 Bin Chen
Bin Chen Ya-fang Liu1
Ya-fang Liu1 Xing Yang
Xing Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 13 September 2023
Sec. Parasitology
Volume 10 - 2023 | https://doi.org/10.3389/fvets.2023.1255017
Fleas (Order Siphonaptera) are common blood-feeding ectoparasites, which have important economic significance. Limited mitochondrial genome information has impeded the study of flea biology, population genetics and phylogenetics. The Ctenophthalmus quadratus and Stenischia humilis complete mt genomes are described in this study. The samples were collected from Jianchuan, Yunnan plague foci, China. The mt genomes of C. quadratus and S. humilis were 15,938 bp and 15,617 bp, respectively. The gene arrangement of mt genome was consistent with that of other fleas, which include 22 tRNA genes, 13 protein-coding genes, and two rRNA genes, with a total of 37 genes. The relationship between C. quadratus and S. humilis in fleas was inferred by phylogenetic analysis of mt genome sequence datasets. Phylogenetic analyzes showed that the C. quadratus and S. humilis belonged to different species in the same family, and were closely related to Hystrichopsylla weida qinlingensis in the same family; and revealed that the family Hystrichopsyllidae is paraphyletic, supporting the monophyly of the order Siphonaptera. This study decodes the complete mt genomes of the C. quadratus and S. humilis for the first time. The results demonstrate that the C. quadratus and S. humilis are distinct species, and fleas are monophyletic. Analysis of mt genome provides novel molecular data for further studying the phylogeny and evolution of fleas.
Fleas (Order Siphonaptera) include about 2,574 species in 238 genera and 16 families, are some of the most common blood-feeding ectoparasites of birds and mammals (1). They are the most economically significant ectoparasites, resulting in spending of more than $15 billion per year in the world to control and prevent of flea infestations in companion animals (2). Flea is of major epidemiological importance because they can transmit various pathogens. They have worldwide distribution and a wide range of host preference, and are vectors for many pathogens, such as Yersinia pestis (plague), Rickettsia spp. (Rickettsia typhi, Rickettsia prowazekii, and Rickettsia rickettsi), and Bartonella henselae (cat-scratch disease) (3–5). Plague is the most serious of these diseases, with multiple outbreaks around the world killing hundreds of millions of people (4). Up to now, China has identified 15 natural plague foci covering an area of more than 1.4 million square kilometers (6). The world’s third plague pandemic originated in Yunnan Province, China (7, 8). Ctenophthalmus quadratus and Stenischia humilis are one of the most common fleas in the focus in Yunnan Province, and Yersinia pestis has been isolated from these two fleas many times (9). Ctenophthalmus quadratus was considered to be the main vector species of plague bacilli, but later experiments showed that it has no vector role (10).
In fundamental research on these important ectoparasites and the diagnosis of the diseases they transmit, accurate classification and identification of fleas are required (11–13). Due to the absence of molecular data, most fleas are identified solely by their essential shape and morphological identification such as the distribution of their setae, spines, and ctenidia (14). However, the morphological identification of related species and variant species has some limitations and is easy to be misidentified (11). Until now, the phylogenetic relationship of fleas has been unclear, and the phylogeny at high taxonomic levels has been controversial (15). With the development of genetic technology, molecular biological methods have been widely used in taxonomy, population genetics, and systematics, to some extent, to supplement the limitations of traditional morphology (13, 16). The mitochondrial (mt) genomes have been extensively used in molecular phylogenetic studies, genetic diversity, subspecies and cryptic species identification of different ectoparasites at various taxonomic levels because of their rapid evolutionary rate, simple structure, maternal inheritance, high mutation rate, and the lack of genetic recombination (17–21). Nevertheless, there is little information about the whole mt genome of the flea, no more than 10 species, which greatly limits the studies on flea genetics and phylogenetics. Hence, more flea mt genomes need to be investigated to obtain more genetic data.
At present, no molecular data are available for the mt genomes of Ctenophthalmus quadratus and Stenischia humilis. In this study, the whole mt genome of these two fleas was annotated and analyzed. The intentions of this study were to: (i) sequence and annotate the complete mitochondrial genomes of C. quadratus and S. humilis; (ii) analyze and compare the structural features of mt genomes of C. quadratus and S. humilis; and (iii) establish phylogenetic relationships with other fleas to reassess the taxonomic status of C. quadratus and S. humilis in Yunnan, China.
Adults of Ctenophthalmus quadratus and Stenischia humilis were collected from the Eothenomys miletus in Jianchuan plague foci (26°12′N, 99°33′E), Yunnan Province, China. The key points of morphological identification of Ctenophthalmus quadratus and Stenischia humilis were recorded in detail in “the Siphonaptera of Yunnan” (22). The fleas were photographed with the Ultra-Depth Three-Dimensional Microscope (VHX-5000), and the basic morphological features were identified by professionals (22). Then, those identified as C. quadratus and S. humilis were stored, respectively, in 75% ethanol and stored at −20°C until use. According to the manufacturer’s instructions, the total genomic DNA was extracted from a single intact female flea individual using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The voucher specimen and genome DNA were deposited at the Parasitological Museum, Dali University, Yunnan, China.
The mitochondrial genomes of C. quadratus and S. humilis were amplified using designed long-PCR primers based on the 12S rRNA and cox1 genes of Ceratophyllus wui (MG886872) and Hystrichopsylla weida qinlingensis (MH259703) using the Primer3 (23), respectively (Table 1). All PCR reactions (volume: 50 μL) were performed in 23 μL ddH2O, 4 μL of dNTP mixture, 4 μL of DNA sample, 4 μL of forward primer, 4 μL of reverse primer, 10 μL of 5X PrimeSTAR GXL Buffer, and 1 μL of PrimeSTAR GXL DNA Polymerase (Takara, Japan). The PCR conditions were as follows: 94°C for 2 min, followed by 35 cycles of 98°C for 10 s, 65–68°C for 30 s, 68°C for 10 min, and with a final extension to 10 min at 68°C. The PCR products were detected in a 1.2% agarose gel electrophoresis upon ethidium-bromide staining. The purified PCR amplicons were sequenced on the Illumina NovaSeq 6,000 platform (Harbin Botai Biotechnology Co., Ltd., Heilongjiang, China).
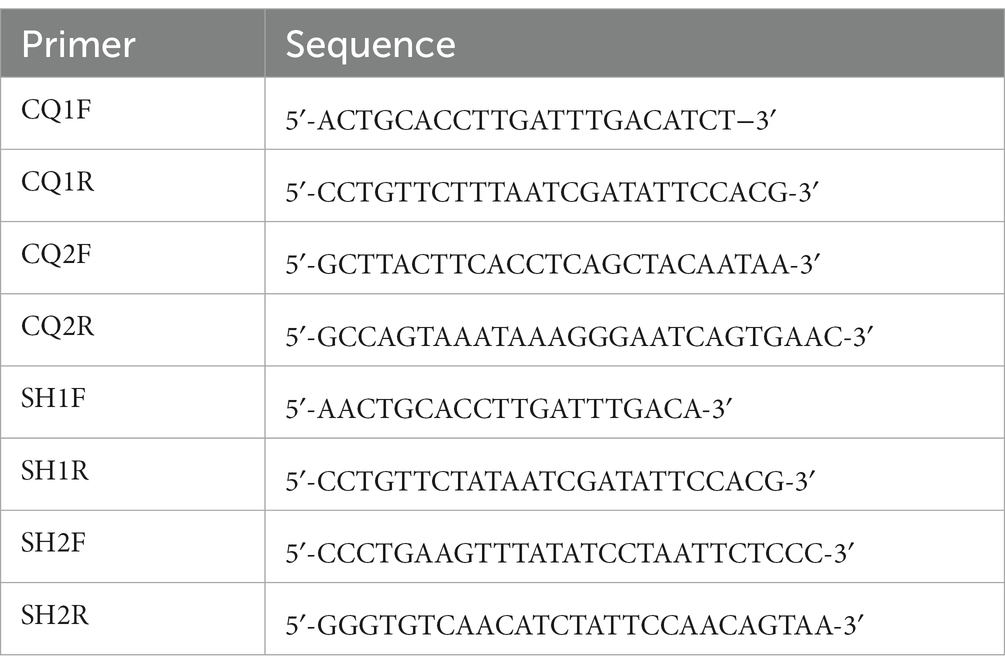
Table 1. The primer sequences used to PCR amplification.
The raw data obtained by sequencing was filtered by AdapterRemoval software (v2.0) to remove the presence of low-quality data. The software FastQC was used to conduct quality control on the clean data filtered in the previous step. Genome assembly was carried out from clean data after sample quality control, and IDBA software was used for assembly (24). The predicted genes were compared with each functional database by BLAST (blastp, evalue ≤1e-5), and the comparison results with the highest score were selected (25). The MITOS webserver was used to annotate the mitochondrial genome (26). The tRNAscan-SE webserver was used to verify transfer RNA (tRNA) genes with secondary structure. The GC skew and AT skew were calculated by the strand asymmetry formulas (27). The amino acid sequences of PCGs, nucleotide composition, and base composition were analyzed using MEGA X (28).
For the phylogenetic relationship analysis, 15 additional mitochondrial genome sequences were downloaded from GenBank, and Philaenus spumarius (GenBank accession number: AY630340) was selected as an outgroup (Table 2). The sequences of amino acids of 13 protein-coding genes (PCGs) were aligned using MAFFT software. All positions containing blank and missing data were eliminated. The General Time Reversible (GTR + G + I) model was selected as the most suitable model of evolution by the MrModeltest 2.3 based on the Akaike information criterion (AIC) (29). The Bayesian inference (BI) phylogenetic tree was reconstructed with 1,000,000 generations and sampled every 100 generations in MrBayes 3.2.5 (30). And the maximum likelihood (ML) phylogenetic tree was constructed on IQ-TREE based on 10,000 ultrafast bootstrap approximations (31). The resulting phylogenetic tree was edited using FigTree v.1.4.2.
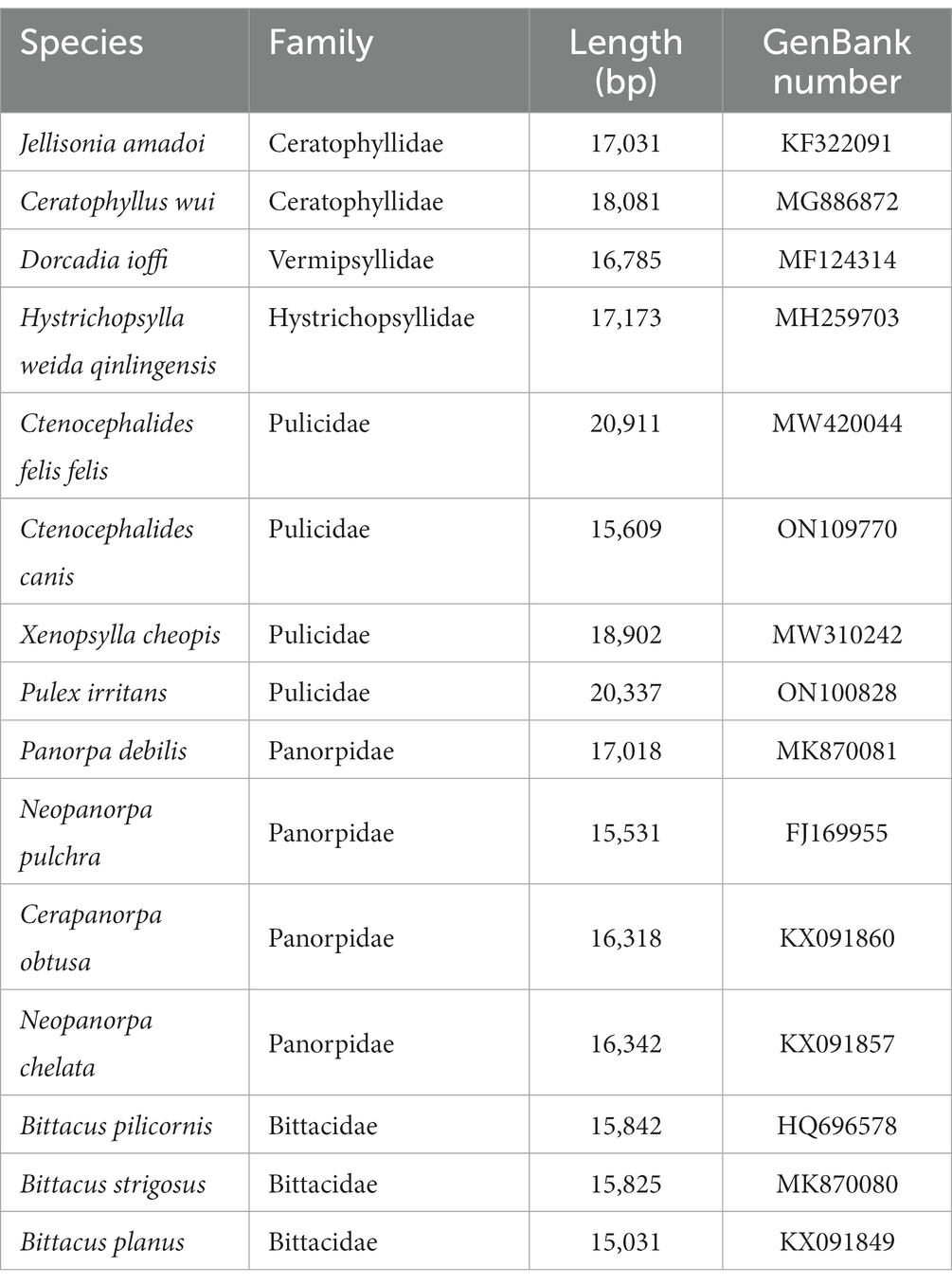
Table 2. The flea species analyzed in the current study with their GenBank numbers.
The complete mitochondrial genomes of Ctenophthalmus quadratus and Stenischia humilis were submitted to GenBank with accession numbers OQ023577 and OQ366410. The sequences of C. quadratus and S. humilis was 15,938 bp and 15,617 bp in size, respectively, and both contained 37 genes, including 22 tRNA genes, 13 PCGs, and two rRNA genes (Figure 1). 23 genes (nad2, cox1, cox2, atp8, atp6, cox3, nad3, nad6, cob, trnI, trnM, trnW, trnL2, trnK, trnD, trnG, trnA, trnR, trnN, trnS1, trnE, trnT, trnS2) were on the heavy strand (H-strand), while the rest 14 genes (nad5, nad4, nad4L, nad1, trnQ, trnC, trnY, trnF, trnH, trnP, trnL1, trnV, rrnL, rrnS) were on the light strand (L-strand). The mitochondrial genome of C. quadratus had 13 overlapping locations, consisting of a total of 36 bp, with each overlap one to eight bp, and had seven intergenic regions comprising 151 bp in total and ranging from one to 73 bp (Table 3). Likewise, the mt genome of S. humilis had 11 overlapping locations with a total length of 33 bp, and had 16 intergenic regions consisting of 183 bp in total (Table 3). The nucleotide composition of C. quadratus was: A = 39.19%, T = 40.26%, G = 7.96%, C = 12.59%, and A + T = 79.45% (Table 4). Similarly, The nucleotide composition of S. humilis was: A = 38.57%, T = 39.43%, G = 8.38%, C = 13.62%, and A + T = 78.00% (Table 4).
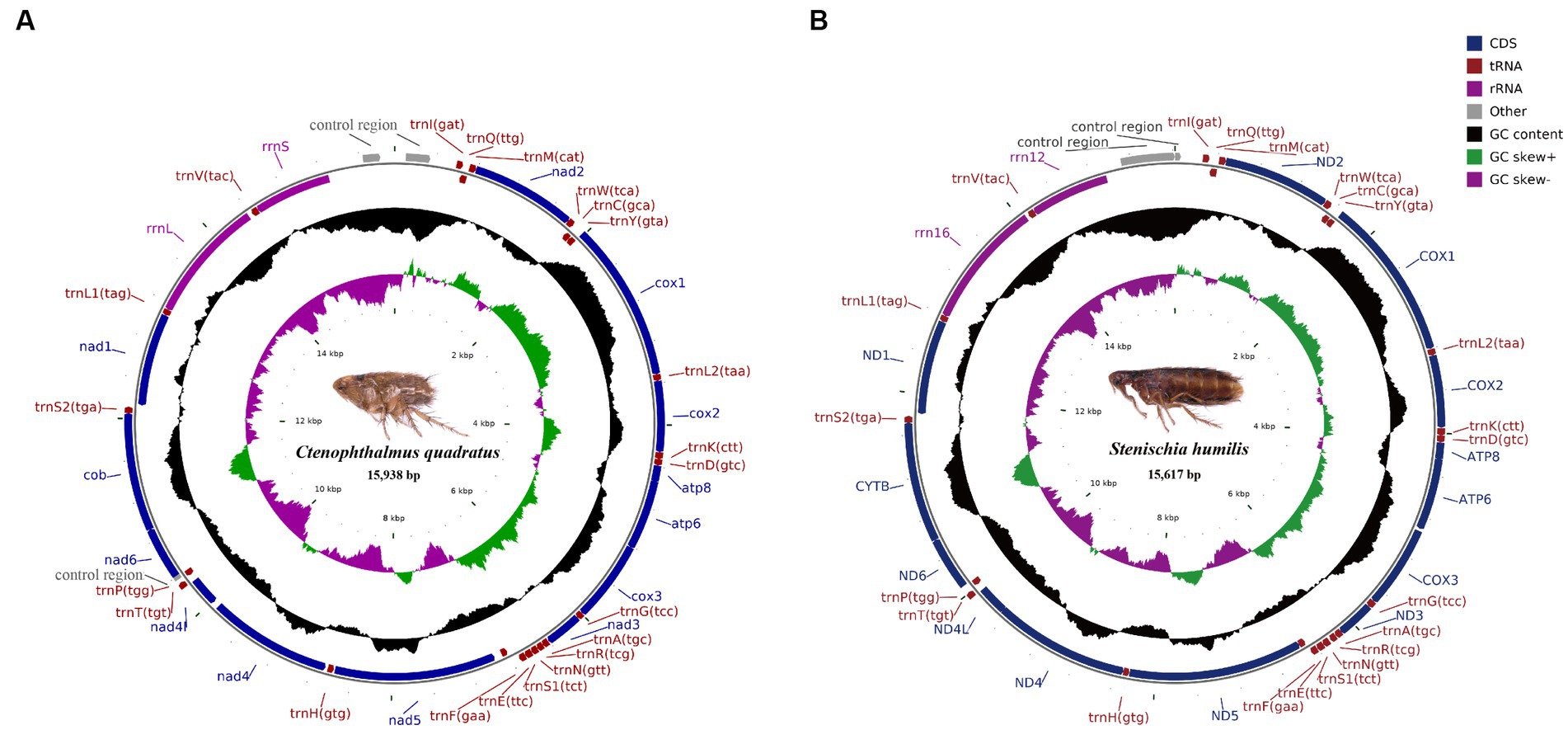
Figure 1. Arrangement of the mitochondrial genome of (A) Ctenophthalmus quadratus and (B) Stenischia humilis. All genes are indicated using standard nomenclature.
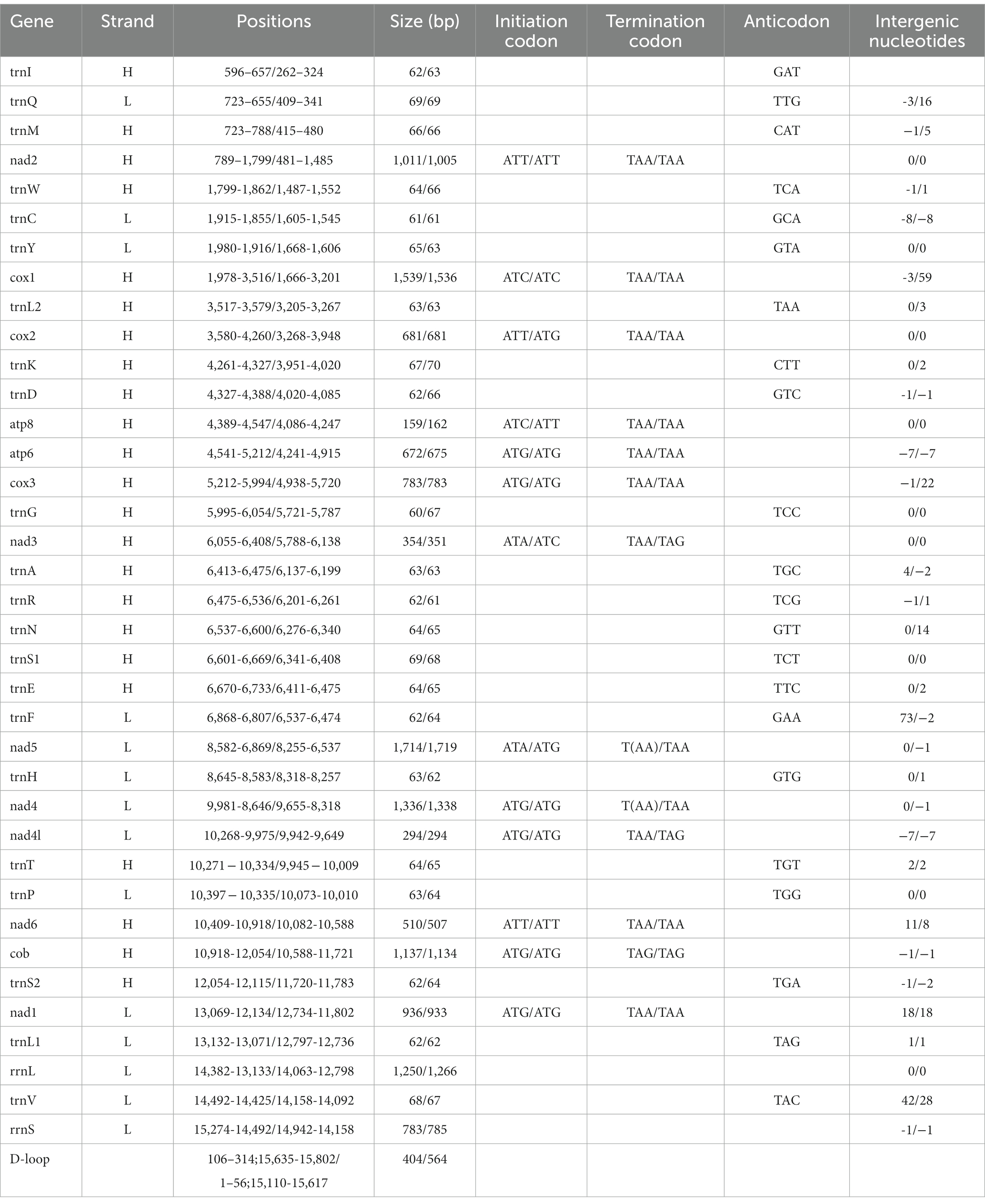
Table 3. Organization of the mitochondrial genomes of Ctenophthalmus quadratus and Stenischia humilis.
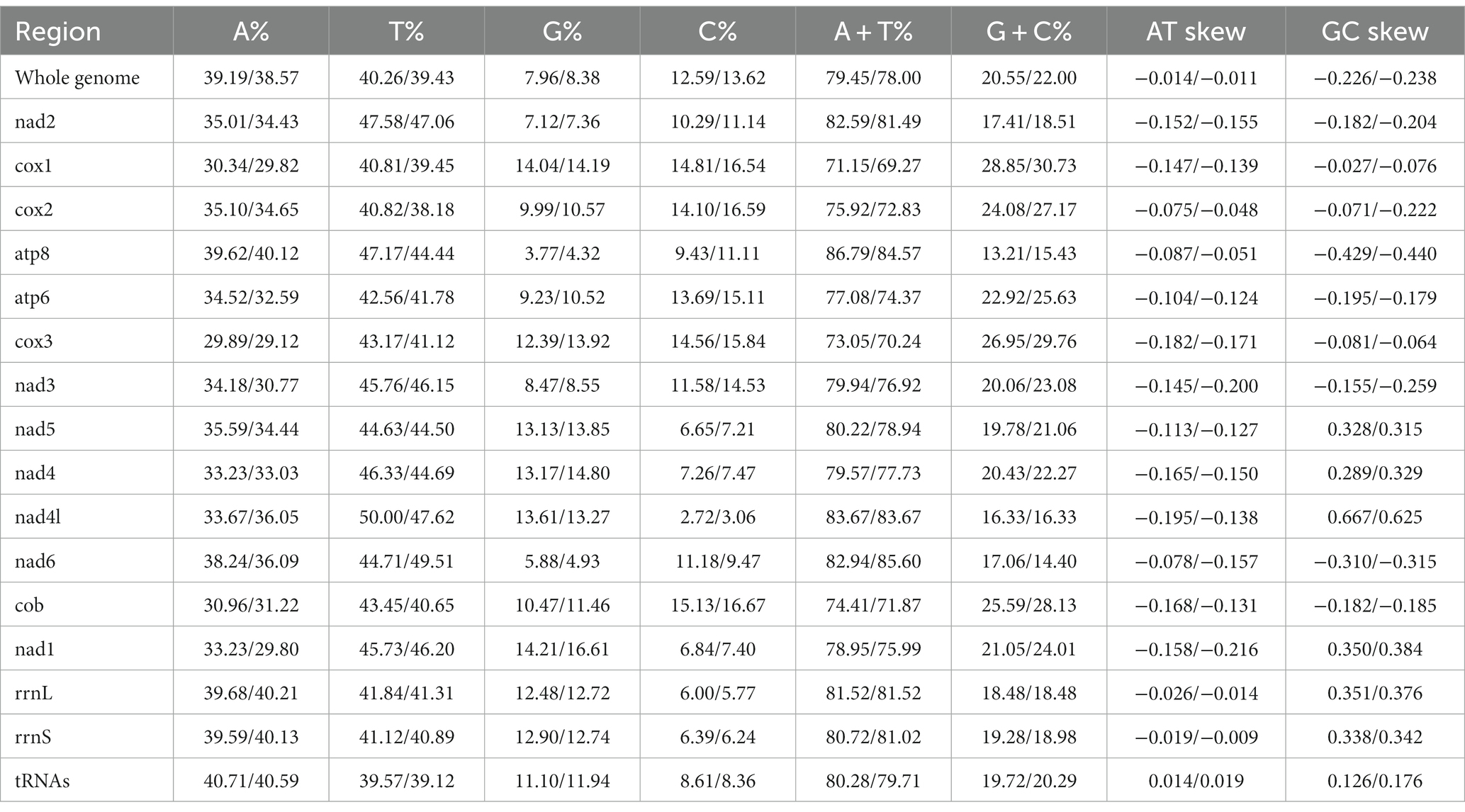
Table 4. Composition of mitochondrial genomes in the Ctenophthalmus quadratus and Stenischia humilis.
In the mt genome of C. quadratus, three genes (nad2, cox2, nad6) use ATT, two genes (cox1, atp8) use ATC, two genes (nad3, nad5) use ATA, and six genes (atp6, cox3, nad4, nad4L, cob, nad1) use ATG as initiation codon, respectively, and TAA as termination codon in addition to nad5, nad4 and cob genes (Table 3). In the S. humilis mt genome, three genes (nad2, atp8, nad6) use ATT, two genes (cox1, nad3) uses ATC and eight genes (cox2, atp6, cox3, nad5, nad4, nad4L, cob, nad1) use ATG as initiation codon, respectively, and TAA as termination codon except for nad3, nad4L and cob genes (Table 3). The relative synonymous codon usage (RSCU) values and codon use patterns were computed for 13 protein-coding genes of the C. quadratus and S. humilis mt genomes. In total, 3,546 and 3,693 amino acids were encoded by the mt genomes of C. quadratus and S. humilis, respectively. Leucine, isoleucine, and phenylalanine were the most frequently utilized amino acids, followed by serine and methionine; arginine and cysteine were the least utilized amino acids (Figure 2).
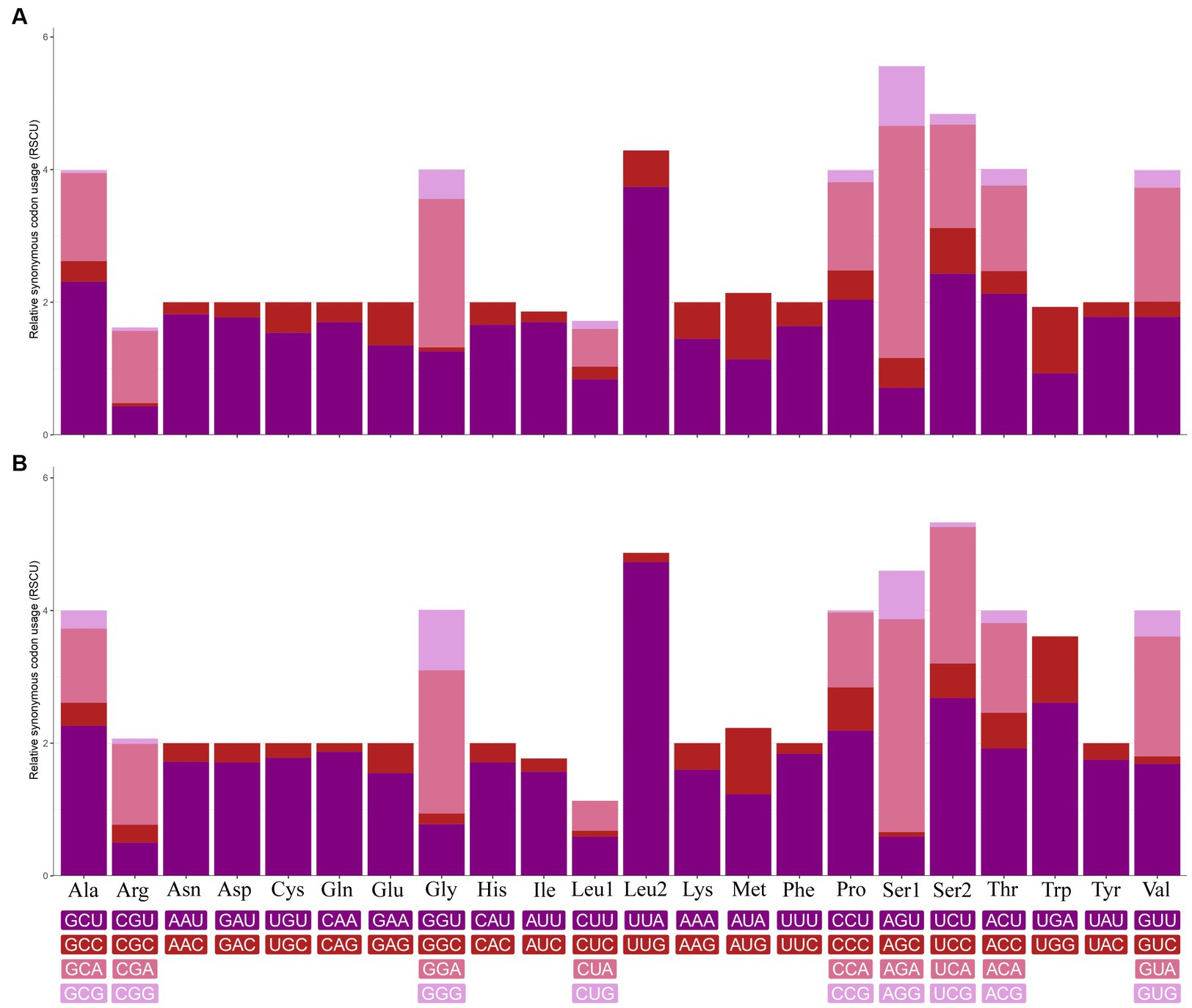
Figure 2. Relative synonymous codon usage (RSCU) of (A) Ctenophthalmus quadratus and (B) Stenischia humilis. All codons coding for each amino acid are represented in the boxes below the bar chart.
The small subunit of rRNA gene (rrnS) was situated next to trnV, and the large subunit of rRNA gene (rrnL) was located between trnL1 and trnV (Table 3). The length of rrnS and rrnL genes in C. quadratus were 783 and 1,250 bp, respectively, and the A + T contents of the rrnS and rrnL were 80.72 and 81.52%, respectively (Tables 3, 4). In the same way, the rrnS and rrnL genes of S. humilis were 785 and 1,266 bp, respectively, and the A + T contents of the rrnS and rrnL were 81.02 and 81.52% (Tables 3, 4). The length of 22 tRNA genes of C. quadratus ranged from 60 to 69 bp, and those of S. humilis ranged from 61 to 70 bp (Table 3). Most of the predicted secondary structures of 22 tRNA genes showed typical cloverleaf structure (Figures 3, 4).
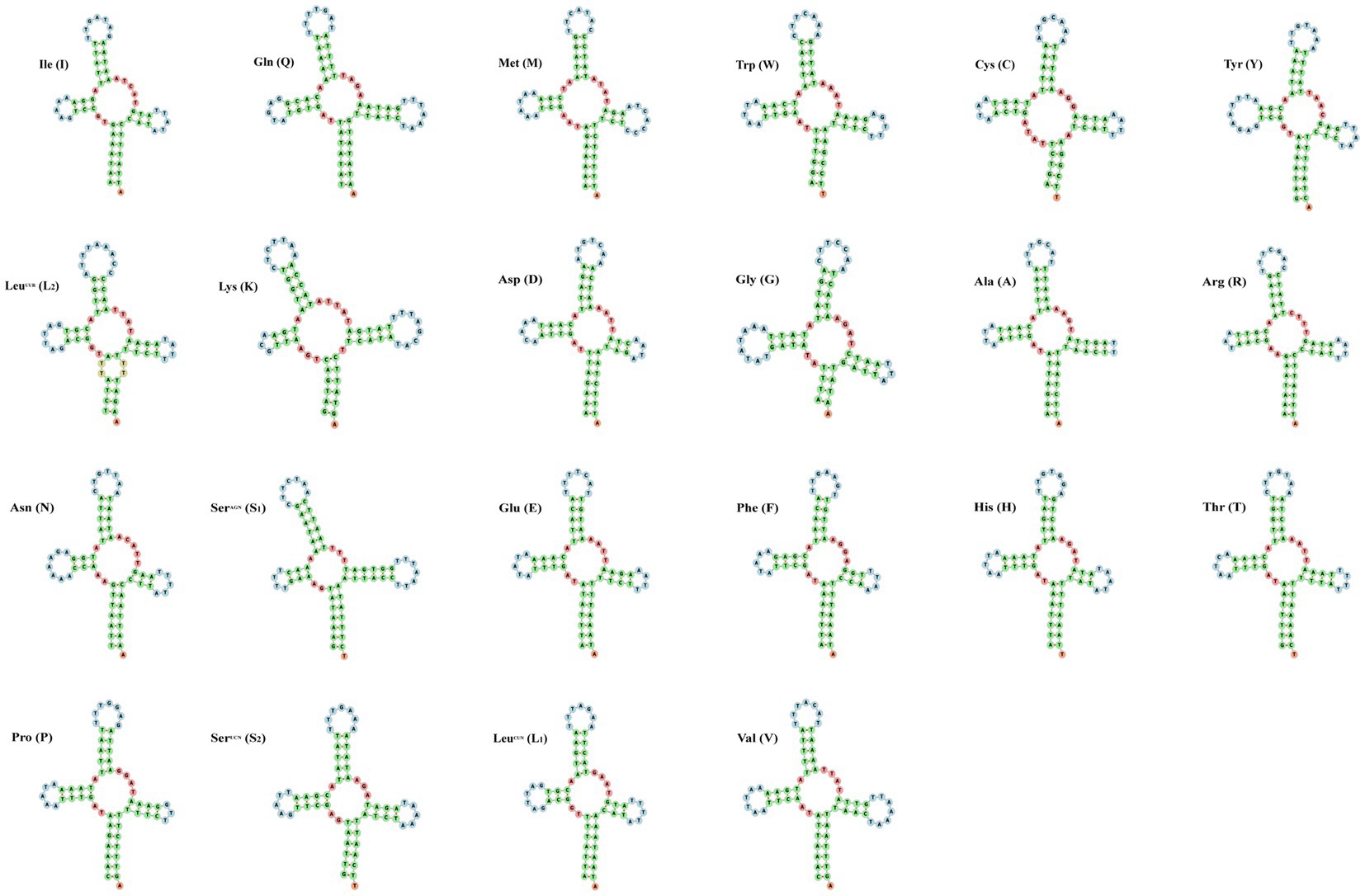
Figure 3. Putative secondary structure of the 22 mt tRNA of Ctenophthalmus quadratus.
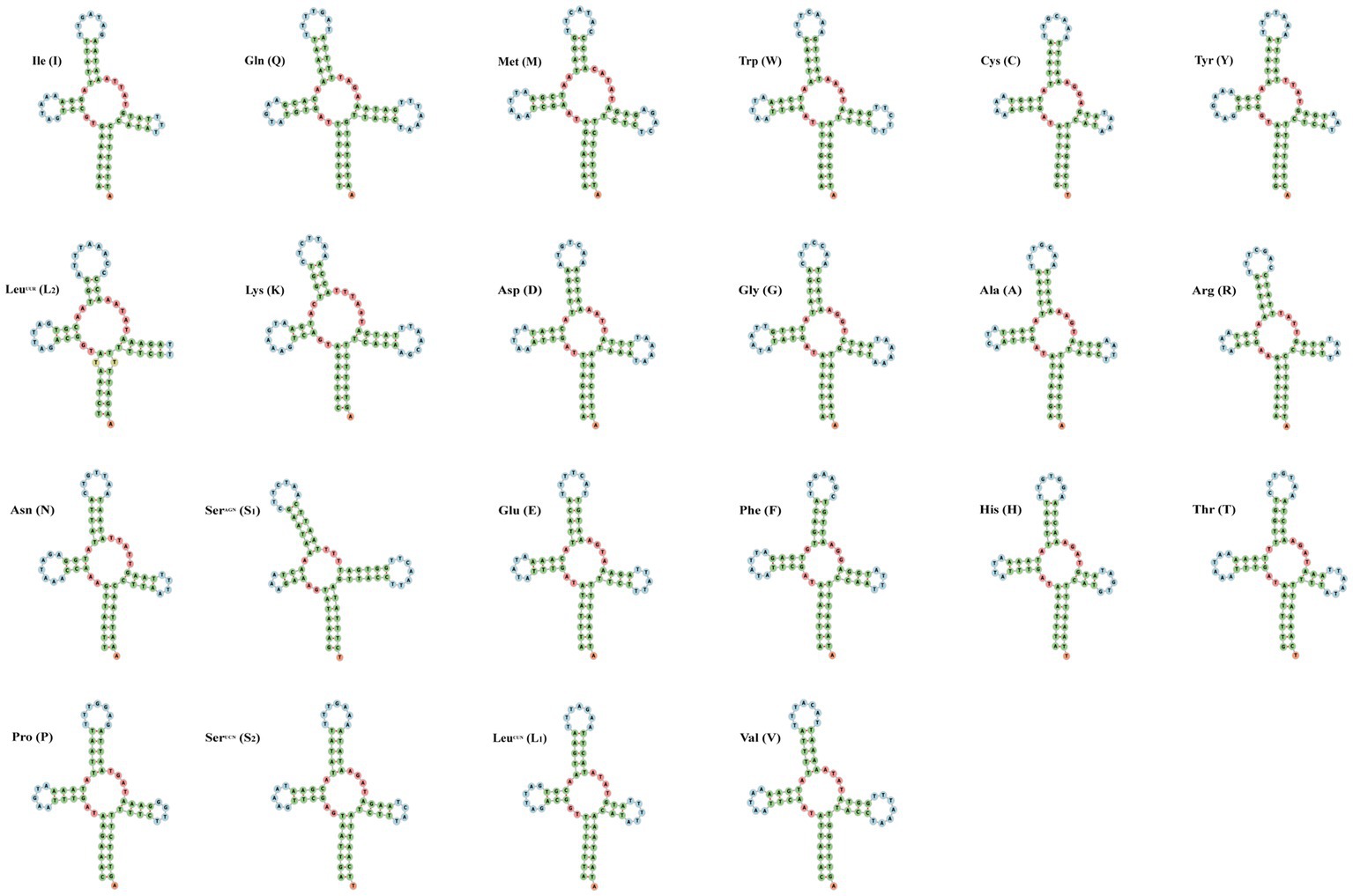
Figure 4. Putative secondary structure of the 22 mt tRNA of Stenischia humilis.
Among the 17 species used for phylogenetic analysis by the BI and ML methods in this study, 10 species belonged to the Siphonaptera and seven species belonged to the Mecoptera (Figure 5). The monophyly of the order Siphonaptera and Mecoptera were strongly supported with the Bayesian posterior probability (Bpp) of 1 and the Ultrafast bootstrap approximation (UFBoot) of 100% in the BI and ML analyzes, respectively. Meanwhile, the family Hystrichopsyllidae may be paraphyletic. The close relationship between C. quadratus and S. humilis was strongly supported in BI analysis (Bpp = 0.96) and moderately supported in ML analysis (UFBoot = 70%). The close relationship between Dorcadia ioffi and Hystrichopsylla weida qinlingensis was strongly supported in BI analysis (Bpp = 0.99) and moderately supported in ML analysis (UFBoot = 77%). Nevertheless, C. quadratus + S. humilis was the sister group of Dorcadia ioffi + Hystrichopsylla weida qinlingensis, with strongly support in the BI and ML analyzes (Bpp = 0.97, UFBoot = 95%).
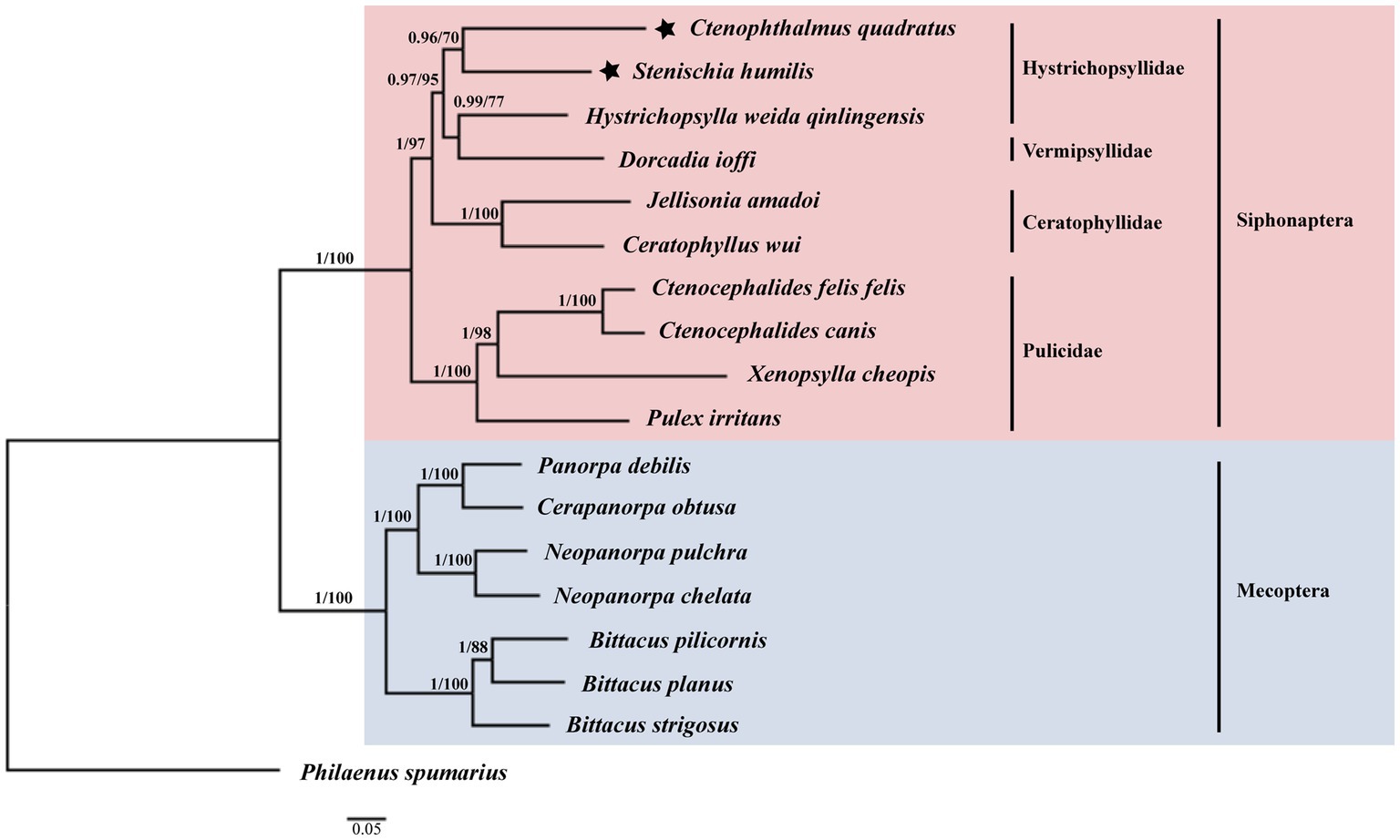
Figure 5. Phylogenetic relationships of 17 species of Siphonaptera and Mecoptera inferred from BI and ML analyzes of deduced nucleotide sequences of 13 PCGs. Bayesian posterior probability (Bpp) and Ultrafast bootstrap approximation (UFBoot) values were indicated at nodes, respectively. Philaenus spumarius (AY630340) was used as the outgroup.
Fleas (Order Siphonaptera) are the most common blood-sucking ectoparasites and vector of many pathogens. People living in plague foci are more likely to be exposed to fleas carrying Yersinia pestis (32). The accurate identification and differentiation of flea species is significance for the control and diagnosis of flea-borne diseases (33). However, morphological identification of related and variant flea species are often challenging (11). Molecular data on fleas is still deficient. In the present study, the whole mt genomes of Ctenophthalmus quadratus and Stenischia humilis were analyzed for the first time to provide additional molecular data for phylogenetic studies of fleas.
In this study, the mt genomes of C. quadratus and S. humilis were consistent with the basic structural characteristics of other fleas, both containing 37 genes, two rRNA genes, 22 tRNA genes, 13 PCGs, and non-coding regions, and the gene arrangement sequences were consistent with that of other fleas (34). Negative AT-skew and GC-skew were found in their mt genomes, showing a bias toward T and C in nucleotide composition. Flea species are abundant, but few flea species have the complete mt genome in NCBI. So far, there are still less than 20 complete mt genomes in fleas, and there is no information on the C. quadratus and S. humilis mt genomes. Thus, the two mt genomes provided in this study will promote future studies on flea phylogeny and population evolution.
The monophyly of the Holometabola is well supported by morphological traits and molecular evidence (35, 36). Among them, the monophyly of Mecoptera remains extremely controversial. Phylogenetic analyzes using the 18S and 28S rRNA sequences suggest that the order Mecoptera may be paraphyletic, and that the order Siphonaptera (fleas) may be subordinate within Mecoptera (36–38). Similarly, the most recent study used the largest molecular dataset of over 1,400 protein-coding genes, as well as the smaller mitochondrial genome of 16 genes, indicate that Siphonaptera is nested within Mecoptera and suggest that Siphonaptera be treated as an infraorder of Mecoptera (39). Nevertheless, phylogenetic relationships inferred from 1,478 protein-coding genes strongly support that the Mecoptera and Siphonaptera are monophyletic (40). Phylogenetic analysis of 11 orders of holometabola using 13 PCGs in mitochondrial genomes supported the monophyly of Siphonaptera and paraphyly of Mecoptera. The results show that the Siphonaptera is an independent order and as a sister group of the family Boreidae, rather than subordinate to Mecoptera (34). The above results show that the phylogenetic position of fleas in holometabolan insects is still unclear and highly controversial.
This study is the first in the world to analyze the mt genomes of Ctenophthalmus quadratus and Stenischia humilis, and to analyze their phylogenetic positions in fleas using 13 PCGs. The results support that the order Siphonaptera is monophyletic. And the family Hystrichopsyllidae is paraphyletic. The seven species of Mecoptera analyzed in this study cluster together to form one clade, and the 10 species of Siphonaptera cluster together to form another clade, and strongly support a sister relationship between the orders Siphonaptera and Mecoptera, consistent with other research findings (34). Up to now, there is no information on the mt genomes of the genera of Ctenophthalmus and Stenischia species. The present study is the first to analyze flea species from the two genera. Due to the lack of mt genome data for all lineages of fleas, which is not fully representative of the overall phylogenetic relationships of fleas. Hence, further acquisition of mt genomes from more flea species is needed to further evaluate the evolutionary relationship between the orders Siphonaptera and Mecoptera, and their phylogenetic position in holometabolous insects with more comprehensive molecular data.
The present study is the first to obtained the complete mitochondrial genomes of Ctenophthalmus quadratus and Stenischia humilis. Phylogenetic analysis of eight other fleas and seven species of Mecoptera demonstrated that the C. quadratus and S. humilis are distinct species in the same family, and provided a sister relationship between the Siphonaptera and Mecoptera, supporting the monophyly of fleas. These mt genomes provide a hint for the phylogenetic position of C. quadratus and S. humilis in fleas, and provide novel genetic information for the phylogeny and evolution of fleas.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, OQ023577; https://www.ncbi.nlm.nih.gov/genbank/, OQ366410.
The animal study was approved by the Administration Committee of Experimental Animals, Dali University. The study was conducted in accordance with the local legislation and institutional requirements.
XY and BC designed the study. D-dJ and X-yL are in charge of data gathering. Y-fL and BC designed the primers and conducted the experiment. BC and XW collated and analyzed the data. G-pY and Q-fZ contribute to the results discussion. BC wrote the manuscript. Y-fL, X-yL, D-dJ, XW, Q-fZ, G-pY, and XY writing--review and editing. All authors read and provided critical viewpoint and determine the final version.
This study was funded by the following organizations and initiatives: Yunnan Natural Science Foundation (2017FD139), and Scientifc Research Fund of Yunnan Education Department (2022 J0687).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bitam, I, Dittmar, K, Parola, P, Whiting, MF, and Raoult, D. Fleas and flea-borne diseases. Int J Infect Dis. (2010) 14:e667–76. doi: 10.1016/j.ijid.2009.11.011
2. Nisbet, AJ, and Huntley, JF. Progress and opportunities in the development of vaccines against mites, fleas and myiasis-causing flies of veterinary importance. Parasite Immunol. (2006) 28:165–72. doi: 10.1111/j.1365-3024.2006.00803.x
3. Hamzaoui, BE, Zurita, A, Cutillas, C, and Parola, P. Fleas and flea-borne diseases of North Africa. Acta Trop. (2020) 211:105627. doi: 10.1016/j.actatropica.2020.105627
4. Eisen, RJ, and Gage, KL. Transmission of flea-borne zoonotic agents. Annu Rev Entomol. (2012) 57:61–82. doi: 10.1146/annurev-ento-120710-100717
5. Iannino, F, Sulli, N, Maitino, A, Pascucci, I, Pampiglione, G, and Salucci, S. Fleas of dog and cat: species, biology and flea-borne diseases. Vet Ital. (2017) 53:277–88. doi: 10.12834/VetIt.109.303.3
6. Li, Y, Dai, E, Cui, Y, Li, M, Zhang, Y, Wu, M, et al. Different region analysis for genotyping Yersinia pestis isolates from China. PLoS One. (2008) 3:e2166. doi: 10.1371/journal.pone.0002166
7. Shi, L, Yang, G, Zhang, Z, Xia, L, Liang, Y, Tan, H, et al. Reemergence of human plague in Yunnan, China in 2016. PLoS One. (2018) 13:e0198067. doi: 10.1371/journal.pone.0198067
8. Han, H, Liang, Y, Song, Z, He, Z, Duan, R, Chen, Y, et al. Epidemiological characteristics of human and animal plague in Yunnan Province, China, 1950 to 2020. Microbiol Spectr. (2022) 10:e0166222. doi: 10.1128/spectrum.01662-22
9. Xing-Yuan, M, Xian-Guo, G, Wen-Ge, D, Ai-Qin, N, Ti-Jun, Q, and Dian, W. Ectoparasites of Chevrier's field mouse, Apodemus chevrieri, in a focus of plague in Southwest China. Med Vet Entomol. (2007) 21:297–300. doi: 10.1111/j.1365-2915.2007.00679.x
10. He, JH, Liang, Y, and Zhang, HY. A study on the transmission of plague though seven kinds of fleas in rat type and wild rodent type plague foci in Yunnan. Zhonghua Liu Xing Bing Xue Za Zhi. (1997) 18:236–40. Chinese.
11. Linardi, PM, and Santos, JL. Ctenocephalides felis felis vs. Ctenocephalides canis (Siphonaptera: Pulicidae): some issues in correctly identify these species. Rev Bras Parasitol Vet. (2012) 21:345–54. doi: 10.1590/S1984-29612012000400002
12. Hornok, S, Beck, R, Farkas, R, Grima, A, Otranto, D, Kontschán, J, et al. High mitochondrial sequence divergence in synanthropic flea species (Insecta: Siphonaptera) from Europe and the Mediterranean. Parasit Vectors. (2018) 11:221. doi: 10.1186/s13071-018-2798-4
13. Lawrence, AL, Webb, CE, Clark, NJ, Halajian, A, Mihalca, AD, Miret, J, et al. Out-of-Africa, human-mediated dispersal of the common cat flea, Ctenocephalides felis: the hitchhiker's guide to world domination. Int J Parasitol. (2019) 49:321–36. doi: 10.1016/j.ijpara.2019.01.001
14. Zurita, A, García-Sánchez, ÁM, and Cutillas, C. Ctenophthalmus baeticus boisseauorum (Beaucournu, 1968) and Ctenophthalmus apertus allani (smit, 1955) (Siphonaptera: Ctenophthalmidae) as synonymous taxa: morphometric, phylogenetic, and molecular characterization. Bull Entomol Res. (2020) 110:663–76. doi: 10.1017/S0007485320000127
15. Whiting, MF, Whiting, AS, Hastriter, MW, and Dittmar, KA. Molecular phylogeny of fleas (Insecta: Siphonaptera): origins and host associations. Cladistics. (2008) 24:677–707. doi: 10.1111/j.1096-0031.2008.00211.x
16. Van der Mescht, L, Matthee, S, and Matthee, CA. New taxonomic and evolutionary insights relevant to the cat flea, Ctenocephalides felis: a geographic perspective. Mol Phylogenet Evol. (2021) 155:106990. doi: 10.1016/j.ympev.2020.106990
17. Tatarenkov, A, and Avise, JC. Rapid concerted evolution in animal mitochondrial DNA. Proc Biol Sci. (2007) 274:1795–8. doi: 10.1098/rspb.2007.0169
18. Shao, R, and Barker, SC. Mitochondrial genomes of parasitic arthropods: implications for studies of population genetics and evolution. Parasitology. (2007) 134:153–67. doi: 10.1017/S0031182006001429
19. Wang, T, Zhang, S, Pei, T, Yu, Z, and Liu, J. Tick mitochondrial genomes: structural characteristics and phylogenetic implications. Parasit Vectors. (2019) 12:451. doi: 10.1186/s13071-019-3705-3
20. Nie, Y, Fu, YT, Zhang, Y, Deng, YP, Wang, W, Tu, Y, et al. Highly rearranged mitochondrial genome in Falcolipeurus lice (Phthiraptera: Philopteridae) from endangered eagles. Parasit Vectors. (2021) 14:269. doi: 10.1186/s13071-021-04776-5
21. Fu, YT, Zhang, Y, Xun, Y, Liu, GH, and Suleman, ZY. Characterization of the complete mitochondrial genomes of six horseflies (Diptera: Tabanidae). Infect Genet Evol. (2021) 95:105054. doi: 10.1016/j.meegid.2021.105054
22. Xie, BQ, and Zeng, JF. The Siphonaptera of Yunnan. Yunnan: Yunnan Science and Technology Press (2000). 223 p.
23. Rozen, S, and Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. (2000) 132:365–86. doi: 10.1385/1-59259-192-2:365
24. Peng, Y, Leung, HC, Yiu, SM, and Chin, FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. (2012) 28:1420–8. doi: 10.1093/bioinformatics/bts174
25. Kearse, M, Moir, R, Wilson, A, Stones-Havas, S, Cheung, M, Sturrock, S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. (2012) 28:1647–9. doi: 10.1093/bioinformatics/bts199
26. Donath, A, Jühling, F, Al-Arab, M, Bernhart, SH, Reinhardt, F, Stadler, PF, et al. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. (2019) 47:10543–52. doi: 10.1093/nar/gkz833
27. Perna, NT, and Kocher, TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. (1995) 41:353–8. doi: 10.1007/BF01215182
28. Kumar, S, Stecher, G, Li, M, Knyaz, C, and Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. (2018) 35:1547–9. doi: 10.1093/molbev/msy096
29. Yamaoka, K, Nakagawa, T, and Uno, T. Application of Akaike's information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet Biopharm. (1978) 6:165–75. doi: 10.1007/BF01117450
30. Ronquist, F, and Huelsenbeck, JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. (2003) 19:1572–4. doi: 10.1093/bioinformatics/btg180
31. Nguyen, LT, Schmidt, HA, von Haeseler, A, and Minh, BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. (2015) 32:268–74. doi: 10.1093/molbev/msu300
32. Glatter, KA, and Finkelman, P. History of the plague: an ancient pandemic for the age of COVID-19. Am J Med. (2021) 134:176–81. doi: 10.1016/j.amjmed.2020.08.019
33. Zurita, A, Callejón, R, García-Sánchez, ÁM, Urdapilleta, M, Lareschi, M, and Cutillas, C. Origin, evolution, phylogeny and taxonomy of Pulex irritans. Med Vet Entomol. (2019) 33:296–311. doi: 10.1111/mve.12365
34. Zhang, Y, Fu, YT, Yao, C, Deng, YP, Nie, Y, and Liu, GH. Mitochondrial phylogenomics provides insights into the taxonomy and phylogeny of fleas. Parasit Vectors. (2022) 15:223. doi: 10.1186/s13071-022-05334-3
35. Whiting, MF. Mecoptera is paraphyletic: multiple genes and phylogeny of Mecoptera and Siphonaptera. Zool Scr. (2002) 31:93–104. doi: 10.1046/j.0300-3256.2001.00095.x
36. Whiting, MF, Carpenter, JC, Wheeler, QD, and Wheeler, WC. The Strepsiptera problem: phylogeny of the holometabolous insect orders inferred from 18S and 28S ribosomal DNA sequences and morphology. Syst Biol. (1997) 46:1–68. doi: 10.1093/sysbio/46.1.1
37. Chalwatzis, N, Hauf, J, Van De Peer, Y, Kinzelbach, R, and Zimmermann, FK. 18S ribosomal Rna genes of insects: primary structure of the genes and molecular phylogeny of the Holometabola. Ann Entomol Soc Am. (1996) 89:788–803. doi: 10.1093/aesa/89.6.788
38. Whiting, MF. Phylogeny of the holometabolous insect orders based on 18S ribosomal DNA: when bad things happen to good data. EXS. (2002) 92:69–83. doi: 10.1007/978-3-0348-8114-2_5
39. Tihelka, E, Giacomelli, M, Huang, D, Pisani, D, Donoghue, PCJ, and Cai, C. Fleas are parasitic scorpionflies. Palaeoentomology. (2020) 3:641–53. doi: 10.11646/palaeoentomology.3.6.16
Keywords: Ctenophthalmus quadratus , Stenischia humilis , mitochondrial genome, phylogenetic analyzes, fleas
Citation: Chen B, Liu Y-f, Lu X-y, Jiang D-d, Wang X, Zhang Q-f, Yang G-p and Yang X (2023) Complete mitochondrial genome of Ctenophthalmus quadratus and Stenischia humilis in China provides insights into fleas phylogeny. Front. Vet. Sci. 10:1255017. doi: 10.3389/fvets.2023.1255017
Edited by:
Vikrant Sudan, Guru Angad Dev Veterinary and Animal Sciences University, IndiaReviewed by:
Qiaocheng Chang, Shantou University, ChinaCopyright © 2023 Chen, Liu, Lu, Jiang, Wang, Zhang, Yang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quan-fu Zhang, MjYyMjA0NTgwN0BxcS5jb20=; Guo-ping Yang, eWFuZ2d1b3BpbmdAZGFsaS5lZHUuY24=; Xing Yang, eWFuZzA4MjIwMDEzQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.