 Min-hao Zeng
Min-hao Zeng Shan Li
Shan Li Qing-bo Lv
Qing-bo Lv Xiao-xu Wang1
Xiao-xu Wang1 Abdul Qadeer
Abdul Qadeer- 1School of Biotechnology, Jiangsu University of Science and Technology, Zhenjiang, China
- 2Jiangxi Provincial Key Laboratory of Cell Precision Therapy, School of Basic Medical Sciences, Jiujiang University, Jiujiang, China
- 3Key Laboratory of Zoonosis Research, Institute of Zoonosis, College of Veterinary Medicine, Ministry of Education, Jilin University, Changchun, China
- 4Department of Cell Biology, School of Life Sciences, Central South University, Changsha, China
- 5Department of Biochemistry, College of Science, King Saud University, Riyadh, Saudi Arabia
Background: Anisakis are globally distributed, marine parasitic nematodes that can cause human health problems, including symptoms such as vomiting, acute diarrhea, and allergic reactions. As parasitic nematodes that primarily affect the patient’s digestive tract, intestinal helminths can interact directly with the host microbiota through physical contact, chemicals, or nutrient competition. It is widely accepted that the host microbiota plays a crucial role in the regulation of immunity.
Materials and methods: Nematodes collected from the abdominal cavity of marine fish were identified by molecular biology and live worms were artificially infected in rats. Infection was determined by indirect ELISA based on rat serum and worm extraction. Feces were collected for 16S rDNA-based analysis of microbiota diversity.
Results: Molecular biology identification based on ITS sequences identified the collected nematodes as A. pegreffii. The success of the artificial infection was determined by indirect ELISA based on serum and worm extraction from artificially infected rats. Microbiota diversity analysis showed that a total of 773 ASVs were generated, and PCoA showed that the infected group was differentiated from the control group. The control group contained five characterized genera (Prevotellaceae NK3B31 group, Turicibacter, Clostridium sensu stricto 1, Candidatus Stoquefichus, Lachnospira) and the infected group contained nine characterized genera (Rodentibacter, Christensenella, Dubosiella, Streptococcus, Anaeroplasma, Lactococcus, Papillibacter, Desulfovibrio, Roseburia). Based on the Wilcoxon test, four processes were found to be significant: bacterial secretion system, bacterial invasion of epithelial cells, bacterial chemotaxis, and ABC transporters.
Conclusion: This study is the first to analyze the diversity of the intestinal microbiota of rats infected with A. pegreffii and to determine the damage and regulation of metabolism and immunity caused by the infection in the rat gut. The findings provide a basis for further research on host-helminth-microbe correlationships.
1 Introduction
Anisakiasis is a parasitic disease caused by the ingestion of larvae of the Anisakis nematode. These larvae are commonly found in marine fish worldwide. Consumption of raw or undercooked fish poses a risk of infection due to the presence of Anisakis larvae within the fish tissue (1). Symptoms of Anisakiasis typically include gastrointestinal disturbances such as vomiting, nausea and diarrhea (2). Additionally, allergic reactions and ectopic parasitism may occur due to the migration of the nematode larvae, presenting a potential health threat (3–6). The infection is prevalent globally and is particularly associated with regions where raw or lightly processed seafood is commonly consumed, such as Japan, Korea, China, Taiwan, Portugal, and Chile (7). In 2010, the European Food Safety Authority (EFSA) estimated that there were around 20,000 reported cases of Anisakiasis worldwide. In Japan alone, the average annual incidence of Anisakiasis in 2018–2019 was 19,737, based on insurance claims records (8, 9). Anisakiasis is widely prevalent in seas around the globe, have been identified by the FAO (Food and Agriculture Organization of the United Nations) as one of the 10 most important parasites affecting humans and fish (10).
As parasitic nematodes that primarily affect the patient’s digestive tract, intestinal helminths can interact directly with the host microbiota through physical contact, chemical compounds, or nutritional competition. It is widely recognized that the host microflora plays a crucial role in regulating immunity (11–13). Parasites can also indirectly impact immunity and other physiological processes by regulating the host gut microbiota (14). Changes in gut microbes can also protect the host from parasites (15). Research has demonstrated that Haemonchus contortus has a significant impact on the gastrointestinal microbiota in lambs. It also reduces the α-diversity of the gastrointestinal microbial community in the rumen, abomasum, and duodenum, and disrupts the partitioning of amino acids for protein digestion and absorption (16). Ascaris lumbricoides, belonging to the superfamily Ascaridoidea along with Anisakis, is also capable of reducing the microbiota diversity of the porcine gut and altering the metabolic potential of the host (17).
It is of great interest to investigate whether Anisakiasis induces changes in the host gut microbiota. Using 16S rRNA sequence-based NGS sequencing and bioinformatics analysis, we researched the response of rat fecal microbiota communities to Anisakiasis to gain insight into how A. pegreffii affects host gut microbiota and metabolism. This statement provides a basis for further studies on the interactions between Anisakis-hosts-microbiota.
2 Materials and methods
2.1 Parasites collection
We obtained fresh, unfrozen marine fish such as Little Yellow Croaker (Larimichthys polyactis) and Largehead hairtail (Trichiurus lepturus) from markets in Shanghai City, China. The fish were dissected using scissors to expose the organs and digestive tract. Nematodes were isolated from tissues including the abdominal cavity, stomach, intestines, gonads, and muscles using forceps or needles. The worms were then washed three times with saline solution to remove any attached fish tissue.
2.2 Identification of parasites
The nematodes were categorized by microscopic observation and genomic DNA was extracted from randomly selected worms for molecular identification. In brief, the nematodes were homogenized in 200 μL of lysis buffer (Tris–HCl 100 mM, EDTA 25 mM, NaCl 500 mM, Protease K 10 μL), then incubated at 56°C for 6 h. After centrifugation at 12,000 rpm for 5 min to remove the precipitate, 200uL of Tris-saturated phenol: chloroform: isoamyl alcohol mixture (25, 24, 1, pH 8.0) were added, and then centrifuged at 12,000 rpm for another 5 min. The supernatant aqueous phase was transferred to a new tube. To extract genomic DNA, 200 μL of pre-cooled anhydrous ethanol was added to the sample, followed by thorough mixing. The mixture was then centrifuged at 12,000 rpm for 10 min, and the supernatant was carefully removed, leaving the precipitate behind. Wash the precipitate twice with 200 μL of 75% ethanol. Finally, dissolve the precipitate with ddH2O. Genomic DNA was used as the template for PCR. The molecular identification by PCR was based on the ITS (Internal Transcribed Spacer) rDNA regions, the sequences were amplified using NC5 (5′- GTA GGT GAA CCT GCG GAA GGA TCA TT −3′) and NC2 (5′- TTA GTT TCT TTT CCT CCG CT −3′) primers (18, 19). PCR reaction (20 μL) contained 10 μL 2 × Hieff PCR Master Mix (Yeasen, Shanghai, China), 0.2 μL of each 10 μM NC5 and NC2 primers, ddH2O up to 20 min were performed on the following conditions: 95°C for 3 min (initial denaturation), then 35 cycles of 95°C for 15 s (denaturation), 55°C for 15 s (annealing), and 72°C for 1 min (extension), and a final extension at 72°C for 5 min. The PCR products were analyzed by PCR-RFLP using Hinf I endonuclease (NEB, Beijing, China) and the PCR products were sequenced (20). For the elongation factor (EF1 α-1 nDNA) nuclear gene, the sequences were amplified using EF-F (5′- TCC TCA AGC GTT GTT ATC TGT T − 3′) and EF-R (5′- AGT TTT GCC ACT AGC GGT TCC -3′). The PCR reaction was essentially the same as above, but with an annealing temperature of 58°C (21). After sequencing, the obtained sequences and peak maps were aligned with the reference of EF1 α-1 nDNA sequences using SnapGene software 4.2.41.
2.3 Artificial infection of rats
With reference to the methodology of previous studies, a total of 6 5-6-week-old Sprague–Dawley (SD) rats were artificially infected with 10 live A. pegreffii using the infant rectal drug delivery tubes (22). Briefly, active worms are inserted into the tube stretching from the mouth of the tubes, the tubes were inserted into the rats’ stomach, and a syringe was used to push the worms into the stomach. Six additional rats were used as negative controls. Serum samples were collected at the 1, 2, 3, and 5 weeks after infection, and feces were collected at 5 weeks after infection. The collected samples were either stored at −80°C or directly used in additional experiments. Five A. pegreffii were homogenized in pre-cold PBS (Phosphate Buffered Saline). The precipitate was removed by centrifugation, and the supernatant protein solution was filtered using a 0.22 μm filter. The concentration was determined using a BCA kit (Beyotime, Shanghai, China). An indirect ELISA was performed using worm extraction proteins and rat serum to diagnose the success of the artificial infection. Briefly, 96-well microtiter plates were coated with 1 μg of worm extraction protein in 100 μL of carbonate buffer (150 mM Na2CO3, 349 mM NaHCO3, pH 9.6) and incubated at 4°C overnight. After washing three times, the plates were blocked with 5% bovine serum albumin (BSA) for 2 h at 37°C. Sbsequently, rat sera diluted in PBST at 1:400 were added for 2 h at 37°C. HRP-conjugated goat anti-rat IgG antibody (Servicebio, Wuhan, China) was used as a secondary antibody at a 1:5000 dilution, and the reaction were revealed by TMB (Beyotime, Shanghai, China) for 15 min at 37°C and stopped by 2 M H2SO4. The reaction was measured at 450 nm with an ELISA reader. Data are represented as the mean ± standard deviation (SD), and the t-test was used for comparisons between the two groups. GraphPad Prism 8.3.1 (GraphPad Software, Inc., San Diego, CA, United States) was utilized for statistical analysis and generating graphs. p-values <0.05 were considered statistically significant.
2.4 Analysis of 16S rRNA gene sequences
The extraction of fecal genomic DNA and the 16S amplification were both provided by Wuhan Wanmo Technology Co., Ltd. The 16S rDNA V3-V4 region of the sample DNA was amplified using common primers 341-F (5′- CCT AYG GGR BGC ASC AG −3′) and 806-R (5′- GGA CTA CNN GGG TAT CTA AT −3′), and the resulting products were used to construct the libraries (21), which were then sequenced using the BGISEQ-500 platform. Poor quality bases of the paired-end sequence data were trimmed using Trimmomatic v0.36 (23) and were merged using FLASH v1.20 (24). The sequences were then imported into QIIME2 (25). Species annotation was performed on the obtained Amplicon Sequence Variants (ASVs) based on the 16S bacterial ribosomal databases SILVA (Release 128) (26), RDP (v1.6) (27), and Greengene (Release 13.5) (28), and the number of annotations and scores were counted at the phylum, family, and genus levels, and beta diversity was analyzed by PCoA based on Bray-Curtis and Jaccard distance. Species differing between groups were screened using Linear discriminant analysis Effect Size (LEfSe), (LDA > 2 as the threshold). The results of the analysis were visualized through the online tool TUTOOL2.
3 Results
3.1 Diagnosis for Anisakis pegreffii infection in rats
The ITS fragment (approximately 1,000 bp) from each sample was amplified (Figure 1A). The PCR-RFLP showed three sharp fragments of 370, 300 and 250 bp consistent with A. pegreffii (Figure 1B). After sequencing, the target sequence showed 99.5% identity with A. simplex and up to 100% identity with A. pegreffii (GenBank Accession: AY821739.1 and MF820020.1, in Supplementary Figure S1). Based on the EF1 α-1 sequence of A. simplex (s. s.) and A. pereffii (GenBank Accession: KP326558.1 and MH443145.1), the sequences of the larvae obtained in this study were consistent with A. pegreffii and did not exist overlapping peaks (Figures 1C,D). Therefore, based on the detection method we used, the larvae used in this study were preliminarily identified as A. pegreffii. Based on the diagnostic results of the indirect ELISA assay, the results of all rat sera in the infection group were significantly higher than those of the control group at 2 weeks post-infection (wpi). As the duration of infection increased, the optical density 450 nm and P/N (Positive group/Negative group) values of the ELISA results for the infection group also increased significantly. At 5 wpi, the mean P/N values for both the infection and control groups were as high as 11.6 (Figure 1E). Therefore, the rats in the infected group were diagnosed with A. pegreffii infection, while those in the negative group were not infected.
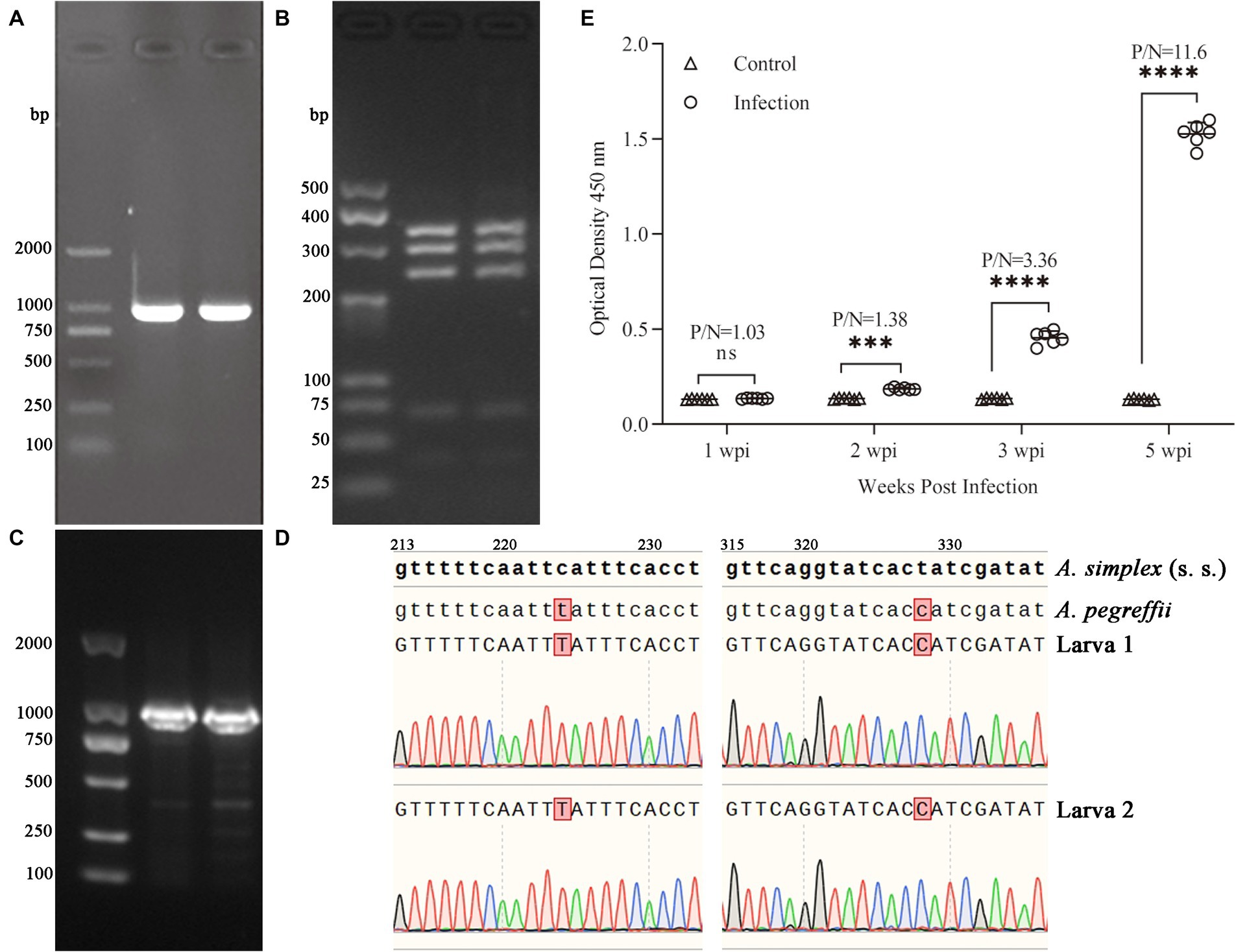
Figure 1. Electrophoresis of PCR products for ITS sequences in 1.0% agarose gel (A) and PCR-RFLP in 3.5% agarose gel (B) of A. pegreffii. The fragment sizes were all as expected. Results of electropherogram in 1.0% agarose gel (C) and alignment (D) of EF1 α-1 sequences. Indirect ELISA results (E) showed that OD450 of the infected group was significantly higher than that of the control group, and the P/N (Positive/Negative) values had reached 3.36 at 3 wpi, indicating that the rats were successfully infected with A. pegreffii.
3.2 Anisakis pegreffii restructure microbiota community in rats
A total of 773 ASVs were generated. The number of ASVs increased gradually with the sequencing depth, which met the analysis requirements (Supplementary Figure S2A). The relative abundance curves of the top 200 ASVs were smooth, indicating an even distribution of ASVs (Supplementary Figure S2B). At more than 3 biological replicates in each of the infection and control groups, the cumulative species curve tended to flatten, and the number of species did not significantly change with additional replicates. The number of replicates met the analysis requirements (Supplementary Figure S2C). Alpha diversity was assessed using Shannon’s index at various levels. The results indicated no significant differences in diversity (Figure 2A). However, beta-diversity analyses revealed a significant separation of clustering profiles of the PCoA indices based on Bray-Curtis and Jaccard (Figures 2B,C). This suggests that A. pegreffii infections have a significant impact on the composition of the gut microbiota.
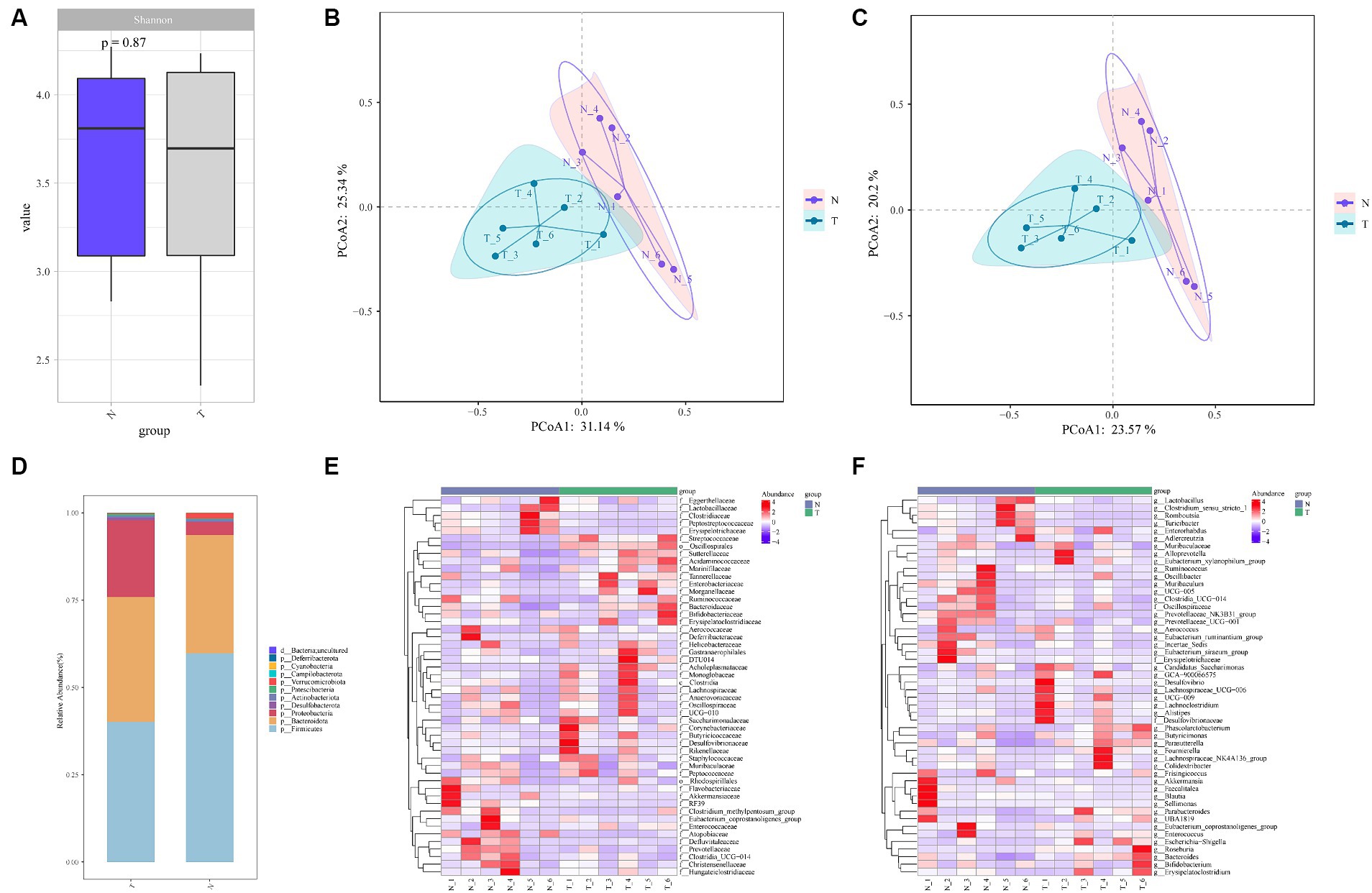
Figure 2. Alpha and beta diversity analyses between feces microbiota of A. pegreffii infection rats and control rats. N-negative control samples, T-infection samples. (A) Shannon Index Box Chart indicated no significant differences in alpha diversity. Beta-diversity analyses of clustering profiles based on Bray-Curtis (B) and Jaccard (C) showed the existence of significantly differences in beta diversity. (D) Bar plot of species composition at Phylum level. (E) Family level heatmap maps. (F) Genus-level heatmap maps.
A total of 11 Phylum were obtained from the annotation of the two groups, with roughly the same composition in both groups (Figure 2D). Among them, Firmicutes and Bacteroidota were the dominant Phylum in both groups. In the control group, 61.93% of the species belonged to Firmicutes, with an abundance of 59.75, and 29.66% of the species belonged to Bacteroidota, with an abundance of 33.89%. In the infected group, 58.82% of the species belonged to the Firmicutes Phylum with an abundance of 40.15 and 32.86% of the species belonged to the Anamorph Phylum with an abundance of 35.76%. The abundance of the Proteobacteria Phylum was higher in the infected group compared to the control group. The top 50 Families and Genera in terms of abundance were selected and analyzed at the Family and Genus level, respectively, and it was evident that there were significant changes between the infected group and the control group at the Family and Genus level (Figures 2E,F).
The relative abundance of the top 30 families and genera in each group was statistically analyzed (Figures 3A,B). The families with more than 10% abundance in the control group were Lactobacillaceae, Lachnospiraceae and Prevotellaceae, and the families with more than 10% abundance in the infected group were Enterobacteriaceae, Bacteroidaceae, Lachnospiraceae, Lactobacillaceae and Muribaculaceae; compared to the control group, the relative abundance of Enterobacteriaceae in the infected group increased by 18.4%, Bacteroidaceae by 7.42%, Lactobacillaceae by 14.05%, Prevotellaceae by 8.46% and Peptostreptococcaceae by 4.28%. The genera with an abundance greater than 10% in the control group were Lactobacillus and Muribaculaceae, and the genera with an abundance greater than 10% in the infected group were Bacteroides, Lactobacillus, and Muribaculaceae; the relative abundance of Escherichia-Shigella genera increased by 19.71%, Bacteroides by 8.07%, and Lachnospiraceae_NK4A136_group by 3.00% compared to the control group increased by 19.71%, Bacteroides by 8.07% and Lachnospiraceae_NK4A136_group by 3.00% compared to the control group (Figures 3C,D).
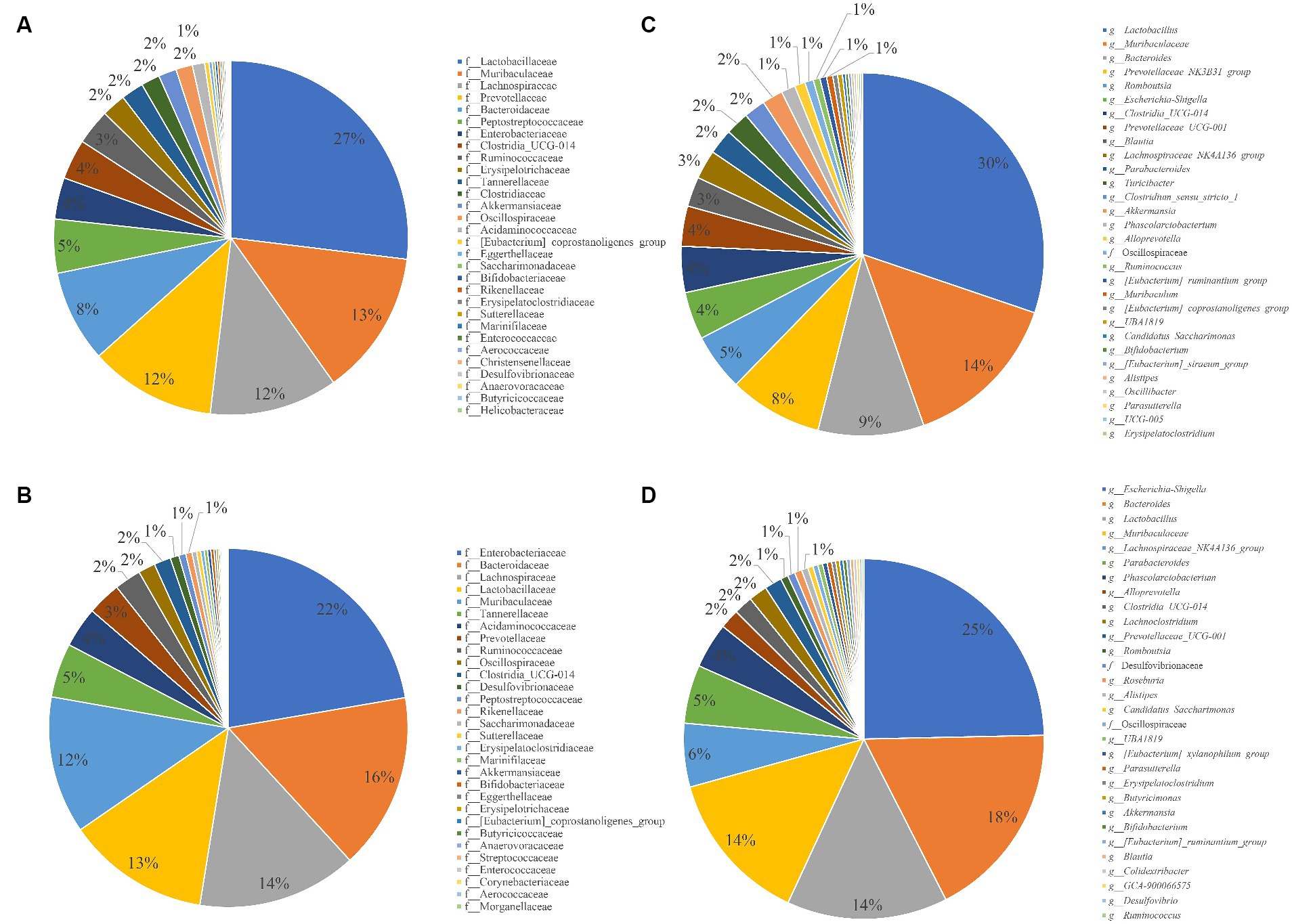
Figure 3. Pie chart for the proportion of species in different groups and taxonomic levels. (A) Composition of gut microbiota at family level in control group. (B) Composition of gut microbiota at family level in Infection group. (C) Control group at the Genus level. (D) Infection group at the Genus level.
For discovering significant biomarkers between different groups, the LEfSe analysis was performed. The characteristic homogeneous genera in each group were screened and the results are shown in Figure 4A, and the intergroup differences present at each genus level are shown in Figure 4B. The control group contained five characterized genera (Prevotellaceae NK3B31 group, Turicibacter, Clostridium sensu stricto 1, Candidatus Stoquefichus, Lachnospira) and the infected group contained nine characterized genera (Rodentibacter, Christensenella, Dubosiella, Streptococcus, Anaeroplasma, Lactococcus, Papillibacter, Desulfovibrio, Roseburia).
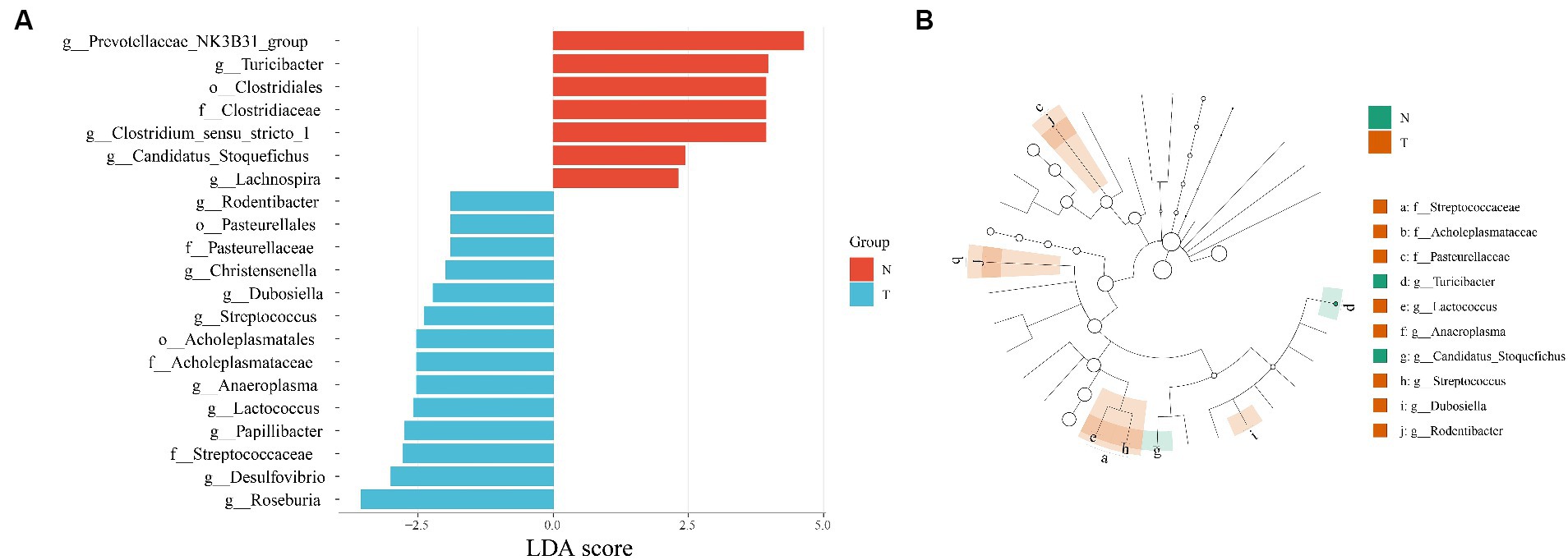
Figure 4. Distribution of LDA values (A) and evolutionary branching maps (B) of divergent species.
3.3 Reorganization for intestinal microbiota correlation
To represent the effect of A. pegreffii infection on rat intestinal microbiota, intragroup abundance correlation network diagrams were constructed at the Genus level, respectively. The correlation coefficients were analyzed based on the Spearman method for the 60 most abundant Genera, with the parameters of R > 0.6 and p < 0.01 (Figure 5). In the control group, the network’ formed two large clusters, while the remaining genera formed smaller connections. The core genera were identified as Butyricimonas, Alloprevotella, Lachnospiraceae_UCG-006, A2, g_Erysipelotrichaceae, and g_Desulfovibrionaceae based on their weighted degree of centrality. The infection group exhibited two significantly large clusters in the network, which were connected to smaller clusters. The core genera identified were Lachnoclostridium, Eubacterium_coprostanoligenes_group, Lachnospiraceae_UCG-006, and Alistipes. Significant changes in abundance and connectivity of differential marker genera in the interactions network are shown in Table 1. Out of all the differential genera, 9 were absent in the network between the control and infection groups, 6 were present only in the interactions network with the control group, and 8 were present only in the interactions network with the infected group. Three genera were present in the interaction network for both the control and infected groups. The Prevotellaceae_NK3B31_group had greater weighted centrality in both groups, while Romboutsia and Lachnoclostridium had greater weighted centrality in the infected group compared to the control group.
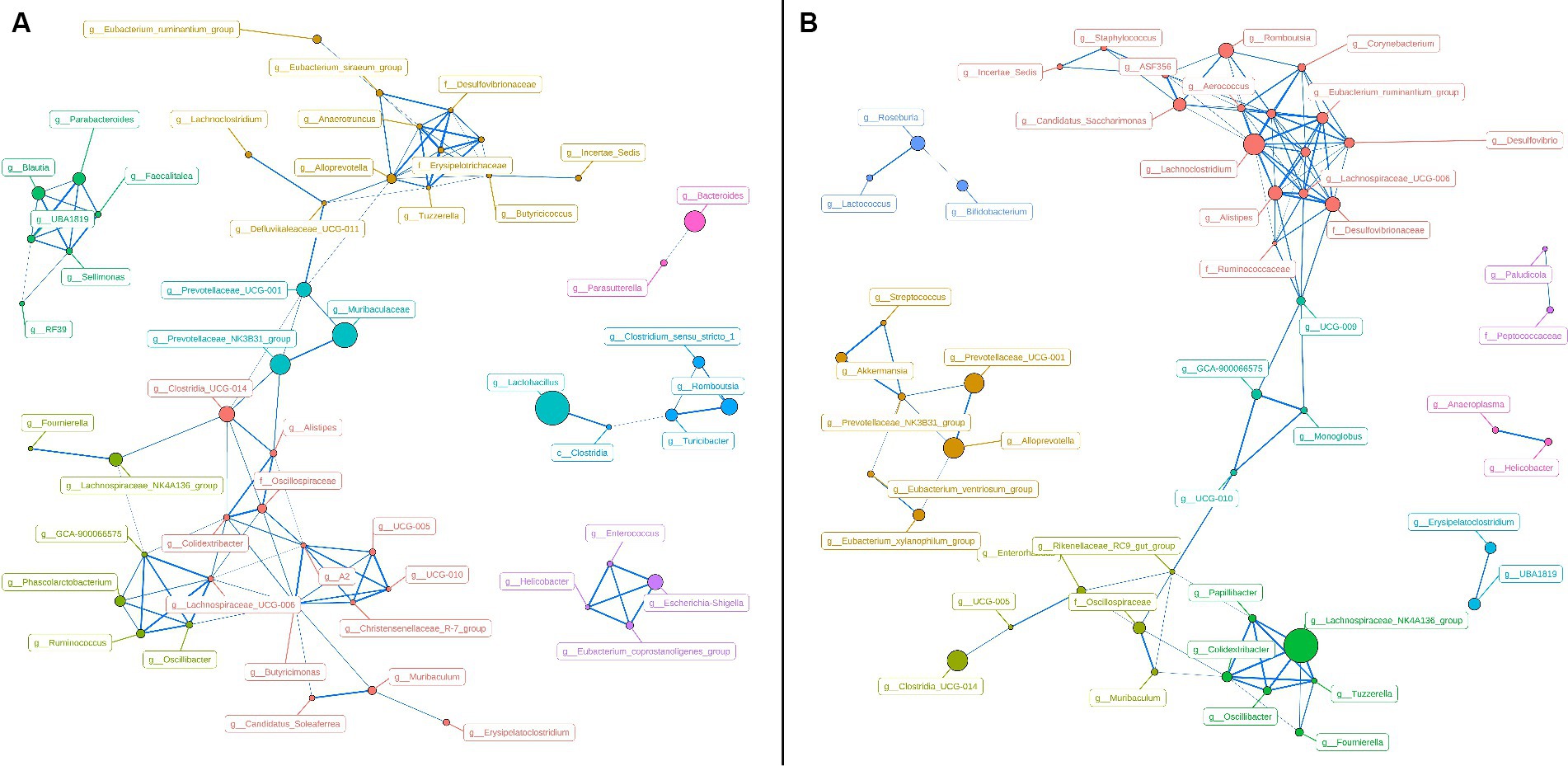
Figure 5. Network analysis chart in control group (A) and Infection group (B).
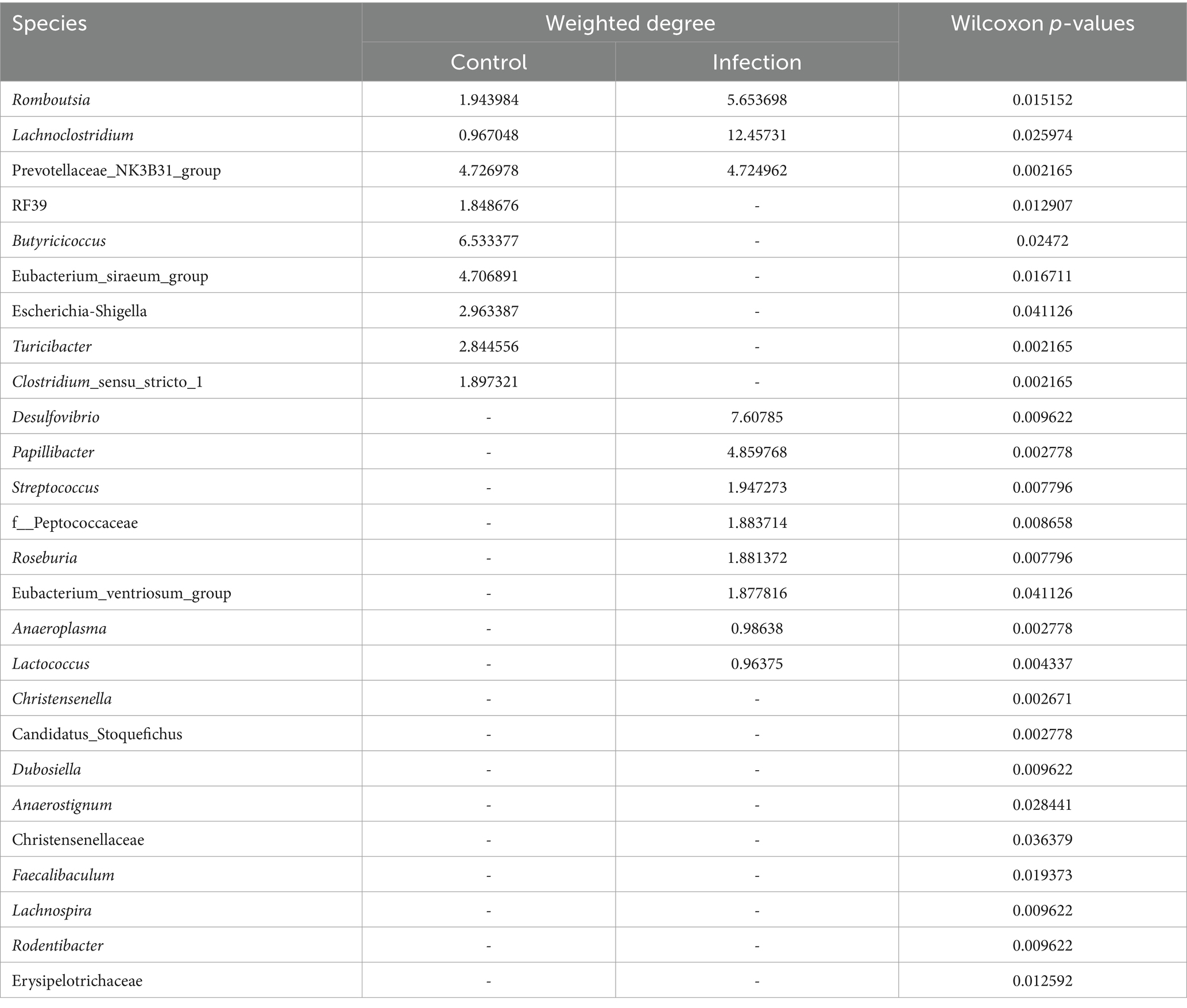
Table 1. Weighted degree of divergent species with significant changes in the relative abundance.
3.4 Functional gene composition of intestinal microbiota in response to Anisakis pegreffi
To further research on metabolic differences in microbiota, based on the ASVs and abundances identified by 16S rDNA, the PICRUSt2 tool was used to predict metagenomic functional information (29). Based on the relative abundance of the top 30 functions, the Bray distance was calculated between samples and hierarchical clustering was performed (Figure 6A). The infection group and control group were not clustered together and did not form two independent clusters. The results of the PCoA analysis showed partial overlap between the infected and control groups (Figure 6B). The analysis of the differences between the within and between groups indicated that the between-group difference was larger than the within-group difference (R > 0) for the infected and control groups, but it was not significant (p > 0.01) (Figure 6C). Based on the Wilcoxon test, four processes were found to be significant: bacterial secretion system, bacterial invasion of epithelial cells, bacterial chemotaxis, and ABC transporters (Figure 6D). All these processes were more abundant in the infection group than in the control group.
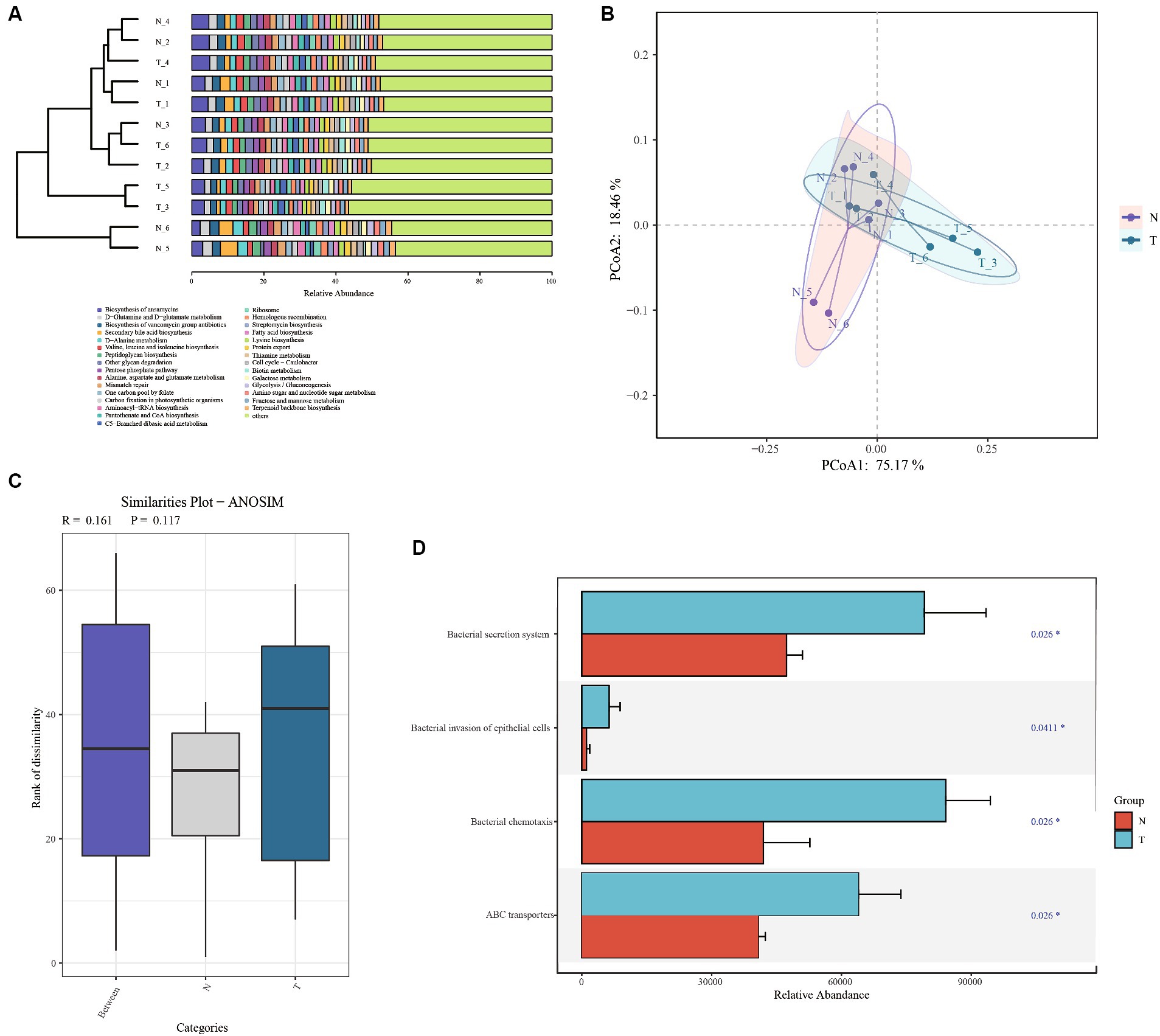
Figure 6. Functional analysis based on PICRUSt predictions. (A) Bray Cluster analysis and functional composition stacking map. (B) Bray Curtis PCoA analysis plots. (C) Anosim analysis based on functional gene abundance. (D) Significantly differential functional genes bar chart.
4 Discussion
Animal models of infection are widely employed to investigate the cause-effect relationships between helminth colonization, shifts in gut microbial composition, and modifications in the immune or metabolic functions of the host (30). However, interactions between helminths and the gut microbiota vary significantly depending on the specific host–parasite combination being studied (31). Consequently, comparing data across different host–parasite systems, even if they involve the same helminth species, could potentially lead to misunderstandings. Conversely, research on naturally infected humans may offer insights into the relationships between gut microbial composition and infection with one or more parasites (32). Moreover, such studies often lack evidence of a causal link between specific gut microbial signatures and susceptibility to or the pathophysiology of infection (33). Therefore, it is important to conduct complementary studies that involve naturally infected human populations in endemic regions alongside experimental animal models to advance our understanding of the role of the gut microbiota in human parasitic diseases (34). The gut microbiota is a crucial element of the immune system and overall health (35). In this study, rats were used to examine the links between A. pegreffii infections and the composition of the host gut microbiota. As far as we know, this is the first-ever study investigating the associations between Anisakiasis and the gut microbiota of the host (rats).
Given the recognized risk posed by Anisakis nematodes, further investigation into their impact on the host is warranted (36). Our current study found that A. pegreffii infection did not lead to significant alterations in the alpha diversity of rat gut microbiota. However, differences in beta diversity were observed, which is in line with previous research (32, 37, 38). Interestingly, we observed a significant reduction in the abundance of the Genus Turicibacter and the Order Clostridiales in the infected group, suggesting potential intestinal inflammation (39). This finding is consistent with the results of research that showed a decrease in Clostridiales due to Toxocara canis infection. Despite the known ability of Firmicutes bacteria to metabolize free fatty acids and bile acids, resulting in the production of butyrate, the reduced abundance of Firmicutes phylum in our study did not reflect anti-inflammatory properties (40, 41).
The Prevotellaceae_NK3B31_group is a key group of intestinal microorganisms, typically considered for their roles as probiotic bacteria producing short-chain fatty acid (SCFA). They are capable of producing various essential salts and fatty acids from dietary fiber, thereby playing a significant role in animal growth and development. Moreover, an increase in the abundance of the population of this group has been associated with the mitigation of gastric ulcer pathology (42–45). The high weighted degree of centrality of this bacterial group in both the control and infected groups indicates its importance in the intestinal tract of both groups of test animals. However, a significant decrease in the abundance (8% in control groups, 0.1% in infection groups) of this genus in the infection group resulted in a substantial alteration in gut microbiota diversity. Bacteria of the genus Turicibacter are also important members of the mammalian gut microbiota with evidence suggesting their involvement in modifying host bile acids and lipids, thereby regulating host lipid metabolism (46, 47). In the infection group, there was a significant decrease in the relative abundance of this genus (2% in control groups, 0.038% in infection groups), leading to a diminished capacity to regulate the overall gut microbiota. Intestinal helminth infections often increase the microbial origin of SCFAs, which can help regulate allergic diseases (48). However, this phenomenon was not observed in the present study. The Genus Clostridium_sensu_stricto_1 is known for its production of butyrate, which plays an important role in the development of intestinal epithelial cells and hosts energy supply (49, 50). In the present study, the relative abundance of this genus was significantly reduced (1.9% in control groups, 0.031% in infection groups), along with its ability to regulate the gut microbiota. The decreased level of Romboutsia (4.9% in control groups, 0.65% in infection groups) can lead to an increase in intestinal pH, subsequently promoting the proliferation of harmful bacteria like Streptococcus (0.007% in control groups, 0.052% in infection groups) (51). In heavily infected individuals with roundworms, Streptococcus was found to be the most prevalent bacteria in the intestines, significantly decreasing the pro-inflammatory activity of the host colonic mucosa (52, 53). The metabolism of butyric acid in the host’s intestines has been significantly altered. Indigestible complex carbohydrates, such as dietary fiber, serve as an energy source for specific bacteria, resulting in the production of various short-chain fatty acid molecules, including butyrate. These microbial compounds play a role in regulating various metabolic pathways in the gut and other parts of the body, such as the liver, adipose tissue, muscle, and brain. It’s now well-established that these metabolites have a significant impact on several physiological processes, including energy balance, glucose, and lipid metabolism, inflammation, as well as immunity, and cancer (54–56). Interestingly, the infection group showed enrichment of Roseburia (0.015% in control groups, 0.64% in infection groups), typically considered a probiotic that produces butyric acid and contributes to asthma prevention in children (57).
Moreover, the presence of Lactococcus was augmented in the infected cohort, aimed at curbing the onset of hypersensitivity reactions (58). The increase in both Desulfovibrio (0.043% in control groups, 0.198% in infection groups) and Streptococcus may indicate issues with bile acid metabolism, which aligns with the previously mentioned decrease in fatty acid synthesis. The Genus Desulfovibrio is commonly regarded as a harmful group of bacteria that produces reduced sulfate, generating endogenous H2S that can be detrimental to the organism (59, 60). The relative abundance of Desulfovibrio spp. is elevated in the intestinal tract of individuals with colitis, inflammatory bowel disease, and irritable bowel syndrome (61–63). The genus of Escherichia-Shigella was more abundant in the infection group (4.104% in control groups, 23.82% in infection groups), which is typically associated with dysentery (64). Additionally, it is positively associated with sulfate toxin metabolism (65). The genus Bacteroides was more abundant in the infection group (9.11% in control groups, 17.19% in infection groups) and can produce toxins that cause secretory diarrhea and colonic epithelial damage, leading to chronic colitis and colorectal cancer (66, 67). Bacteria have the potential to harm the host’s health by invading tissue sites and evading the immune system. One of the ways they achieve this is through the secretion of virulent proteins, which can be released into the host cell or surrounding environment (68). Nevertheless, a more in-depth comprehension of the alterations in the intestinal microbiota in infected states can facilitate the treatment by improving the microbiological environment. For instance, the regulation of the composition of gut microbes may be employed to alleviate systemic inflammation and cardiovascular disease caused by chronic nephritis (69). Such research is also frequently employed in the investigation of neurological disorders (70–72).
In this study, based on the analysis of microbiota diversity and functional prediction, the infected group exhibited notable increases in the Bacterial secretion system, invasion of epithelial cells, and ABC transporter functions, implying significant microbiota-induced impact to the host gut.
5 Conclusion
In this study, rats infected with A. pegreffii were obtained by artificial infection and infection was determined by indirect ELISA diagnosis based on worm extraction. For the first time, the microbiota diversity analysis of host feces was performed using 16S sequencing. The results indicate that rats infected with A. pegreffii exhibited significant changes in microbiota diversity. The rats’ SCFAs metabolism was affected, which in turn affected their nutrient metabolism and immune function. The increase in the abundance of pathogenic enteric bacteria indicated that the gut flora had been invaded by pathogens. It provides a basis for future research on host-helminth-microbiota correlations and microbiota metabolism.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA1088275.
Ethics statement
The animal study was approved by Medical Science and Technology Ethics Committee of Jiujiang University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
M-hZ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. SL: Methodology, Supervision, Validation, Writing – review & editing. Q-bL: Data curation, Software, Writing – review & editing. X-xW: Conceptualization, Data curation, Investigation, Writing – review & editing. AQ: Investigation, Methodology, Writing – review & editing. MM: Funding acquisition, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors would like to extend their gratitude to King Saud University (Riyadh, Saudi Arabia) for funding this research through Researchers supporting Project number (RSP-2024-R406).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2024.1403920/full#supplementary-material
Footnotes
References
1. Buchmann, K, and Mehrdana, F. Effects of Anisakid nematodes Anisakis Simplex (S.L.), Pseudoterranova Decipiens (S.L.) and Contracaecum Osculatum (S.L.) on Fish and consumer health. Food Waterborne Parasitol. (2016) 4:13–22. doi: 10.1016/j.fawpar.2016.07.003
2. Marcelino, L, Garfias, C, Zaheer, T, Maqsood, A, Bamarni, S, Abdullah, B, et al. Potential of Anisakiasis in foodborne zoonosis. Pak Vet J. (2022) 42:433–44. doi: 10.29261/pakvetj/2022.080
3. Villazanakretzer, DL, Napolitano, PG, Cummings, KF, and Magann, EF. Fish parasites: a growing concern during pregnancy. Obstet Gynecol Surv. (2016) 71:253–9. doi: 10.1097/ogx.0000000000000303
4. Takano, K, Okuni, T, Murayama, K, and Himi, T. A case study of Anisakiasis in the palatine tonsils. Adv Otorhinolaryngol. (2016) 77:125–7. doi: 10.1159/000441903
5. Groudan, K, Martins, T, and Schmelkin, IJ. Gastric Anisakiasis masquerading as gastroesophageal reflux disease. Case Rep Gastrointest Med. (2023) 2023:8635340–4. doi: 10.1155/2023/8635340
6. Ramanan, P, Blumberg, AK, Mathison, B, and Pritt, BS. Parametrial Anisakidosis. J Clin Microbiol. (2013) 51:3430–4. doi: 10.1128/jcm.01398-13
7. Mattiucci, S. Molecular epidemiology of Anisakis and Anisakiasis: an ecological and evolutionary road map. Adv Parasitol. (2018) 99:93–263. doi: 10.1016/bs.apar.2017.12.001
8. Ahuir-Baraja, AE, Llobat, L, and Garijo, MM. Effectiveness of gutting blue whiting (Micromesistius Poutassou, Risso, 1827), in Spanish supermarkets as an Anisakidosis safety measure. Food Secur. (2021) 10:862. doi: 10.3390/foods10040862
9. Sugiyama, H, Shiroyama, M, Yamamoto, I, Ishikawa, T, and Morishima, Y. Anisakiasis annual incidence and causative species, Japan, 2018-2019. Emerg Infect Dis. (2022) 28:2105–8. doi: 10.3201/eid2810.220627
10. Huss, HH, Ababouch, L, and Gram, L. Assessment and management of seafood safety and quality. Rome: FAO (2003).
11. Ruff, WE, Greiling, TM, and Kriegel, MA. Host-microbiota interactions in immune-mediated diseases. Nat Rev Microbiol. (2020) 18:521–38. doi: 10.1038/s41579-020-0367-2
12. El-Sayed, A, Aleya, L, and Kamel, M. Microbiota's role in health and diseases. Environ Sci Pollut Res Int. (2021) 28:36967–83. doi: 10.1007/s11356-021-14593-z
13. Martel, J, Chang, SH, Ko, YF, Hwang, TL, Young, JD, and Ojcius, DM. Gut barrier disruption and chronic disease. Trends Endocrinol Metab. (2022) 33:247–65. doi: 10.1016/j.tem.2022.01.002
14. Hooper, LV, Littman, DR, and Macpherson, AJ. Interactions between the microbiota and the immune system. Science. (2012) 336:1268–73. doi: 10.1126/science.1223490
15. Jaenike, J, Unckless, R, Cockburn, SN, Boelio, LM, and Perlman, SJ. Adaptation via Symbiosis: recent spread of a Drosophila defensive symbiont. Science. (2010) 329:212–5. doi: 10.1126/science.1188235
16. Xiang, H, Fang, Y, Tan, ZL, and Zhong, RZ. Haemonchus Contortus infection alters gastrointestinal microbial community composition, protein digestion and amino acid allocations in lambs. Front Microbiol. (2022) 12:15. doi: 10.3389/fmicb.2021.797746
17. Wang, Y, Liu, F, Urban, JF Jr, Paerewijck, O, Geldhof, P, and Li, RW. Ascaris Suum infection was associated with a worm-independent reduction in microbial diversity and altered metabolic potential in the porcine gut microbiome. Int J Parasitol. (2019) 49:247–56. doi: 10.1016/j.ijpara.2018.10.007
18. Gasser, RB, Bott, NJ, Chilton, NB, Hunt, P, and Beveridge, I. Toward practical, DNA-based diagnostic methods for parasitic nematodes of livestock — bionomic and biotechnological implications. Biotechnol Adv. (2008) 26:325–34. doi: 10.1016/j.biotechadv.2008.03.003
19. Gasser, RB, Rossi, L, and Zhu, X. Identification of Nematodirus species (Nematoda: Molineidae) from wild ruminants in Italy using ribosomal DNA markers. Int J Parasitol. (1999) 29:1809–17. doi: 10.1016/s0020-7519(99)00123-x
20. D'Amelio, S, Mathiopoulos, KD, Santos, CP, Pugachev, ON, Webb, SC, Picanço, M, et al. Genetic markers in ribosomal DNA for the identification of members of the genus Anisakis (Nematoda: Ascaridoidea) defined by polymerase-chain-reaction-based restriction fragment length polymorphism. Int J Parasitol. (2000) 30:223–6. doi: 10.1016/s0020-7519(99)00178-2
21. Mattiucci, S, Acerra, V, Paoletti, M, Cipriani, P, Levsen, A, Webb, SC, et al. No more time to stay 'Single' in the detection of Anisakis Pegreffii, A. simplex (S. S.) and hybridization events between them: a multi-marker nuclear genotyping approach. Parasitology. (2016) 143:998–1011. doi: 10.1017/s0031182016000330
22. Figueiredo, I, Cardoso, L, Teixeira, G, Lopes, L, São Clemente, SC, and Vericimo, MA. A technique for the intra-gastric Administration of Live Larvae of Anisakis Simplex in mice. Exp Parasitol. (2012) 130:285–7. doi: 10.1016/j.exppara.2012.01.004
23. Bolger, AM, Lohse, M, and Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
24. Magoč, T, and Salzberg, SL. Flash: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. (2011) 27:2957–63. doi: 10.1093/bioinformatics/btr507
25. Bolyen, E, Rideout, JR, Dillon, MR, Bokulich, NA, Abnet, CC, Al-Ghalith, GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using Qiime 2. Nat Biotechnol. (2019) 37:852–7. doi: 10.1038/s41587-019-0209-9
26. Yilmaz, P, Parfrey, LW, Yarza, P, Gerken, J, Pruesse, E, Quast, C, et al. The Silva and “all-species living tree project (Ltp)” taxonomic frameworks. Nucleic Acids Res. (2013) 42:D643–8. doi: 10.1093/nar/gkt1209
27. Cole, JR, Wang, Q, Fish, JA, Chai, B, McGarrell, DM, Sun, Y, et al. Ribosomal database project: data and tools for high throughput Rrna analysis. Nucleic Acids Res. (2014) 42:D633–42. doi: 10.1093/nar/gkt1244
28. DeSantis, TZ, Hugenholtz, P, Larsen, N, Rojas, M, Brodie, EL, Keller, K, et al. Greengenes, a chimera-checked 16s Rrna gene database and workbench compatible with arb. Appl Environ Microbiol. (2006) 72:5069–72. doi: 10.1128/aem.03006-05
29. Douglas, GM, Maffei, VJ, Zaneveld, JR, Yurgel, SN, Brown, JR, Taylor, CM, et al. Picrust 2 for prediction of metagenome functions. Nat Biotechnol. (2020) 38:685–8. doi: 10.1038/s41587-020-0548-6
30. Cortés, A, Toledo, R, and Cantacessi, C. Classic models for new perspectives: delving into helminth-microbiota-immune system interactions. Trends Parasitol. (2018) 34:640–54. doi: 10.1016/j.pt.2018.05.009
31. Cortés, A, Peachey, L, Scotti, R, Jenkins, TP, and Cantacessi, C. Helminth-microbiota cross-talk - a journey through the vertebrate digestive system. Mol Biochem Parasitol. (2019) 233:111222. doi: 10.1016/j.molbiopara.2019.111222
32. Cortés, A, Peachey, LE, Jenkins, TP, Scotti, R, and Cantacessi, C. Helminths and microbes within the vertebrate gut-not all studies are created equal. Parasitology. (2019) 146:1371–8. doi: 10.1017/s003118201900088x
33. Arrieta, MC, Walter, J, and Finlay, BB. Human microbiota-associated mice: a model with challenges. Cell Host Microbe. (2016) 19:575–8. doi: 10.1016/j.chom.2016.04.014
34. Cortés, A, Clare, S, Costain, A, Almeida, A, McCarthy, C, Harcourt, K, et al. Baseline gut microbiota composition is associated with Schistosoma Mansoni infection burden in rodent models. Front Immunol. (2020) 11:593838. doi: 10.3389/fimmu.2020.593838
35. Lloyd-Price, J, Mahurkar, A, Rahnavard, G, Crabtree, J, Orvis, J, Hall, AB, et al. Strains, functions and dynamics in the expanded human microbiome project. Nature. (2017) 550:61–6. doi: 10.1038/nature23889
36. Bao, M, Pierce, GJ, Pascual, S, González-Muñoz, M, Mattiucci, S, Mladineo, I, et al. Assessing the risk of an emerging zoonosis of worldwide concern: Anisakiasis. Sci Rep. (2017) 7:43699. doi: 10.1038/srep43699
37. Scotti, R, Southern, S, Boinett, C, Jenkins, TP, Cortés, A, and Cantacessi, C. Michelindb: a web-based tool for Mining of Helminth-Microbiota Interaction Datasets, and a Meta-analysis of current research. Microbiome. (2020) 8:10. doi: 10.1186/s40168-019-0782-7
38. Stracke, K, Adisakwattana, P, Phuanukoonnon, S, Yoonuan, T, Poodeepiyasawat, A, Dekumyoy, P, et al. Field evaluation of the gut microbiome composition of pre-school and school-aged children in Tha Song Yang, Thailand, following Oral Mda for Sth infections. PLoS Negl Trop Dis. (2021) 15:e0009597. doi: 10.1371/journal.pntd.0009597
39. Sun, XM, Hao, CY, Wu, AQ, Luo, ZN, El-Ashram, S, Alouffi, A, et al. Trichinella spiralis-induced immunomodulation signatures on gut microbiota and metabolic pathways in mice. PLoS Pathog. (2024) 20:e1011893. doi: 10.1371/journal.ppat.1011893
40. Zhong, H, Penders, J, Shi, Z, Ren, H, Cai, K, Fang, C, et al. Impact of early events and lifestyle on the gut microbiota and metabolic phenotypes in Young school-age children. Microbiome. (2019) 7:2. doi: 10.1186/s40168-018-0608-z
41. Kupritz, J, Angelova, A, Nutman, TB, and Gazzinelli-Guimaraes, PH. Helminth-induced human gastrointestinal Dysbiosis: a systematic review and Meta-analysis reveals insights into altered taxon diversity and microbial gradient collapse. MBio. (2021) 12:e0289021. doi: 10.1128/mBio.02890-21
42. Wang, J, Ji, H, Wang, S, Liu, H, Zhang, W, Zhang, D, et al. Probiotic Lactobacillus Plantarum promotes intestinal barrier function by strengthening the epithelium and modulating gut microbiota. Front Microbiol. (2018) 9:1953. doi: 10.3389/fmicb.2018.01953
43. Feng, L, Bao, T, Bai, L, Mu, X, Ta, N, Bao, M, et al. Mongolian medicine formulae Ruda-6 alleviates indomethacin-induced gastric ulcer by regulating gut microbiome and serum metabolomics in rats. J Ethnopharmacol. (2023) 314:116545. doi: 10.1016/j.jep.2023.116545
44. Wang, D, Tang, G, Zhao, L, Wang, M, Chen, L, Zhao, C, et al. Potential roles of the rectum keystone microbiota in modulating the microbial community and growth performance in goat model. J Anim Sci Biotechnol. (2023) 14:55. doi: 10.1186/s40104-023-00850-3
45. Hu, W, Huang, L, Zhou, Z, Yin, L, and Tang, J. Diallyl disulfide (dads) ameliorates intestinal Candida Albicans infection by modulating the gut microbiota and metabolites and providing intestinal protection in mice. Front Cell Infect Microbiol. (2021) 11:743454. doi: 10.3389/fcimb.2021.743454
46. Lynch, JB, Gonzalez, EL, Choy, K, Faull, KF, Jewell, T, Arellano, A, et al. Gut microbiota Turicibacter strains differentially modify bile acids and host lipids. Nat Commun. (2023) 14:3669. doi: 10.1038/s41467-023-39403-7
47. Li, H, Liu, C, Huang, S, Wang, X, Cao, M, Gu, T, et al. Multi-omics analyses demonstrate the modulating role of gut microbiota on the associations of unbalanced dietary intake with gastrointestinal symptoms in children with autism Spectrum disorder. Gut Microbes. (2023) 15:2281350. doi: 10.1080/19490976.2023.2281350
48. Zaiss, MM, Rapin, A, Lebon, L, Dubey, LK, Mosconi, I, Sarter, K, et al. The intestinal microbiota contributes to the ability of helminths to modulate allergic inflammation. Immunity. (2015) 43:998–1010. doi: 10.1016/j.immuni.2015.09.012
49. Pryde, SE, Duncan, SH, Hold, GL, Stewart, CS, and Flint, HJ. The microbiology of butyrate formation in the human Colon. FEMS Microbiol Lett. (2002) 217:133–9. doi: 10.1111/j.1574-6968.2002.tb11467.x
50. Pei, Y, Chen, C, Mu, Y, Yang, Y, Feng, Z, Li, B, et al. Integrated microbiome and metabolome analysis reveals a positive change in the intestinal environment of Myostatin edited large White pigs. Front Microbiol. (2021) 12:12. doi: 10.3389/fmicb.2021.628685
51. Sieng, S, Chen, P, Wang, N, Xu, JY, and Han, Q. Toxocara Canis-induced changes in host intestinal microbial communities. Parasit Vectors. (2023) 16:462. doi: 10.1186/s13071-023-06072-w
52. Klomkliew, P, Sawaswong, V, Chanchaem, P, Nimsamer, P, Adisakwattana, P, Phuphisut, O, et al. Gut Bacteriome and metabolome of Ascaris Lumbricoides in patients. Sci Rep. (2022) 12:19524. doi: 10.1038/s41598-022-23608-9
53. Vitetta, L, Llewellyn, H, and Oldfield, D. Gut Dysbiosis and the intestinal microbiome: Streptococcus Thermophilus a key probiotic for reducing uremia. Microorganisms. (2019) 7:228. doi: 10.3390/microorganisms7080228
54. de Vos, WM, Tilg, H, Van Hul, M, and Cani, PD. Gut microbiome and health: mechanistic insights. Gut. (2022) 71:1020–32. doi: 10.1136/gutjnl-2021-326789
55. Frost, G, Sleeth, ML, Sahuri-Arisoylu, M, Lizarbe, B, Cerdan, S, Brody, L, et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun. (2014) 5:3611. doi: 10.1038/ncomms4611
56. Cani, PD, and Jordan, BF. Gut microbiota-mediated inflammation in obesity: a link with gastrointestinal Cancer. Nat Rev Gastroenterol Hepatol. (2018) 15:671–82. doi: 10.1038/s41575-018-0025-6
57. Depner, M, Taft, DH, Kirjavainen, PV, Kalanetra, KM, Karvonen, AM, Peschel, S, et al. Maturation of the gut microbiome during the first year of life contributes to the protective farm effect on childhood asthma. Nat Med. (2020) 26:1766–75. doi: 10.1038/s41591-020-1095-x
58. Zhang, JD, Liu, J, Zhu, SW, Fang, Y, Wang, B, Jia, Q, et al. Berberine alleviates visceral hypersensitivity in rats by altering gut microbiome and suppressing spinal microglial activation. Acta Pharmacol Sin. (2021) 42:1821–33. doi: 10.1038/s41401-020-00601-4
59. Verstreken, I, Laleman, W, Wauters, G, and Verhaegen, J. Desulfovibrio Desulfuricans bacteremia in an immunocompromised host with a liver graft and ulcerative colitis. J Clin Microbiol. (2012) 50:199–201. doi: 10.1128/jcm.00987-11
60. Murros, KE, Huynh, VA, Takala, TM, and Saris, PEJ. Desulfovibrio Bacteria are associated with Parkinson's disease. Front Cell Infect Microbiol. (2021) 11:652617. doi: 10.3389/fcimb.2021.652617
61. Gobert, AP, Sagrestani, G, Delmas, E, Wilson, KT, Verriere, TG, Dapoigny, M, et al. The human intestinal microbiota of constipated-predominant irritable bowel syndrome patients exhibits anti-inflammatory properties. Sci Rep. (2016) 6:39399. doi: 10.1038/srep39399
62. He, P, Zhang, Y, Chen, R, Tong, Z, Zhang, M, and Wu, H. The Maca protein ameliorates Dss-induced colitis in mice by modulating the gut microbiota and production of Scfas. Food Funct. (2023) 14:10329–46. doi: 10.1039/d3fo03654e
63. Singh, SB, Coffman, CN, Varga, MG, Carroll-Portillo, A, Braun, CA, and Lin, HC. Intestinal alkaline phosphatase prevents sulfate reducing Bacteria-induced increased tight junction permeability by inhibiting snail pathway. Front Cell Infect Microbiol. (2022) 12:882498. doi: 10.3389/fcimb.2022.882498
64. Fung, CC, Octavia, S, Mooney, AM, and Lan, R. Virulence variations in Shigella and Enteroinvasive Escherichia Coli using the Caenorhabditis Elegans model. FEMS Microbiol Lett. (2015) 362:1–5. doi: 10.1093/femsle/fnu045
65. Hayashi, T, Yamashita, T, Watanabe, H, Kami, K, Yoshida, N, Tabata, T, et al. Gut microbiome and plasma microbiome-related metabolites in patients with decompensated and compensated heart failure. Circ J. (2018) 83:182–92. doi: 10.1253/circj.CJ-18-0468
66. Chung, L, Thiele Orberg, E, Geis, AL, Chan, JL, Fu, K, DeStefano Shields, CE, et al. Bacteroides Fragilis toxin coordinates a pro-carcinogenic inflammatory Cascade via targeting of colonic epithelial cells. Cell Host Microbe. (2018) 23:203–14.e5. doi: 10.1016/j.chom.2018.01.007
67. Valguarnera, E, and Wardenburg, JB. Good gone bad: one toxin away from disease for Bacteroides Fragilis. J Mol Biol. (2020) 432:765–85. doi: 10.1016/j.jmb.2019.12.003
68. Green, ER, and Mecsas, J. Bacterial secretion systems: an overview. Microbiol Spectr. (2016) 4:2015. doi: 10.1128/microbiolspec.VMBF-0012-2015
69. Mafra, D, Lobo, JC, Barros, AF, Koppe, L, Vaziri, ND, and Fouque, D. Role of altered intestinal microbiota in systemic inflammation and cardiovascular disease in chronic kidney disease. Future Microbiol. (2014) 9:399–410. doi: 10.2217/fmb.13.165
70. Berer, K, Mues, M, Koutrolos, M, Rasbi, ZA, Boziki, M, Johner, C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. (2011) 479:538–41. doi: 10.1038/nature10554
71. Bravo, JA, Forsythe, P, Chew, MV, Escaravage, E, Savignac, HM, Dinan, TG, et al. Ingestion of Lactobacillus strain regulates emotional behavior and central Gaba receptor expression in a mouse via the Vagus nerve. Proc Natl Acad Sci USA. (2011) 108:16050–5. doi: 10.1073/pnas.1102999108
Keywords: Anisakis pegreffii , rat, microbiota, diversity analysis, host effects
Citation: Zeng M-h, Li S, Lv Q-b, Wang X-x, Qadeer A and Mahmoud MH (2024) Modulation of the rat intestinal microbiota in the course of Anisakis pegreffii infection. Front. Vet. Sci. 11:1403920. doi: 10.3389/fvets.2024.1403920
Edited by:
Hussam Askar, Al Azhar University, EgyptReviewed by:
Bernardo Pereira Moreira, University of Giessen, GermanyMahmoud Abdelhamid, Aswan University, Egypt
Copyright © 2024 Zeng, Li, Lv, Wang, Qadeer and Mahmoud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min-hao Zeng, emVuZ21pbmhhb0BzdHUuanVzdC5lZHUuY24=; Shan Li, c2xvdmUwNDA4QDE2My5jb20=
†These authors have contributed equally to this work