 Sun-Hak Lee1
Sun-Hak Lee1 Heesu Lee1
Heesu Lee1 Andrew Y. Cho1
Andrew Y. Cho1 Tae-Hyeon Kim1
Tae-Hyeon Kim1 Yun-Jeong Choi1
Yun-Jeong Choi1 Nyamsuren Otgontogtokh1
Nyamsuren Otgontogtokh1 Ye-Ram Seo1
Ye-Ram Seo1 Dong-Yeop Lee2
Dong-Yeop Lee2 Erdene-Ochir Tseren-Ochir3
Erdene-Ochir Tseren-Ochir3 Temuulen Myagmarsuren3
Temuulen Myagmarsuren3 Dong-Hun Lee2
Dong-Hun Lee2 Ji-Yeon Hyeon1*
Ji-Yeon Hyeon1* Chang-Seon Song1
Chang-Seon Song1- 1Avian Disease Laboratory, College of Veterinary Medicine, Konkuk University, Seoul, Republic of Korea
- 2Wildlife Health Laboratory, College of Veterinary Medicine, Konkuk University, Seoul, Republic of Korea
- 3Department of Infectious Diseases and Microbiology, School of Veterinary Medicine, Mongolian University of Life Sciences, Ulaanbaatar, Mongolia
1 Introduction
Infectious bronchitis virus (IBV) causes considerable economic losses in poultry industry due to respiratory diseases, decreased egg production, and increased mortality in chickens (1). The virus, a member of the genus Gammacoronaviruses and family Coronaviridae, has a highly mutable RNA genome that facilitates the rapid emergence of new variants (2). Its genome consists of single-stranded positive-sense RNA (~27.6 kb in size), which encodes four structural proteins [spike (S), envelope (E), membrane (M), and nucleocapsid (N)] (3) and several non-structural proteins, including two polyproteins (pp1a and pp1ab), and accessory proteins (3a, 3b, 5a, and 5b) (4, 5).
The IBV strains were categorized into six genotypes encompassing 32 distinct viral lineages based on the full S1 gene sequence (6). The geographic distribution and diversity of IBV lineages varied significantly. Some lineages, such as the Mass-type, 4/91 (793 B or CR88)-like, D274-like, D3128, QX-like, and Italy02, are globally widespread (7). Conversely, the GI-23 lineage, which includes Variant-2 viruses, is predominantly found in the Middle East (6).
Despite the distinct origins, evolution, and transmission dynamics of each genotype, several common factors including live poultry trade and use of exogenous live vaccines contribute to their widespread distribution across various countries (8). For example, the GI-5 and GI-6 genotypes of IBV, originating from the Australian vaccine strains JAAS and J9, respectively, introduced into China, have contributed to the genetic diversity observed in IBV populations (9).
The global spread of IBV underscores the critical need for comprehensive surveillance. Early detection through active surveillance of poultry flocks or products can identify novel IBVs before it becomes widespread, aiding in virus control and contributing to global efforts to track its evolution and transmission dynamics (10).
In this study, we detected a novel IBV strain in retail chicken meat, sampled in a retail grocery market in Mongolia in 2023. For whole-genome sequencing of the IBV isolate, we utilized the Sequence-Independent Single-Primer Amplification (SISPA) protocol for next-generation sequencing. We conducted a comprehensive phylogenetic analysis of the S1 gene to determine the genetic relatedness of the isolated IBVs to the globally distributed IBVs. Additionally, comparative genome analysis revealed recombination events in the IBV isolate, highlighting its genetic diversity and evolutionary complexity.
2 Materials and methods
2.1 Sample collection and virus isolation
On August 21, 2023, six chicken meat samples were purchased from a retail grocery market in Ulaanbaatar, Mongolia. Four of these chickens were locally sourced from Mongolia, whereas the remaining two were imported from Russia, processed in April 2023, and distributed in a frozen state. Each chicken meat sample was placed separately in a sterile plastic bag containing 400 mL of phosphate buffered saline (PBS) and shaken for 2 min. The resulting rinse was clarified using centrifugation at 3,000 rpm for 10 min and filtered through a sterile 0.45 μm Minisart® Syringe Filter (Sartorius, Göttingen, Germany). Viruses were isolated via the allantoic route in 10-day-old specific-pathogen-free (SPF) chicken embryos, with each embryo receiving a 0.2 mL inoculation of the supernatant from the rinse solution, followed by incubation at 37°C for 72 h. The allantoic fluid harvested from the eggs was tested for hemagglutination (HA) assay to screen viruses including avian influenza virus (11). RNA was extracted from the allantoic fluid using a Qiagen RNeasy Mini Kit (Hilden, Germany) and IBV was detected using real-time quantitative reverse transcription PCR (qRT-PCR) as previously described (12), using Qiagen Quantitect RT-PCR reagents (Qiagen, Manchester, UK).
2.2 NGS preparation using SISPA
The SISPA method was employed for amplification of the RNA viral genomes utilizing random priming, as previously described (13). Briefly, cDNA was synthesized from DNase I-treated RNA using the SuperScript™ IV First-Strand Synthesis System (Thermo Fisher Scientific, Waltham, MA, USA) with primer K-8N. The complementary strand of cDNA was synthesized using DNA Polymerase I, Large (Klenow) Fragment [New England Biolabs (NEB), Ipswich, MA, USA], and the primer K-8 N, resulting in dsDNA. The resulting products were purified using a QIAquick PCR Purification Kit (QIAGEN, Hilden, Germany). Purified dsDNA was amplified using a Phusion High-Fidelity PCR kit (New England Biolabs, Ipswich USA, MA) with primer K, followed by purification using the same method. All procedures were performed according to manufacturer's instructions. Library preparation and sequencing were performed at Bionics (Seoul, Republic of Korea) using an Illumina MiSeq next-generation sequencing (NGS) system (San Diego, California, USA).
2.3. Assembly and phylogenetic analysis
The resulting raw reads were trimmed to remove adapters and low-quality bases using BBDuk v35.49. After de novo assembly of the trimmed reads using Geneious assembler, reference mapping was performed using Geneious mapper in Geneious G9.0.5(Biomatters, Auckland, New Zealand). A reference sequence (GenBank accession no. KJ135013) was selected based on BLAST results of contigs.
To determine the genotype and lineage based on the S1 phylogeny-based classification system (6), S1 gene sequences, including representative sequences for each genotype, were obtained from GenBank (Supplementary Table 1). To investigate the genetic relationship with global IBV lineages, the S1 gene sequences of the determined lineage and sequences with high nucleotide sequence identities (based on BLASTn results) were downloaded from the GenBank database (Supplementary Table 2). All sequences were aligned using MAFFT v7.308 for multiple sequence alignment (14). A maximum-likelihood phylogenetic tree was constructed using RAxML with a general time-reversible (GTR) nucleotide model and gamma distribution, along with 1,000 rapid bootstrap replicates (15).
2.4 Recombination analysis
Potential recombination events were investigated using Recombination Detection Program (RDP) 5 software (v.5.56) employing multiple detection methods, including RDP, GENECONV, BootScan, MaxChi, Chimera, SiScan, Phylpro, LARD, and 3Seq, as previously described (16). Recombination events were considered significant when at least five detection methods yielded a p-value < 0.01. A confirmatory analysis of potential recombination events and breakpoints was conducted using the SimPlot Program (version 3.5.1) and BootScan analyses (17). Nucleotide identity comparisons were performed using the Kimura 2-parameter method with a transition-transversion ratio of 2, a window size of 500 bp, and a step size of 50 bp. BootScan analyses were conducted using the neighbor-joining method with 100 replicates to ensure robust validation of recombination events. Recombination breakpoints were analyzed by maximization of χ2 using the program Findsites included in the SimPlot. The similarity plot displays the percentage of permuted trees (%) among the queried strain and parental strains. Strains were considered as recombinants if any crossover event took place between three putative parental strains.
3 Descriptive results
Among the six samples, we isolated the MR23/6/Mongolia/2023 (MR23/6) virus from a chicken sample imported from Russia using chicken embryo inoculation and confirmed the presence of IBV by real-time qRT-PCR, while none of the samples showed positive for HA activity in the HA assay. A total of 211,210 NGS reads were produced, resulting in a contig length of 27,526 bp and 14 coding sequences (CDSs). The complete genome was deposited in the GenBank database under accession no. PP871397.
Classification based on the S1 gene confirmed that the virus belonged to the GI-19 lineage (Figure 1A, Supplementary Figure 1A). Phylogenetic analysis of GI-19 lineage viruses revealed that the isolate clustered with other GI-19 lineage viruses from Asia (Korea and China) and Europe (Poland, Ukraine, the United Kingdom, the Netherlands, Italy, and Spain; Figure 1B, Supplementary Figure 1B). It formed a distinct long branch, indicating that no highly similar viruses have been identified, suggesting the need for additional genomic surveillance.
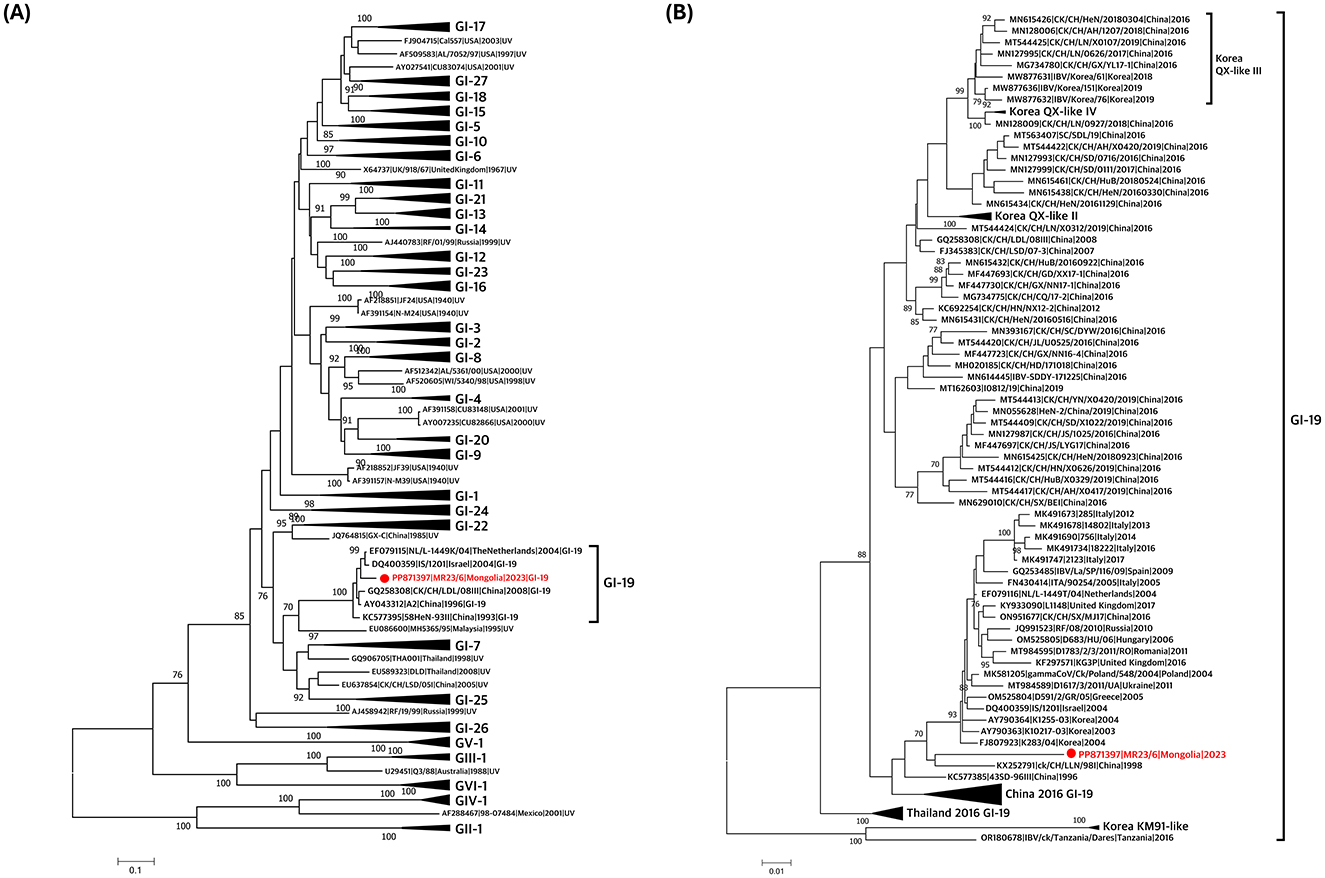
Figure 1. Phylogenetic trees based on alignment of S1 gene sequences of the Mongolian infectious bronchitis virus (IBV) isolate (red) and reference strains. The trees were created by the maximum-likelihood method in RAxML using Tamura-Nei model and 1,000 bootstrap replicates. The IBVs newly isolated in this study are shown in red with a circle. (A) S1 gene-based phylogenetic tree was constructed using 191 reference sequences. All lineages except for GI-19 lineage were compressed. (B) S1 gene-based phylogenetic tree was constructed using 101 IBV strains previously reported as GI-19 lineage. Korean GI-19 genotypes, Chinese GI-19 lineage in 2016, and Thailand GI-19 lineage in 2016 were compressed.
BLAST search of the complete genome revealed the highest similarity to the CK/CH/MY/2020 strain identified in China, with a sequence identity of 92.66%. BLAST search of each CDS identified genetically similar IBVs in a range of 90.07%−100% sequence identity (Supplementary Table 3). Based on these results, 22 reference strains were selected for the analysis of potential recombination events. The RDP analysis confirmed five recombination events (Supplementary Table 4). The IBV strains YX10, CK/CH/MY/2020, and gammaCoV/Ck/Poland/G052/2016, derived from the RDP analysis, were used as putative parental strains for SimPlot analysis when the MR23/6 was queried (Figure 2). The similarity plot also identified three putative parental strains, the YX10 and CK/CH/MY/2020 originated from China, belonging to the GI-19 and GI-28 lineage, respectively (18, 19) and the gammaCoV/Ck/Poland/G052/2016 belonging to the GI-23 (Var2-like) lineage, which traces its origin back to the Middle East (20). Reassortant IBVs produced by recombination between IBVs of Chinese and European origin also have been reported in Italy (21, 22) and Spain (22), highlighting frequent reassortment of IBVs in Eurasia.
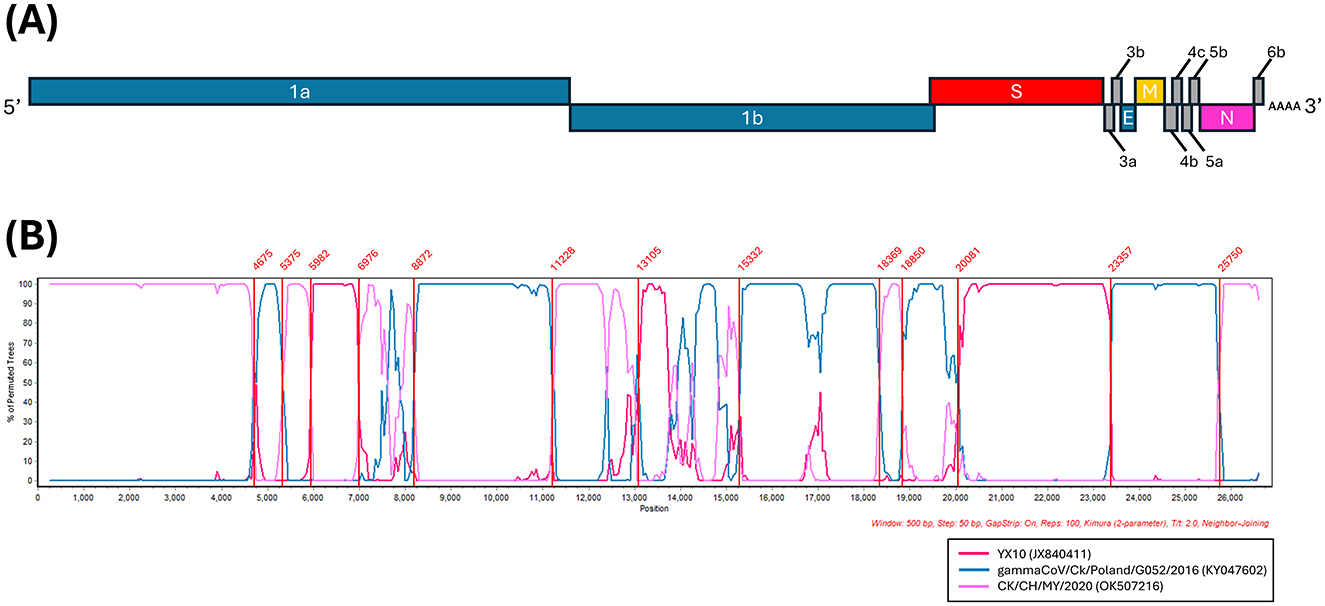
Figure 2. Multiple recombination events detected in the Mongolian IBV isolate. (A) Genome organization of the Mongolian IBV isolate (MR23/6). (B) Simplot analysis was performed to detect recombination in the whole genome from two Chinese IBV isolates, CK/CH/MY/2020 (JX840411) and YX10 (JX840411), and one Poland IBV isolate, gammaCoV/Ck/Poland/G052/2016 (KY047602). Recombination breakpoints analyzed by maximization of χ2 using the program Findsites included in the SimPlot were indicated within the plot (lines and numbers in red). Bootscan evidence for the recombination origin analyzed with Simplot based on pairwise distance, modeled with a window size of 500 bp and a step size of 50 bp, and 100 Bootstrap replicates.
In conclusion, we report the first isolation and genome sequences of reassortant GI-19 IBV identified from retail chicken meat in Mongolia. Our findings also highlight the practicality of utilizing retail chicken meat samples from grocery markets for pathogen surveillance, circumventing the necessity for direct sampling of live animals.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because Ethical approval was not required for this study because it involved the use of retailed chicken meats purchased from a grocery store, rather than live animals. The research did not involve any procedures on living animals or the collection of samples from live animals.
Author contributions
S-HL: Data curation, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. HL: Data curation, Formal Analysis, Writing – review & editing. AC: Formal Analysis, Writing – review & editing. T-HK: Formal Analysis, Writing – review & editing. Y-JC: Writing – review & editing. NO: Writing – review & editing. Y-RS: Formal Analysis, Writing – review & editing. D-YL: Formal Analysis, Writing – review & editing. E-OT-O: Project administration, Writing – review & editing. TM: Formal Analysis, Writing – review & editing. D-HL: Supervision, Validation, Writing – review & editing. J-YH: Project administration, Supervision, Validation, Writing – review & editing. C-SS: Conceptualization, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the ZODIAC in Asia and the Pacific project (CRP D32039) of International Atomic Energy Agency.
Acknowledgments
This research was supported by the KU Research Professor Program of the Konkuk University.
Conflict of interest
All authors declare that this research was conducted without any commercial or financial relationships that could be interpreted as potential conflicts of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2024.1465342/full#supplementary-material
Supplementary Table 1 | Metadata for infectious bronchitis virus sequences (n = 190) used in S1 gene based phylogenetic analysis (Supplementary Figure 1A).
Supplementary Table 2 | Metadata for GI-19 lineage infectious bronchitis virus sequences (n = 102) used in S1 gene based phylogenetic analysis (Supplementary Figure 1B).
Supplementary Table 3 | Pairwise comparison of nucleotide homology of coding sequences (CDS) between the Mongolian infectious bronchitis virus (MR23-6) and other IBV strains.
Supplementary Table 4 | Recombination events detected in whole genome of the Mongolian infectious bronchitis virus isolate (MR23/6).
Supplementary Figure 1 | (A) Phylogenetic trees based on alignment of S1 gene sequences of the Mongolian infectious bronchitis virus (IBV) isolate (red) and reference strains. The trees were created by the maximum-likelihood method in RAxML using Tamura-Nei model and 1,000 bootstrap replicates. The IBVs newly isolated in this study are shown in red with a circle. S1 gene-based phylogenetic tree was constructed using 191 reference sequences. (B) Phylogenetic trees based on alignment of S1 gene sequences of the Mongolian infectious bronchitis virus (IBV) isolate (red) and reference strains. The trees were created by the maximum-likelihood method in RAxML using Tamura-Nei model and 1,000 bootstrap replicates. The IBVs newly isolated in this study are shown in red with a circle. S1 gene-based phylogenetic tree was constructed using 101 IBV strains previously reported as GI-19 lineage.
References
1. Jackwood MW, de Wit S. Infectious bronchitis. In: Swayne DE, Boulianne M, Logue CM, McDougald LR, Nair V, Suarez DL, et al., , editors. Diseases of Poultry. Hoboken, NJ: John Wiley and Sons (2013), p. 139–59. doi: 10.1002/9781119421481.ch4
2. Gonzalez J, Gomez-Puertas P, Cavanagh D, Gorbalenya A, Enjuanes L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch Virol. (2003) 148:2207–35. doi: 10.1007/s00705-003-0162-1
3. Jackwood MW, Hall D, Handel A. Molecular evolution and emergence of avian gammacoronaviruses. Infect Genet Evol. (2012) 12:1305–11. doi: 10.1016/j.meegid.2012.05.003
4. Laconi A, van Beurden SJ, Berends AJ, Krämer-Kühl A, Jansen CA, Spekreijse D, et al. Deletion of accessory genes 3a, 3b, 5a or 5b from avian coronavirus infectious bronchitis virus induces an attenuated phenotype both in vitro and in vivo. J Gen Virol. (2018) 99:1381–90. doi: 10.1099/jgv.0.001130
5. Hodgson T, Britton P, Cavanagh D. Neither the RNA nor the proteins of open reading frames 3a and 3b of the coronavirus infectious bronchitis virus are essential for replication. J Virol. (2006) 80:296–305. doi: 10.1128/JVI.80.1.296-305.2006
6. Valastro V, Holmes EC, Britton P, Fusaro A, Jackwood MW, Cattoli G, et al. S1 gene-based phylogeny of infectious bronchitis virus: an attempt to harmonize virus classification. Infect Genet Evol. (2016) 39:349–64. doi: 10.1016/j.meegid.2016.02.015
7. Bande F, Arshad SS, Omar AR, Hair-Bejo M, Mahmuda A, Nair V, et al. Global distributions and strain diversity of avian infectious bronchitis virus: a review. Anim Health Res Rev. (2017) 18:70–83. doi: 10.1017/S1466252317000044
8. Abozeid HH. Global emergence of infectious bronchitis virus variants: evolution, immunity, and vaccination challenges. Transbound Emerg Dis. (2023) 2023:1144924. doi: 10.1155/2023/1144924
9. Liu S, Chen J, Han Z, Zhang Q, Shao Y, Kong X, et al. Infectious bronchitis virus: S1 gene characteristics of vaccines used in China and efficacy of vaccination against heterologous strains from China. Avian Pathol. (2006) 35:394–9. doi: 10.1080/03079450600920984
10. Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet Res. (2007) 38:281–97. doi: 10.1051/vetres:2006055
11. Killian, M. L. Hemagglutination assay for influenza virus. In: Spackman E, , editor. Animal Influenza Virus. Cham: Springer (2014), p. 3–9. doi: 10.1007/978-1-4939-0758-8_1
12. Callison SA, Hilt DA, Boynton TO, Sample BF, Robison R, Swayne DE, et al. Development and evaluation of a real-time Taqman RT-PCR assay for the detection of infectious bronchitis virus from infected chickens. J Virol Methods. (2006) 138:60–5. doi: 10.1016/j.jviromet.2006.07.018
13. Chrzastek K, Segovia K, Torchetti M, Killian ML, Pantin-Jackwood M, Kapczynski DR, et al. Virus adaptation following experimental infection of chickens with a domestic duck low pathogenic avian influenza isolate from the 2017 USA H7N9 outbreak identifies polymorphic mutations in multiple gene segments. Viruses. (2021) 13:1166. doi: 10.3390/v13061166
14. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
15. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. (2014) 30:1312–3. doi: 10.1093/bioinformatics/btu033
16. Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. (2015) 1:vev003. doi: 10.1093/ve/vev003
17. Robertson DL, Hahn BH, Sharp PM. Recombination in AIDS viruses. J Mol Evol. (1995) 40:249–59. doi: 10.1007/BF00163230
18. Xue Y, Xie Q, Yan Z, Ji J, Chen F, Qin J, et al. genome sequence of a recombinant nephropathogenic infectious bronchitis virus strain in China. Am Soc Microbiol. (2012) 86:13812–3. doi: 10.1128/JVI.02575-12
19. Gong H, Ni R, Qiu R, Wang F, Yan W, Wang K, et al. Evaluation of a novel recombinant strain of infectious bronchitis virus emerged from three attenuated live vaccine strains. Microb Pathog. (2022) 164:105437. doi: 10.1016/j.micpath.2022.105437
20. Lisowska A, Sajewicz-Krukowska J, Fusaro A, Pikula A, Domanska-Blicharz K. First characterization of a Middle-East GI-23 lineage (Var2-like) of infectious bronchitis virus in Europe. Virus Res. (2017) 242:43–8. doi: 10.1016/j.virusres.2017.09.010
21. Franzo G, Listorti V, Naylor CJ, Lupini C, Laconi A, Felice V, et al. Molecular investigation of a full-length genome of a Q1-like IBV strain isolated in Italy in 2013. Virus Res. (2015) 210:77–80. doi: 10.1016/j.virusres.2015.07.008
Keywords: infectious bronchitis virus (IBV), retailed chicken meat, grocery market, whole genome sequencing, recombination
Citation: Lee S-H, Lee H, Cho AY, Kim T-H, Choi Y-J, Otgontogtokh N, Seo Y-R, Lee D-Y, Tseren-Ochir E-O, Myagmarsuren T, Lee D-H, Hyeon J-Y and Song C-S (2024) First report and complete genome analysis of infectious bronchitis virus from retailed chicken meat in Mongolia in 2023. Front. Vet. Sci. 11:1465342. doi: 10.3389/fvets.2024.1465342
Received: 16 July 2024; Accepted: 29 November 2024;
Published: 13 December 2024.
Edited by:
Ying Liao, Chinese Academy of Agricultural Sciences, ChinaCopyright © 2024 Lee, Lee, Cho, Kim, Choi, Otgontogtokh, Seo, Lee, Tseren-Ochir, Myagmarsuren, Lee, Hyeon and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Yeon Hyeon, c2g3MDJAa29ua3VrLmFjLmty