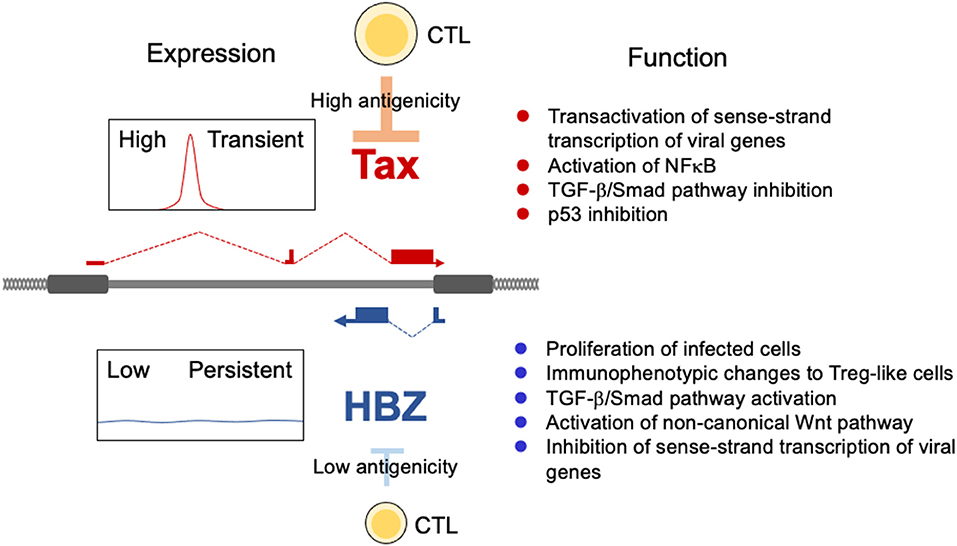
Figure 1. Expression patterns and functions of tax and HBZ. Tax is intermittently transcribed from the 5′LTR, whereas HBZ is constantly expressed from the 3′LTR. Tax function is essential for de novo infection. HBZ promotes the proliferation of expressing T cells and drives infected cells toward a Treg-like immunophenotype.
HTLV-1 is susceptible to APOBEC3G (A3G). Non-sense mutations caused by A3G are frequently observed in the tax gene (13, 14). HTLV-1 infected cells and ATL cases with mutated tax genes were also reported (14). Clonal proliferation of infected T cells with non-sense mutations of tax is found in carriers and ATL cases (15–17). These findings indicate that HBZ can induce clonal proliferation of HTLV-1 infected cells and cause ATL even without Tax (18). HBZ promotes proliferation of T cells in vitro and in vivo (19). Conversely, a burst of Tax expression (see below) suppresses cell cycling of T cells rather than inducing their proliferation (20).
Tax is essential for de novo infection by HTLV-1 (5). However, Tax is a highly immunogenic viral protein (21, 22). Cytotoxic T lymphocytes (CTLs) against Tax are frequently detected in HTLV-1 infected individuals (23). Tax expression is thus the Achilles heel of HTLV-1: it is necessary for transmission, but it renders the expressing cells vulnerable to the host immune response. Ex vivo culture of peripheral blood mononuclear cells (PBMCs) induces Tax expression, indicating that Tax expression is largely suppressed in vivo (24). HTLV-1 minimizes Tax expression by intermittent transcription (Tax burst) (20, 25). Stresses like low pH or oxidative stress can induce Tax expression (20). This Tax burst is strongly associated with the activation of p38 MAP kinase (p38 MAPK). p38 MAPKs sense extracellular stress in vivo, including heat shock, ultraviolet light, and hypoxia (5). Activation of plus-strand viral transcription is associated with an increase in the tri-methylation at the 4th lysine residue of the histone H3 protein (H3K4me3) at the HTLV-1 5′LTR promoter, and reduced levels of histone H2A monoubiquitylated at lysine 119 (H2AK119ub1) (26). The duration of Tax expression in PBMCs from an ATL patient with one dominant clone is estimated to be <1 h using single-molecule RNA FISH (27).
Transient Tax expression induces dramatic changes in the transcriptome of expressing cells. In particular, NFkB is strongly activated and anti-apoptotic genes are upregulated by the Tax burst (20). Transient Tax expression generates vigorously proliferating cells, and may be a viral mechanism for maintaining the infected cell population. This type of Tax expression is observed in about half of ATL cases (18).
In contrast to Tax, HBZ is constantly expressed in ATL cells and HTLV-1 infected cells (19, 28). Transcription of HBZ is driven by the cellular transcription factor SP1 (29). Since the immunogenicity of HBZ protein is low (30), the CTL response to HBZ is weak in vivo: although CTLs to HBZ are critical for determining the provirus load in HTLV-1 carriers (31). This is a reason why infected cells and ATL cells can express HBZ in vivo. Such low immunogenicity of viral proteins is observed in other oncogenic viruses including Epstein-Barr virus (EBV) and human papilloma virus (HPV). The necessity for persistent expression selects for low immunogenicity of these viral proteins (32).
HBZ expression affects the host cell in myriad ways, some of which are summarized in Figure 2. Of particular interest is the fact that HBZ induces transcription of the Foxp3 gene by activating the TGF-b/Smad pathway (33). Indeed, most ATL cells express Foxp3 and ~30–40% of infected T cells express Foxp3 (34). Foxp3 is the master gene of regulatory T (Treg) cells for their differentiation and functions. Therefore, HBZ-expressing T cells acquire Treg-like immunophenotypes. Furthermore, HBZ induces the expression of other Treg-associated molecules, including CCR4 and T cell immunoglobulin and ITIM domain (TIGIT) (35, 36). Treg cells express immunosuppressive molecules on their surfaces and produce immunoinhibitory cytokines like TGF-b and IL-10. These attributes of Treg cells benefit the survival of infected cells in vivo.
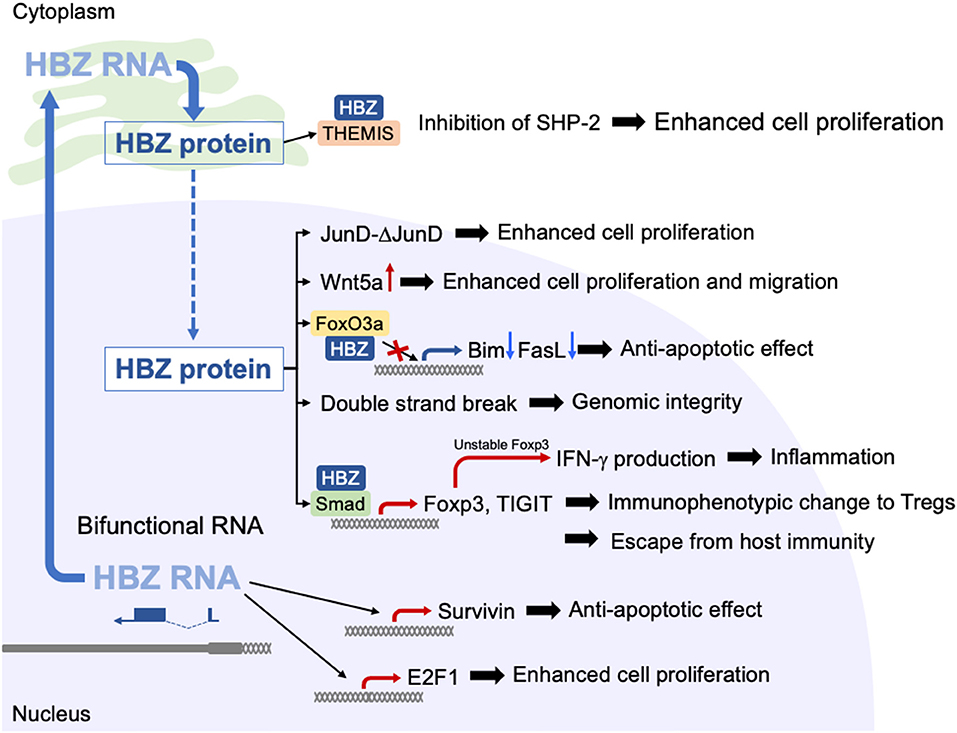
Figure 2. Functions of HBZ protein and HBZ RNA. HBZ protein and HBZ RNA have a variety of functions in expressing T cells. HBZ RNA is mainly present in the nucleus, where it promotes the proliferation of T cells and suppresses apoptosis. HBZ protein induces transcription of the Foxp3 gene by activation of TGF-b/Smad pathway.
HBZ contains bZIP domain that is similar to that of c-Fos (12). Therefore, HBZ interacts with the transcription factors of AP-1 family, such as c-Jun, JunD and ATF3 (37, 38).
Interaction between JunD and HBZ promotes proliferation of ATL cells by the following mechanism (38, 39). JunD mRNA produces two protein isoforms using alternative translation initiation sites: full-length JunD (JunD-FL) and Δ-JunD that is an N-terminal truncated form of JunD-FL. HBZ promotes translation of Δ-JunD by depleting the ribosomal protein S25 (39), which is unable to bind to a tumor suppressor, menin. Thus, enhanced Δ-JunD expression by HBZ results in promoted proliferation. In addition, HBZ protein interacts with Rb/E2F1 complex and activates the transcription of E2F-target genes associated with cell cycle progression (40).
HBZ strongly inhibits canonical Wnt pathway by interacting with lymphoid enhancer-binding factor 1 (LEF-1), and upregulates expression of non-canonical Wnt ligand, Wnt5a (41). Since knocking down of Wnt5a in ATL cells repressed cellular proliferation, activated non-canonical Wnt pathway by HBZ plays an important role in the pathogenesis of ATL.
Different from protein, RNA itself is not recognized by CTLs. Therefore, functional RNAs are of advantage for viral replication and survival of infected cells. Epstein-Barr virus (EBV) and Kaposi sarcoma herpes virus (KSHV) encode viral microRNAs (42, 43). In addition, viral microRNAs of bovine leukemia virus (BLV) are critical for proliferation of infected cells and oncogenesis (44, 45). HBZ is a unique viral gene in that HBZ mRNA functions not only to produce the protein but also as mRNA itself, in a manner resembling that of long non-coding RNAs (19, 46). Such mRNAs are named coding non-coding RNAs (cncRNAs) or bifunctional RNAs (47). Using RNA FISH, HBZ mRNA is found to be mainly present in the nucleus. When HBZ is expressed by its native promoter, the 3′LTR, HBZ mRNA is mainly present in the nucleus, but it resides in the cytoplasm when expressed by the exogenous strong promoter. The difference between the HBZ mRNAs in these two scenarios is the length of the poly A tail: poor polyadenylation is the cause of the nuclear localization of HBZ mRNA (48). HBZ mRNA expressed by the 3′LTR can promote the proliferation of T cells, whereas HBZ mRNA expressed by a strong promoter did not promote T-cell proliferation, indicating that nuclear localization is involved in this function. Interestingly, the anti-sense transcript of human immunodeficiency virus type 1 (HIV-1), ASP, is also chiefly localized in the nucleus with poor polyadenylation, indicating that this nuclear localization is common to anti-sense transcripts of the retrovirus.
The 5′ region of HBZ mRNA is responsible for its functions in the nucleus (46). This region forms a strong stem-loop structure, which is likely involved in interaction of HBZ mRNA with cellular factors. HBZ mRNA promotes the proliferation of T cells and enhances transcription of anti-apoptotic genes including survivin (46). Furthermore, HBZ mRNA interferes with the basal transcription machinery, leading to suppression of sense-transcription from the LTR (49). It is reported that HBZ protein also suppresses sense-transcription from the LTR (11). Thus, both HBZ mRNA and protein are involved in the suppression of sense transcription of viral genes (11, 49). Viral proteins that are encoded in the plus strand are well-recognized by CTLs. Silencing of transcription of sense-strand viral genes helps infected cells to escape from the host immune response.
Since HBZ is critical for survival of ATL cells, its knockdown strongly suppresses proliferation of ATL cells (19). Therefore, HBZ is the ideal therapeutic target of ATL. HBZ functions as RNA and protein. Targeting HBZ RNA is the best choice although delivery to ATL cells is very difficult. Immunization by HBZ protein can suppress ATL cells (50) although method of strong immunization with adjuvants or mRNA should be established to overcome low immunogenicity of HBZ protein.
HTLV-1 largely depends on clonal proliferation of infected cells to persist in vivo. It is critical for infected cells to evade the host immune system. One mechanism for this evasion is that HBZ causes infected cells to acquire a Treg-like phenotype. Treg cells express immunosuppressive surface molecules and produce immunosuppressive cytokines like TGF-b and IL-10, which enable the virus to evade host immunosurveillance. IL-10 secretion is elevated in HTLV-1 infected cells of carriers and ATL patients (51). IL-10 is an immunomodulating cytokine that is critical for suppressing excessive immune activation and consequent tissue damage (52). IL-10 suppresses the antigen presenting capacity of dendritic cells (DCs) and leads to the exhaustion of T cells, which allows viruses to persist (52, 53). Several viruses utilize the immunosuppressive function of IL-10 to establish persistent infection (54). For HTLV-1, HBZ-mediated enhanced expression of the co-inhibitory receptor TIGIT is thought to be a mechanism of increased IL-10 production (Figure 3) (36). TIGIT-mediated signaling increases IL-10 production from not only DCs but also T cells. Since TIGIT is a co-inhibitory receptor, its signaling normally inhibits the proliferation of T cells. However, HBZ impairs this inhibitory signaling from TIGIT via interaction with THEMIS, which forms a complex with Grb2 and SHP-2 (55). Thus, HBZ induces TIGIT expression but impairs its inhibitory function within infected cells.
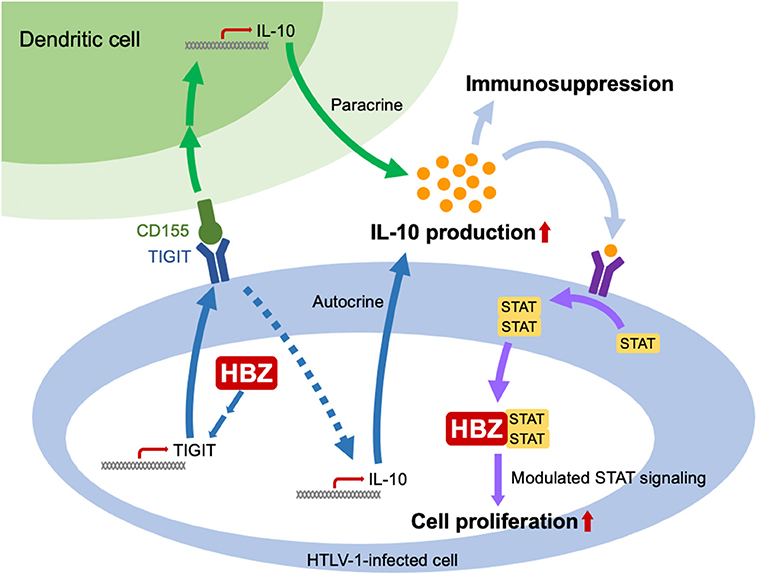
Figure 3. HBZ increases production of IL-10 and alters intracellular signaling from the IL-10 receptor. HBZ induces expression of TIGIT. When TIGIT binds to its ligand, CD155, IL-10 production by DC's is enhanced. In addition, the reverse signal from TIGIT to the infected T cell increases its IL-10 production. TGF-b production is also enhanced by HBZ (not shown). Thus, the immunosuppressive cytokines TGF-b and IL-10 inhibit the host immune response. HBZ also binds to STAT1 and 3 and modulates intracellular signaling from the IL-10 receptor, enhancing proliferation of the infected cell even while the response of neighboring uninfected cells is suppressed.
IL-10 does not promote the proliferation of normal T cells. However, it is reported to promote the proliferation of ATL cells (56). This difference is thought to be due to another activity of HBZ: HBZ modulates intracellular signaling from the IL-10 receptor by interacting with STAT1 and STAT3 (Figure 3) (34). Combining HBZ mediated enhancement of IL-10 production with modulated signaling from the IL-10 receptor seems to be a clever strategy of HTLV-1 – a strategy that enables both proliferation of infected T cells and suppression of host immune responses.
An accessory protein p12 interferes with the intracellular trafficking of major histocompatibility complex class I heavy chain (MHC-I-Hc) of HLA-A2, -B7, and -Cw4, resulting in downmodulates its cell surface expression (57). Down-modulated MHC-I-Hc impairs recognition of HTLV-1 infected cells by CTLs. It is noteworthy that p12 is expressed from the plus-strand of the provirus by Tax-dependent transcription. Immunosuppressive effect of p12 enables Tax expressing cells to escape from CTLs. Loss of MHC-I allows attack from natural killer (NK) cells. However, p12 also down-modulates expression of intercellular adhesion molecule 1 (ICAM-1) and ICAM-2, and K cell activating receptors, NCR and NKG2D (58), which confers resistance of HTLV-1 infected cells to NK cells.
High throughput sequencing enables us to identify a wide variety of HTLV-1 provirus integration sites (17). The presence of identical integration sites among cells of different hematopoietic lineages (CD4 T cells, CD8 T cells, B cells, monocytes and neutrophils) in the same HTLV-1 infected individuals (59). This is also demonstrated by the report of two cases with ATL clones that had different T-cell receptor gene rearrangements and identical proviral integration sites (60). These data suggest that HTLV-1 infects hematopoietic stem cells and that infected cells differentiate in vivo. It is possible that HBZ directs the differentiation toward Treg cells. What is the advantage for HTLV-1 to infect hematopoietic stem cells? Since the bone marrow (BM) is under hypoxic conditions, immune responses are suppressed (61), which likely allows infected cells to express Tax. Indeed, an unexpectedly high frequency of tax mRNA-expressing cells was reported in the BM of HAM patients (62). Newly infected hematopoietic stem cells at the BM can differentiate without Tax expression. HBZ directs differentiation of infected cells to Treg cells and promotes their proliferation. Since the actions of HBZ are specialized to Treg cells, it is unlikely that HBZ increases the number of other hematopoietic cells. Thus, most infected cells only have to express the HBZ gene and the necessity to express more immunogenic Tax is not essential in the periphery.
Latently infected cells lead to development of ATL in some HTLV-1 carriers. Although ATL is caused by HTLV-1, the requirements of ATL cells for viral genes are not uniform. Tax is not expressed in approximately half of ATL cases, whereas HBZ is expressed in all (63). Importantly, mutations that abrogate Tax expression can occur very early, before proviral integration, and ATL can still develop. Non-sense mutations of the tax gene are formed by APOBEC3G, which means that they are generated before the proviral integration (14). Furthermore, deletion of the 5′LTR also occurs before the integration of HTLV-1 provirus, since the genomic regions adjacent to the LTR retain six bp repeats (64). Since Tax is not expressed before proviral integration, these findings indicate that leukemogenesis of these ATL cases depends on HBZ alone (18).
In the other half of ATL cases, the HTLV-1 provirus retains the structure to express Tax (intact tax gene and 5′LTR, unmethylated 5′LTR). In these cases, the level of tax transcription is low, indicating that ATL cells in these cases are similar to the MT-1 cell line, which expresses Tax intermittently (20). It is noteworthy that only in vitro cultured HTLV-1 infected cell lines produce abundant Tax. Most of these cell lines are established only in vitro and do not reflect ATL cells in vivo.
HTLV-1 has existed in humans for a long time since its interspecies transmission from monkeys. STLV-1 has existed in monkeys for even longer. These viruses have acquired strategies to evade the host immune response and to increase their chances of transmission. Treg like cells are suitable resident cells for HTLV-1 to escape from CTLs. Treatments that intervene in these strategies could be useful in preventing the development of ATL.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This research was supported by a grant from the Project for Cancer Research and Therapeutic Evolution (P-CREATE) (20cm0106306h0005 to MM), the Research Program on Emerging and Re-emerging Infectious Diseases (20fk0108088h0002 to MM) from the Japan Agency for Medical Research and Development (AMED), and JSPS KAKENHI (19H03689 to MM and 20K22904 to YH). This study was also supported in part by the JSPS Core-to-Core Program A, Advanced Research Networks.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Matsuoka M, Jeang KT. Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene. (2011) 30:1379–89. doi: 10.1038/onc.2010.537
2. Bangham CRM, Matsuoka M. Human T-cell leukaemia virus type 1: parasitism and pathogenesis. Philos Trans R Soc Lond B Biol Sci. (2017) 372:20160272. doi: 10.1098/rstb.2016.0272
3. Van Dooren S, Salemi M, Vandamme AM. Dating the origin of the African human T-cell lymphotropic virus type-i (HTLV-I) subtypes. Mol Biol Evol. (2001) 18:661–71. doi: 10.1093/oxfordjournals.molbev.a003846
4. Slattery JP, Franchini G, Gessain A. Genomic evolution, patterns of global dissemination, and interspecies transmission of human and simian T-cell leukemia/lymphotropic viruses. Genome Res. (1999) 9:525–40. doi: 10.1101/gr.9.6.525
5. Bangham CRM, Miura M, Kulkarni A, Matsuoka M. Regulation of latency in the human T cell leukemia virus, HTLV-1. Annu Rev Virol. (2019) 6:365–85. doi: 10.1146/annurev-virology-092818-015501
6. Mazurov D, Ilinskaya A, Heidecker G, Lloyd P, Derse D. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. (2010) 6:e1000788. doi: 10.1371/journal.ppat.1000788
7. Matsuoka M, Jeang KT. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer. (2007) 7:270–80. doi: 10.1038/nrc2111
8. Matsuoka M, Yasunaga J. Human T-cell leukemia virus type 1: replication, proliferation and propagation by Tax and HTLV-1 bZIP factor. Curr Opin Virol. (2013) 3:684–91. doi: 10.1016/j.coviro.2013.08.010
9. Satou Y, Yasunaga J, Zhao T, Yoshida M, Miyazato P, Takai K, et al. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. (2011) 7:e1001274. doi: 10.1371/journal.ppat.1001274
10. Tanaka A, Matsuoka M. HTLV-1 Alters T cells for viral persistence and transmission. Front Microbiol. (2018) 9:461. doi: 10.3389/fmicb.2018.00461
11. Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol. (2002) 76:12813–22. doi: 10.1128/JVI.76.24.12813-12822.2002
12. Matsuoka M, Mesnard JM. HTLV-1 bZIP factor: the key viral gene for pathogenesis. Retrovirology. (2020) 17:2. doi: 10.1186/s12977-020-0511-0
13. Furukawa Y, Kubota R, Tara M, Izumo S, Osame M. Existence of escape mutant in HTLV-I tax during the development of adult T-cell leukemia. Blood. (2001) 97:987–93. doi: 10.1182/blood.V97.4.987
14. Fan J, Ma G, Nosaka K, Tanabe J, Satou Y, Koito A, et al. APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J Virol. (2010) 84:7278–87. doi: 10.1128/JVI.02239-09
15. Wattel E, Vartanian JP, Pannetier C, Wain-Hobson S. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J Virol. (1995) 69:2863–8. doi: 10.1128/jvi.69.5.2863-2868.1995
16. Etoh K, Tamiya S, Yamaguchi K, Okayama A, Tsubouchi H, Ideta T, et al. Persistent clonal proliferation of human T-lymphotropic virus type I-infected cells in vivo. Cancer Res. (1997) 57:4862–7.
17. Gillet NA, Malani N, Melamed A, Gormley N, Carter R, Bentley D, et al. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood. (2011) 117:3113–22. doi: 10.1182/blood-2010-10-312926
18. Nosaka K, Matsuoka M. Adult T-cell leukemia-lymphoma as a viral disease: subtypes based on viral aspects. Cancer Sci. (2021) 112:1688–94. doi: 10.1111/cas.14869
19. Satou Y, Yasunaga J, Yoshida M, Matsuoka M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA. (2006) 103:720–5. doi: 10.1073/pnas.0507631103
20. Mahgoub M, Yasunaga JI, Iwami S, Nakaoka S, Koizumi Y, Shimura K, et al. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc Natl Acad Sci USA. (2018) 115:E1269–78. doi: 10.1073/pnas.1715724115
21. Jacobson S, Shida H, Mcfarlin DE, Fauci AS, Koenig S. Circulating CD8+ cytotoxic T lymphocytes specific for HTLV-I pX in patients with HTLV-I associated neurological disease. Nature. (1990) 348:245–8. doi: 10.1038/348245a0
22. Kannagi M, Harada S, Maruyama I, Inoko H, Igarashi H, Kuwashima G, et al. Predominant recognition of human T cell leukemia virus type I (HTLV-I) pX gene products by human CD8+ cytotoxic T cells directed against HTLV-I-infected cells. Int Immunol. (1991) 3:761–7. doi: 10.1093/intimm/3.8.761
23. Hilburn S, Rowan A, Demontis MA, Macnamara A, Asquith B, Bangham CR, et al. In vivo expression of human t-lymphotropic virus type 1 basic Leucine-Zipper protein generates specific CD8+ and CD4+ T-lymphocyte responses that correlate with clinical outcome. J Infect Dis. (2011) 203:529–36. doi: 10.1093/infdis/jiq078
24. Hanon E, Hall S, Taylor GP, Saito M, Davis R, Tanaka Y, et al. Abundant tax protein expression in CD4+ T cells infected with human T- cell lymphotropic virus type I (HTLV-I) is prevented by cytotoxic T lymphocytes. Blood. (2000) 95:1386–92. doi: 10.1182/blood.V95.4.1386.004k22_1386_1392
25. Billman MR, Rueda D, Bangham CRM. Single-cell heterogeneity and cell-cycle-related viral gene bursts in the human leukaemia virus HTLV-1. Wellcome Open Res. (2017) 2:87. doi: 10.12688/wellcomeopenres.12469.2
26. Kulkarni A, Taylor GP, Klose RJ, Schofield CJ, Bangham CR. Histone H2A monoubiquitylation and p38-MAPKs regulate immediate-early gene-like reactivation of latent retrovirus HTLV-1. JCI Insight. (2018) 3:e123196. doi: 10.1172/jci.insight.123196
27. Miura M, Dey S, Ramanayake S, Singh A, Rueda DS, Bangham CRM. Kinetics of HTLV-1 reactivation from latency quantified by single-molecule RNA FISH and stochastic modelling. PLoS Pathog. (2019) 15:e1008164. doi: 10.1371/journal.ppat.1008164
28. Saito M, Matsuzaki T, Satou Y, Yasunaga J, Saito K, Arimura K, et al. In vivo expression of the HBZ gene of HTLV-1 correlates with proviral load, inflammatory markers and disease severity in HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP). Retrovirology. (2009) 6:19. doi: 10.1186/1742-4690-6-19
29. Yoshida M, Satou Y, Yasunaga J, Fujisawa J, Matsuoka M. Transcriptional control of spliced and unspliced human T-cell leukemia virus type 1 bZIP factor (HBZ) gene. J Virol. (2008) 82:9359–68. doi: 10.1128/JVI.00242-08
30. Sugata K, Yasunaga J, Miura M, Akari H, Utsunomiya A, Nosaka K, et al. Enhancement of anti-STLV-1/HTLV-1 immune responses through multimodal effects of anti-CCR4 antibody. Sci Rep. (2016) 6:27150. doi: 10.1038/srep27150
31. Macnamara A, Rowan A, Hilburn S, Kadolsky U, Fujiwara H, Suemori K, et al. HLA class I binding of HBZ determines outcome in HTLV-1 infection. PLoS Pathog. (2010) 6:e1001117. doi: 10.1371/journal.ppat.1001117
32. Stuber G, Dillner J, Modrow S, Wolf H, Szekely L, Klein G, et al. HLA-A0201 and HLA-B7 binding peptides in the EBV-encoded EBNA-1, EBNA-2 and BZLF-1 proteins detected in the MHC class I stabilization assay. Low proportion of binding motifs for several HLA class I alleles in EBNA-1. Int Immunol. (1995) 7:653–63. doi: 10.1093/intimm/7.4.653
33. Zhao T, Satou Y, Sugata K, Miyazato P, Green PL, Imamura T, et al. HTLV-1 bZIP factor enhances TGF-{beta} signaling through p300 coactivator. Blood. (2011) 118:1865–76. doi: 10.1182/blood-2010-12-326199
34. Higuchi Y, Yasunaga JI, Mitagami Y, Tsukamoto H, Nakashima K, Ohshima K, et al. HTLV-1 induces T cell malignancy and inflammation by viral antisense factor-mediated modulation of the cytokine signaling. Proc Natl Acad Sci USA. (2020) 117:13740–9. doi: 10.1073/pnas.1922884117
35. Sugata K, Yasunaga J, Kinosada H, Mitobe Y, Furuta R, Mahgoub M, et al. HTLV-1 viral factor HBZ induces CCR4 to promote T-cell migration and proliferation. Cancer Res. (2016) 76:5068–79. doi: 10.1158/0008-5472.CAN-16-0361
36. Yasuma K, Yasunaga J, Takemoto K, Sugata K, Mitobe Y, Takenouchi N, et al. HTLV-1 bZIP factor impairs anti-viral immunity by inducing co-inhibitory molecule, T cell immunoglobulin and ITIM domain (TIGIT). PLoS Pathog. (2016) 12:e1005372. doi: 10.1371/journal.ppat.1005372
37. Hagiya K, Yasunaga J, Satou Y, Ohshima K, Matsuoka M. ATF3, an HTLV-1 bZip factor binding protein, promotes proliferation of adult T-cell leukemia cells. Retrovirology. (2011) 8:19. doi: 10.1186/1742-4690-8-19
38. Gazon H, Lemasson I, Polakowski N, Cesaire R, Matsuoka M, Barbeau B, et al. Human T-cell leukemia virus type 1 (HTLV-1) bZIP factor requires cellular transcription factor JunD to upregulate HTLV-1 antisense transcription from the 3′ long terminal repeat. J Virol. (2012) 86:9070–8. doi: 10.1128/JVI.00661-12
39. Terol M, Gazon H, Lemasson I, Duc-Dodon M, Barbeau B, Cesaire R, et al. HBZ-mediated shift of JunD from growth suppressor to tumor promoter in leukemic cells by inhibition of ribosomal protein S25 expression. Leukemia. (2017) 31:2235–43. doi: 10.1038/leu.2017.74
40. Kawatsuki A, Yasunaga JI, Mitobe Y, Green PL, Matsuoka M. HTLV-1 bZIP factor protein targets the Rb/E2F-1 pathway to promote proliferation and apoptosis of primary CD4(+) T cells. Oncogene. (2016) 35:4509–17. doi: 10.1038/onc.2015.510
41. Ma G, Yasunaga J, Fan J, Yanagawa S, Matsuoka M. HTLV-1 bZIP factor dysregulates the Wnt pathways to support proliferation and migration of adult T-cell leukemia cells. Oncogene. (2013) 32:4222–30. doi: 10.1038/onc.2012.450
42. Qin J, Li W, Gao SJ, Lu C. KSHV microRNAs: tricks of the devil. Trends Microbiol. (2017) 25:648–61. doi: 10.1016/j.tim.2017.02.002
43. Iizasa H, Kim H, Kartika AV, Kanehiro Y, Yoshiyama H. Role of viral and host microRNAs in immune regulation of Epstein-Barr virus-associated diseases. Front Immunol. (2020) 11:367. doi: 10.3389/fimmu.2020.00367
44. Kincaid RP, Burke JM, Sullivan CS. RNA virus microRNA that mimics a B-cell oncomiR. Proc Natl Acad Sci USA. (2012) 109:3077–82. doi: 10.1073/pnas.1116107109
45. Safari R, Jacques JR, Brostaux Y, Willems L. Ablation of non-coding RNAs affects bovine leukemia virus B lymphocyte proliferation and abrogates oncogenesis. PLoS Pathog. (2020) 16:e1008502. doi: 10.1371/journal.ppat.1008502
46. Mitobe Y, Yasunaga J, Furuta R, Matsuoka M. HTLV-1 bZIP Factor RNA and protein impart distinct functions on T-cell proliferation and survival. Cancer Res. (2015) 75:4143–52. doi: 10.1158/0008-5472.CAN-15-0942
47. Hube F, Francastel C. Coding and non-coding RNAs, the frontier has never been so blurred. Front Genet. (2018) 9:140. doi: 10.3389/fgene.2018.00140
48. Ma G, Yasunaga JI, Shimura K, Takemoto K, Watanabe M, Amano M, et al. Human retroviral antisense mRNAs are retained in the nuclei of infected cells for viral persistence. Proc Natl Acad Sci USA. (2021) 118:e2014783118. doi: 10.1073/pnas.2014783118
49. Gazon H, Chauhan PS, Porquet F, Hoffmann GB, Accolla R, Willems L. Epigenetic silencing of HTLV-1 expression by the HBZ RNA through interference with the basal transcription machinery. Blood Adv. (2020) 4:5574–9. doi: 10.1182/bloodadvances.2020001675
50. Sugata K, Yasunaga J, Mitobe Y, Miura M, Miyazato P, Kohara M, et al. Protective effect of cytotoxic T lymphocytes targeting HTLV-1 bZIP factor. Blood. (2015) 126:1095–105. doi: 10.1182/blood-2015-04-641118
51. Kagdi H, Demontis MA, Ramos JC, Taylor GP. Switching and loss of cellular cytokine producing capacity characterize in vivo viral infection and malignant transformation in human T- lymphotropic virus type 1 infection. PLoS Pathog. (2018) 14:e1006861. doi: 10.1371/journal.ppat.1006861
52. Rojas JM, Avia M, Martin V, Sevilla N. IL-10: a multifunctional cytokine in viral infections. J Immunol Res. (2017) 2017:6104054. doi: 10.1155/2017/6104054
53. Blackburn SD, Wherry EJ. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. (2007) 15:143–6. doi: 10.1016/j.tim.2007.02.006
54. Ouyang P, Rakus K, Van Beurden SJ, Westphal AH, Davison AJ, Gatherer D, et al. IL-10 encoded by viruses: a remarkable example of independent acquisition of a cellular gene by viruses and its subsequent evolution in the viral genome. J Gen Virol. (2014) 95:245–62. doi: 10.1099/vir.0.058966-0
55. Kinosada H, Yasunaga JI, Shimura K, Miyazato P, Onishi C, Iyoda T, et al. HTLV-1 bZIP factor enhances T-cell proliferation by impeding the suppressive signaling of co-inhibitory receptors. PLoS Pathog. (2017) 13:e1006120. doi: 10.1371/journal.ppat.1006120
56. Sawada L, Nagano Y, Hasegawa A, Kanai H, Nogami K, Ito S, et al. IL-10-mediated signals act as a switch for lymphoproliferation in Human T-cell leukemia virus type-1 infection by activating the STAT3 and IRF4 pathways. PLoS Pathog. (2017) 13:e1006597. doi: 10.1371/journal.ppat.1006597
57. Johnson JM, Nicot C, Fullen J, Ciminale V, Casareto L, Mulloy JC, et al. Free major histocompatibility complex class I heavy chain is preferentially targeted for degradation by human T-cell leukemia/lymphotropic virus type 1 p12(I) protein. J Virol. (2001) 75:6086–94. doi: 10.1128/JVI.75.13.6086-6094.2001
58. Banerjee P, Feuer G, Barker E. Human T-cell leukemia virus type 1 (HTLV-1) p12I down-modulates ICAM-1 and−2 and reduces adherence of natural killer cells, thereby protecting HTLV-1-infected primary CD4+ T cells from autologous natural killer cell-mediated cytotoxicity despite the reduction of major histocompatibility complex class I molecules on infected cells. J Virol. (2007) 81:9707–17. doi: 10.1128/JVI.00887-07
59. Furuta R, Yasunaga JI, Miura M, Sugata K, Saito A, Akari H, et al. Human T-cell leukemia virus type 1 infects multiple lineage hematopoietic cells in vivo. PLoS Pathog. (2017) 13:e1006722. doi: 10.1371/journal.ppat.1006722
60. Rauch DA, Conlon KC, Janakiram M, Brammer JE, Harding JC, Ye BH, et al. Rapid progression of adult T-cell leukemia/lymphoma as tumor-infiltrating Tregs after PD-1 blockade. Blood. (2019) 134:1406–14. doi: 10.1182/blood.2019002038
61. Vito A, El-Sayes N, Mossman K. Hypoxia-driven immune escape in the tumor microenvironment. Cells. (2020) 9:992. doi: 10.3390/cells9040992
62. Levin MC, Krichavsky M, Fox RJ, Lehky T, Jacobson S, Fox C, et al. Extensive latent retroviral infection in bone marrow of patients with HTLV-I-associated neurologic disease. Blood. (1997) 89:346–8. doi: 10.1182/blood.V89.1.346
63. Takeda S, Maeda M, Morikawa S, Taniguchi Y, Yasunaga J, Nosaka K, et al. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int J Cancer. (2004) 109:559–67. doi: 10.1002/ijc.20007
Keywords: HTLV-1, HBZ, tax, regulatory T cell, IL-10
Citation: Higuchi Y, Yasunaga J-i and Matsuoka M (2022) HTLV-1's Foxy Strategy for Survival and Transmission. Front. Virol. 1:792659. doi: 10.3389/fviro.2021.792659
Received: 10 October 2021; Accepted: 07 December 2021;
Published: 03 January 2022.
Edited by:
Masaaki Miyazawa, Kindai University, JapanReviewed by:
Yuetsu Tanaka, University of the Ryukyus, JapanCopyright © 2022 Higuchi, Yasunaga and Matsuoka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masao Matsuoka, bWFtYXRzdUBrdW1hbW90by11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.