 Yafang Liu1
Yafang Liu1 Bin Chen1Xinyan Lu1Shuang Liu1Dandan Jiang2Xuan Wang3Lin Yi4Rongyu Li4Quanfu Zhang5*Lixian Wu1*
Bin Chen1Xinyan Lu1Shuang Liu1Dandan Jiang2Xuan Wang3Lin Yi4Rongyu Li4Quanfu Zhang5*Lixian Wu1* Xing Yang1*
Xing Yang1*- 1Integrated Laboratory of Pathogenic Biology, College of Preclinical Medicine, Dali University, Dali, China
- 2School of Public Health, Dali University, Dali, China
- 3Queen Mary School, Nanchang University, Nanchang, China
- 4College of Preclinical Medicine, Dali University, Dali, China
- 5Department of Gastroenterology, Clinical Medical College and the First Affiliated Hospital of Chengdu Medical College, Sichuan, China
Fleas represent a group of paramount medical significance, subsisting on blood and acting as vectors for an array of naturally occurring diseases. These pathogens constitute essential elements within the plague biome, exerting deleterious effects on both human and livestock health. In this study, we successfully assembled and sequenced the whole mitochondrial genome of Frontopsylla spadix and Neopsylla specialis using long-range PCR and next-generation sequencing technologies. The mitogenomes of F. spadix and N. specialis both have 37 genes with full lengths of 15,085 bp and 16,820 bp, respectively. The topology of the phylogenetic tree elucidates that species F. spadix is clustered in a branch alongside other members of the family Leptopsyllidae, whereas species N. specialis is a sister taxon to Dorcadia ioffi and Hystrichopsylla weida qinlingensis. It also suggests that Pulicidae form a monophyletic clade, Ctenopthalmidae, Hystrichopsyllidae, Vermipsyllidae form a sister group to Ceratophyllidae/Leptopsyllidae group. The mitochondrial genomes of F. spadix and N. specialis were sequenced for the first time, which will contribute to a more comprehensive phylogenetic analysis of the Siphonaptera order. The foundation for subsequent systematic studies, and molecular biology of fleas was established.
Introduction
Fleas (Order Siphonaptera) are small, wingless insects with laterally compressed bodies undergo holometaboliam and parasitize mammals and birds (1, 2). Over, 2500 species of fleas have been identified, with approximately 200 species capable of harboring epidemic bacteria (3, 4). These serve as vectors for a range of pathogens, including Rickettsia, Bartonella, bubonic plague, and Tularemia, thereby functioning as both disease-causing vectors and reservoir hosts (5, 6). As a result of changes in the environment and human behavior, the vector-host ecology has changed, increasing human exposure to flea vectors and the pathogens they transmit, and flea-borne diseases may re-emerge as epidemics (7). Fleas are an early warning indicator of the plague epidemic, which has important significance in medicine and veterinary medicine (8). Fleas and flea-borne diseases are increasingly threatening human and animal health and causing serious economic losses, so flea identification is of great practical importance for flea-borne disease prevention and control.
Both Frontopsylla spadix and Neopsylla specialis are found in the Siphonaptera order, belonging to the families Leptopsyllidae and Ctenophthalmidae, to the Amphipsyllinae and Neopsyllinae, and Frontopsylla and Neopsylla, respectively. Species F. spadix are parasitic on wild rodents, specifically Apodemus chevrieri and Rattus flavipectus, found in regions such as Yunnan, Gansu, and Tibet in China, and extending into Nepal. This geographic distribution is considered a conduit for the transference of plague from wild to domestic rodents (9). Species N. specialis, parasitizing mammals like Apodemus chevrieri, Apodemus draco, and Apodemus latronum, is in China and acts as a principal vector of the plague in Yunnan's natural foci, akin to species F. spadix (10). Morphological characteristics of F. spadix and N. specialis have been described previously, the identification resolution of traditional flea classification methods is low, which may have certain limitations (2).
Mitochondria are placed in cells that produce energy and have a separate set of genetic material called mitochondrial DNA (11). Mitochondrial DNA is one of the most commonly used molecular markers in systematics and is widely used in phylogenetic studies of different organisms because of its simple structure, maternal inheritance, and rapid evolutionary rate (12). The analysis of mitochondrial genome structure and sequence is helpful to clarify the classification, genetic evolution, and phylogenetic relationship of fleas more clearly (13). However, at present, the mitochondrial genome data of fleas is very limited, resulting in a huge obstacle to fleas and flea-borne diseases. Therefore, we need to continuously increase and improve the flea mitochondrial gene database to lay the foundation for flea taxonomy, population genetics, and phylogeny.
In this study, we provide the first complete description of the mitochondrial genomes of F. spadix and N. specialis, analyze mitogenome structures to address the lack of mitochondrial gene resources in fleas, and construct the phylogenetic relationships of known mitochondrial genomes in the order Siphonaptera, while providing molecular information for flea prevention and control.
Materials and methods
Sample collection and DNA extraction
Adult specimens of F. spadix (one female and one male) were collected in July 2020 from Luoping Mountain, eryuan City, Dali Bai Autonomous Prefecture, Yunnan Province, China (26°07′N, 99°85′E). Three females and one male adult specimens of N. specialis were found in June 2022 from Laojun Mountain, Lijiang City, Yunnan Province of China (26°53′N, 99°58′E). Species identification was conducted based on morphological characteristics with F. spadix and N. specialis samples extracted from an adult female using the TIANamp Genomic DNA Kit (TIANGEN, Beijing, China) following the manufacturer's instructions.
PCR amplification
The study design included the development of two sets of overlapping long fragment PCR primers to amplify the mitochondrial genomes of F. spadix and N. specialis. This was conducted using cox1 and 12S rRNA genes of Ctenophthalmus quadratus (OQ023577) and Leptopsylla segnis (OQ023576), with primer design achieved through Primer 5.0 software, as delineated in Table 1. The PCR was performed in the 50 μl system, including 10 μl 5× PrimerSTAR GXL Buffer (Takara, Japan), 4 μl of each primer, 4 μl of dNTPs, 1 μl of PrimerSTAR GXL DNA Polymerase (Takara, Japan), 4 μl of DNA template and 23 μl of ddH2O under the following reaction conditions: 92°C for 2 min for initial denaturation, followed by 35 cycles of denaturation at 92°C for 10 s, annealing at 68°C for 30 s and extension at 68°C for 10 min and the final 68°C extension time of 10 min. PCR amplification products were detected by electrophoresis on 1% agarose gels, purified, and sequenced by Sangon Biotech Company (Shanghai, China).

Table 1. PCR primers for sequencing the mitogenome of F. spadix and N. specialis.
Gene annotation
Sequencing employed next-generation sequencing technology (NGS) on the Illumina NovaSeq platform. Annotations were facilitated via the MITOS WebServer (http://mitos.bioinf.uni-leipzig.de/index.py), with the A5-miseq v20150522 program utilized for the construction of the complete mitochondrial genome (14, 15). Alignment with closely related species in the NCBI database were performed to ascertain the location of protein-coding, tRNA, and rRNA genes. Predictive analyses of tRNA genes secondary structure were conducted on the tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/) online platform (16), and the CGView Server (https://paulstothard.github.io/cgview/) was employed for mapping mitochondrial genome circles. Analytical tools included DNAStar V7.1 for nucleotide composition analysis and CodonW 1.4.2 the relative synonymous codon usage (RSCU) computation.
Phylogenetic analysis
The 13 protein-coding gene sequences from 15 flea species were independently aligned using MUSCLE nucleotide mode, and datasets were manually concatenated. Positions containing gaps and incomplete data were excluded through Bioedit v7.0.5.3 software. Phylogenetic relationships were analyzed using Casmara patrona as an outgroup (Table 2), and trees were constructed with MEGA 7.0 software and Mrbayes v.3.2.7 software. The ML tree was formulated using GTR+G+I as the optimal model based on the Akaike Information Criterion (AIC) (17), employing the maximum likelihood method across 1,000 bootstrap datasets. The BI tree underwent 10,000,000 generations, sampled every 1,000 generations. The evolutionary relationships among flea species were visually depicted using the software Figtree v1.4.2.
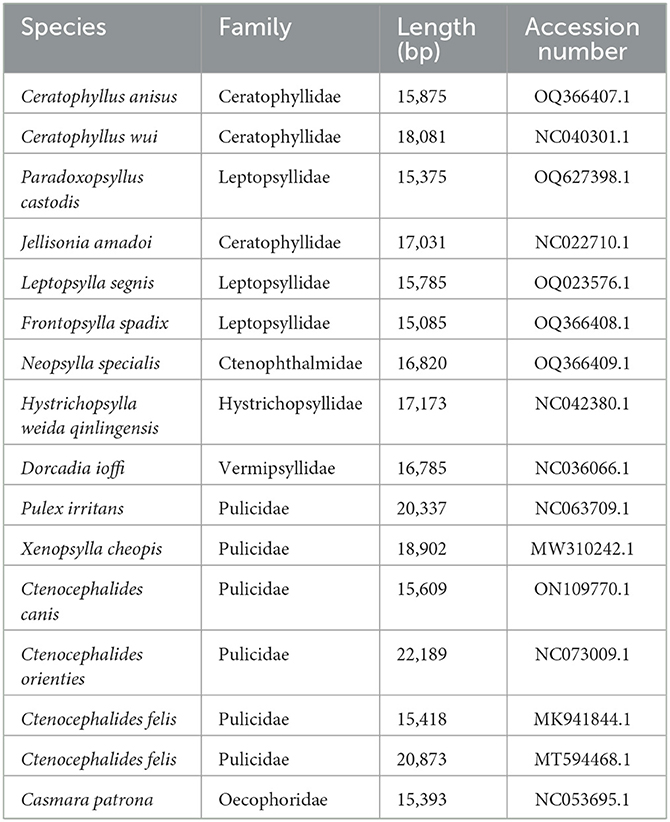
Table 2. Mitochondrial genome sequence information used in this paper.
Results
Organization of mitochondrial genome
The mitochondrial genomes of F. spadix and N. specialis, which are typically closed double-stranded molecular structures, were uploaded to Genbank in TBL format and obtained accession numbers OQ366408 and OQ366409, respectively. The length of the mitochondrial genome was 15,085 bp and 16,820 bp, respectively (Figure 1), with differences in length mainly determined by the length of the control region. Most of the genes including 14 tRNAs and 9 PCGs are distributed on the positive strand, the same as the other fleas (18, 19). Both intergenic regions and overlapping domains are present within the mitochondrial genome (Table 3). The mitogenomes of F. spadix and N. specialis had a significant AT preference with AT content of 78.83% and 77.27%, respectively, and the base content was 37.99% (38.64%) A, 40.84% (38.63%) T, 12.85 (14.22%) C, and 8.31 (8.51%) G (Table 4).
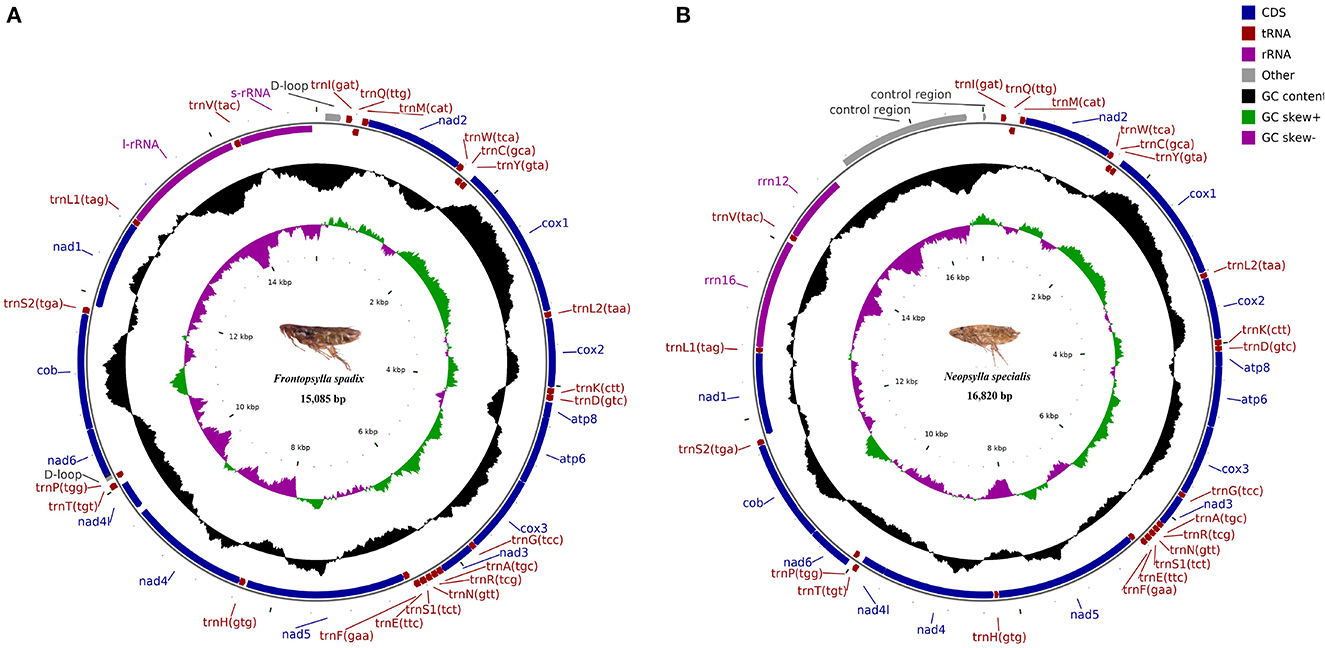
Figure 1. Map of the mitochondrial genome of Frontopsylla spadix (A) and Neopsylla specialis (B). Genes in the inner circle plot are located on the reverse strand, and the rest of the genes are located on the forward strand.
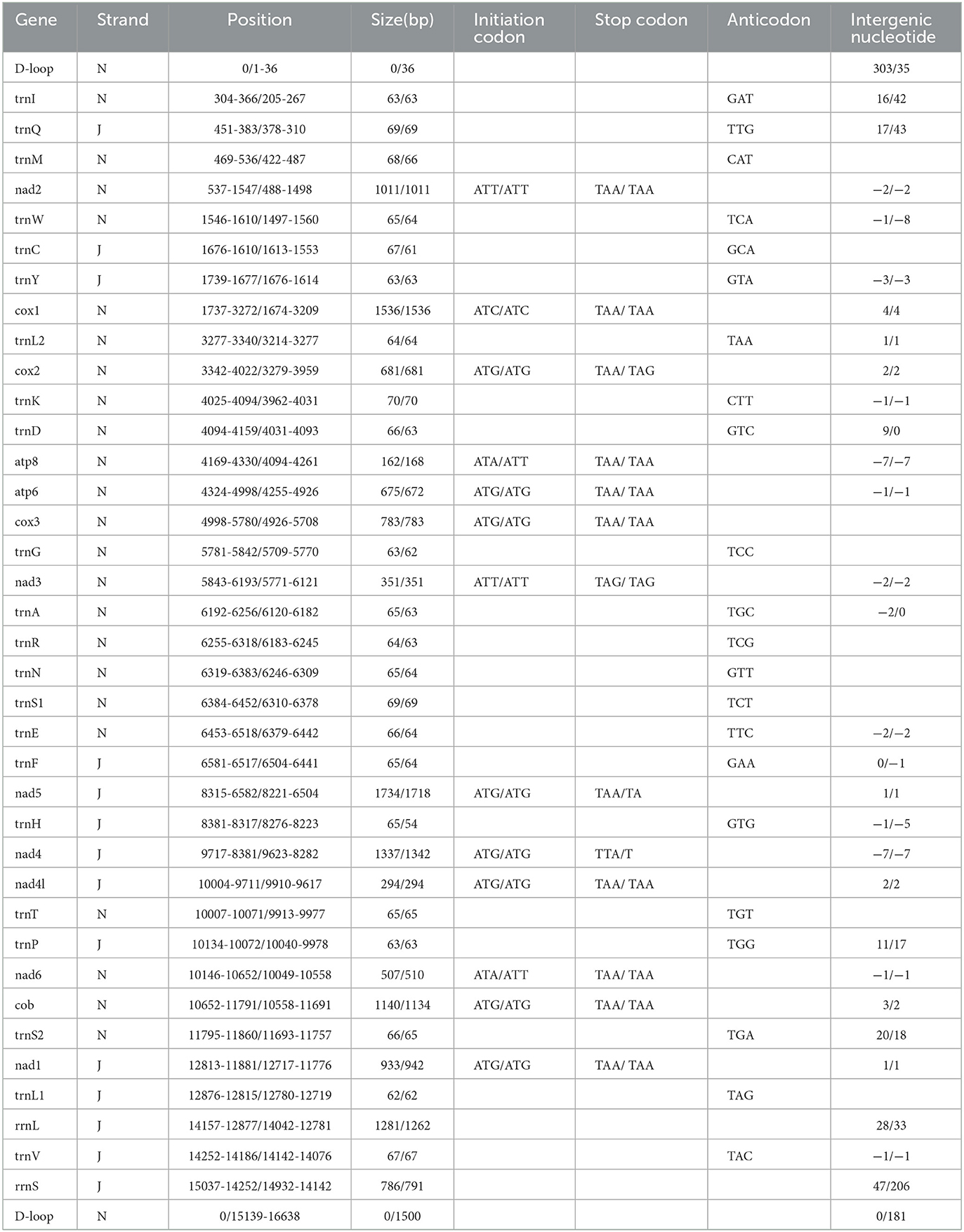
Table 3. Summary of the mitogenome of F. spadix and N. specialis.
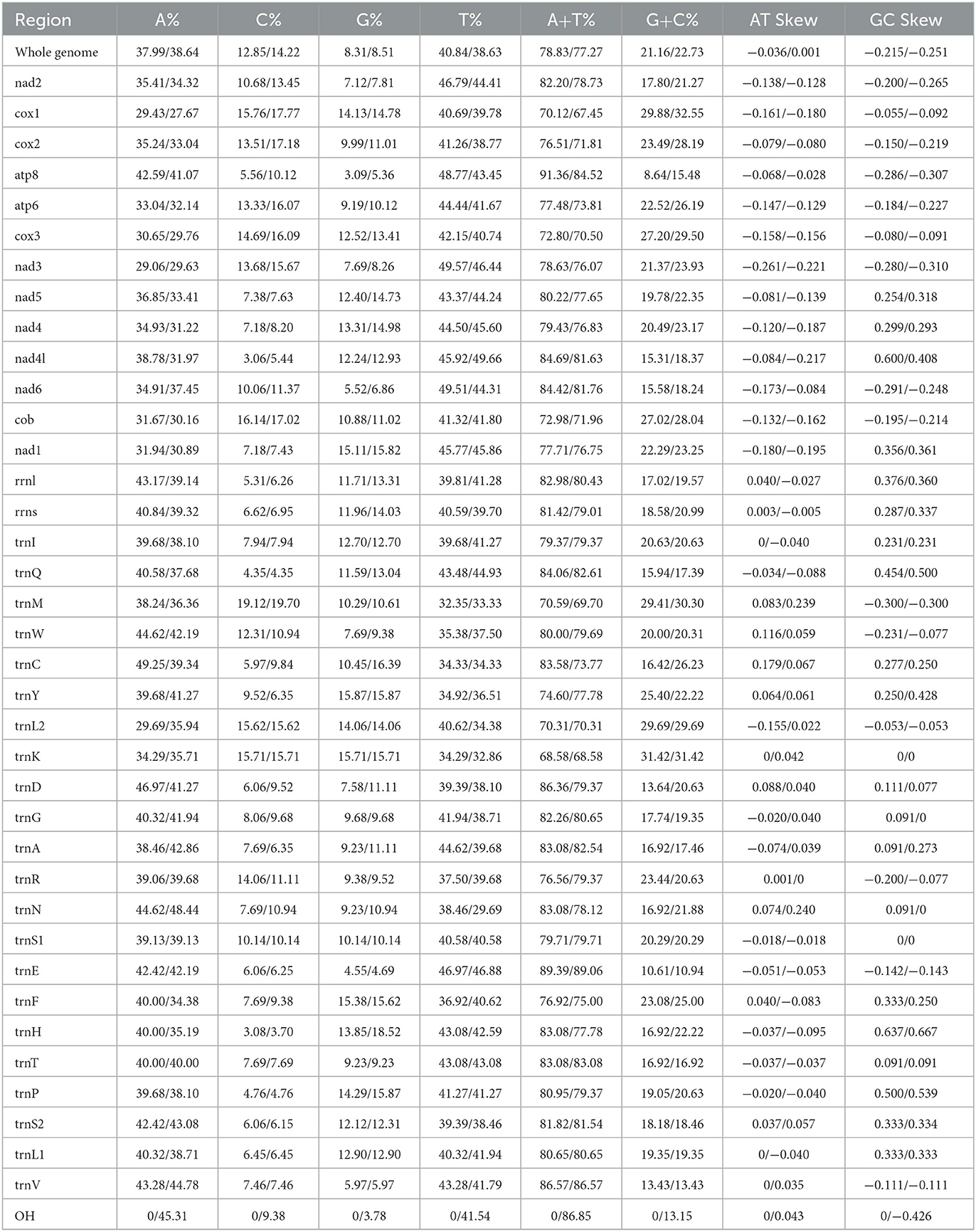
Table 4. Composition and skewness of F. spadix and N. specialis mitogenome.
Protein-coding genes
The PCGs of F. spadix and N. specialis were 11,144 bp and 11,142 bp long, accounting for 73.87% and 66.24% of the complete mitochondrial genome length, respectively. Of the 13 protein-coding genes (PCGs) of F. spadix, which encode a total of 3713 codons, the initiation codon is the standard codon ATN, with TAA as the termination codon except for NAD3 (TAG). N. specialis encodes a total of 3714 codons, with incomplete termination codons occurring in NAD5 and NAD4, and NAD3 with TAG as a stop codon. Leucine is the dominant amino acid and cysteine is the rarest amino acid (Figure 2). The mitochondrial genomes of F. spadix and N. specialis are mostly nonpolar amino acid groups with 1851 (49.85%) and 1933 (52.05%), respectively, and the remaining polar, basic, and acidic amino acid groups are 1209 (32.51%) and 1206 (32.47%), 235 (6.33%) and 230 (6.19%), 194 (5.22%) and 157 (4.23%).
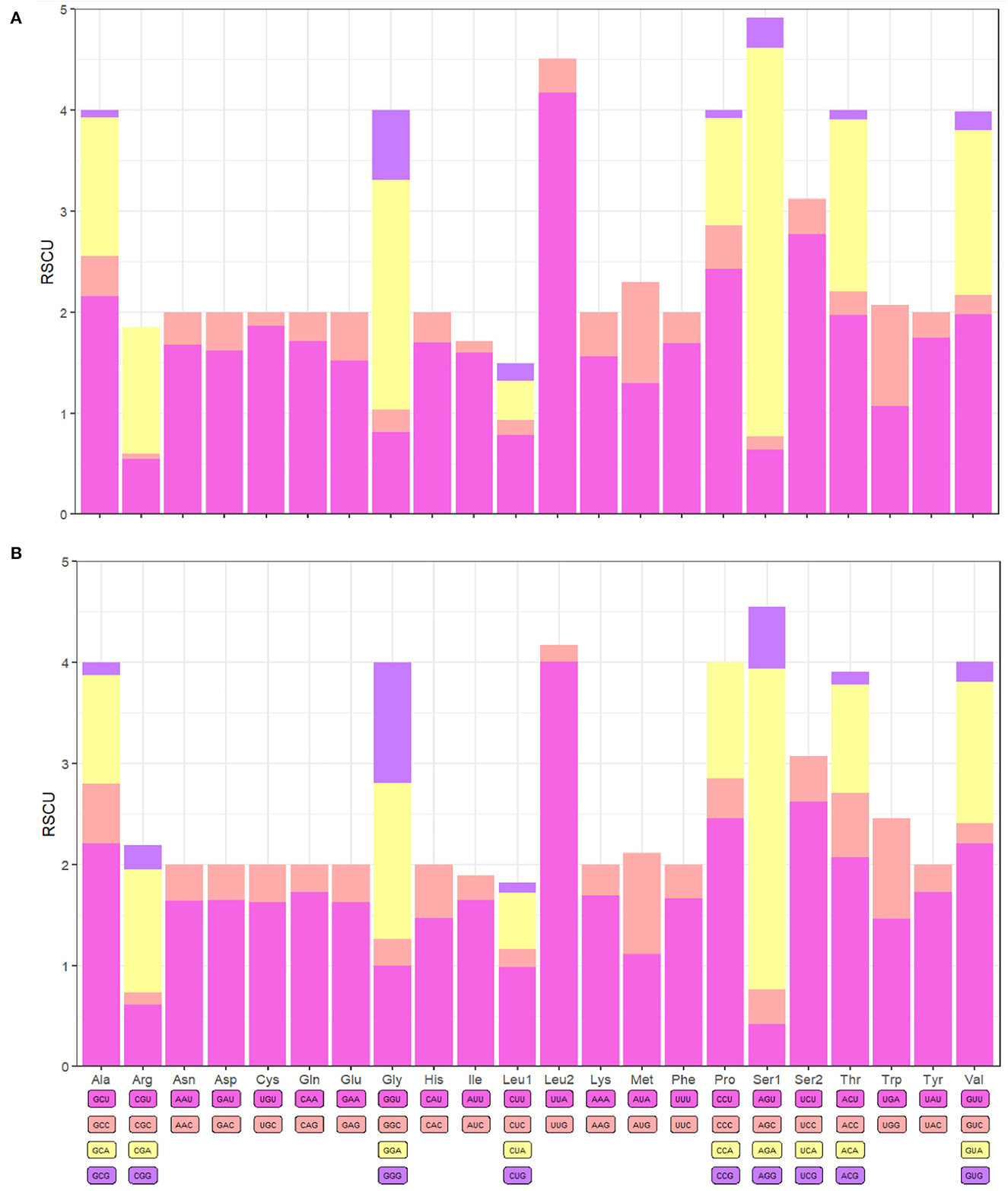
Figure 2. F. spadix (A) and N. specialis (B) protein-coding gene relative synonymous codon usage (RSCU).
Transfer RNA genes and ribosomal RNA genes
The mitogenomes of F. spadix and N. specialis have 14 tRNAs located in the positive strand with full lengths of 1439 bp and 1408 bp, respectively. Among the 22 tRNA genes of F. spadix, the length of tRNA genes ranged from 62 bp (trnL1) to 70 bp (trnK), with the shortest amino acid of 61 bp (trnC) in N. specialis. The mitochondrial genome was conjured according to the special genetic code so that all the 22 tRNA genes could be identified. G-U oscillating bases appear as a common mismatch in most tRNA genes to maintain tRNA secondary structure (20). The relationship between base mismatch and evolution needs further consideration. With a length of 7 bp, which is typical of arthropods, ATP8 and ATP6 overlap (21). The 16S rRNA and 12S rRNA of F. spadix and N. specialis are both located in the reverse strand, separated by Valine, with AT contents of 82.98% (80.43%) and 81.42% (79.01%), respectively (Table 4).
Phylogenetic analysis
We harnessed available flea genomic data from the NCBI database pertaining to fleas and amalgamated this with our successfully sequenced F. spadix and N. specialis, thus facilitating a refined exploration of the topology of flea phylogenetic relationships. Utilizing the maximum likelihood method, we constructed a phylogenetic tree anchored on the concatenated nucleotide sequences of 13 PCG genes, thereby providing an insightful perspective into flea evolutionary trajectories. The ML and BI trees show identical topologies. According to ML and BI analysis, the families Ctenopthalmidae, Hystrichopsyllidae, Vermipsyllidae, and Pulicidae form a monophyletic clade, while the family Ceratophyllidae and Leptopsyllidae are paraphyletic. A principal clade encompasses species of the family Pulicidae, crystallizing into a definitive monophyletic clade. In juxtaposition, Ctenopthalmidae, Hystrichopsyllidae, and Vermipsyllidae form a sister aggregation to the Ceratophyllidae and Leptopsyllidae group. Notably, F. spadix and Leptopsylla segnis belonging to the Leptopsyllidae family emerge as the most phylogenetically congruent entities, bolstered by robust node support values. N. specialis resides solitarily on a branch, constituting a strongly endorsed linkage with Dorcadia ioffi and Hystrichopsylla weida qinlingensis and fostering sister group affiliations (Figure 3).
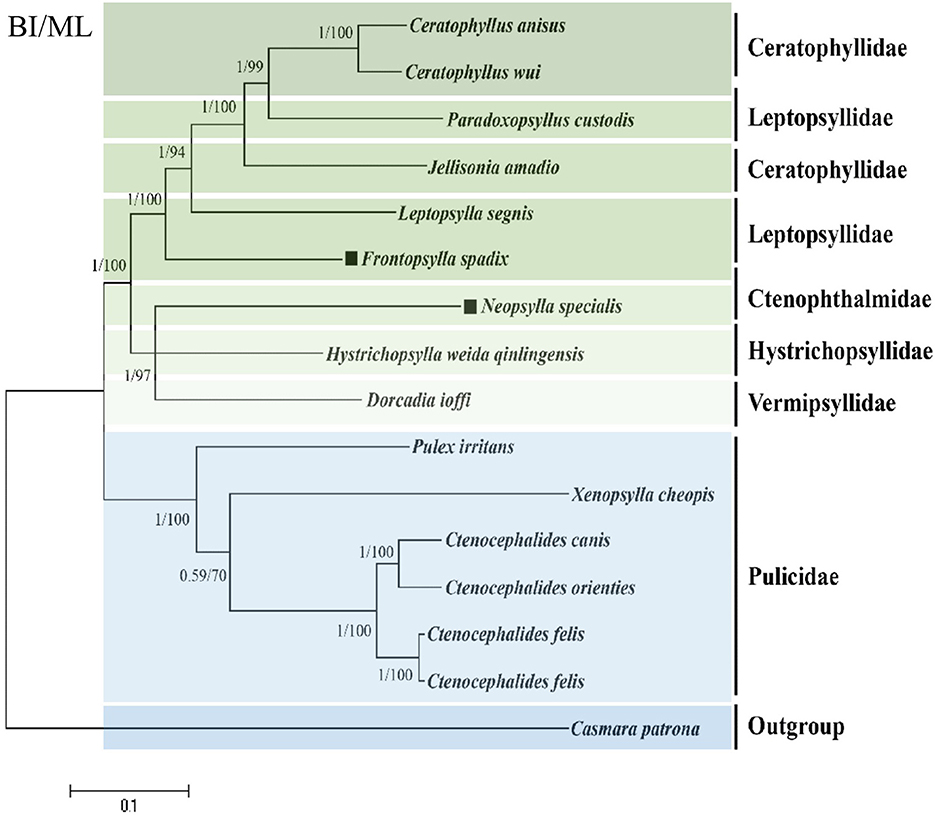
Figure 3. Phylogenetic analysis based on the nucleotide sequences of the 13 PCGs in the mitogenome. The number beside the nodes are posterior probabilities (BI) and bootstrap (ML). The black square markers represent the species in this study.
Discussion
The endeavor to taxonomically identify and comprehend the ecological proclivities of fleas stands as an integral foundation in the mitigation and management of a plethora of naturally transpiring diseases. Fleas, being a medically salient insect group, partake substantially in perpetuating major plague epidemics and preserving natural epidemiological origin (22). Their abundance and widespread dissemination render them instrumental in the animal-mediated propagation of diseases during epidemic occurrences. Species F. spadix and N. specialis, as principal flea vectors, are posited to serve as conduits for the transmission of wild rodent plagues into domestic rats, thereby instigating epidemics.
In an examination of mitogenomes of F. spadix and N. specialis a pronounced predilection for AT bases was discerned, with an AT content surpassing that of GC, a feature congruent with arthropods (23). An anomalous stop codon was detected in N. specialis, subsequently rectified to TAA by PolyA complementation to terminate translation (24). The control region, harboring initiation sites that govern the replication and transcription of the mitochondrial genome, evolves at a rate three to five times that of other regions, accounting for its employment in population genetics and origin evolution studies (25, 26). Variability in the number and location of non-coding regions is evidenced across species, with singular, dual, and triple D-Loops manifesting in different species. Within this study's purview, species F. spadix was devoid of D-Loop, while species N. specialis harbored two, spanning 1,536 base pairs.
The six families are split into two large clades, as shown by phylogenetic clustering, except the family Pulicidae, where the remaining five families cluster in the other clade, with Paradoxopsyllus custodis located in the Superfamily Ceratophylloidae as a member of the family Leptopsyllidae, which is a discovery that also indicates that expanded sequencing of mitochondrial genomic data is beneficial for more intensive phylogenetic studies of the species. However, individual mitochondrial genes are less informative than the whole mitogenome which may bias the reflection of phylogenetic relationships (27), and in order to make the phylogenetic relationships of the flea more convincing, we need to sequence the whole mitochondrial genome of the flea more frequently (11).
Accurate differentiation and identification of flea species are essential in the diagnosis of disease and basic and applied research on these important ectoparasites. The mitochondrial genome is frequently used in phylogenetic and phylogenetic studies of different ectoparasites at various taxonomic levels due to its matrilineal inheritance, lack of recombination, and rapid rate of evolution. The in-depth analysis of the mitochondrial genomes of F. spadix and N. specialis augments the data corpus, fortifying further phylogenetic inquiry within the Siphonaptera order. This enhances both the resolution at the family echelon and the informativeness of the phylogenetic tree. The entire mitochondrial genome sequence has also been demonstrated to proffer elevated phylogenetic precision, rendering it an apt molecular marker for elucidating the evolutionary interconnections amongst flea species. Nevertheless, the sequencing of additional flea mitochondrial genomes is requisite to facilitate a more systematized and encompassing analysis of flea evolutionary relationships.
Conclusion
In this study, the mitochondrial genomes of F. spadix and N. specialis were successfully sequenced based on the combination of long-range PCR technology and next-generation sequencing technology. In both F. spadix and N. specialis, the mitochondrial genomes are circular with the same genetic composition and arrangement as other fleas, which provides the basis for further understanding of the molecular evolution, and phylogeny of fleas, as well as providing useful molecular markers for studying the taxonomy and systematics of the flea species.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The animal study was approved the Laboratory Animal Management Committee of Dali University and First Affiliated Hospital of Chengdu Medical College. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
YL conceived the study and wrote the manuscript. BC, XL, SL, and DJ collected specimens and participated in experimental operations. XW, LY, and RL analyzed the experimental data. QZ, LW, and XY are responsible for the interpretation of experimental data, critical revision of important knowledge content, and final approval of the version to be published. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Yunnan Natural Science Foundation (2017FD139) and Scientific Research Fund of Yunnan Education Department (2022J0687).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang Y, Nie Y, Li LY, Chen SY, Liu GH, Liu W. Population genetics and genetic variation of Ctenocephalides felis and Pulex irritans in China by analysis of nuclear and mitochondrial genes. Parasit Vectors. (2022) 215:266. doi: 10.1186/s13071-022-05393-6
2. Marrugal A, Callejón R, de Rojas M, Halajian A, Cutillas C. Morphological, biometrical, and molecular characterization of Ctenocephalides felis and Ctenocephalides canis isolated from dogs from different geographical regions. Parasitol Res. (2013) 112:2289–98. doi: 10.1007/s00436-013-3391-6
3. Hornok S, Beck R, Farkas R, Grima A, Otranto D, Kontschán J, et al. High mitochondrial sequence divergence in synanthropic flea species (Insecta: Siphonaptera) from Europe and the Mediterranean. Parasit Vectors. (2018) 211:221. doi: 10.1186/s13071-018-2798-4
4. Lewis RE. Résumé of the Siphonaptera (Insecta) of the world. J Med Entomol. (1998) 35:377–89. doi: 10.1093/jmedent/35.4.377
5. Zeppelini CG, de Almeida AM, Cordeiro-Estrela P. Zoonoses as ecological entities: a case review of plague. PLoS Negl Trop Dis. (2016) 10:e0004949. doi: 10.1371/journal.pntd.0004949
6. Bitam I, Dittmar K, Parola P, Whiting MF, Raoult D. Fleas and flea-borne diseases. Int J Infect Dis. (2010) 14:e667–76. doi: 10.1016/j.ijid.2009.11.011
7. Friggens MM, Beier P. Anthropogenic disturbance and the risk of flea-borne disease transmission. Oecologia. (2010) 164:809–20. doi: 10.1007/s00442-010-1747-5
8. Andrianaivoarimanana V, Kreppel K, Elissa N, Duplantier JM, Carniel E, Rajerison M, et al. Understanding the persistence of plague foci in Madagascar. PLoS Negl Trop Dis. (2013) 7:e2382. doi: 10.1371/journal.pntd.0002382
9. Cai W, Luo J, Su L, Shao Z, Li S, Zhang S. Rodent nest fleas in plague foci in Jianchuan County, Yunnan, 2008-2017. China Tropical Medicine. (2018) 18:1236–8. doi: 10.13604/j.cnki.46-1064/r.2018.12.16
10. Liu Z, Guo XG, Yang ZH. Status of research on the flea Neopsylla specialis. Pathogen Biol. (2018) 13:1416–9. doi: 10.13350/j.cjpb.181228
11. Lu XY, Zhang QF, Jiang DD, Du CH, Xu R, Guo XG, et al. Characterization of the complete mitochondrial genome of Ixodes granulatus (Ixodidae) and its phylogenetic implications. Parasitol Res. (2022) 121:2347–58. doi: 10.1007/s00436-022-07561-0
12. Lu XY, Zhang QF, Jiang DD, Liu YF, Chen B, Yang SP, et al. Complete mitogenomes and phylogenetic relationships of Haemaphysalis nepalensis and Haemaphysalis yeni. Front Vet Sci. (2022) 9:1007631. doi: 10.3389/fvets.2022.1007631
13. Zhang Y, Fu YT, Yao C, Deng YP, Nie Y, Liu GH. Mitochondrial phylogenomics provides insights into the taxonomy and phylogeny of fleas. Parasit Vectors. (2022) 15:223. doi: 10.1186/s13071-022-05334-3
14. Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. (2013) 69:313–9. doi: 10.1016/j.ympev.2012.08.023
15. Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. (2015) 31:587–9. doi: 10.1093/bioinformatics/btu661
16. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. (1997) 25:955–64. doi: 10.1093/nar/25.5.955
17. Yamaoka K, Nakagawa T, Uno T. Application of Akaike's information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet Biopharm. (1978) 6:165–75. doi: 10.1007/BF01117450
18. Verhoeve VI, Plumer ML, Driscoll TP, Macaluso KR, Azad AF, Gillespie JJ. The complete mitochondrial genome of the cat flea, Ctenocephalides felis. Mitochondrial DNA B Resour. (2020) 5:3422–4. doi: 10.1080/23802359.2020.1823259
19. Zhang Y, Nie Y, Deng YP, Liu GH, Fu YT. The complete mitochondrial genome sequences of the cat flea Ctenocephalides felis felis (Siphonaptera: Pulicidae) support the hypothesis that C. felis isolates from China and USA were the same C f felis subspecies. Acta Trop. (2021) 217:105880. doi: 10.1016/j.actatropica.2021.105880
20. Watanabe Y, Kawai G, Yokogawa T, Hayashi N, Kumazawa Y, Ueda T, et al. Higher-order structure of bovine mitochondrial tRNA(SerUGA): chemical modification and computer modeling. Nucleic Acids Res. (1994) 22:5378–84. doi: 10.1093/nar/22.24.5378
21. Sun ET Li CP, Nie LW, Jiang YX. The complete mitochondrial genome of the brown leg mite, Aleuroglyphus ovatus (Acari: Sarcoptiformes): evaluation of largest non-coding region and unique tRNAs. Exp Appl Acarol. (2014) 64:141–57. doi: 10.1007/s10493-014-9816-9
22. Liu YF, Chen B, Lu XY, Jiang DD, Wang T, Geng L, et al. Complete mitogenomes characterization and phylogenetic analyses of Ceratophyllus anisus and Leptopsylla segnis. Front Vet Sci. (2023) 10:1218488. doi: 10.3389/fvets.2023.1218488
23. Yuan ML, Wei DD, Zhang K, Gao YZ, Liu YH, Wang BJ, et al. Genetic diversity and population structure of Panonychus citri (Acari: Tetranychidae), in China based on mitochondrial COI gene sequences. J Econ Entomol. (2010) 103:2204–13. doi: 10.1603/EC09392
24. Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. (1981) 290:470–4. doi: 10.1038/290470a0
25. Taanman TW. The mitochondrial genome: structure, transcription, translation and replication. BBA Bioenergetics. (1999) 1410:103–23. doi: 10.1016/S0005-2728(98)00161-3
26. Muchadeyi FC, Eding H, Simianer H, Wollny CB, Groeneveld E, Weigend S. Mitochondrial DNA D-loop sequences suggest a Southeast Asian and Indian origin of Zimbabwean village chickens. Anim Genet. (2008) 39:615–22. doi: 10.1111/j.1365-2052.2008.01785.x
Keywords: Frontopsylla spadix, Neopsylla specialis, flea, mitochondrial genome, phylogenetic
Citation: Liu Y, Chen B, Lu X, Liu S, Jiang D, Wang X, Yi L, Li R, Zhang Q, Wu L and Yang X (2023) Analysis of complete mitogenomes and phylogenetic relationships of Frontopsylla spadix and Neopsylla specialis. Front. Vet. Sci. 10:1250381. doi: 10.3389/fvets.2023.1250381
Received: 30 June 2023; Accepted: 28 August 2023;
Published: 07 September 2023.
Edited by:
Alireza Sazmand, Bu-Ali Sina University, IranReviewed by:
Nabil Amor, Tunis El Manar University, TunisiaMichael Hastriter, Brigham Young University, United States
Copyright © 2023 Liu, Chen, Lu, Liu, Jiang, Wang, Yi, Li, Zhang, Wu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quanfu Zhang, MjYyMjA0NTgwN0BxcS5jb20=; Lixian Wu, d19saXhpYW5AMTYzLmNvbQ==; Xing Yang, eWFuZzA4MjIwMDEzQDE2My5jb20=