 Chaoliang Leng
Chaoliang Leng Xiang Tian
Xiang Tian Jun Ji
Jun Ji Lunguang Yao
Lunguang Yao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Vet. Sci., 01 May 2024
Sec. Veterinary Infectious Diseases
Volume 11 - 2024 | https://doi.org/10.3389/fvets.2024.1375948
Chaphamaparvovirus carnivoran1 (canine Chaphamaparvovirus, also known as Cachavirus [CachaV]) is a novel parvovirus first reported in dog feces collected from the United States in 2017 and China in 2019. To continuously track its infection and evolution status, 276 canine anal swabs were obtained from pet hospitals in central, northern, and eastern China between 2021 and 2023 and screened via polymerase chain reaction; subsequently, a systematic study was conducted. Of these samples, nine (3.3%) were positive for CachaV. Using polymerase chain reaction, whole genome sequences of the nine CachaV-positive strains were amplified. The NS1 amino acid sequence identity between CachaV strains from China and other countries was 96.23–99.85%, whereas the VP1 protein sequence identity was 95.83–100%. CHN230521 demonstrated the highest identity for NS1 amino acids (99.85%) and VP1 amino acids (100%) with NWT-W88 and CP-T015. According to the model prediction of CHN220916-VP1 protein, Met64Thr, Thr107Ala, and Phe131Ser mutations may cause tertiary structural changes in VP1 protein. Interestingly, each of the nine CachaV strains harbored the same site mutations in NS1 (Ser252Cys, Gly253Leu, and Gly254Thr). Although no explicit recombination events were predicted, the clustering and branching of the phylogenetic tree were complicated. Based on the evolution trees for VP1 and NS1, the nine CachaV strains identified from 2021 to 2023 were closely related to those identified in gray wolves and cats. This study may be beneficial for evaluating the prevalence of CachaVs in China, thereby understanding the evolution trend of CachaVs.
Parvoviruses are nonenveloped single-stranded DNA viruses with a small genome (4–5 kbp) (1, 2). Historically, parvoviruses infecting both vertebrate and invertebrate hosts were categorized into two subfamilies, namely, Densovirinae and Parvovirinae, respectively, which were further divided into eight genera (3). Recent research has identified new parvovirus variations in animals, directly leading to the reclassification of Parvoviridae (3–5). Currently, the International Committee on Taxonomy of Viruses has included a third subfamily into the family Parvoviridae, comprising Parvovirinae, Densovirinae, and Hamaparvovirinae, which further comprises five novel genera—Penstylhamaparvovirus, Brevihamaparvovirus, Hepanhamaparvovirus, Ichthamaparvovirus, and Chaphamaparvovirus (6–8).
Owing to advancements in detection and sequencing technologies in recent years, chaphamaparvoviruses has been identified in numerous hosts, including bat (Eidolon helvum) parvovirus 2 (EHPV2) (9), Cachavirus (CachaV) (10), Chaphamaparvovirus carnivoran2 (feline ChPV, termed as fechavirus) (11), chicken parvovirus 1 and 2 (12), porcine parvovirus 7 (13), murine chapparvovirus (14), simian parvovirus (15), murine kidney parvovirus (16), and Hedgehog chapparvovirus (17). Until recently, CachaV was considered to be originally reported in American dog feces in 2019, which raised concerns among researchers about the virus (10). Similar to other parvoviruses, the CachaV genome mainly consists of two open reading frames that encode a capsid protein (VP1) and a nonstructural replication protein (NS1) (18). In the United States, CachaV was detected in 80 of 2,053 fecal samples (3 of 203 stool samples from healthy dogs and 77 of 1767 stool samples from dogs with diarrhea), with an infection rate of 4.35%, marking the first report of the novel parvovirus (10). Subsequently, researchers in China identified two samples that were positive for CachaV from 171 cats with diarrhea, whereas the 378 samples from healthy cats were all negative (19). In a study conducted in Thailand, among five dead dogs, three tested positive for CachaV in the lung tissue sample and one in the intestinal tissue sample (20). These similar reports indicate that CachaV is mainly related to the intestinal tract or diarrhea syndrome. To understand the recent prevalence and evolution status of CachaV, we investigated dogs from central, eastern, and northern China; sequenced and analyzed their genome sequences to facilitate epidemiological studies and mutation analysis; and then assessed their prevalence with possible clinical significance.
Anal swabs (cotton swabs soaked in physiological saline and inserted 2–3-cm deep into the anus of the sampling dog; these swabs were gently rotated, applied inside the anus, and then inserted into a clean test tube) from 276 dogs (182 with diarrhea and 94 healthy) were obtained from pet hospitals in five provinces (Henan, Inner Mongolia, Jiangsu, Hubei, and Anhui) of China between 2020 and 2023.
Each swab was washed with 1 mL of phosphate-buffered saline solution (0.01 mol/L), and 200 μL of suspensions were separated for viral DNA/RNA extraction using Simply Viral DNA/RNA Coextraction Kit (Bioer Biotechnology, Inc., Hangzhou, China) according to the manufacturer’s instructions. The extracted DNA and RNA were stored at −80°C for future use.
As previously reported, CachaV detection was performed using nested polymerase chain reaction (nt-PCR) as follows: the first PCR used the outer primer set IF (5′- CAACTAGCCGAATGCAGGGA-3′) and IR (5′-CGATAACATCCCCGGACTGG-3′) and the nested PCR used the inner primer set IF (5′-AGCTCAGTTTGGCCCAGATC-3′) and IR (5′-AGAGGGATCGCTGGATCTGT-3′) (10). In addition, canine parvovirus (CPV-2) (21), canine distemper virus (CDV) (22), canine coronavirus (CCoV) (23), and canine bufavirus (CBuV) (24) were screened in these collected samples using PCR/RT–PCR as previously described. The infection and co-infection statuses of the samples were summarized and displayed using UpSet plot packages (25). The prevalence of CachaV was compared between healthy and diarrheal dogs using Fisher’s exact test via GraphPad Prism 9.5 (San Diego, California, United States). Statistics were deemed significant at p < 0.05.
The nearly complete genome sequences of the nine CachaVs were amplified using specific primers as described previously (19). Overlapped amplification of CachaV genome segments was performed using PCR in a 20-μL reaction system containing a template (>100 ng/L), 10 pmol forward/reverse primer sets, and Ex-Taq polymerase (TaKaRa Biotechnology Co., Ltd., Dalian, China). The reaction procedure was as follows: predenaturation at 94°C for 5 min; followed by 35 cycles of combined denaturation at 94°C for 45 s, annealing at 56°C for 45 s, extension of 72°C for 70 s; and final extension at 72°C for 10 min. After ligation of the PCR amplicons using ClonExpress Ultra One Step Cloning Kit (Vazyme Biotechnology Co. Ltd., Nanjing China), positive clones were sent for sequencing to Generalbiol -Biotechnology, Chuzhou, China.
To analyze the genome sequence of CachaV and demonstrate its phylogenetic relationship, the sequence segments were assembled using DNAStar7.0 software (DNASTAR Inc., Madison, WI, United States) and aligned with all chaphamaparvoviruses. We used BioAider kit for the differential analysis of the 9 studied CachaV strains and 24 CachaV strains. A phylogenetic evolutionary tree was constructed using the genomes of the 9 studied CachaV strains; 24 CachaV strains detected in dogs, wolves, or cats in China or other countries (Supplementary Table S1). According to the MODELS program in MEGA 11 software, the HKY + G + I model, JTT + G model, and JTT + G model were used for the whole genome phylogenetic tree, NS1 phylogenetic tree, and VP1 phylogenetic tree, respectively. Evolutionary trees were further constructed based on the amino acid (aa) sequences of VP1 and NS1 using 1,000 guided replicates and the maximum-likelihood method via MEGA 11 software (26). Furthermore, recombination was predicted in the strains evaluated in this study using RDP4. Potential recombination events detected via three or more programs along with the identity analysis of the parents were considered potential events, with the highest acceptable p-value cutoff of 0.05 (27). RDP4 is a computer program typically used for recombination prediction; it includes guided scan, MAXCHI, mosaic, 3SEQ, gene cloning, LARD, and SISCAN in addition to the traditional RDP method that can predict recombination (28).
Based on the Meg-Align results, the aa sequences of NS1 and VP1 of the nine studied strains were compared with the reference CachaV strains identified in dogs or cats. Antigenic epitope prediction was performed using DNAMAN 5.2.2 for the resulting and reference strains (LynnonBiosoft, America) (Supplementary Table S2). To understand the molecular characteristics of the coding proteins, IDEXX1 (accession number: MH893826) and two variant CachaV strains (CHN230216 and CHN220916) were subjected to structural homology modeling for NS1 and VP1 using SWISS-MODEL2 and were visualized using PyMOL Molecular Graphics System 2.3 (DeLano Scientific LLC, America).
Among the 276 samples, 9 (3.3%) tested positive for CachaV after viral screening, including 8 (8/182, 4.4%) from dogs with diarrhea in Henan and Jiangsu provinces and 1 (1/94, 1.1%) from a healthy dog in Anhui province. Notably, more than half of the total positive cases were detected in Henan and Jiangsu provinces. Table 1 shows the clinical information of the CachaV-positive dogs. Statistical analysis revealed no association between CachaV infection and clinical symptoms (p > 0.05). Meanwhile, two dogs with diarrhea were quadruply infected (CachaV + CPV-2 + CCoV + CDV), one dog with diarrhea was triply infected (CachaV + CDV + CBuV), and one dog with diarrhea was dually infected (CachaV + CPV-2). The infection and co-infection statuses of each pathogen screened are shown via UpSet and Venn plots in Figure 1.
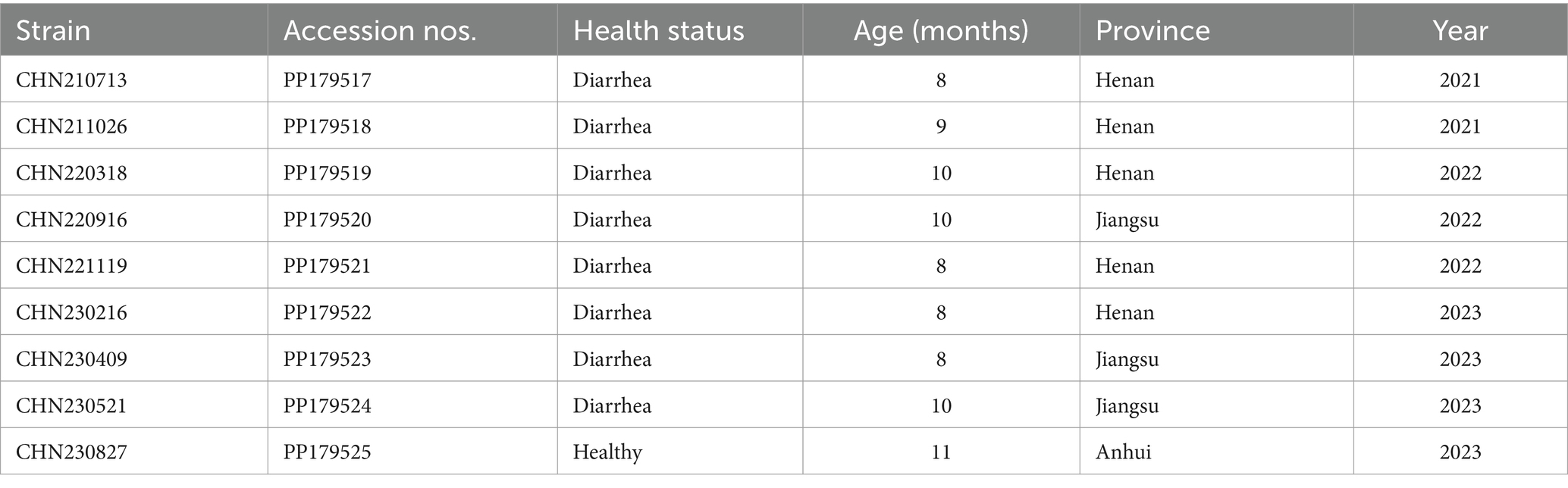
Table 1. Details of the Cachavirus strains identified in this study.
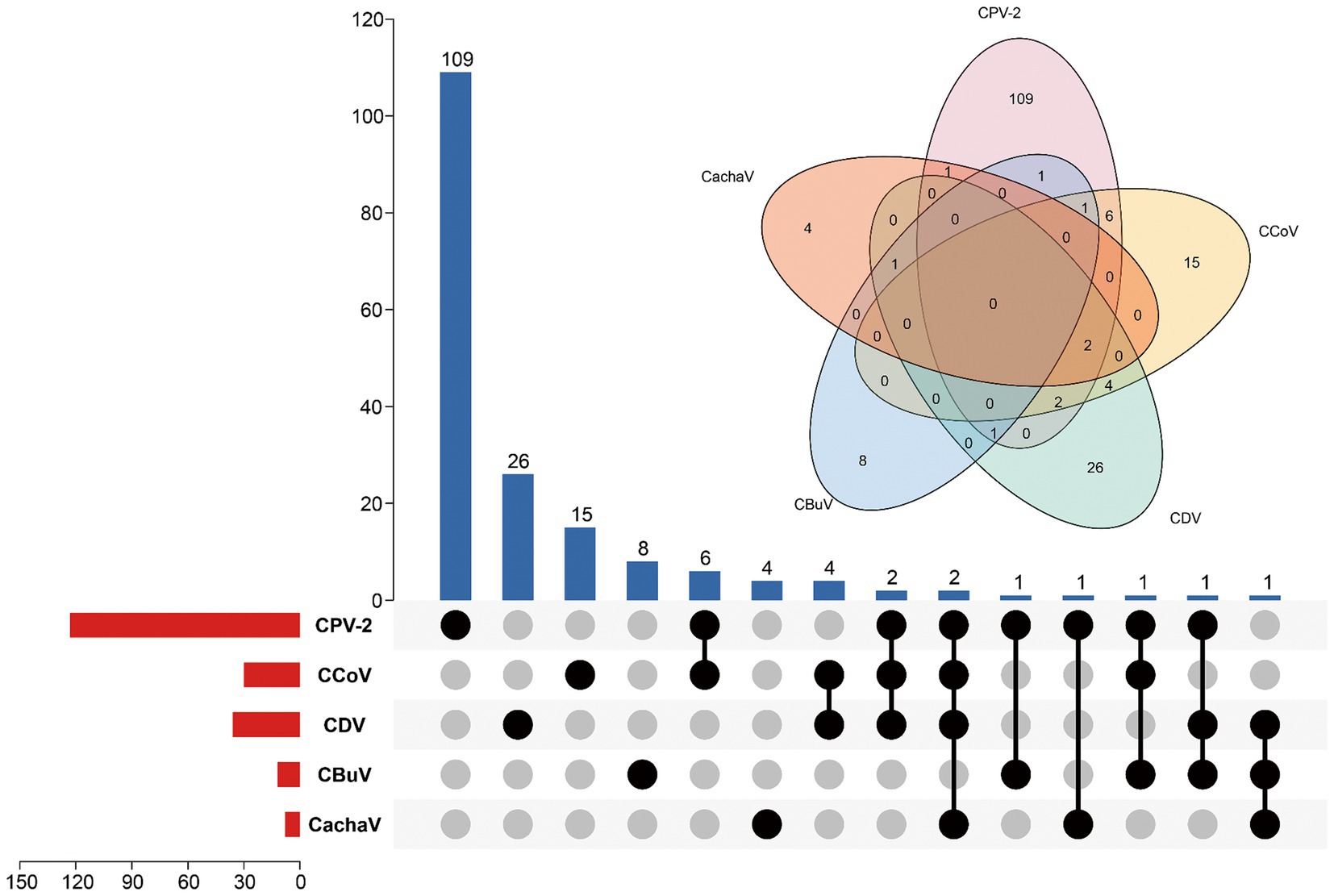
Figure 1. Infection status for CachaV, CPV-2, CCoV, CDV, and CBuV in samples from dogs with diarrhea. The upper bars indicate the numbers of positive samples in each group. The lower bars indicate the numbers of positive samples for each virus. The dotted line on the lower right indicates the types of infections.
The nine identified strains had the same genome structure as IDEXX1, each with two main codes: NS1 (663 aa) and VP1 (504 aa). Notably, the aa sequence identities of NS1 and VP1 in the nine CachaV strains with IDEXX1 were 97.89–98.94% and 97.42–99.8%, respectively. Compared with the 24 previously reported CachaV strains, the aa sequence identities of NS1 and VP1 in the 9 obtained CachaV strains were 97.59–99.55% and 96.23–99.8%, respectively. Moreover, the NS1 of CHN230521 showed the highest identity (99.85%) with NWT-W88 and CP-T015, whereas CHN220318 showed the lowest identity (96.23%) with OM640109. For the VP1 protein, CHN230521 exhibited the highest identity (100%) with NWT-W88 (accession no.: OK546101, originated from wolf, Canada, 2009), CP-R107C (accession no.: OP225937), CP-T015 (accession no.: OP225942), and CP-T046 (accession no.: OP225944), whereas CHN220916 showed the lowest identity (95.83%) with CY56 (accession no.: OM640109).
Phylogenetic analysis was performed using the genome sequences and aa sequences of NS1 and VP1 in the 9 identified strains and 24 CachaV strains (Figure 2). As shown in the evolution trees, CHN230521were closely related to some CachaV strains collected in Thailand, Canada and the United States, and the remain eight obtained strains mainly clustered with Chinese CachaV strains and displayed different branching, respectively. Most strains obtained in this study belonged to the same branch with the two cat-originated strains previously reported in China. Notably, no obvious recombination signal was observed in our comparative analysis with all chaphamaparvoviruses, despite the complex whole genome evolution of these nine strains.
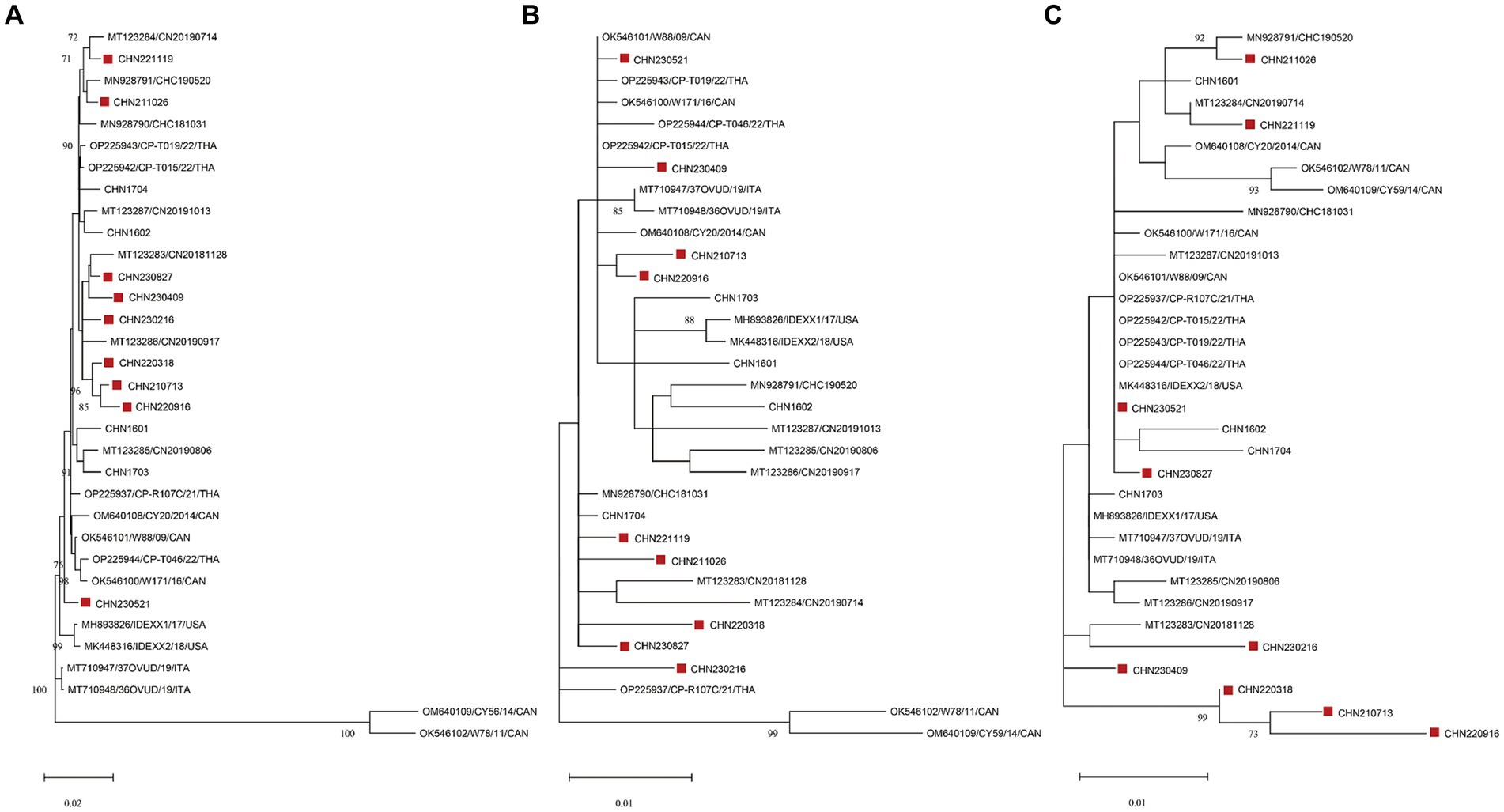
Figure 2. Maximum-likelihood trees based on genome sequences (A) deduced from NS1 (B) and VP1 (C) amino acid sequences of CachaVs. The CachaV strains obtained in this study are marked with red rectangles.
Through alignment of the derived aa sequences of the NS1 protein between the obtained CachaV and 24 CachaV strains, the following mutation sites were detected only in the nine obtained strains: Met104Leu, Asn207Asp., His220Arg, Gln469Arg, and Gly519Ser (Supplementary Table S3). The CHN210713 and CHN220916 strains showed unique site mutations at 99 (Phe → Ser), whereas the remaining seven strains had undergone aa changes at 456 (Leu → Thr). By comparing the aa sequences of CachaV-NS1, continuous mutations of SerGlyGlyTyr252-255CysLeuThrPhe were identified, which were common in other CachaV reference strains detected in China (Supplementary Table S4). For NS1, the CHN210713 strain identified in this study had mutations at loci 131 (Phe → Ser), 238 (Asp→Gly), and 247 (Val → Ala). Notably, the mutations generated at N9D in VP1 were present only in the CHN220916 and CHN230216 strains. To better understanding these mutation sites, mutations related to the tertiary structure change in NS1 were predicted and are displayed in Figure 3. The NS1 tertiary structure is predicted to vary at S252C which caused the change ofα-helix and random coil region, whereas these sites in VP1 have not been predicted to undergo structural changes.
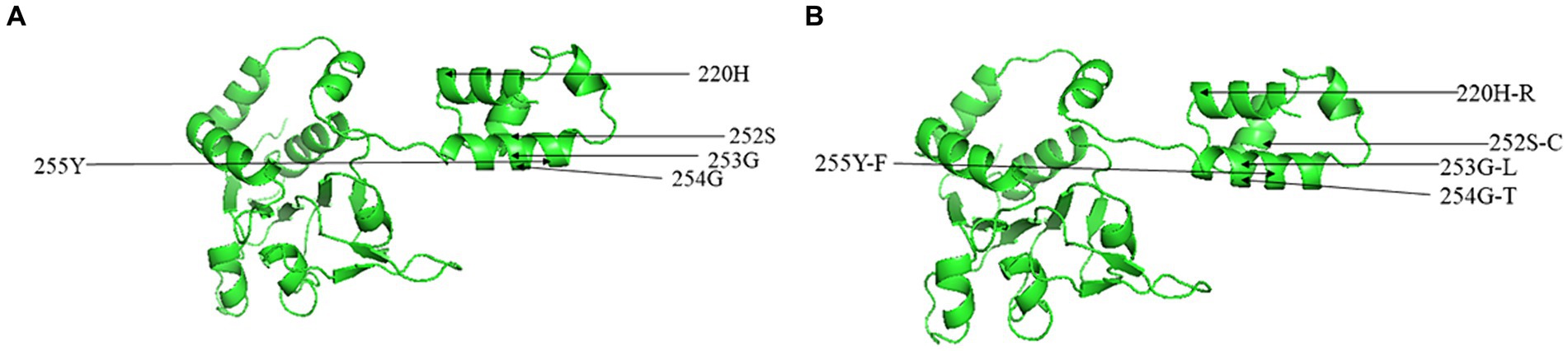
Figure 3. Predicted three-dimensional model of NS1 for CachaV strain of IDEXX1 (A) and CHN230216 (B).
The first report related to CachaV detected in healthy dogs from the United States in 2017 revealed that dogs can serve as reservoir hosts for this virus without clinical symptoms (10). Currently, CachaV infection is mainly related to clinical symptoms of diarrhea, and the virus has been mainly reported in the United States, Italy, Canada, China, and Thailand (10, 20, 21, 29, 30). According to a report related to CachaV in northeast China, the positive rate of CachaV in samples from healthy dogs and dogs with diarrhea was 10% (4/40) and 6.3% (18/285) (31). In this study, nine samples tested positive for CachaV; the positive rate of CachaV in healthy dogs was (1/94, 1.1%), whereas the positive rate of CachaV in dogs with diarrhea was (8/182, 4.4%). Our statistical analysis showed no correlation (p > 0.05) between the presence of viruses and diarrhea and no statistical difference. In two studies in Canada, (i) 8 of 303 (2.6%) spleen samples from wolves tested positive for CachaV (30) and (ii) 3 of 87 (3.5%) spleen samples from coyotes tested positive for CachaV (32). In the past few years, we have tested positive samples from nine dogs and two cats, all of which showed symptoms of diarrhea (19, 33, 34). The global spread of CachaV has increased concerns worldwide. However, the evolution characteristics of CachaV remain relatively limited.
To further explore the evolutionary trend of CachaV strains, we performed phylogenetic analyses. In this study, according to the evolution trees, eight of nine identified strains belonged to the same clade as the dog–cat CachaV already reported in China, which was also observed in wolves, indicating the genetic relationship and distance between ChPV strains detected in other organisms (7). Based on the evolution tree constructed using genome sequences, the CHN230521 strain was closely related to the strain isolated from dogs in Canada (NWT-W171 and NWT-W88) and the United States (IDEXX2); however, the reasons for the similarity in these strains are not clear and need to be verified in future studies. Furthermore, no obvious recombination signal was observed in our comparative analysis with the reference strains, suggesting that site mutations remain the main evolutionary driver.
The NS1 protein in the nine studied strains exhibited unique mutations at the G253L locus compared with the IDEXX1 strain and Chinese dog strains. Compared with the antigenic site predictions of VP1 protein, we identified several site mutations of antigenic significance that may provide evidence for vaccine-strain selection in the future. Similar to the finding of a previous study in China, NS1 mutations occurred at both G254T and Y255F loci of Chinese strains, indicating that the determining region might have a common origin. For VP1, the CHN230513 strain differed from the IDEXX1 strain only at V265L, whereas CHN220916 varied significantly from the IDEXX1 strain and harbored the highest number of mutation sites and consecutive mutations at D278G–S279G. Notably, the site mutations at S77F, T152A, and D238G were only detected in the dog-originated strains. We found that the tertiary structure models varied in strains with more mutated sites. However, these are speculations, and further studies are warranted to demonstrate whether these mutations change the protein function. In addition, the reasons for these occurrences remain unknown. Mutational analysis revealed no significant differences between the VP1 model structure of the CHN220916 strain and IDEXX1 strain. A broader and more systematic study on the patterns and specific structural characteristics of CachaV mutations is warranted.
In conclusion, one and eight CachaV strains were identified in healthy and diarrheic dogs, respectively. No recombination events were predicted, and special mutation sites were detected in both VP1 and NS1 proteins. This study may contribute to the research on the monitoring and evolution of CachaV worldwide.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The animal studies were approved by sample collection was approved by the pet owner and Nanyang Normal University Animal Care Committee (No. 14027). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
CL: Data curation, Investigation, Writing – review & editing. XT: Investigation, Software, Writing – original draft. HZ: Investigation, Methodology, Writing – review & editing. JJ: Conceptualization, Supervision, Writing – review & editing. LY: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (Grant no. 31870917), the Program for Science & Technology Innovation Talents in Universities of Henan Province (Grant no. 22HASTIT042), and the Technological Project of Nanyang Normal University (Grant no. 2024QN012).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2024.1375948/full#supplementary-material
1. Conteville, LC, Zanella, L, Marín, MA, Filippis, AMBD, Nogueira, RMR, Vicente, ACP, et al. Mendonça: Parvovirus B19 1A complete genome from a fatal case in Brazil. Mem Inst Oswaldo Cruz. (2015) 110:820–1. doi: 10.1590/0074-02760150261
2. Qiu, J, Lee, Q, Padula, MP, Pinello, N, Williams, SH, O'Rourke, MB, et al. Murine and related chapparvoviruses are nephro-tropic and produce novel accessory proteins in infected kidneys. PLoS Pathog. (2020) 16:e1008262. doi: 10.1371/journal.ppat.1008262
3. Cotmore, SF, Agbandje-McKenna, M, Chiorini, JA, Mukha, DV, Pintel, DJ, Qiu, J, et al. The family Parvoviridae. Arch Virol. (2013) 159:1239–47. doi: 10.1007/s00705-013-1914-1
4. Hoelzer, K, and Parrish, CR. The emergence of parvoviruses of carnivores. Vet Res. (2010) 41:39. doi: 10.1051/vetres/2010011
5. Cotmore, SF, Agbandje-McKenna, M, Canuti, M, Chiorini, JA, Eis-Hubinger, A-M, Hughes, J, et al. ICTV virus taxonomy profile: Parvoviridae. J Gen Virol. (2019) 100:367–8. doi: 10.1099/jgv.0.001212
6. Chung, HC, Kim, SJ, Nguyen, VG, Shin, S, Kim, JY, Lim, SK, et al. New genotype classification and molecular characterization of canine and feline parvoviruses. J Vet Sci. (2020) 21:e43. doi: 10.4142/jvs.2020.21.e43
7. Pénzes, JJ, de Souza, WM, Agbandje-McKenna, M, and Gifford, RJ. An ancient lineage of highly divergent parvoviruses infects both vertebrate and invertebrate hosts. Viruses. (2019) 11:525. doi: 10.3390/v11060525
8. Pénzes, JJ, Söderlund-Venermo, M, Canuti, M, Eis-Hübinger, AM, Hughes, J, Cotmore, SF, et al. Reorganizing the family Parvoviridae: a revised taxonomy independent of the canonical approach based on host association. Virology Division News. (2020) 165:2133–46. doi: 10.1007/s00705-020-04632-4
9. Baker, KS, Leggett, RM, Bexfield, NH, Alston, M, Daly, G, Todd, S, et al. Metagenomic study of the viruses of African straw-coloured fruit bats: detection of a chiropteran poxvirus and isolation of a novel adenovirus. Virology. (2013) 441:95–106. doi: 10.1016/j.virol.2013.03.014
10. Fahsbender, E, Altan, E, Seguin, MA, Young, P, Estrada, M, Leutenegger, C, et al. Chapparvovirus DNA found in 4% of dogs with diarrhea. Viruses. (2019) 11:398. doi: 10.3390/v11050398
11. Li, Y, Gordon, E, Idle, A, Altan, E, Seguin, MA, Estrada, M, et al. Virome of a feline outbreak of diarrhea and vomiting includes bocaviruses and a novel chapparvovirus. Viruses. (2020) 12:506. doi: 10.3390/v12050506
12. Lima, DA, Cibulski, SP, Tochetto, C, Varela, APM, Finkler, F, Teixeira, TF, et al. The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics. Virus Res. (2019) 261:9–20. doi: 10.1016/j.virusres.2018.12.005
13. Palinski, RM, Mitra, N, and Hause, BM. Discovery of a novel Parvovirinae virus, porcine parvovirus 7, by metagenomic sequencing of porcine rectal swabs. Virus Genes. (2016) 52:564–7. doi: 10.1007/s11262-016-1322-1
14. Williams, SH, Che, X, Garcia, JA, Klena, JD, Lee, B, Muller, D, et al. Denison: viral diversity of house mice in new York city. MBio. (2018) 9:e01354–17. doi: 10.1128/mBio.01354-17
15. Kapusinszky, B, Ardeshir, A, Mulvaney, U, Deng, X, and Delwart, E. Case-control comparison of enteric viromes in captive rhesus macaques with acute or idiopathic chronic diarrhea. J Virol. (2017) 91:e00952-17. doi: 10.1128/jvi.00952-17
16. Roediger, B, Lee, Q, Tikoo, S, Cobbin, JCA, Henderson, JM, Jormakka, M, et al. An atypical parvovirus drives chronic tubulointerstitial nephropathy and kidney fibrosis. Cell. (2018) 175:530–543.e24. doi: 10.1016/j.cell.2018.08.013
17. D. GLanave, G, Pellegrini, F, Lombardi, R, Prioletti, M, Circella, E, et al. Novel parvovirus in an outbreak of fatal enteritis in European hedgehogs (Erinaceus europaeus), Italy, 2022. Microbiol Spectr. (2023) 11:e0249423. doi: 10.1128/spectrum.02494-23
18. G D, A, Stutika, C, Botschen, L, Mietzsch, M, Weger, S, Feldkamp, M, et al. A comprehensive RNA sequencing analysis of the adeno-associated virus (AAV) type 2 transcriptome reveals novel AAV transcripts, splice variants, and derived proteins. J Virol. (2015) 90:1278–89. doi: 10.1128/JVI.02750-15
19. Ji, J, Hu, W, Liu, Q, Zuo, K, Zhi, G, Xu, X, et al. Genetic analysis of cachavirus-related prvoviruses detected in pet cats: the first report from China. Front Vet Sci. (2020) 7:580836. doi: 10.3389/fvets.2020.580836
20. Piewbang, C, Lohavicharn, P, Nguyen, TV, Punyathi, P, Kasantikul, T, and Techangamsuwan, S. Carnivore chaphamaparvovirus-1 (CaChPV-1) infection in diarrheic dogs reveals viral endotheliotropism in intestine and lung. Vet Q. (2023) 43:1–10. doi: 10.1080/01652176.2023.2185696
21. Han, SC, Guo, HC, Sun, SQ, Shu, L, Wei, YQ, Sun, DH, et al. Full-length genomic characterizations of two canine parvoviruses prevalent in Northwest China. Arch Microbiol. (2015) 197:621–6. doi: 10.1007/s00203-015-1093-4
22. Wang, J, Wang, J, Li, R, Liu, L, and Yuan, W. Rapid and sensitive detection of canine distemper virus by real-time reverse transcription recombinase polymerase amplification. BMC Vet Res. (2017) 13:241. doi: 10.1186/s12917-017-1180-7
23. van Nguyen, D, Terada, Y, Minami, S, Yonemitsu, K, Nagata, N, Le, TDH, et al. Characterization of canine coronavirus spread among domestic dogs in Vietnam. J Vet Med Sci. (2017) 79:343–9. doi: 10.1292/jvms.16-0538
24. Martella, V, Lanave, G, Mihalov-Kovács, E, Marton, S, Varga-Kugler, R, Kaszab, E, et al. Novel parvovirus related to primate bufaviruses in dogs. Emerg Infect Dis. (2018) 24:1061–8. doi: 10.3201/eid2406.171965
25. Chen, C, Chen, H, Zhang, Y, Thomas, HR, Frank, MH, He, Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. (2020) 13:1194–202. doi: 10.1016/j.molp.2020.06.009
26. Kumar, S, Stecher, G, Li, M, Knyaz, C, Tamura, K, and Battistuzzi, FU. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. (2018) 35:1547–9. doi: 10.1093/molbev/msy096
27. Meng, Z, Geng, Y, Guo, D, Li, C, Wang, E, Wei, S, et al. Co-circulation of the rare CPV-2c with unique Gln370Arg substitution, new CPV-2b with unique Thr440Ala substitution, and new CPV-2a with high prevalence and variation in Heilongjiang province, Northeast China. Plos One. (2015) 10:e0137288. doi: 10.1371/journal.pone.0137288
28. Martin, DP, Murrell, B, Golden, M, Khoosal, A, and Muhire, B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. (2015) 1:vev003. doi: 10.1093/ve/vev003
29. Canuti, M, Fry, K, Cluff, HD, Mira, F, Fenton, H, and Lang, AS. Co-circulation of five species of dog parvoviruses and canine adenovirus type 1 among gray wolves (Canis lupus) in northern Canada. Transbound Emerg Dis. (2022) 69:e1417–33. doi: 10.1111/tbed.14474
30. Palombieri, A, Di Profio, F, Lanave, G, Capozza, P, Marsilio, F, Martella, V, et al. Molecular detection and characterization of carnivore chaphamaparvovirus 1 in dogs. Vet Microbiol. (2020) 251:108878. doi: 10.1016/j.vetmic.2020.108878
31. Li, N, Bai, Y, Yan, X, Guo, Z, Xiang, K, Yang, Z, et al. The prevalence, genetic diversity and evolutionary analysis of cachavirus firstly detected in northeastern China. Front Vet Sci. (2023) 10:123372. doi: 10.3389/fvets.2023.1233972
32. Canuti, M, Mira, F, Sorensen, RG, Rodrigues, B, Bouchard, É, Walzthoni, N, et al. Distribution and diversity of dog parvoviruses in wild, free-roaming and domestic canids of Newfoundland and Labrador, Canada. Transbound Emerg Dis. (2022) 69:e2694–705. doi: 10.1111/tbed.14620
33. Hu, W, Liu, Q, Chen, Q, and Ji, J. Molecular characterization of Cachavirus firstly detected in dogs in China. Infect Genet Evol. (2020) 85:104529. doi: 10.1016/j.meegid.2020.104529
Keywords: Cachavirus, molecular epidemiological investigation, recombination analysis, Chaphamaparvovirus, diarrhea
Citation: Leng C, Tian X, Zhai H, Ji J and Yao L (2024) Molecular epidemiological investigation and recombination analysis of Cachavirus prevalent in China. Front. Vet. Sci. 11:1375948. doi: 10.3389/fvets.2024.1375948
Edited by:
Marta Canuti, University of Copenhagen, DenmarkReviewed by:
Chutchai Piewbang, Chulalongkorn University, ThailandCopyright © 2024 Leng, Tian, Zhai, Ji and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Ji, amlqdW4wMjBAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.