 Faisal Almathen1,2*†
Faisal Almathen1,2*†- 1Department of Public Health, College of Veterinary Medicine, King Faisal University, Al-Hofuf, Saudi Arabia
- 2Camel Research Center, King Faisal University, Al-Hofuf, Saudi Arabia
Introduction: The Awarik camel population in southwestern Saudi Arabia exhibits unique genetic and phenotypic traits compared to other domestic camel populations. This study aims to explore the genomic signatures of positive selection in Awarik camels to understand their evolutionary history and identify genetic adaptations potentially shared with East African camel populations.
Methods: Whole genome sequencing data from nine Awarik camels were analyzed using two robust intra-population haplotype-based statistical methods: integrated haplotype score (iHS) and number of segregating sites by length (nSL). These analyses were conducted to identify candidate regions under positive selection within the Awarik camel genome.
Results and discussion: These analyses identified 66 and 53 candidate selection regions, encompassing 185 and 123 genes, respectively. The iHS analysis revealed significant selection signals on chromosomes 15 and 16, including a robust overlap on chromosome 15 (10 regions) involving the TRNAI-AAU gene, suggesting its critical role in adaptive processes. Additionally, chromosome 3 exhibited the highest number of candidate regions totaling 10. The nSL analysis highlighted statistically significant regions on chromosomes 2 and 7, as well as a high concentration of candidate regions on chromosome 14, totaling five regions. Notably, large candidate regions were also identified on chromosome 11 (200 kb: 51.750–51.950 kb) and chromosome 9 (325 kb: 45.825–46.150 kb). Functional annotation of these genes revealed involvement in diverse biological processes including olfactory activity, immune regulation, metabolism, insulin secretion, reproductive performance, kidney function, and cellular signaling, with specific genes like BAG5, septin 7, SLC13A1, PCED1B, BMPR1B, ZAR1, JAKMIP2, and NOTCH2 highlighted. These findings contribute to our understanding of the adaptive mechanisms of Awarik camels and have important implications for breeding and conservation strategies. Further research on these genetic adaptations, particularly those affecting immune response, is crucial to mitigate the impacts of climate change on camel populations.
1 Introduction
The Awarik camel population in Saudi Arabia is distinguished by its unique geographical distribution, phenotypic traits, and genetic characteristics. Studies have consistently demonstrated that the Awarik camels, particularly those from the western and southwestern regions, possess distinct genetic traits when compared to other camel populations, particularly those in the northern and central regions of the country (1). The uniqueness. The uniqueness of the Awarik camel population has been highlighted in various studies, indicating that these camels represent a genetically distinct group within Saudi Arabia (2–5). Recent research, including the study by Bahbahani et al. (6) which analyzed the whole genomes of 40 dromedary camels from across the Arabian Peninsula, has provided pivotal insights into the geographical genetic distinctions and regions under positive selection in dromedaries. This research has identified specific regions and haplotype blocks associated with adaptive physiological traits that are crucial for conservation. Building upon these comprehensive findings, this study focuses on exploring the unique genomic signatures of positive selection in the Awarik population. By examining these specific genetic adaptations, w aim to further understand the evolutionary pressures and adaptations that have uniquely shaped the Awarik camels, emphasizing the necessity for focused research on this distinct population.
The consistency of the Awarik camel population is evidenced by both genetic and demographic data. Genetically, the Awarik camels exhibit significant differentiation from other regional populations, reflecting a high degree of genetic homogeneity and stability within this group. This is supported by specific alleles and haplotypes that are consistent across generations, indicative of strong selective pressures and limited gene flow with other populations. Demographically, the population size has been relatively stable, as suggested by historical and recent surveys. This stability is crucial for preserving genetic integrity and ensures the reliability of evolutionary and adaptive studies focused on this unique group. Together, these genetic and demographic aspects confirm the Awarik population's consistency, underscoring its suitability for detailed genomic and adaptive analyses.
These one-humped dromedary camels are primarily located in the southwestern region of the country, which features a semi-arid climate with variable humidity levels, particularly high along the western coasts and mountains. Historically, Awarik camels share genetic ties with camel populations from the Horn of Africa, including Kenya, Somalia, and Sudan, regions that collectively host the largest camel populations in Africa.
Awarik camels have adapted to thrive in the harsh desert environment and humid conditions of the Red Sea coastal areas. Traditionally bred by local tribes for their milk and meat, these camels are highly valued for their resilience, endurance, and adaptability to coastal and mountainous terrains. They graze on the arak plant (Salvadora persica), which is abundant in their natural habitat. With unique physiological and behavioral traits, Awarik camels exhibit efficient temperature regulation and heat tolerance, enabling them to flourish in their coastal and mountainous environments (2).
Named after the arak plant, a significant part of their diet, the Awarik camel population is primarily found near the Red Sea coast of Saudi Arabia and is colloquially known as “Beach camels.” Predominantly concentrated in the Jazan region, these camels are characterized by a light brown coat, almost white, and short hair. They typically have a well-developed udder, medium neck circumference, pointed ears, and a hump positioned toward the hind of the back, giving them a shorter stature compared to other desert camels (7, 18). Awarik camels exhibit moderate milk production, with total lactation yields averaging 1,047.5 ± 11 liters (8).
This study aims to analyze the genomic signatures of positive selection in Awarik camels, seeking to uncover their evolutionary history and identify the genetic factors associated with their adaptation. Insights from this research can inform camel breeding programs, enhance conservation efforts, and develop strategies to mitigate the impact of climate change on camel populations.
2 Materials and methods
2.1 Sample collection and whole genome sequencing
Blood samples were obtained from nine unrelated female Awarik camels residing in the southwestern regions (Jazan) of Saudi Arabia, selected based on stringent phenotypic criteria. Genomic DNA was extracted from these samples using the Puregene® Blood Core Kit C (Qiagen) according to the manufacturer's instructions. Subsequently, the extracted DNA underwent sequencing on the Illumina NovaSeq 6000 platform, employing a 150 bp paired-end approach. This sequencing was conducted at the Beijing Genomics Institute in China. The sequences data were deposited at European Nucleotide Archive Bioproject number: PRJEB47650.
2.2 Whole genome sequence read processing and variant calling
Whole genome sequences from the Awarik camels were aligned to the African dromedary reference genome (CamDro3) (9) using the bwa-mem algorithm of Burrows-Wheeler Aligner version 0.7.17 (10). Post alignment, reads were organized by coordinates using the SortSam and PCR duplicates were removed using the MarkDuplicates tool with (REMOVE_DUPLICATES=true) from Picard tools version 3.0.0 (http://broadinstitute.github.io/picard/index.html). SNPs calling was performed on Awarik autosomes using the HaplotypeCaller algorithm in GVCF mode of Genome Analysis Toolkit (GATK) version 4.2.5.0 (19). Autosomal SNPs were then filtered based on the criteria outlined by Bahbahani et al. (6), retaining those with a depth of coverage within ten reads and three standard deviations from the mean across all samples, for subsequent selection signature analyses.
2.3 Single nucleotide polymorphism quality control and pruning
A total of 5,720,174 autosomal SNPs were subjected to quality control pruning using PLINK v1.9 (11). SNPs were excluded based on the following criteria: a call rate below 100% of the genotyped samples, deviation from Hardy-Weinberg equilibrium (P < 1 × 10−6), or a minor allele frequency (MAF) of 5% or less. After filtering, 3,364,881 SNPs remained for selection signature analysis (Table 1). Samples were also evaluated and would be excluded for a genotyping call rate under 100% or a maximum pairwise identity-by-state (IBS) of 95% or greater; however, no samples met these exclusion criteria.
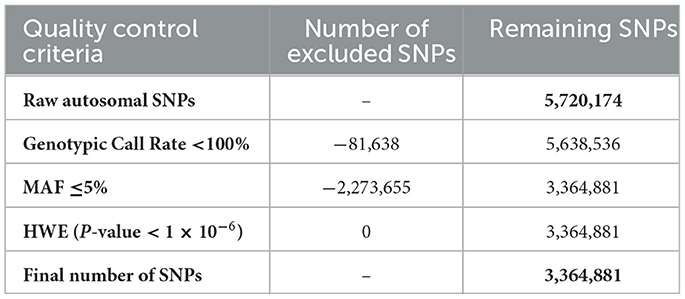
Table 1. Quality control criteria and the number of excluded and remaining SNPs for the signatures of selection analysis.
2.4 Signatures of selection analysis
Signatures of selection analyses were conducted on nine Awarik dromedary camels from southwestern Saudi Arabia using two intra-population haplotype-based statistics, integrated haplotype score (iHS) (12) and number of segregating sites by length (nSL) (13). These statistics were converted to P-values based on fractional ranks using the stat_to_pvalue based on the fractional ranks. These P-values were further transformed into rank-based values using two-tailed tests. Windows displaying–log10 (P-values) ≥ 4 (equivalent to P ≤ 0.0001) were defined as candidate windows with signatures of selection.
2.5 Functional annotation and enrichment analysis
The coordinates of the candidate regions were cross-referenced against the dromedary camel reference genome assembly CamDro3 gene list using the GenomicRanges package (14) in R. Functional profiling of the overlapping genes was conducted using the g: GOSt function of gProfiler (15), which identified functionally enriched terms for gene ontology biological processes and molecular functions. The gProfiler g: SCS algorithm was employed to compute multiple testing corrections for P-values from the gene ontology and pathway enrichment analyses. All identified genes were also analyzed using the functional annotation tool in the Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics resource version 6.7 (16, 17) to determine enriched functional terms. An enrichment score of 1.3, equivalent to a Fisher exact test P-value of 0.05, was used as the threshold to define significantly enriched functional terms compared to the dromedary reference genome background. The genes were then cross-referenced with the literature to evaluate their relevance to the dromedaries' environmental adaptations and physiological traits.
3 Results
3.1 Summary statistics of the mapped sequence reads
The depth of coverage for the mapped sequence reads among the dromedary samples ranged from 19X to 22X, with an mean of 21X. On average, 99.7% of the sequence reads were mapped to the dromedary reference genome, and 95% of these were properly paired. The mapped reads covered ~94.7% of the reference genome.
3.2 Signatures of selection analysis of the two analysis
In the iHS analysis, the most significant regions were identified on chromosomes 15 and 16 as depicted in Figure 1. These regions displayed the strongest signals of selection, indicating that they may play a crucial role in the genetic adaptation of Awarik camels. Notably, a region on chromosome 15 was identified by both the iHS and nSL analyses, with the TRNAI-AAU gene located within this overlapping region, indicating a robust selection signal that may be significant for adaptive processes in camels. Additionally, chromosome 3 exhibited the highest number of candidate regions, with a total of 10 identified (see Supplementary Tables S1, S5).
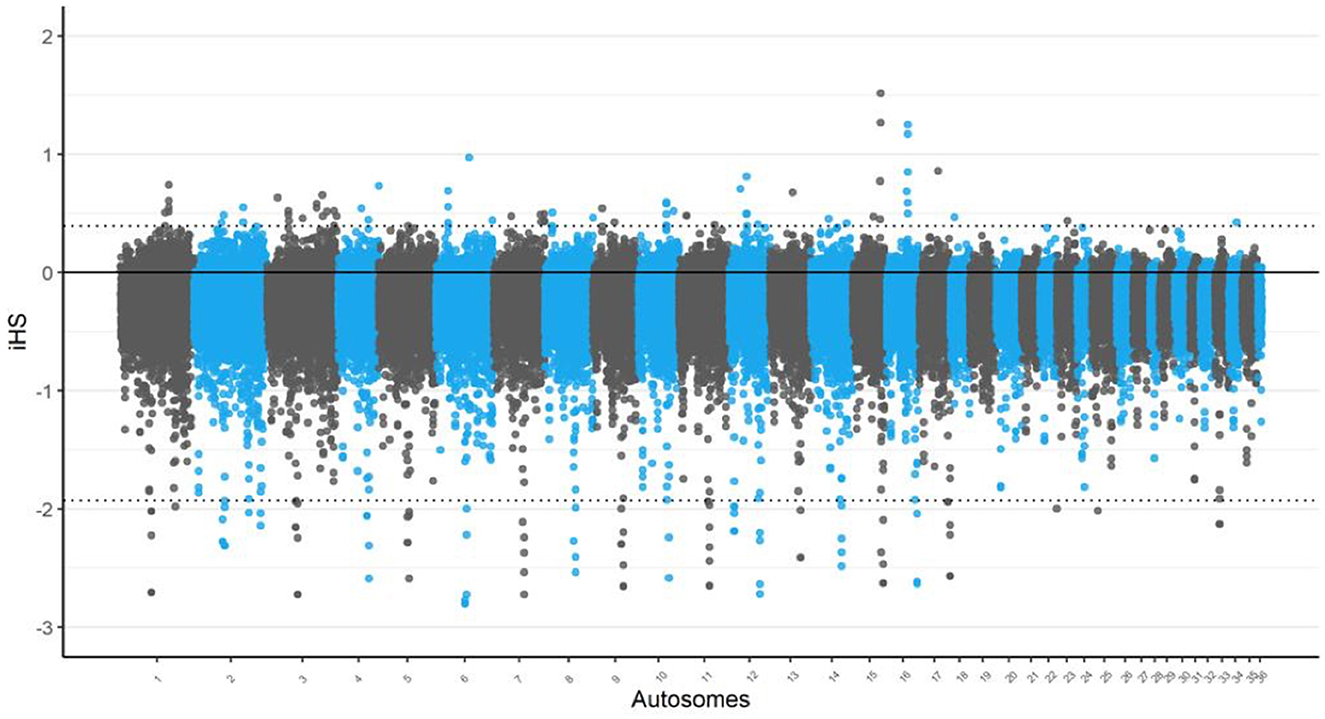
Figure 1. Signatures of selection analysis on autosomes of Awarik dromedary camels. Manhattan plots of genome-wide iHS analysis with a two-tailed Z-test applied and the significance threshold is set at–log10 (two-tailed P-value) = 4.
Conversely, the nSL analysis, chromosomes 2 and 7 exhibited the most statistically significant selection signals as shown in Figure 2. Although these regions were not the largest in terms of selection signal size, their high statistical significance makes them noteworthy. In particular, chromosome 2 showed a pronounced peak that warrants further investigation to identify potential candidate genes related to adaptive traits in Awarik camels. The nSL analysis also revealed a high concentration of candidate regions on chromosome 14, totaling five regions. Furthermore, the largest candidate regions were located on chromosome 11 (spanning 200 kb: 51.750–51.950 kb) and chromosome 9 (spanning 325 kb: 45.825–46.150 kb) in the iHS and nSL analyses, respectively (see Supplementary Tables S1, S5).
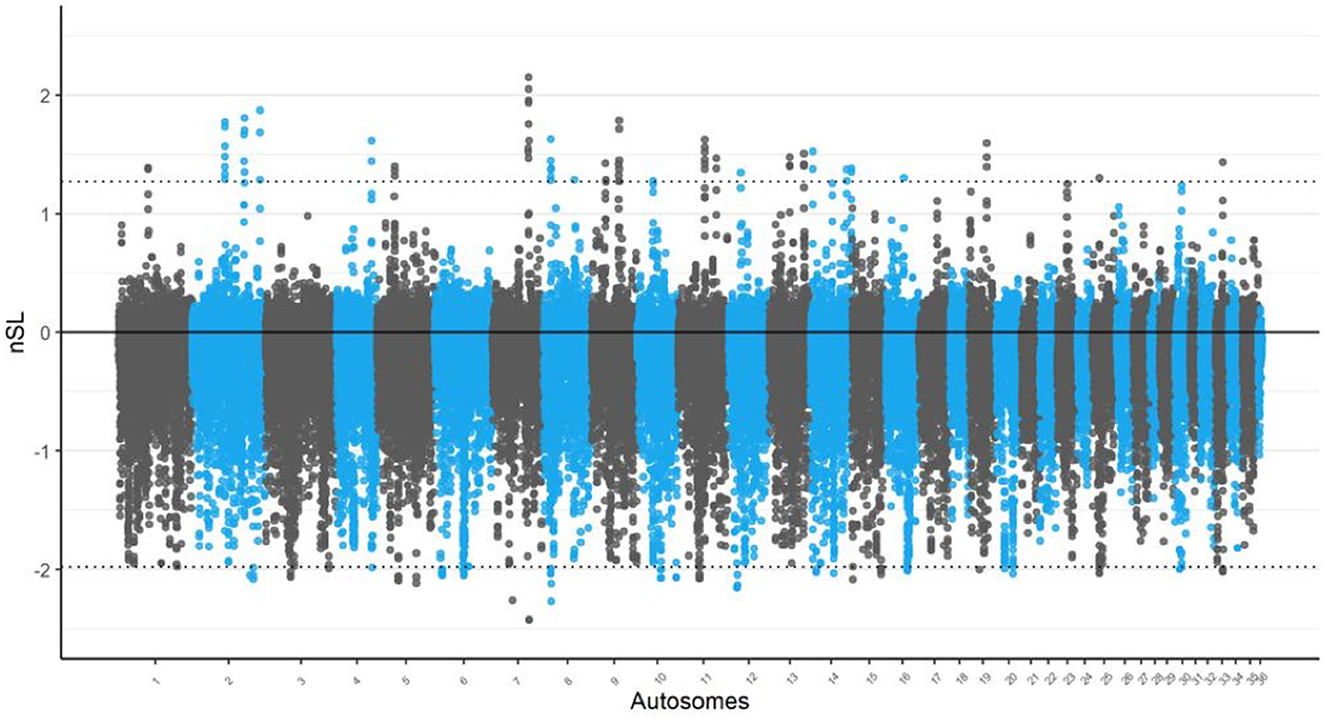
Figure 2. Signatures of selection analysis on autosomes of Awarik dromedary camels. Manhattan plots of genome-wide nSL analysis with a two-tailed Z-test applied and the significance threshold is set at–log10 (two-tailed P-value) = 4.
3.3 Functional annotation of candidate regions and enrichment analysis
3.3.1 Integrated haplotype score (iHS)
The iHS analysis identified 185 genes within the 66 candidate selection regions (Supplementary Table S1). Functional profiling of these genes revealed several enriched molecular and biological processes, including the Wnt signaling pathway (see selected results in Table 1, Supplementary Tables S1, S2). However, none of these processes were significantly enriched. DAVID analysis identified six functional clusters, showing enrichment for functions related to olfactory activity (enrichment score = 4.65), immunoglobulin subtype (enrichment score = 0.65), ATP binding (enrichment score = 0.41), basic and acidic residues (enrichment score = 0.24), and zinc finger C2H2-type/integrase DNA-binding domain (enrichment score = 0.13). Literature review highlighted candidate genes involved in immune regulation and inflammatory responses, metabolism, cell signaling and receptor regulation, neuronal development and function, and cytoskeleton and cellular structure (Table 2).
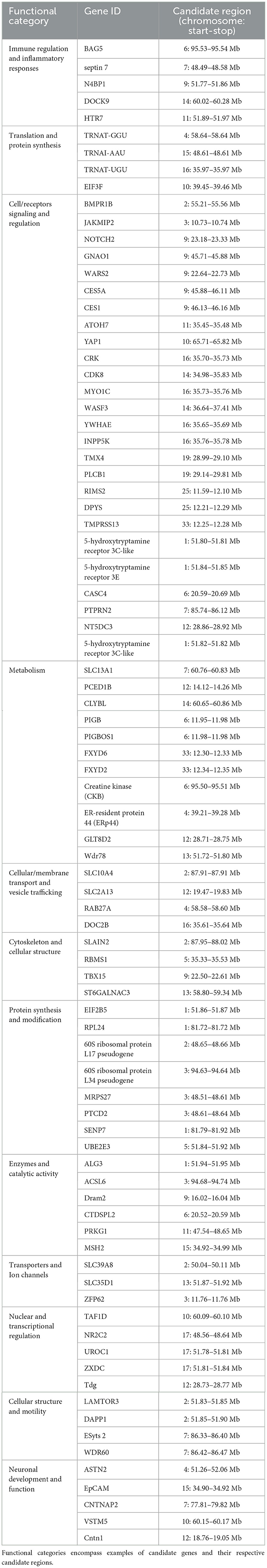
Table 2. Biological functions of candidate genes under selection using iHS analysis on Awarik dromedary camel autosomes.
3.3.2 Number of segregating sites by length (nSL)
The nSL analysis identified 123 genes within the 53 candidate selection regions (Supplementary Tables S4, S5). Functional profiling of these genes revealed several enriched molecular and biological processes, including the Wnt signaling pathway (see selected results in Table 2, Supplementary Tables S4, S5). However, none of these processes were significantly enriched. DAVID analysis identified five functional clusters, showing enrichment for functions related to transmembrane helices (enrichment score = 0.52), olfactory and transducer activity (enrichment score = 0.39), immunoglobulin-like domains (enrichment score = 0.38), cell and plasma membranes (enrichment score = 0.38), and ion and zinc binding (enrichment score = 0.20). Literature review highlighted several candidate genes associated with key biological processes such as reproductive performance, immune response, neuroplasticity, insulin secretion and signaling, as well as kidney absorption and reabsorption (Table 3, Supplementary Table S6). These genes are of particular interest due to their roles in physiological adaptations that are critical for the survival and reproductive success of Awarik camels in challenging environments.
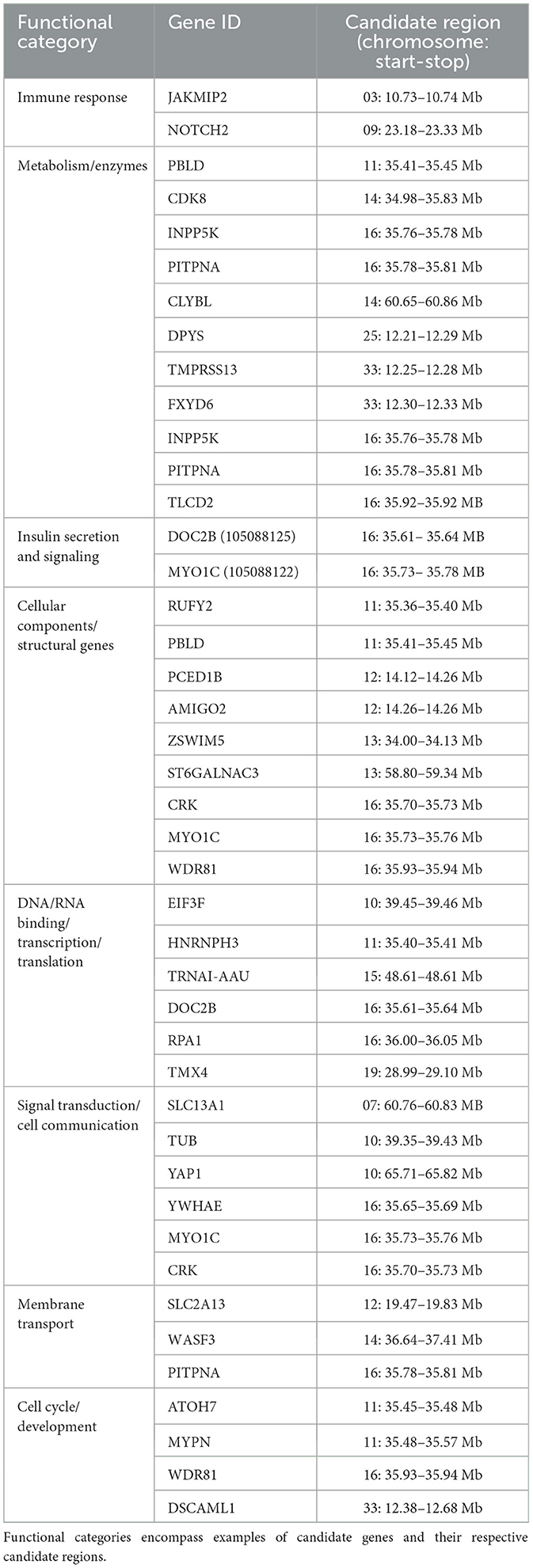
Table 3. Biological functions of candidate genes under selection using nSL analysis on Awarik dromedary camel autosomes.
4 Discussion
This study provides a comprehensive genomic analysis the Awarik camel population, indigenous to the southwestern region of Saudi Arabia, offering significant insights into the genetic mechanisms underlying their adaptation to challenging environments (2, 3). Through high-depth whole genome sequencing, we identified key genomic regions under positive selection, shedding light on the evolutionary strategies that have enabled these camels to thrive in semi-arid and coastal habitats (6). The identification of significant selection regions on chromosomes 2 and 7 in the nSL analysis, and on chromosomes 15 and 16 in the iHS analysis, emphasizes the complexity of the selection landscape in Awarik camels. The statistically significant signals on chromosomes 2 and 7 suggest that these regions may harbor genes of adaptive significance, potentially influencing traits that are critical for survival in the harsh environments inhabited by these camels. In particular, the overlap identified on chromosome 15, where the TRNAI-AAU gene was detected in both the iHS and nSL analyses, strengthens the case for importance of this gene. which likely plays a pivotal role in the genetic adaptation of Awarik camels. Further functional analysis is needed to elucidate its specific role in camel physiology and adaptation.
Our analysis highlighted significant selection signatures on chromosomes 3, 14, 11, and 9, each pointing to different aspects of genetic adaptation. Chromosomes 3 and 14, which showed the highest number of candidate regions in the iHS and nSL analyses, respectively, are particularly important as they likely host genes conferring adaptive advantages in response to the specific selection pressures faced by the Awarik population (Supplementary Tables S1, S5). This pattern suggests a tailored response to the unique selection pressures faced by the Awarik population, potentially reflecting localized adaptations not as pronounced in other regional camel populations.
Moreover, the notable presence of large candidate regions on chromosomes 9 and 11 underscores the importance of these loci in the adaptation of the Awarik camels. Previous studies such as those by Bahbahani et al. (6) and Al Abri et al. (5) have reported similar high-frequency haplotypes on these chromosomes in mixed groups of dromedaries, including Awarik camels. The consistency of these findings across studies supports the hypothesis that these genomic regions may hold unique significance for the Awarik population, necessitating further research to fully understand their biological relevance.
Functional annotation of these regions revealed involvement in diverse biological processes critical to survival in extreme environments, such as immune regulation, metabolism, and reproductive performance. Although no significant enrichment was found in pathways like Wnt signaling, the recurring identification of genes associated with this pathway across different analyses suggest its potential role in developmental and adaptive processes (15). The DAVID analysis further supported this by identifying functional clusters related to sensory functions, immune responses, and metabolic processes, all of which are vital for thriving in the variable climates of the Red Sea coastal areas (16, 17). These findings imply a complex and multifaceted genetic basis for the adaptation Awarik camel, involving various physiological and cellular processes.
The genes uncovered in this study, particularly those involved in olfactory activity, immune response, and kidney function, highlight the Awarik camel's genetic specialization for their diet and environmental stressors. These adaptations appear to be deeply embedded in the genetic fabric of the population, suggesting a complex evolutionary history that has finely tuned these animals to their specific ecological niches (5).
Furthermore, the implications of these genomic insights extend beyond academic understanding to practical applications in breeding and conservation. The detailed genetic markers identified here can guide selective breeding programs aimed at enhancing desirable traits like resilience to climate variability and disease resistance. Additionally, the genetic diversity revealed through this study underscores the importance of conservation strategies that preserve these unique genetic resources, which are invaluable for the Awarik camel's continued adaptation and survival (1).
Future research should build on on these findings by increasing the sample sizes and incorporating comparative genomic studies with other camel populations, including those from East Africa with whom the Awarik camels share historical ties (9). Such studies could illuminate both shared and unique adaptations, providing a broader understanding of camel evolution across different environments. Integrating environmental and phenotypic data could refine the connections between genetic adaptations and specific environmental challenges, enhancing the predictive power of genomic studies in conservation and management practices.
In conclusion, the genomic signatures of positive selection identified in the Awarik camels not only deepen our understanding of their unique adaptations but also provide a foundational knowledge base for developing targeted interventions in breeding and conservation. These interventions are critical for sustaining the Awarik camel population amid the escalating pressures of climate change and habitat loss, ensuring their resilience and productivity for future generations.
Data availability statement
The data presented in the study are deposited at European Nucleotide Archive Bioproject number: PRJEB47650.
Ethics statement
The animal studies were approved by Ethics Research Committee approval at the King Faisal University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
FA: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
I would like to acknowledge the Deanship of Scientific Research at King Faisal University for providing financial support under Research Grant No. KFU241689, as well as the Research, Development, and Innovation Authority for additional support (Project No. 12977-KFU-2023-KFU-R-3-1-HW-Molecular Genetics and Genomics Laboratory). I also extend my gratitude to the camel owners for their generous cooperation in allowing us to sample their animals for this research. Special thanks go to Prof. Bashir Salim, Dr. Hussain Bahbahani, and Dr. Abdullah Sheikh for their invaluable assistance.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2024.1443748/full#supplementary-material
References
1. Bahbahani H, Almathen F. Homogeneity of Arabian Peninsula dromedary camel populations with signals of geographic distinction based on whole genome sequence data. Sci Rep. (2022) 12:130. doi: 10.1038/s41598-021-04087-w
2. Almathen F, Charruau P, Mohandesan E, Mwacharo JM, Orozco-terWengel P, Pitt D, et al. Ancient and modern DNA reveal dynamics of domestication and cross-continental dispersal of the dromedary. Proc Nat Acad Sci. (2016) 113:6707–12. doi: 10.1073/pnas.1519508113
3. Porter V, Alderson L, Hall SJ, Sponenberg DP. Mason's world encyclopedia of livestock breeds and breeding, 2 Volume Pack. Wallingford: CABI. (2016).
4. AlAskar H, Alhajeri BH, Almathen F, Alhaddad H. Genetic diversity and population structure of dromedary camel-types. J Heredity. (2020) 111:405–13. doi: 10.1093/jhered/esaa016
5. Al Abri M, Alfoudari A, Mohammad Z, Almathen F, Al-Marzooqi W, Al-Hajri S, et al. Assessing genetic diversity and defining signatures of positive selection on the genome of dromedary camels from the southeast of the Arabian Peninsula. Front Vet Sci. (2023) 10:1296610. doi: 10.3389/fvets.2023.1296610
6. Bahbahani H, Alfoudari A, Al-Ateeqi A, Al Abri M, Almathen F. Positive selection footprints and haplotype distribution in the genome of dromedary camels. Animal. (2024) 18:101098. doi: 10.1016/j.animal.2024.101098
7. Abdallah HR, Faye B. Phenotypic classification of Saudi Arabian camel (Camelus dromedarius) by their body measurements. Emir J Food Agric. (2012) 24:272–280.
8. Gaili ESE, Al-Eknah MM, Sadek MH. Comparative milking performance of three types of Saudi camels (Camelus dromedarius). J Camel Pract Res. (2000) 7:73–6.
9. Elbers JP, Rogers MF, Perelman PL, Proskuryakova AA, Serdyukova NA, Johnson WE, et al. Improving Illumina assemblies with Hi-C and long reads: an example with the North African dromedary. Mol Ecol Resour. (2019) 19:1015–26. doi: 10.1111/1755-0998.13020
10. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
11. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. (2015) 4:s13742–015. doi: 10.1186/s13742-015-0047-8
12. Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. (2006) 4:e72. doi: 10.1371/journal.pbio.0040072
13. Ferrer-Admetlla A, Liang M, Korneliussen T, Nielsen R. On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol Biol Evol. (2014) 31:1275–91. doi: 10.1093/molbev/msu077
14. Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. (2013) 9:e1003118. doi: 10.1371/journal.pcbi.1003118
15. Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. (2019). g: Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47:W191–W198. doi: 10.1093/nar/gkz369
16. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. (2009) 37:1–13. doi: 10.1093/nar/gkn923
17. Sherman BT, Huang da W, Tan Q, Guo Y, Bour S, Liu D, et al. DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformat. (2007) 8:1–11. doi: 10.1186/1471-2105-8-426
18. Almathen F, Elbir H, Bahbahani H, Mwacharo J, Hanotte O. Polymorphisms in MC1R and ASIP genes are associated with coat color variation in the Arabian camel. J Hered. (2018) 109:700–6. doi: 10.1093/jhered/esy024
Keywords: dromedary, positive selection, evolutionary adaptation, haplotype-based statistics, camels
Citation: Almathen F (2024) Genomic signatures of positive selection in Awarik dromedary camels from southwestern of Saudi Arabia. Front. Vet. Sci. 11:1443748. doi: 10.3389/fvets.2024.1443748
Received: 04 June 2024; Accepted: 28 August 2024;
Published: 18 September 2024.
Edited by:
Peter Dovc, University of Ljubljana, SloveniaReviewed by:
Kwan-Suk Kim, Chungbuk National University, Republic of KoreaMaria Cristina Cozzi, University of Milan, Italy
Copyright © 2024 Almathen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Faisal Almathen, ZmFsbWF0aGVuQGtmdS5lZHUuc2E=
†ORCID: Faisal Almathen orcid.org/0000-0001-6970-9155