 Nikita Krasnikov
Nikita Krasnikov Anton Yuzhakov
Anton Yuzhakov- Laboratory of Biochemistry and Molecular Biology, Federal State Budget Scientific Institution “Federal Scientific Center VIEV” (FSC VIEV), Moscow, Russia
During the last decade, porcine rotavirus H was detected in the USA, Asian regions, South Africa, Brazil, and a couple of European countries. In the presented study, the virus was identified in piglets on a farrow-to-finish farm in Russia during metagenomic surveillance. Currently, it is the first identification of this species in the country. As a diagnostic method, nanopore-based metagenomic sequencing was applied. The obtained nanopore reads allowed for the assembly of 10 genome segments out of 11. The phylogenetic analysis revealed the virus belonged to the porcine cluster and had GX-P3-I3-R3-C3-M8-A7-N1-T5-E3-H3 genome constellation. Moreover, three potential new genotype groups for VP3, NSP1, and NSP3 genes were determined. Additionally, a recombination between RVH and RVC in the NSP3 gene was detected. The study provides significant information about a novel RVH strain.
Introduction
Rotaviruses (RVs) are the most common cause of diarrhea in humans and animals, including pigs (1). The rotavirus genome is represented by linear double-stranded RNA (dsRNA) and consists of 11 segments that encode six structural proteins (VP1–VP4, VP6, and VP7) and five to six nonstructural proteins (NSP1–NSP5/6). RVs belong to the Sedoreoviridae family and are currently classified into nine species, A to J, based on the sequence diversity of the inner capsid protein (VP6) (2).
RV infections are quite common on swine farms and frequently associated with post-weaning and suckling diarrhea, which cause significant financial losses for the pork industry. Among the nine RV species, RVA, RVB, RVC, and RVH have been identified in fecal samples from pigs of different ages. Besides, Rotavirus E stands out, but it was revealed only once in 1986 and has not been studied genetically (3), so it has been excluded from the RV classification.
RVA, RVB, and RVC are widely spread in swine herds worldwide (1). RVA mostly affects piglets between 3 and 6 weeks of age (4, 5). RVB infections are generally associated with pigs >3 weeks old and occasionally cause diarrhea in neonatal piglets (5), while RVC is more commonly seen in young piglets (< 7 days of age) and is often associated with neonatal diarrhea (5, 6). At the same time, there is still no evidence of RVH pathogenic influence on pig health.
There is limited information on the geographical distribution and genetic characterization of RVH. The virus was originally identified in humans with symptoms of gastroenteritis in China in 1997 and named “adult diarrhea rotavirus” (ADRV-N) (7). Afterward, a similar virus was detected in Bangladesh in 2002 (the B219 strain) (8). These strains were genetically related to group B rotaviruses to some extent and close to them ecologically, but then were separated into a distinct branch based on VP6 sequence analysis (9, 10).
Subsequently, the RVH circulation was confirmed in pigs of different ages in the USA (11, 12), Japan (13, 14), Brazil (15, 16), South Africa (17), Vietnam (18), and some European countries, such as Spain (19) and Italy (20). A recent large-scale study in China has also indicated a high prevalence of RVH in the local swine herds (21). Furthermore, the virus has also been observed in bats in west-central Africa (22) and Korea (23). There were also some studies that demonstrated detection in bat metagenomes (24, 25). In this study, we characterize the first identified isolate of porcine RVH in Russia and discuss its phylogenetic relationships with known RVH strains.
Materials and methods
Sample collection
During metagenomic surveillance in the fall of 2022, within the framework of the project “TechPEPCon,” we collected nasal and rectal swabs from three piglets on the 10th week of life. Finally, three distinct samples of one type from three different piglets were pooled into one. The case farm was located in the Moscow region of Russia and positioned as healthy. According to the herd veterinarian, the animals had no clinical manifestations of infections. For taking swabs and further sample filtering, a specially designed sampler was used (PathoSense BV, Belgium). The samples were transported in Dulbecco's phosphate-buffered saline (dPBS) at 4°C to the lab for immediate subsequent analysis.
Nanopore sequencing
The lab diagnostic procedure was performed according to a protocol developed by PathoSense BV (Merelbeke, Belgium). Briefly, it included nucleic acid extraction using the Quick-DNA/RNA viral kit (Zymo Research, CA, USA), reverse transcription using the SuperScript IV Reverse Transcriptase (ThermoFisher Scientific, USA) with following enrichment by PCR (KAPA HiFi HotStart ReadyMix; Roche, Switzerland). Then amplicons were cleaned with the magnetic beads (AMPure XP; Beckman Coulter, USA) in a ratio 1:1. Final quantity and quality were verified using NanoDrop OneC spectrophotometer (ThermoFisher Scientific, USA). For the library preparation, the Rapid Barcoding Kit (RBK-004; ONT, Oxford, UK) was used. After that, nanopore sequencing was performed on a MinION flow cell (R9.4.1; ONT, Oxford, UK) for 6 h.
Metagenomic analysis and genome assembly
FASTQ files were generated and demultiplexed using Guppy basecaller (v. 21.10.4) in the super-accurate basecalling setting. Quality filtering was performed using NanoFilt (v. 2.6.0) (26). Reads with a q-score <7 were omitted. Swine host reads were removed after alignment to the Sus scrofa genome (GCA_000003025.6) using graphmap (v. 0.5.2) (27) and samtools (v. 1.6) (28). For metagenomic analysis, filtered sequences were scanned using the BLASTn method (BLAST v. 2.13.0) with customized nucleotide databases. The hits with lowest e-value were visualized using KronaTools (v. 2.8.1) (29).
The nanopore segment assembly was performed as follows: minimap2 (v. 2.24) (30) was used for mapping filtered reads according to their genome segment, and samtools (v. 1.6) (28) was used to generate an individual segment FASTQ file. For each segment, the corresponding FASTQ file was taken to perform de novo segment assembly using Canu (v. 2.2) (31), minimap2 (v. 2.24) (30), and several rounds of medaka polishing (v. 1.7.2; ONT) for generating final consensus sequences. The sequence coverage was different between the segments from 20× to 150× (Supplementary Table 1). The quality of the assembled segments was assessed by comparison with publicly available sequences in NCBI GenBank.
Phylogenetic and recombination analyzes
The obtained sequences were aligned by the ClustalW algorithm. Genetic distances were calculated using the Kimura two-parameter correction at the nucleotide level. Phylogenetic dendrograms were plotted using the maximum likelihood (ML) method and the GTR (G+I) model (MEGA 7.0) (32). The robustness of the topology was evaluated by 1000 bootstrap replications. The visualization of the Newick files of phylogenetic dendrograms was performed using iTOL v6 (33). Recombination events were evaluated using the Recombination Detection Program (RDP) (v. 4.101) with RDP, GENECOV, Bootscan, MaxChi, Chimera, SiSscan, and 3Seq algorithms (34). Similarity plot analysis was conducted using the SimPlot software (v. 3.5.1) with a sliding window of 200 bp (step: 20 bp).
Results
Metagenomic nanopore-based analysis
In the presented study, RVH (isolate KLM-22) was detected in rectal samples of ten-week-old piglets while conducting nanopore-based metagenomic surveillance in the Moscow region of Russia. The rectal sample was composed of three distinct rectal samples from three different piglets. Taking into account other swine viruses, the metagenomic composition also included porcine torovirus, porcine sapelovirus, porcine kobuvirus, porcine enterovirus 9, and porcine astrovirus 4, composed of 1–3% each. Among bacterial species, the metagenomic approach revealed Prevotella sp., Campilobacter sp., Bacteriodes sp., and Chlamydia suis in low quantities.
As a result of the nanopore sequencing, complete RVH segment sequences were achieved for VP2, VP3, VP6, NSP1, NSP2, NSP3, NSP4, and NSP5 and partial sequences for VP1 and VP4. There were some reads of VP7, but not enough to assemble the sequence consensus properly. All obtained sequences of the detected isolate KLM-22 were deposited in the NCBI GenBank with the accession numbers: OQ603399–OQ603403 and OR589274-OR589278.
Phylogenetic analysis
The obtained RVH segment sequences were used for phylogenetic analysis, including complete and partial genome sequences of RVH isolates from NCBI GenBank. According to the constructed ML trees, the sequences were related to a porcine RVH cluster (Figures 1, 2, Supplementary Figures 2–6). The pairwise identity among the presented RVH isolate and GenBank sequences varied between 71 and 86% for VP4, 78–90% for VP6, 78–86% for VP1, 82–88% for VP2, 78–85% for VP3, 74–82% for NSP1, 85–88% for NSP2, 74–83% for NSP3, 70–84% for NSP4, and 83–93% for NSP5 (Supplementary Data Sheet 2). Analysis of VP3, NSP1, and NSP3, based on cut-off values of 86, 84, and 87%, revealed the presence of three potentially new RVH genotype groups: M8, A7, and T5, respectively. Apart from that, the sequences of these segments were related to the distinct clades according the corresponding constructed phylogenetic dendrograms (Figure 1).
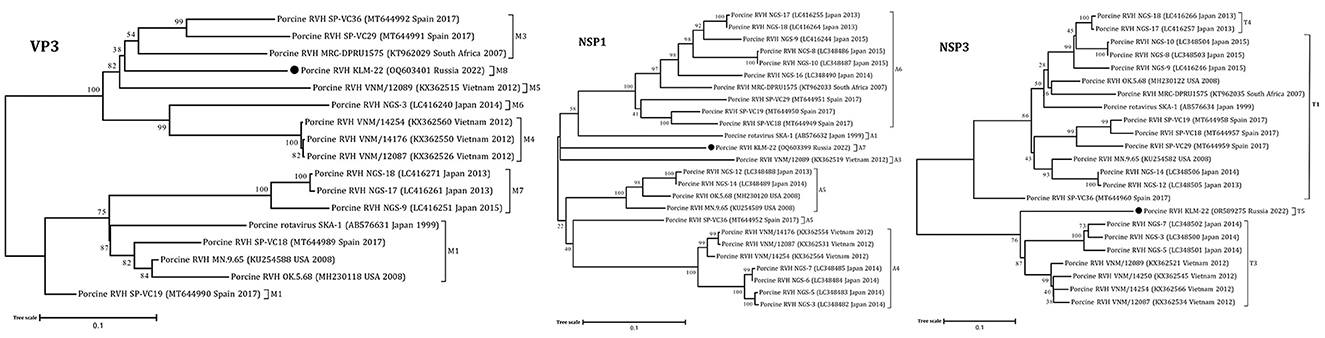
Figure 1. Phylogenetic dendrograms constructed with the GTR model for the VP3, NSP1, and NSP3 RVH segments. Three novel genotype groups M8, A7, and T5 were determined in this study. The filled circles above the strains indicate the RVH strain identified in this study. Scale bars indicate nucleotide substitutions per site. GenBank accession number, country, and collection year of each sample are also shown. Genotypes are specified in accordance with previous studies (13, 19) and indicated on the right of the bracket.
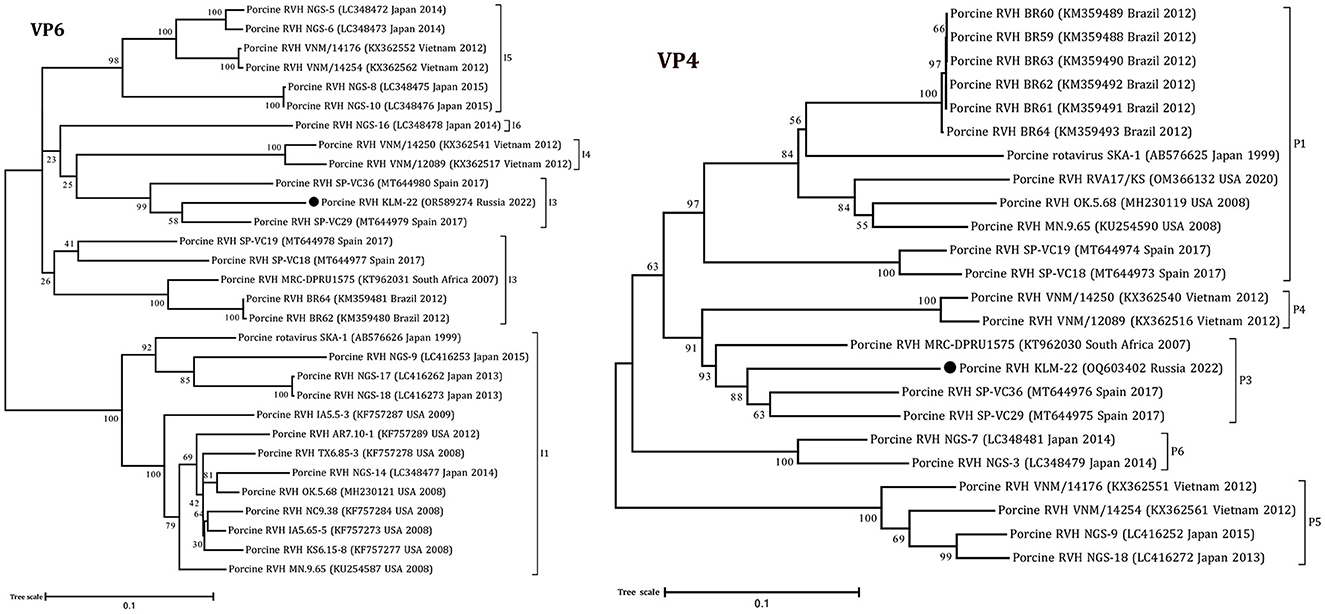
Figure 2. Phylogenetic dendrograms constructed with the GTR model for the VP4 and VP6 RVH segments. The filled circles above the strains indicate the RVH strain identified in this study. Scale bars indicate nucleotide substitutions per site. GenBank accession number, country, and collection year of each sample are also shown. Genotypes are specified in accordance with previous studies (13, 19) and indicated on the right of the bracket.
According to the ML trees and phylogenetic analysis of VP6 and VP4 segments, the KLM-22 RVH strain was closer to the strains from Spain and South Africa (Figure 2). Considering the ORF variability of the NSP1 segment, it was characterized as a long type and was 1206 bp in length.
Finally, taking into account the full genome-base genotyping system for RVH with cut-off nucleotide identity values, advanced by Suzuki and Inoue (13), we classified the detected strain as follows: GX-P3-I3-R3-C3-M8-A7-N1-T5-E3-H3.
Recombination in NSP3 gene
The NSP3 segment sequence was related to a long type, as its ORF length exceeded the standard (around 801 bp) and equaled 1248 bp. To investigate the origin of the extra nucleotide sequence in long-type NSP3, a BLAST search method was performed. The 448 nucleotide sequence (position 815–1262) from the NSP3 sequence displayed a high degree of resemblance with the middle regions (positons 546–987 bp) of two porcine RVC strains from the USA: RVC/Pig-wt/USA/IA46/2012 (GenBank: MG451244.1) - 86.65%, and RVC/Pig-wt/USA/MN_1233/2013 (GenBank: MT761794.1) - 85.97%, using the BLASTn method (Supplementary Figure 7). The recombination event was additionally verified by RDP analysis with a set of the genetically closest sequences by BLASTn (Supplementary Table 2), and following tests with corresponding p-values proved it (Table 1).
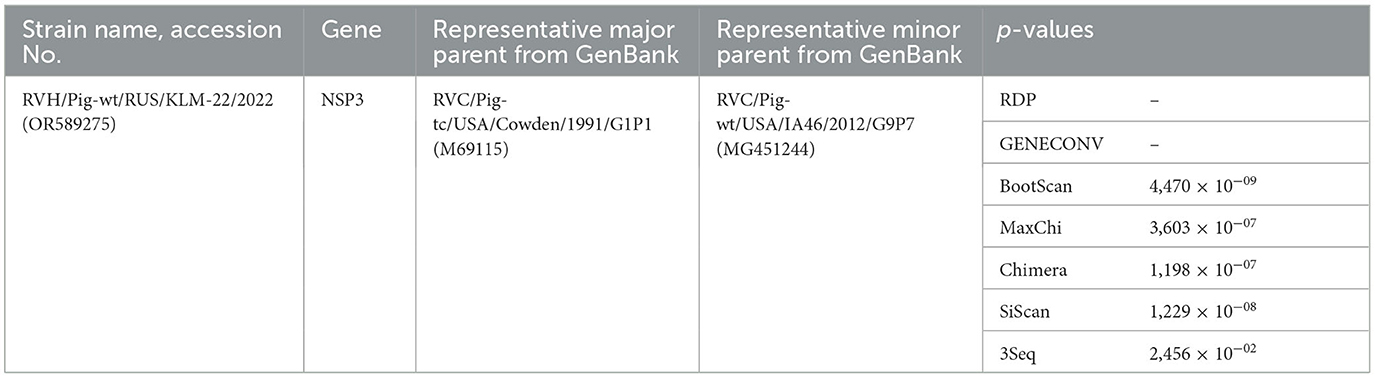
Table 1. Recombination event identified using the Recombination Detection Program (RDP) v.4.101 for rotavirus H NSP3 gene.
The similarity plot analysis also showed a high identity level with RVC strains in the alignment interval from 800 to 1250 bp (Supplementary Figure 1). Additionally, to understand the phylogenetic relationship of the recombinant site from RVC, the corresponding dendrogram was constructed. For comparison, partial NSP3 nucleotide sequences from different rotavirus C strains, including insertions from three Japanese RVH strains (LC348500, LC348501, and LC348502), were selected (Figure 3).
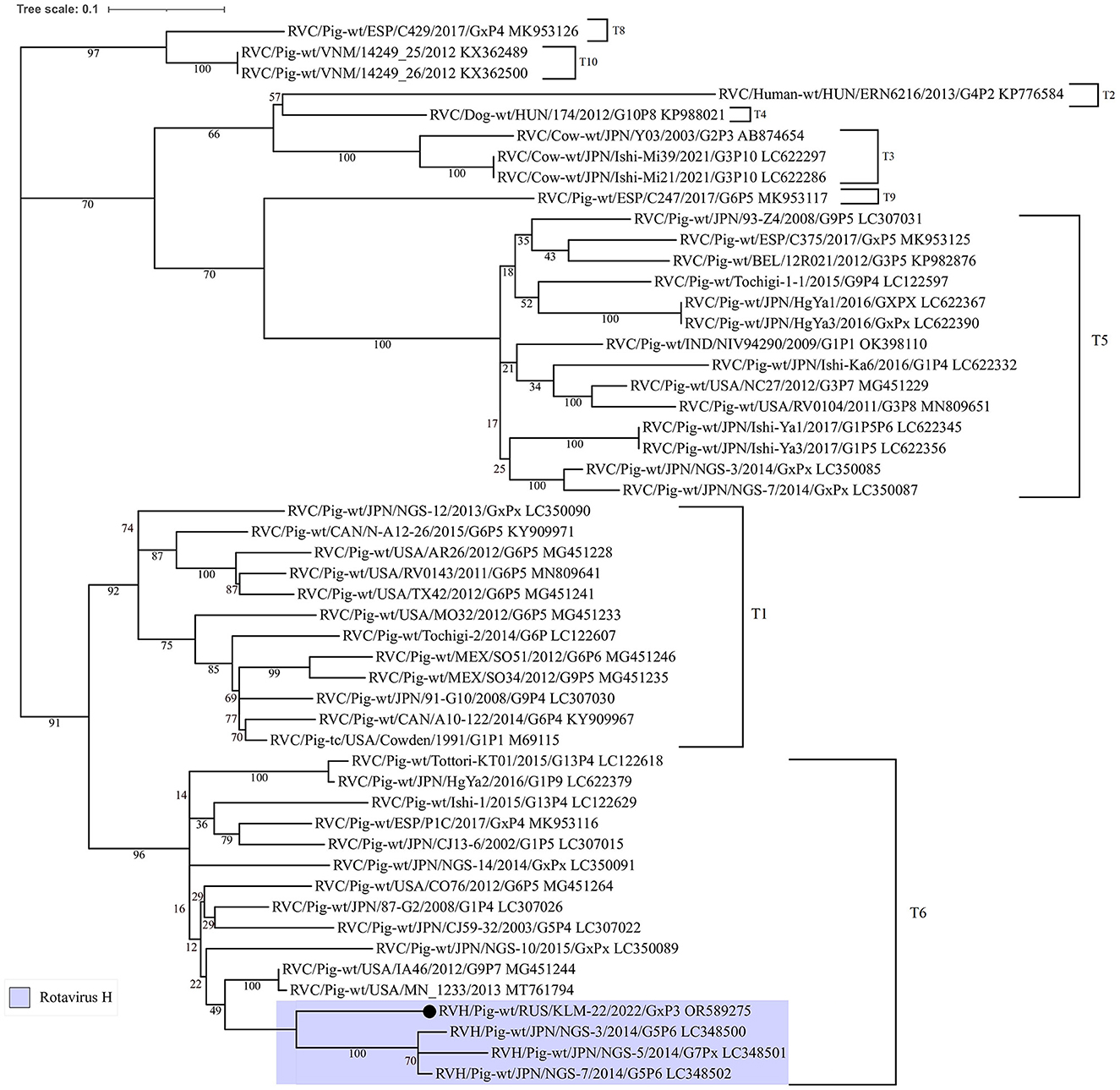
Figure 3. Phylogenetic dendrogram constructed from partial NSP3 open reading frame nucleotide sequences (448 bp) from RVC strains, including three porcine RVH strains from Japan and the studied one. The dendrogram was constructed using the maximum-likelihood method with MEGA 7 software and visualized using iTOL v6. The number near each node displays the percentage of bootstrap support (for 1000 replicates) for the cluster. Genotypes are indicated on the right of the bracket.
According to the ML tree, the RVC site was closer to the insertions from the Japanese RVH strains and related to the T6 genotype by the RVC classification system (35). Moreover, the insertion in question was longer, than Japenese in 27 bp.
Discussion
RVH is a poorly studied species among porcine RVs. Previously, it was detected in swine herds only in eight countries worldwide. In our study, the virus was identified in piglets on the 10th week of life, which correlates with the work by Nyaga et al. (17) in South Africa. Numerous studies also indicate that the virus was more often circulated among weaning and fattening classes, in age groups of 21–70 days, than during the suckling period (12, 19, 20).
RVs, including RVH, can be detected in pigs with and without diarrhea (15). According to the study of Marthaler et al. (12), RVH was most frequently detected in co-infection with A, B, and C species. During the large-scale epidemiological study in Italy, RVH was also found mostly in combinations with other RVs and was predominant in only 0.9% of the samples among single RV infections (20). The recent monitoring study in China demonstrated a high positivity rate for single RVH cases in the clinical samples: 13.68% (190/1447). Otherwise, in co-infections, RVH was noted more rarely: RVA+RVH (2.14%), RVB+RVH (0.83%), and RVC+RVH (1.66%). It is also interesting that the RVA + RVB + RVC + RVH infection showed a positivity rate of 2.07% (30/1447) (21).
The studies likewise specify that the virus is more of an opportunistic agent than a severe pathogen (12, 36). Despite the fact that RVH was occasionally detected in pigs with diarrhea, the virus role in the infection development has yet to be proven experimentally, and the question of RVH pathogenicity is still open. In our case, there were no clinical signs in the studied piglets on the 10th week, and among RV species, RVH was the only one detected. The presented observation demonstrates that RVH can freely circulate in a herd without causing any clinical signs.
The metagenomic approach, in conjunction with ONT sequencing, is a convenient pathogen diagnostic instrument (37). Regarding RVs, the method allows identifying species and subtypes and generating segment assemblies (38, 39). In the given case, a nanopore-based metagenomic approach allowed us to obtain 10 segments out of 11. The phylogenetic analysis suggested an absence of interspecies transmission between pigs, humans, and bats in the presented case. Additionally, the study expanded the current RVH genotype classification and revealed potentially new genotypes for VP3, NSP1, and NSP3.
Gene rearrangements between homologous and heterologous strains are crucial driving forces in the evolution of RVs. There are plenty of studies evidencing reassortation and recombination events among strains of the same rotavirus species (40–42). On the other hand, between different species, such genetic changes happen much less frequently. During the analysis, we identified interspecies recombination in the NSP3 gene between rotavirus H and C. A similar recombination finding has already been noted by Suzuki and Inoue (13) in Japanese RVH strains. This fact leads us to think that such genetic events can occur not accidentally but periodically between these species and deserve further investigation. It is commonly known that rotavirus NSP3 is a surrogate of the Poly(A) Binding Protein-Poly(A) Complex (PABP) (43). The protein evicts PABP from its binding site on eIF4G and binds the 3′-end of viral mRNA and cellular eIF4G, which inhibits host translation and thereby promotes viral one. Simultaneously, the complete functional role of NSP3 in porcine RVH and RVC remains unstudied, and future analyses are required.
Conclusion
In this work, we report the genome sequences of porcine RVH, the first RVH strain identified in Russia. In our case, the virus was detected in piglets during the 10th week of life, between the nursery and fattening periods. We also present a phylogenetic analysis of the 10 segment sequences that showed their porcine RVH cluster membership. Additionally, we demonstrate the gene recombination between rotavirus H and C in NSP3.
The study also emphasizes the importance of epidemiological surveillance at later stages of growth, including the fattening period. Such a procedure can provide full insight regarding animal health and reveal previously unknown pathogens for the exact herd. It is also of great value to conduct phylogenetic and recombination studies to evaluate genetic transformations and predict the rotavirus evolution, and nanopore-based metagenomic sequencing copes with all of the above tasks.
Our data confirm that RVH presently circulates among domestic pigs in Russia. However, detailed monitoring of swine herds on farms is required to fully understand RVH diversity and the currently existing epidemiological situation.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The animal studies were approved by the Local Ethical and Animal Welfare Committee of the Federal State Budget Scientific Institution Federal Scientific Center VIEV (Moscow, Russia). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
NK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft. AY: Data curation, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Ministry of Science and Higher Education of the Russian Federation, grant number 075-15-2021-948; 4 October 2021. The work has received funding through the Horizon 2020 ERA-NET Cofound International Coordination of Research on Infectious Animal Diseases (ICRAD), project TechPEPCon: use of frontline technologies to screen pathogens, environment, and pigs for a better disease control in swine herds.
Acknowledgments
We appreciate the collaboration with the herd veterinarian and farmers and thank them for the opportunity to visit the farm and take samples for this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1302531/full#supplementary-material
References
1. Vlasova AN, Amimo JO, Saif LJ. Porcine rotaviruses: epidemiology, immune responses and control strategies. Viruses. (2017) 9:48. doi: 10.3390/v9030048
2. Current ICTV Taxonomy Release. ICTV [WWW Document]. (2023). Available online at: https://ictv.global/taxonomy (accessed November 7, 2023).
3. Bridger JC, Brown JF. Prevalence of antibody to typical and atypical rotaviruses in pigs. Vet Record. (1985). 116:50–50. doi: 10.1136/vr.116.2.50
4. Homwong N, Diaz A, Rossow S, Ciarlet M, Marthaler D. Three-level mixed-effects logistic regression analysis reveals complex epidemiology of swine rotaviruses in diagnostic samples from North America. PLoS ONE. (2016) 11:e0154734. doi: 10.1371/journal.pone.0154734
5. Zimmerman JJ, Karriker LA, Ramirez A, Schwartz KJ, Stevenson GW, Zhang J. Diseases of Swine. New York, NY: John Wiley & Sons. (2019).
6. Marthaler D, Rossow K, Culhane M, Collins J, Goyal S, Ciarlet M, et al. Identification, phylogenetic analysis and classification of porcine group C rotavirus VP7 sequences from the United States and Canada. Virology. (2013) 446:189–98. doi: 10.1016/j.virol.2013.08.001
7. Yang H, Chen S, Ji S. A novel rotavirus causing large scale of adult diarrhea in Shi Jiazhuang. Zhonghua liu Xing Bing Xue za Zhi Zhonghua Liuxingbingxue Zazhi. (1998) 19:336–8.
8. Alam MM, Kobayashi N, Ishino M, Ahmed MS, Ahmed MU, Paul SK, et al. Genetic analysis of an ADRV-N-like novel rotavirus strain B219 detected in a sporadic case of adult diarrhea in Bangladesh. Arch Virol. (2007) 152:199–208. doi: 10.1007/s00705-006-0831-y
9. Nagashima S, Kobayashi N, Ishino M, Alam MM, Ahmed MU, Paul SK, et al. Whole genomic characterization of a human rotavirus strain B219 belonging to a novel group of the genus Rotavirus. J Med Virol. (2008) 80:2023–33. doi: 10.1002/jmv.21286
10. Matthijnssens J, Otto PH, Ciarlet M, Desselberger U, Van Ranst M, Johne R. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch Virol. (2012) 157:1177–82. doi: 10.1007/s00705-012-1273-3
11. Hull JJ, Marthaler D, Rossow S, Ng TFF, Montmayeur AM, Magana L, et al. Genomic sequence of the first porcine rotavirus group H strain in the United States. Genome Announc. (2016) 4:10–1128. doi: 10.1128/genomeA.01763-15
12. Marthaler D, Rossow K, Culhane M, Goyal S, Collins J, Matthijnssens J, et al. Widespread rotavirus H in commercially raised pigs, United States. Emerg Infect Dis. (2014) 20:1203. doi: 10.3201/eid2007.140034
13. Suzuki T, Inoue D. Full genome-based genotyping system for rotavirus H and detection of potential gene recombination in nonstructural protein 3 between porcine rotavirus H and rotavirus C. Journal of General Virology. (2018) 99:1582–9. doi: 10.1099/jgv.0.001162
14. Wakuda M, Ide T, Sasaki J, Komoto S, Ishii J, Sanekata T, et al. Porcine rotavirus closely related to novel group of human rotaviruses. Emerg Infect Dis. (2011) 17:1491. doi: 10.3201/edi1708.101466
15. Molinari BL, Lorenzetti E, Otonel RA, Alfieri AF, Alfieri AA. Species H rotavirus detected in piglets with diarrhea, Brazil, 2012. Emerg Infect Dis. (2014) 20:1019. doi: 10.3201/eid2006.130776
16. Molinari BLD, Alfieri AF, Alfieri AA. Genetic variability of VP6, VP7, VP4, and NSP4 genes of porcine rotavirus group H detected in Brazil. Virus Res. (2015) 197:48–53. doi: 10.1016/j.virusres.2014.12.003
17. Nyaga MM, Peenze I, Potgieter CA, Seheri LM, Page NA, Yinda CK, et al. Complete genome analyses of the first porcine rotavirus group H identified from a South African pig does not provide evidence for recent interspecies transmission events. Inf Genet Evol. (2016) 38:1–7. doi: 10.1016/j.meegid.2015.11.032
18. Phan MV, Anh PH, Cuong NV, Munnink BBO, van der Hoek L, My PT. Unbiased whole-genome deep sequencing of human and porcine stool samples reveals circulation of multiple groups of rotaviruses and a putative zoonotic infection. Virus Evol. (2016) 2:vew027. doi: 10.1093/ve/vew027
19. Puente H, Cortey M, de Nova PJ, Mencía-Ares Ó, Gómez-García M, Díaz I, et al. First identification and characterization of rotavirus H in swine in Spain. Transbound Emerg Dis. (2021) 68:3055–69. doi: 10.1111/tbed.13992
20. Ferrari E, Salogni C, Martella V, Alborali GL, Scaburri A, Boniotti MB. Assessing the epidemiology of rotavirus a, B, C and H in diarrheic pigs of different ages in northern Italy. Pathogens. (2022) 11:467. doi: 10.3390/pathogens11040467
21. Shi K, Zhou H, Feng S, He J, Li B, Long F, et al. Development of a Quadruplex RT-qPCR for the detection of porcine rotaviruses and the phylogenetic analysis of porcine RVH in China. Pathogens. (2023) 12:1091. doi: 10.3390/pathogens12091091
22. Yinda CK, Ghogomu SM, Conceicao-Neto N, Beller L, Deboutte W, Vanhulle E, Matthijnssens J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. (2018) 4:vey008. doi: 10.1093/ve/vey008
23. Kim HK, Yoon SW, Kim DJ, Koo BS, Noh JY, Kim JH, et al. Detection of severe acute respiratory syndrome-like, middle east respiratory syndrome-like Bat coronaviruses and group H rotavirus in faeces of Korean bats. Transbound Emerg Dis. (2016) 63:365–72. doi: 10.1111/tbed.12515
24. Hardmeier I, Aeberhard N, Qi W, Schoenbaechler K, Kraettli H, Hatt JM, et al. Metagenomic analysis of fecal and tissue samples from 18 endemic bat species in Switzerland revealed a diverse virus composition including potentially zoonotic viruses. PLoS ONE. (2021) 16:e0252534. doi: 10.1371/journal.pone.0252534
25. Lazov CM, Belsham GJ, Bøtner A, Rasmussen TB. Full-genome sequences of alphacoronaviruses and astroviruses from myotis and pipistrelle bats in Denmark. Viruses. (2021) 13:1073. doi: 10.3390/v13061073
26. De Coster W, D'hert S, Schultz DT, Cruts M, Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. (2018) 34:2666–9. doi: 10.1093/bioinformatics/bty149
27. Sović I, Šikić M, Wilm A, Fenlon SN, Chen S, Nagarajan N. Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nat Commun. (2016) 7:11307. doi: 10.1038/ncomms11307
28. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Li H. Twelve years of SAM tools and BCF tools. Gigascience. (2021) 10:giab008. doi: 10.1093/gigascience/giab008
29. Ondov BD, Bergman NH, Phillippy AM. Interactive metagenomic visualization in a Web browser. BMC Bioinf. (2011) 12:1–10. doi: 10.1186/1471-2105-12-385
30. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. (2018) 34:3094–100. doi: 10.1093/bioinformatics/bty191
31. Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. (2017) 27:722–36. doi: 10.1101/gr.215087.116
32. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. (2016) 33:1870–4. doi: 10.1093/molbev/msw054
33. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. (2021) 49:W293–6. doi: 10.1093/nar/gkab301
34. Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. (2015) 1:vev003. doi: 10.1093/ve/vev003
35. Suzuki T, Hasebe A. A provisional complete genome-based genotyping system for rotavirus species C from terrestrial mammals. J Gen Virol. (2017) 98:2647–62. doi: 10.1099/jgv.0.000953
36. Molinari BLD, Possatti F, Lorenzetti E, Alfieri AF, Alfieri AA. Unusual outbreak of post-weaning porcine diarrhea caused by single and mixed infections of rotavirus groups A, B, C, and H. Vet Microbiol. (2016) 193:125–32. doi: 10.1016/j.vetmic.2016.08.014
37. Vereecke N, Zwickl S, Gumbert S, Graaf A, Harder T, Ritzmann M, et al. Viral and bacterial profiles in endemic influenza A virus infected swine herds using nanopore metagenomic sequencing on tracheobronchial swabs. Microbiology Spectrum. (2023) 11:e00098–23. doi: 10.1128/spectrum.00098-23
38. Hull JJ, Qi M, Montmayeur AM, Kumar D, Velasquez DE, Moon SS, et al. Metagenomic sequencing generates the whole genomes of porcine rotavirus A, C, and H from the United States. PLoS ONE. (2020) 15:e0244498. doi: 10.1371/journal.pone.0244498
39. Faizuloev E, Mintaev R, Petrusha O, Marova A, Smirnova D, Ammour Y, et al. New approach of genetic characterization of group A rotaviruses by the nanopore sequencing method. J Virol Methods. (2021) 292:114114. doi: 10.1016/j.jviromet.2021.114114
40. Hoxie I, Dennehy JJ. Intragenic recombination influences rotavirus diversity and evolution. Virus Evol. (2020) 6:vez059. doi: 10.1093/ve/vez059
41. Marthaler D, Suzuki T, Rossow K, Culhane M, Collins J, Goyal S, et al. VP6 genetic diversity, reassortment, intragenic recombination and classification of rotavirus B in American and Japanese pigs. Vet Microbiol. (2014) 172:359–66. doi: 10.1016/j.vetmic.2014.05.015
42. Oki H, Masuda T, Hayashi-Miyamoto M, Kawai M, Ito M, Madarame H, et al. Genomic diversity and intragenic recombination of species C rotaviruses. J Gen Virol. (2022) 103:001703. doi: 10.1099/jgv.0.001703
Keywords: rotavirus H, swine diseases, nanopore sequencing, phylogenetics, metagenomics, viral recombination
Citation: Krasnikov N and Yuzhakov A (2023) Interspecies recombination in NSP3 gene in the first porcine rotavirus H in Russia identified using nanopore-based metagenomic sequencing. Front. Vet. Sci. 10:1302531. doi: 10.3389/fvets.2023.1302531
Received: 26 September 2023; Accepted: 14 November 2023;
Published: 05 December 2023.
Edited by:
Levon Abrahamyan, Montreal University, CanadaReviewed by:
Tara G. McDaneld, Agricultural Research Service (USDA), United StatesTohru Suzuki, National Agriculture and Food Research Organization, Japan
Copyright © 2023 Krasnikov and Yuzhakov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikita Krasnikov, bmljay5rcmFzbmlja29mZjIwMTFAeWFuZGV4LnJ1
†ORCID: Nikita Krasnikov orcid.org/0000-0001-8148-3080
Anton Yuzhakov orcid.org/0000-0002-0426-9678