 Andrew J. McNair
Andrew J. McNair Greg R. Markby
Greg R. Markby Qiyu Tang
Qiyu Tang Vicky E. MacRae
Vicky E. MacRae Brendan M. Corcoran
Brendan M. Corcoran
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci., 16 October 2023
Sec. Comparative and Clinical Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fvets.2023.1202001
Background: TGFβ signaling appears to contribute to the pathogenesis of myxomatous mitral valve disease (MMVD) in both dogs and humans. However, little is known about the extent of the downstream signaling changes that will then affect cell phenotype and function in both species.
Objective: Identify changes in downstream signals in the TGFβ pathway in canine MMVD and examine the effects of antagonism of one significant signal (SMAD2 was selected).
Materials and methods: Canine cultures of normal quiescent valve interstitial cells (qVICs) and disease-derived activated myofibroblasts (aVICs) (n = 6) were examined for TGFβ signaling protein expression using a commercial antibody array. Significant changes were confirmed, and additional proteins of interest downstream in the TGFβ signaling pathway and markers of cell phenotype were examined (PRAS40, S6K, elF4E IRS-1, αSMA, and VIM), using protein immunoblotting. RT-PCR examined expression of gene markers of VIC activation (ACTA2, TAGLN, and MYH10; encoding the proteins αSMA, SM22, and Smemb, respectively). Attenuation of pSMAD2 in aVICs was examined using a combination of RNA interference technology (siRNA) and the SMAD7 (antagonizes SMAD2) agonist asiaticoside.
Results: The antibody array identified significant changes (P < 0.05) in 19 proteins, of which six were phosphorylated (p). There was increased expression of pSMAD2 and pRAC1 and decreased expression of pmTOR, pERK1/2, and pAKT1. Expression of pPRAS40 and pIRS-1 was increased, as was the mTOR downstream transcription factor pS6K, with increased expression of peIF4E in aVICs, indicating negative feedback control of the PI3K/AKT/mTOR pathway. SMAD2 antagonism by siRNA and the SMAD7 agonist asiaticoside decreased detection of pSMAD by at least 50%, significantly decreased expression of the aVIC gene markers ACTA2, TAGLN, and MYH10, and pαSMA, pAKT2, and pERK1, but had no effect on pS6K, pERK2, or pVIM expression in aVICs. SMAD2 antagonism transitioned diseased aVICs to normal qVICs, while maintaining a mesenchymal phenotype (VIM+) while concurrently affecting non-canonical TGFβ signaling.
Conclusion: MMVD is associated with changes in both the canonical and non-canonical TGFβ signaling pathway. Antagonism of SMAD2 transitions diseased-activated myofibroblasts back to a normal phenotype, providing data that will inform studies on developing novel therapeutics to treat MMVD in dogs and humans.
Myxomatous mitral valve disease (MMVD) is the single most important acquired cardiovascular disease in dogs and shares close similarities with analogous human conditions (1, 2). It is a major cause of morbidity and mortality in affected dogs and causes significant financial and emotional stress for owners. The disease is so prevalent that most elderly dogs show some evidence of the disease, but it is predominantly seen in small breed dogs, with a greater preponderance in certain predisposed breeds, the Cavalier King Charles Spaniel being the best example (2, 3). The development and progression of MMVD in terms of the pathological changes in the mitral valve, the hemodynamic consequences including cardiac remodeling, the clinical progression, and the therapeutic options when congestive heart failure develops are well-described. In addition, much is known about valve changes at the ultrastructural and cell level. Briefly, the disease involves the gradual development of myxomatous degeneration over several years with disorganization of collagen bundles, reduction in collagen content, and excess production of glycosaminoglycans (4–7). This results in distorted valve architecture and geometry with poor coaptation of leaflets, allowing mitral valve regurgitation and development of a characteristic murmur. The main cell changes include the transition of quiescent valve interstitial cells (qVICs) to an activated myofibroblast phenotype (aVICs), as evidenced by increased expression of αSMA, SM22, and Smemb, and valve endothelial cell damage and loss (8–11). The appearance of αSMA positive VICs is a cardinal feature of the disease, and this activated myofibroblast cell type is believed to control the aberrant extra cellular matrix (ECM) remodeling characteristic of the disease.
At the molecular level, transcriptomic profiling in both valve tissue and cultured aVICs has identified a range of gene changes, most notably in the TGFβ signaling pathway and ECM genes (12–15). In addition, there is up-regulation of the 5HT2B receptor gene, but since serotonin (5HT) itself has not been shown to transition qVICs, this likely reflects the activity of TGFβ (16). 5HT itself can induce VIC proliferation and ECM production through the activation of ERK1/2, a downstream component of the TGFβ signaling pathway (17, 18). Strong evidence suggests the TGFβ pathway is one of the most important in the pathogenesis of MMVD and in the control of the VIC phenotype. For example, TGFβ antagonism by SB431542 transitions canine aVICs back to a more normal qVIC phenotype (16). Examining further changes in the downstream parts of the TGFB pathway will give insight into the molecular mechanisms controlling disease and allow the identification of potential novel therapeutic targets to control disease development and progression.
Evidence for TGFβ involvement in MMVD can also be seen in the analogous human disease (Barlow's Disease) and various animal models (19–22). This involves both canonical (SMAD2/3) and non-canonical downstream signaling pathways, as demonstrated by several knockout mouse models of MMVD, including the Fbn-1 (fibrillin-1; Marfan syndrome), FLN-A (filamin-A X-linked), and Fstl1 (follistatin) mouse models (23–25). All these models show myxomatous degeneration, changes in TGFβ signaling and downstream signals, and expression of the aVIC marker αSMA. Most of these models have clarified the contribution of canonical SMAD2/3 signaling to MMVD. However, there is evidence that non-canonical signaling including the mitogen-activated protein kinases TAK1, JNK, and ERK1/2 and the PI3K kinase can also contribute to MMVD pathogenesis, and through molecular cross-talk affect SMAD2/3 signaling (24, 26). Furthermore, transcriptomic profiling has found changes in ERK1/2 gene expression in canine valves, mouse models, and human aVICs, and the effects on and interaction between canonical and non-canonical pathways can be clearly seen.
However, the interplay between these pathways is only partially understood and to what extent one alone might dominate to affect aVIC phenotype, and by extension ECM remodeling, is unknown. In this study, the aim was to examine expression of key molecules in the downstream TGFβ signaling pathway in canine aVICs using an antibody bio-array approach, measuring the expression of both total and phosphorylated proteins. This would be followed by confirming such changes using protein immunoblotting and then selecting one target of interest for antagonism, examining the effects on aVIC phenotype.
Mitral valves were collected with full informed owner consent from canine patients presented for euthanasia at the Hospital for Small Animals, R(D)SVS, the University of Edinburgh and with full ethical approval for valve tissue collection from the Veterinary Ethics in Research Committee of the R(D)SVS (VERC# 96/21). Valves were processed for the culture of valve interstitial cells (VICs) using our previously reported protocols, using a validated low serum (2% FBS) culture media method and not used beyond passage eight (16). Cells were grown on 75 cm3 plates and were harvested when they reached ~80% confluence.
VICs isolated from six normal (quiescent phenotype) and diseased (activated phenotype) dog mitral valves were grown in T75 flasks before being split into 100 mm2 cell culture dishes and grown to confluence. Despite confirmed disease status (canine valves are graded normal 0 to severe 4), we have previously identified heterogeneity in valve interstitial cell phenotype. To confirm the consistent phenotype for both groups we examined cells for the expression of ACTA2 (αSMA), TAGLN (SM22), and MYH10 (SMemb) (standard markers of VIC phenotype) from our total archive of 50 until the requisite six per group were identified. The details of the 12 dogs are shown in Table 1.
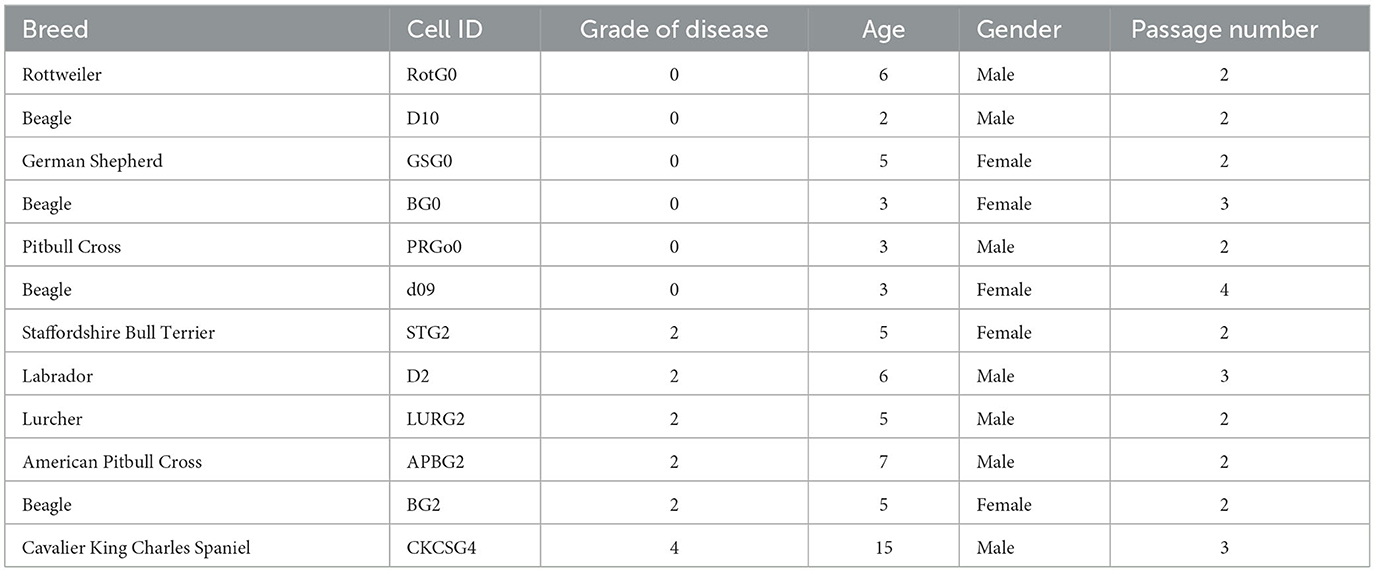
Table 1. Summary of cells used in array analysis.
To examine a large set of phosphorylated proteins in the TGFβ pathway, a MAP kinase signaling antibody array was used (Full Moon Biosystems, Sunnyvale, CA, USA). Full details can be found at this site https://www.fullmoonbio.com/product/tgfb-phospho-antibody-array/). The array includes 63 antibodies with six replicates per antibody; details of the antibodies are show in Table 2. All procedures with the array followed the manufacturer's instructions (Supplementary material 1) and example of the microarray slide shown in Supplementary Figure 1.
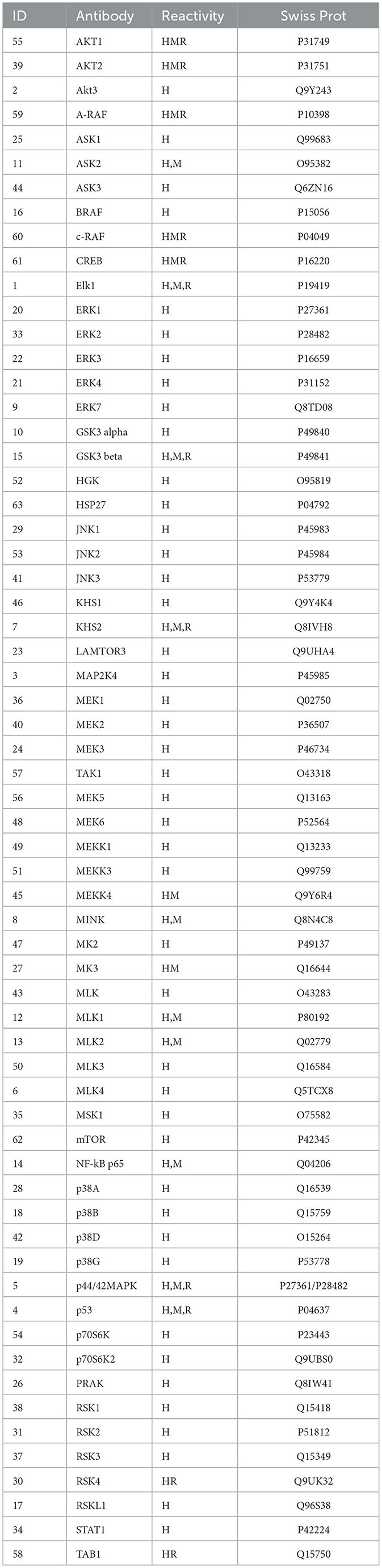
Table 2. Full Moon TGFβ MAP kinase signaling antibody array.
To confirm the array data findings, and to examine other proteins of interest, samples were examined for total and phosphorylated protein expression using protein immuno-blotting (Western Blotting, WB) using a standard protocol. Details of the methodology are shown in Supplementary material 2. All antibodies used for WB are shown in Supplementary Table 2. We used the total expression of each individual phospho experiment as the loading control, as per previous work. In the experiments where we had no totals, β-actin was used as a loading control. In terms of exposure times, we used an automated system (GeneGnome, Syngene, Cambridge, UK) that set the optimal exposure timepoints; it was not something that could be edited by us.
To extract RNA from VICs, lysis was performed using QIAzol lysis reagent and extracted with the miRNEASY extraction kit according to manufacturer's instructions. Initially, 700 μl QIAzol was added to the cells, and they were scraped as previously described. For RNA extraction, chloroform was added and tubes shaken vigorously for separation of RNA from DNA, proteins, and lipids. The RNA was then precipitated with 100% RNA-free ethanol. The sample was transferred onto a spin column to undergo spin column-based nucleic acid purification. Samples were analyzed by a NanoDrop, ND-1000 spectrophotometer (Thermo Scientific, UK) to determine RNA concentrations.
Extracted RNA samples were reverse transcribed using TaqMan® reverse transcription reagents (Applied Biosystems, CA, USA) as per manufacturers' instructions. Approximately 1 μg RNA was transcribed per sample in a cocktail of Reverse Transcriptase buffer, 25 mM magnesium chloride, deoxynucleotide triphosphates, random hexamers, RNase inhibitors, and Multiscribe. Reverse transcription was performed using the Veriti® Thermal Cycler (Life Technologies, Paisley, UK) under the following cycling conditions: 10 min at 25°C to maximize primer RNA template binding, 30 min at 48°C for reverse transcription, and 5 min at 95°C to deactivate reverse transcription.
Quantitative real-time polymerase chain reaction was used to assess mRNA expression. Taqman® PCR master mix and fluorescently tagged Taqman® primers (Supplementary Table 1) (Primer design, Southampton, UK) were used. Fluorescence was measured using a real-time PCR System (Life Technologies, Paisley, UK). The cycle conditions are 50°C for 2 min, 95°C for 10 min and 40 cycles of 95°C for 15 s, and 60°C for 1 min. Primer sequences used can be found in our previous paper [(16), p. 1841].
siRNA duplex sequences targeted to SMAD2 (TriFECTa®RNAi Kit Integrated DNA Technologies; Tyne & Wear, UK) were used to knockdown SMAD2 gene and protein expression. A scrambled siRNA was used as control. siRNA transfection was carried out using lipofectamine 3000 transfection reagent (Invitrogen, Paisley, UK). Lipofectamine 3000 was mixed with the siRNA and incubated for 15 min at room temperature, allowing complexes to form. VICs were then transfected with a final concentration of 5 nM siRNA. Transfection of VICs with siRNA targeting SMAD2 resulted in at least a 50% reduction in SMAD2 protein expression. The siRNA sequence was 5′ TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCCGCCGCGATCGCC 3′.
The SMAD7 agonist asiaticoside (AST; Cambridge Bioscience Limited, Cambridge, UK) was used to increase SMAD7 expression, leading to an inhibition of pSMAD2. AST was prepared in DMSO, and the vehicle was used as control. AST was added to the VICs at a concentration of 500 mg/l for an incubation period of 3 days (27). Incubation with AST reduced SMAD2 protein phosphorylation expression by at least 50%. The concentration and incubation period for AST and the siRNA were determined using both preliminary dose response and time course experiments as well as using published methodology using the same reagents or siRNA (28, 29).
Considering the dataset as a whole, significant (P < 0.05) changes in total protein in 21 antibodies targeted against 19 different proteins were found (Table 3). This included six antibodies targeted against phosphorylated proteins where a constituent decrease in phosphorylated protein was shown. Overall changes in various different downstream TGFβ pathway molecules were detected, including members of the PI3K/AKT/mTOR, JNK, ERK, and SMAD2 pathways.
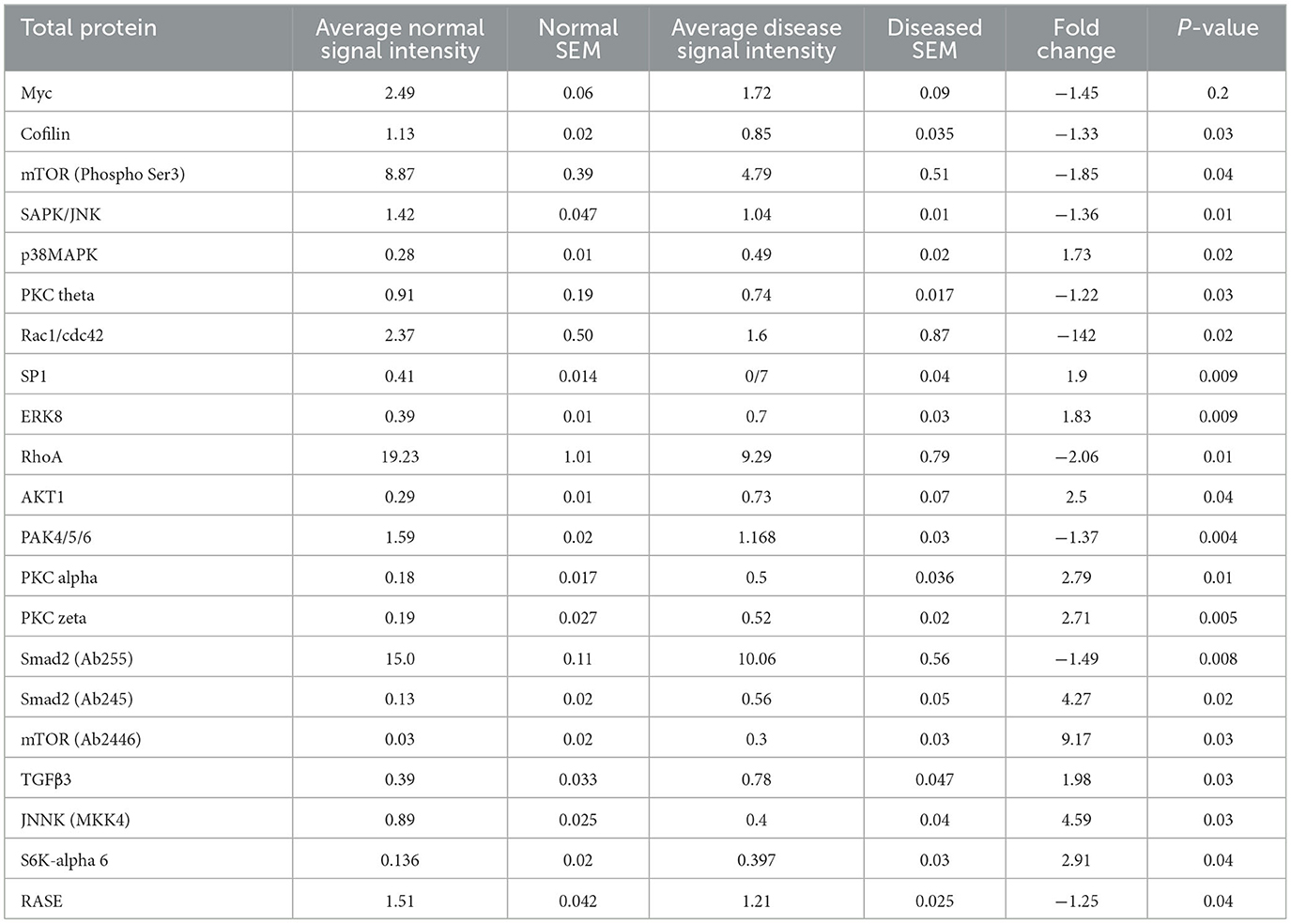
Table 3. Significantly (P < 0.05) altered total protein changes in the TGFβ pathway.
Significant changes were also detected in six phosphorylation states with two increased and four decreased in the diseased cells. The phosphorylation changes detected also match with many of the pathways detected to change at the total protein level in the diseased cells, indicating that these pathways may be of some importance in pathogenesis.
Since the P < 0.05 cut off value is somewhat arbitrary in an experiment where hundreds of comparisons are being performed simultaneously, and to reduce missing proteins of interest by chance, by expanding the inclusion criteria to P < 0.1 in the total protein dataset, changes in an additional 22 antibodies against 17 different proteins were identified (Supplementary Table 1). Examining these proteins more closely, there tended to be a larger variance in the diseased group than the normal (as exampled by the SEMs), with typically 1–3 of the samples in this group showing a large difference to other members of the group.
Conversely, when expanding the same inclusion criteria in the phosphorylated protein dataset, only two more proteins (SP1 and PI3-kinase p85-alpha both downregulated in disease) were identified (Supplementary Table 2). However, throughout the dataset there were several proteins that showed a large fold change (all increasing in disease) but did not reach significance set at P < 0.05. Again, this appears to be due to a larger variance in the diseased group. In general, there appeared to be a greater variation within groups in the phosphorylated dataset than in the total protein dataset.
To confirm the antibody array data for the significantly altered phosphorylated proteins targeted, protein immunoblotting was undertaken for pERK1/2, pRAC1, pSMAD2, pAKT2, and pmTOR. For a loading control, the total expression of each of the targeted proteins were used and then a phosphorylation/total expression ratio was calculated for analysis. Results for all five proteins matched the results observed in the array. Phosphorylation of SMAD2 and RAC1 was increased in canine aVICs and phosphorylation of ERK1/2, AKT2, and mTOR protein was more decreased in the aVICs compared to the qVICs (Figure 1).
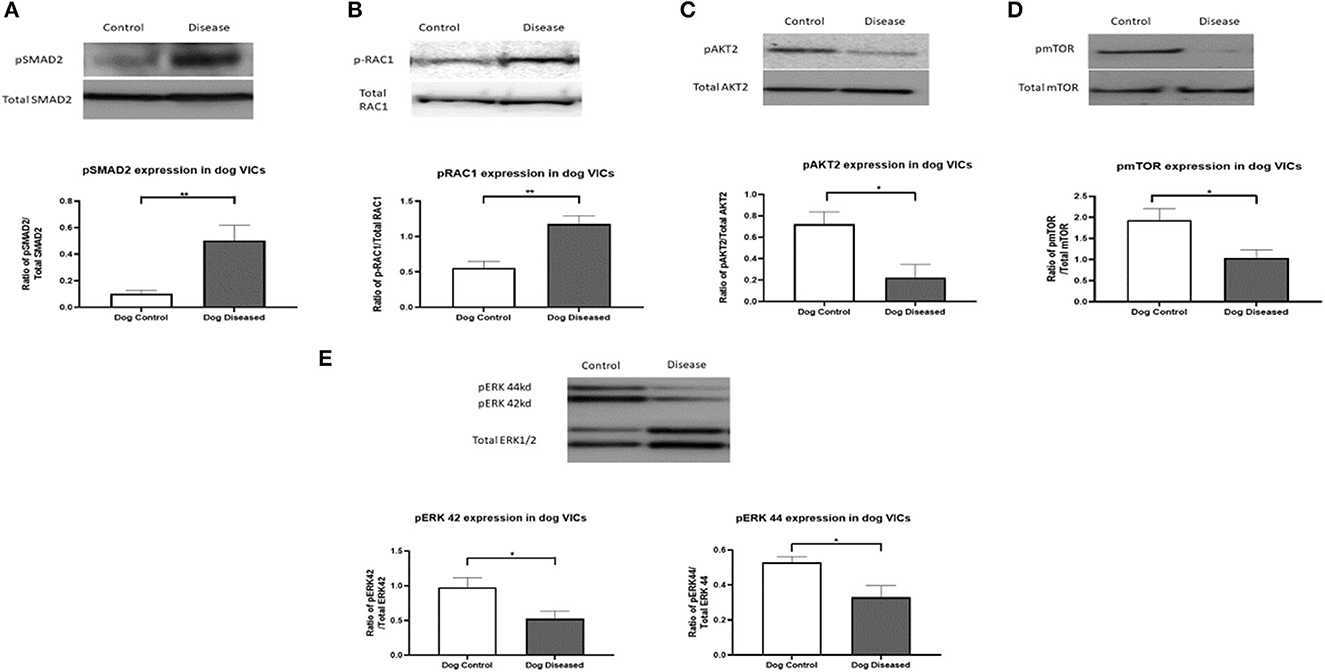
Figure 1. Protein expression of pRAC1 (28 kDa), pSMAD2 (52 kDa), pAKT2 (60 kDa), pmTOR (289 kDa), and pERK1/2 (42/44 kDa) in canine valve interstitial cells. Western blot analysis was used to measure protein expression in qVICs and aVICs. In aVICs pSMAD (A), pRAC1 (B) was increased, whilst pAKT (C), pmTOR (D), and pERK1/2 (E) were decreased compared to the qVICs. Data expressed as mean + SEM and analysis carried out by t-test. *P < 0.05 (n = 6), **p < 0.01.
For further analysis, and since a novel finding that the PI3K/Akt/mTOR pathway appeared to be affected in aVICs, we decided to analyze this pathway in more detail using WB examining the expression of the downstream transcription factors S6K and elF4E, and the negative feedback controllers of this pathway, IRS-1 and PRAS40. In aVICs detection of pS6K, pPRAS40, and pIRS-1 was significantly increased compared to qVICs while peIF4E was significantly decreased (Figure 2).
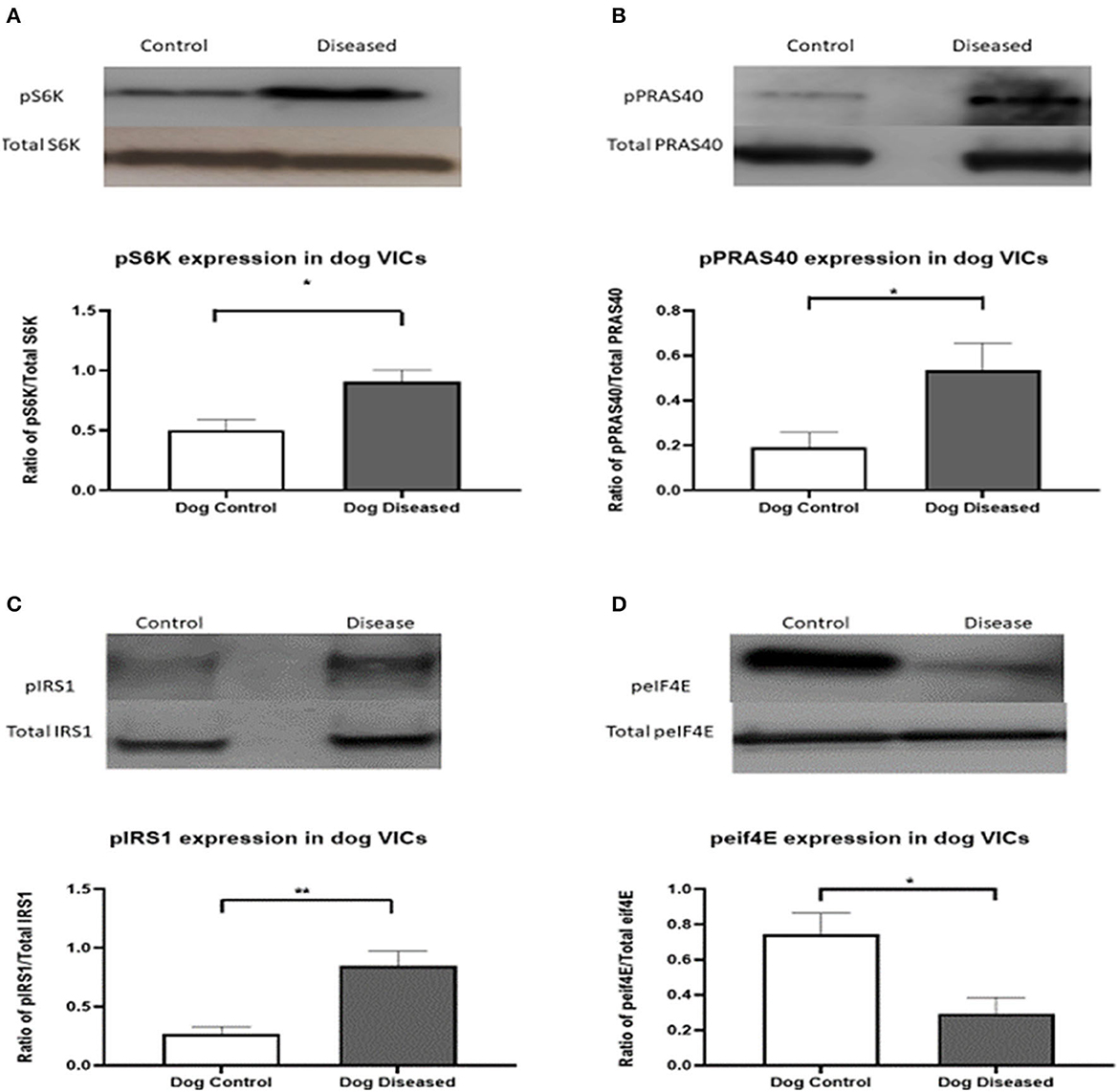
Figure 2. Protein expression of pS6K (70 kDa), pPRAS40 (40 kDa), pIRS-1, and peIF4E (25 kDA) in canine valve interstitial cells. Western blot analysis was used to measure protein expression in qVICs and aVICs. In aVICs pS6K (A), pPRAS40 (B), and pIRS-1 (C) was increased, whilst peIF4E (D) was decreased compared to the qVICs. Data expressed as mean + SEM and analysis carried out by t-test. *P < 0.05, **P < 0.01 (n = 6).
Since our secondary aim was to identify methods to modify disease cell phenotype and we had previously shown TGFβRII receptor antagonism will switch aVICs to a more normal qVIC phenotype, we decided to examine the effects of SMAD2 inhibition on cell phenotype, protein, and gene expression using a combination of SMAD2 siRNA and the SMAD7 agonist asiaticoside (16). In the cells treated with SMAD2 siRNA and AST, there was a significant reduction in the expression of ACTA2, TAGLN, and MHY10 compared to both the diseased control and the scramble siRNA control, indicating revision to a more normal quiescent phenotype (qVIC) (Figure 3).
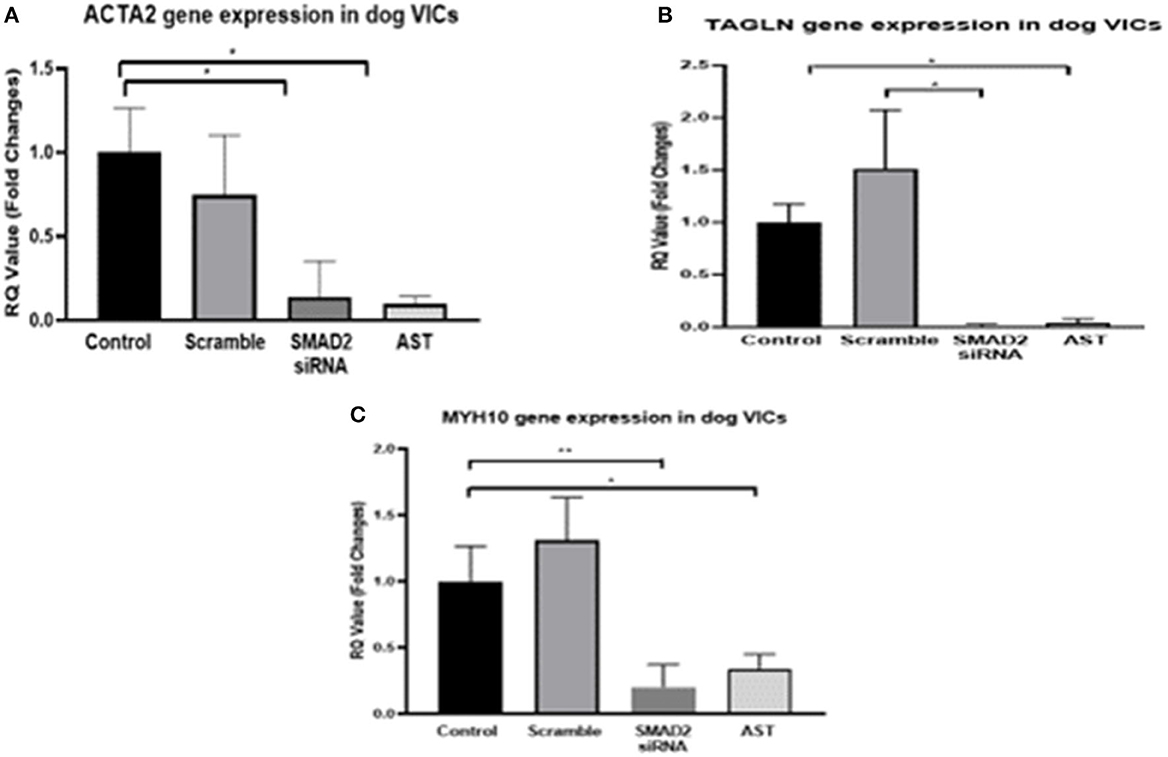
Figure 3. Gene expression of MMVD markers in aVICs. Cells were quiesced for 24 h, 10 nM siRNA or 500 mg/L AST was added to the cells and incubated for 3 days, and then mRNA was extracted for q-PCR. Pharmacological inhibition of SMAD2 resulted in a decreased expression of ACTA2 (A), TAGLN (B), and MYH10 (C) in the aVICs. Data expressed as mean + SEM and analysis carried out by two-way ANOVA with a Tukey post-hoc test. *P < 0.05, **P < 0.01 (n = 6).
SMAD2 siRNA and AST decreased the expression of SMAD2 and the corresponding phosphorylation of SMAD2 compared to the diseased controls and the scramble siRNA groups. There was also a decrease in α-SMA protein expression in both SMAD2 siRNA and AST groups and a decrease in phosphorylation of AKT2, however this occurred in the AST group only. Finally, pS6K and vimentin (a stable marker of mesenchymal cell phenotype) protein expression remained unchanged comparing all groups. For ERK1/2, the 44 kD band (ERK1) was decreased in the siRNA and AST cells, however the 42 kD band (ERK2) remained unchanged (Figure 4).
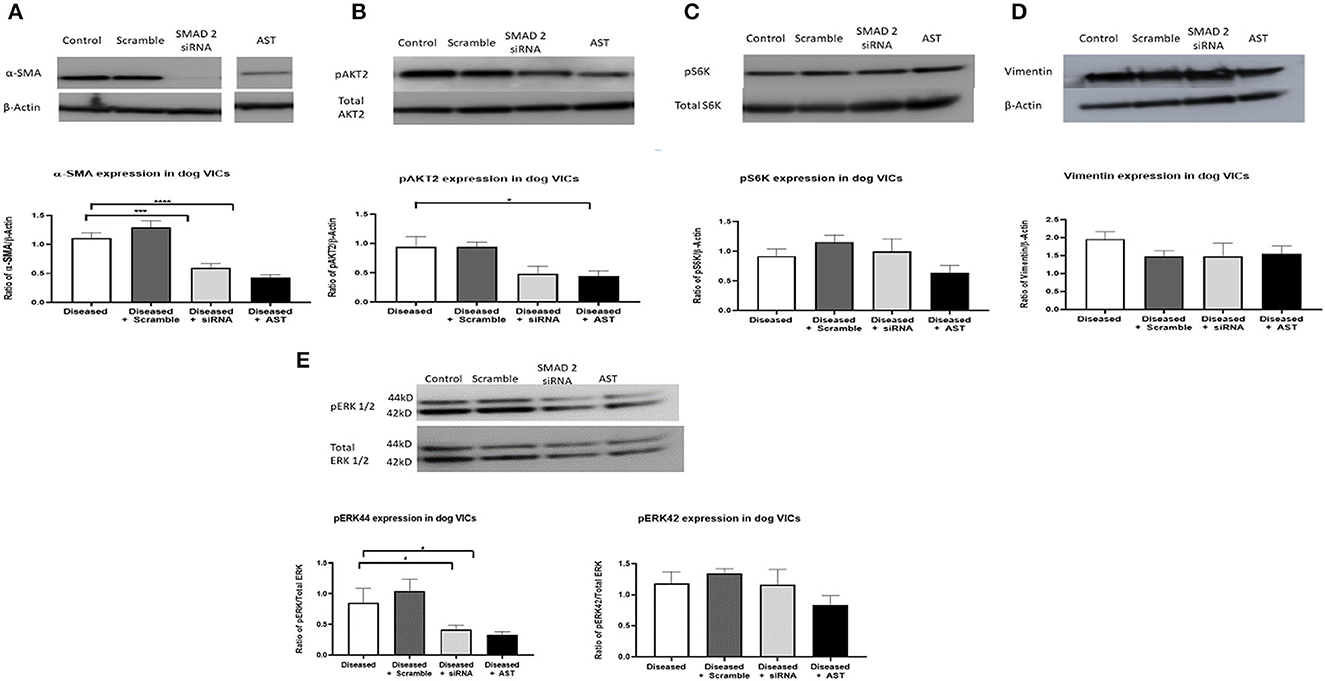
Figure 4. Protein expression of MMVD markers and downstream TGF-β molecules in activated valve interstitial cells. Cells were quiesced for 24 h, and then 10 nM siRNA or 500 mg/L AST was added to the cells and incubated for 3 days. Protein was extracted for Western blotting. Pharmacological inhibition of SMAD2 resulted in a decreased expression of pSMAD2 and αSMA (A). pAKT2 protein expression was decreased in cells treated with AST (B) while expression of pS6K (C) and Vimentin (D) remained unchanged. The 44 kD band of ERK was decreased, however those changes were not observed for the 42 kD band (E). Data expressed as mean + SEM and analysis carried out by two-way ANOVA with a Tukey post-hoc test. *P < 0.05, ***p < 0.001, and ****p < 0.0001. α-SMA (42 kDa), β-actin (42 kDa).
This study aimed to investigate the signaling downstream of TGFβ1 in diseased VICs compared to normal quiescent VICs in canine MMVD. By interrogating diseased valve interstitial cells using a commercially available protein antibody array, we were able to identify changes in SMAD2 expression and to show antagonism would revert cells back to a normal phenotype. We have also shown significant differences in both total and phosphorylated proteins for several of the non-canonical components of the TGFβ signaling pathway. These results can be used to inform future studies and interrogation of these pathways to confirm their role in disease pathogenesis and examine for potential novel therapeutics. One dog included had advanced disease but had a similar response to that from dogs with milder disease. This would suggest the changes are consistent irrespective of disease status, but further studies would be required to confirm this.
One pathway that consistently changed in all forms of analysis (both increased and decreased) was non-canonical PI3K/AKT/mTOR. This pathway has been associated with various context-specific effects on cells including transformation into a more mesenchymal phenotype, inducing and inhibiting apoptosis, and matrix protein expression (30–33). In the data found here there appears to be a downregulation in the phosphorylation of components of this pathway. It may be the case that a general decrease in phosphorylation is indicative of the change in phenotype between diseased and healthy cells, however what this means is difficult to conclude from the data. One explanation for this finding is negative feedback in diseased cells being constitutively active for prolonged periods. By further examining changes in the PI3K/AKT/mTOR pathway looking at the downstream transcription factors S6K and eIF4E, and PRAS40 (inhibits mTOR and IRS-1), we could identify their role in the negative feedback of the PI3K pathway, an effect commonly seen in various cancers and cancer models (27, 34–36). Reduced elF4E, in combination with increased pPRAS40, will repress apoptosis, and by PRAS40 limiting mTOR activation will also reduce autophagy (37, 38). This negative feedback loop involves S6K phosphorylating insulin receptor substrate 1 (IRS-1). IRS-1 then inhibits PI3K, leading to a decreased expression of the downstream molecules. However, considering what is found in cancer models, this negative feedback is not activated in early-stage disease, permitting eIF4E and S6K to act on the downstream regulators of mesenchymal cell transition, apoptosis, autophagy, senescence, cell growth, and motility (35). The context-specificity of this feedback mechanism appears to be related to disease stage and this warrants further study, especially with regards to apoptosis and autophagy, since subtle changes in the ratio of pAKT to pSMAD can have both pro- or anti-apoptotic effects (39, 40). A proposed model of this pathway is shown in Supplementary Figure 2. In a parallel study we have reported the effects of modification of this pathway using pharmacological antagonism and genomic techniques and found VIC phenotype transition can be controlled by the PI3K/AKT/mTOR pathway (41). This suggests both canonical and non-canonical components of the TGFβ signaling pathway can control VIC phenotype.
The RAS-MEK-ERK pathway was also found to be altered in the datasets. ERK1/2 signaling has been shown to be associated with MMVD in both dogs and humans, either through TGFβ-related or serotonergic signaling (42–47). The data presented here indicates some level of both activation and inhibition of this pathway with the decrease in ERK1-p44/42 MAP kinase phosphorylation, increase in total ERK8, and increased MAP3K1/MEKK1 phosphorylation in some diseased cells. Previously, we have shown that activation or inhibition of serotonin-induced ERK1/2 signaling has no effect on quiescent or diseased cells (16). Phosphorylation of ERK1/2 is known to result in the activation of a variety of transcription factors including CREB and c-fos, as well as cell cycle regulatory transcription factors such as Elk-1 and Sep-1a (48, 49). The pathway can also activate transcription factors that regulate cell survival such as Bim and FasL for inducing apoptosis (50). Inhibition of the MAPK-ERK pathway in vivo has been shown to attenuate aortic valve disease progression in Emilin1-deficient mice (51). However, looking at this pathway in the context of TGFβ-induced signaling has not been performed and studies using more specific ERK1/2 inhibitors or investigating phosphorylation changes in this pathway in quiescent cells treated with TGFβ1 would be beneficial.
Changes in phosphorylated Rac1/cdc42 were also identified with a significant increase in diseased cells. Much is known about Rac1/cdc42 in cancers such as breast and pancreatic. Interacting with TGFβ, Rac1 drives endothelial-to-mesenchymal transition (EndoMT) in cancer, in which endothelial cells lose their polarity and cohesiveness and acquire the morphology and migratory properties of fibroblasts (52). To what extent Rac1 might contribute to MMVD is unknown, but EndoMT is recognized to occur and may be an additional source of aVICs (14, 53). Use of inhibitors such as ML141 or a more targeted approach using Rac1 siRNA could be used to further investigate the role of this pathway in MMVD, in particular the effects on expression of key EndoMT transcription factors SNAIL, TWIST, ZEB, and AP-1. On the canonical side of the TGFβ signaling pathway, phosphorylated SMAD2 protein expression was found to be increased, which is also seen, with concurrent increased TGFβ protein expression, in human MMVD (21). SMAD2 expression is thought to be one of the pathways that drive VICs into their activated myofibroblast phenotype. While the downstream events of SMAD2 in MMVD are not fully understood, a pathway has been suggested by Thalji et al. (26) (Supplementary Figure 3).
While we have shown modification of PI3K/AKT/mTOR will affect cell phenotype, with pSMAD2 increased in aVICs, we decided to target it for inhibition using siRNA and the SMAD7 agonist asiaticoside. SMAD7 inhibits SMAD2 phosphorylation and prevents the formation of a complex with SMAD4, stopping SMAD2 from entering the cell nucleus (26). The genes examined as outputs encoded for α-SMA, SM-22, and MHY10, as these markers are commonly expressed at high levels in aVICs (11, 16, 54). We observed the three genes encoding for these myofibroblast markers were significantly decreased after SMAD2 antagonism to an expression level similar to that found in qVICs, suggesting phenotype reversal. Antagonism reduced phosphorylated SMAD2 and αSMA expression while maintaining VIM expression, confirming retention of a mesenchymal phenotype (10). Furthermore, the effects on expression of pERK1/2, pS6K, and pAKT identified levels of cross-talk between the SMAD, MAPK, and PI3K signaling pathways. These data confirm the effectiveness of AST and the SMAD2 siRNA at the dose used in reducing, but not in abolishing, pSMAD2 protein expression, likely preserving normal VIC function. In cardiac models of myocardial disease, SMAD2 inhibition protects against cardiac dysfunction, preventing cardiac fibrosis and cardiomyocyte hypertrophy, raising the possible therapeutic options for treating myocardial and valvular disease by targeting canonical TGFβ signaling (55, 56).
However, the complexity of TGFβ signaling needs to be considered in the development of novel therapeutics. Changes in expression of proteins in the MAPK and PI3K pathways in aVICs shows there is a link between these non-canonical pathways and canonical SMAD signaling, and as we have previously reported, antagonism of PI3K will control disease phenotype (41). The intricacies of this cross talk in disease are best understood in cancer and cancer models, with little information on MMVD. Cross-talk between the canonical SMAD pathway and PI3K pathway has been reported for various cell types including stem cells and cancer cells. This signaling is found to be complex and can either inhibit or stimulate depending on the circumstances. For example, in human embryonic stem cells when PI3K is in abundance, SMAD2 and SMAD3 activate the expression of the pluripotency gene NANOG to maintain self-renewal. However, low PI3K activity switches SMAD2/3 signaling to direct cell differentiation (57). Normal mitral valve function itself is dependent on the balance between the MEK/ERK1/2 and SMAD, which is further regulated by Filamin-A (24). Mutations in the Filamin-A gene cause congenital valvular defects and progression to myxomatous mitral valve disease in human subjects and mouse models. Phosphorylated ERK1/2 inactivates the SMAD pathway, preventing build-up of SMAD2/3 in the nucleus, permitting a balance in transcriptional activities (24). This mutation causes a decrease in ERK1/2 activation alongside a marked increase in activation of SMAD2/3. The changes in protein expression for ERK1/2 and SMAD found in the Filamin-A knockout mice do match the changes observed in the canine aVICs in the current study but inhibiting SMAD did not increase expression of ERK1/2. Further work is now needed to examine these relationships in MMVD in more detail.
Myxomatous mitral valve disease is associated with changes in both the canonical SMAD and several of the non-canonical parts of the TGFβ signaling pathway, particularly PI3K/AKT/mTOR, indicating likely contribution to disease pathogenesis. Antagonizing SMAD2 expression has a beneficial effect on transitioning disease cells back to a normal phenotype, while maintaining their inherent mesenchymal characteristics, and also affects protein expression in the non-canonical components of the TGFβ pathway. Further studies are required to identify additional downstream targets that may then have future novel therapeutic value. In addition, in vitro studies using appropriate rodent models would be needed.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The animal studies were approved by VERC R(D)SVS University of Edinburgh. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
BC and VM secured the funding. BC, GM, and VM contributed to the conception and design. GM, AM, and QT carried out the experimental work. BC, GM, and AM wrote the first draft. GM, AM, QT, and VM revised the manuscript. All authors contributed to manuscript revision and editing and approved the submitted version.
GM and AM and laboratory consumables were funded from the Dogs Trust. VM was supported by the Roslin Institute Strategic Programme Grant [Biotechnology and Biological Sciences Research Council (BBSRC); BB/J004316/1]. QT was funded by the China Scholarship Council.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1202001/full#supplementary-material
Supplementary material 1. Full moon Biosystems TGFB Array Protocol.
Supplementary material 2. Protein immunoblotting protocol (western blotting).
Supplementary Figure 1. Example of the Fullmoon TGFβ phospho array profiler indicating individual antibody spots, sets of replicates of the same antibody, and blocks of different antibody.
Supplementary Figure 2. Theorized model of negative feedback loop in MMVD. In early stages of MMVD, there is an increased expression throughout the PI3K pathway leading to the activation of transcription factors involved in apoptosis and autophagy. In the later stages of MMVDS6K, expression is increased and phosphorylates IRS-1, targeting it for degradation. This then prevents IRS-1 from acting on the downstream PI3K pathway, leading to decreased expression throughout the PI3K pathway.
Supplementary Figure 3. Simplified working model of alterations in TGF-β signaling in MMVD affecting canonical SMAD. Red, increased expression and green = reduced expression; BAMBI, BMP and activin membrane-bound inhibitor homolog (Xenopus laevis); CREB5, Cycle AMP-responsive element-binding protein 5; DAB2, Disabled homolog 2; FOS, FBJ murine osteosarcoma viral oncogene homolog JUN, Jun proto-oncogene; SIK1, Salt-inducible kinase 1; TGF-β2, transforming growth factor-beta; TGF-βR, transforming growth factor-beta receptor; TGIF1, TGF-β-induced factor homeobox 1 [adapted from (26)].
Supplementary Table 1. OD, 260/280, and amount of sample used for analysis for each of the samples used in the Full Moon TGFβ MAP kinase signaling antibody array.
Supplementary Table 2. Antibodies used for protein immuno-blotting.
1. Borgarelli M, Buchanan JW. Historical review, epidemiology and natural history of degenerative mitral valve disease. J Vet Cardiol. (2012) 14:93–101. doi: 10.1016/j.jvc.2012.01.011
2. Beardow AW, Buchanan JW. Chronic mitral valve disease in cavalier King Charles spaniels: 95 cases (1987-1991). J Am Vet Med Assoc. (1993) 203:1023–9.
3. Lu CC, Liu MM, Culshaw G, French A, Corcoran B. Comparison of cellular changes in Cavalier King Charles spaniel and mixed breed dogs with myxomatous mitral valve disease. J Vet Cardiol. (2016) 18:100–9. doi: 10.1016/j.jvc.2015.12.003
4. Aupperle H, Thielebein J, Kiefer B, Marz I, Dinges G, Schoon HA. An immunohistochemical study of the role of matrix metalloproteinases and their tissue inhibitors in chronic mitral valvular disease (valvular endocardiosis) in dogs. Vet J. (2009) 180:88–94. doi: 10.1016/j.tvjl.2007.11.011
5. Hadian M, Corcoran BM, Bradshaw JP. Molecular changes in fibrillar collagen in myxomatous mitral valve disease. Cardiovasc Pathol. (2010) 19:e141–8. doi: 10.1016/j.carpath.2009.05.001
6. Han RI, Black A, Culshaw G, French AT, Corcoran BM. Structural and cellular changes in canine myxomatous mitral valve disease: an image analysis study. J Heart Valve Dis. (2010) 19:60–70.
7. Han RI, Clark CH, Black A, French A, Culshaw GJ, Kempson SA, et al. Morphological changes to endothelial and interstitial cells and to the extra-cellular matrix in canine myxomatous mitral valve disease (endocardiosis). Vet J. (2013) 197:388–94. doi: 10.1016/j.tvjl.2013.01.027
8. Black A, French AT, Dukes-McEwan J, Corcoran BM. Ultrastructural morphologic evaluation of the phenotype of valvular interstitial cells in dogs with myxomatous degeneration of the mitral valve. Am J Vet Res. (2005) 66:1408–14. doi: 10.2460/ajvr.2005.66.1408
9. Disatian S, Ehrhart EJ III, Zimmerman S, Orton EC. Interstitial cells from dogs with naturally occurring myxomatous mitral valve disease undergo phenotype transformation. J Heart Valve Dis. (2008) 17:402–11; discussion: 412.
10. Han RI, Black A, Culshaw GJ, French AT, Else RW, Corcoran BM. Distribution of myofibroblasts, smooth muscle-like cells, macrophages, and mast cells in mitral valve leaflets of dogs with myxomatous mitral valve disease. Am J Vet Res. (2008) 69:763–9. doi: 10.2460/ajvr.69.6.763
11. Markby G, Summers KM, MacRae VE, Del-Pozo J, Corcoran BM. Myxomatous degeneration of the canine mitral valve: from gross changes to molecular events. J Comp Pathol. (2017) 156:371–83. doi: 10.1016/j.jcpa.2017.01.009
12. Lu CC, Liu MM, Culshaw G, Clinton M, Argyle DJ, Corcoran BM. Gene network and canonical pathway analysis in canine myxomatous mitral valve disease: a microarray study. Vet J. (2015) 204:23–31. doi: 10.1016/j.tvjl.2015.02.021
13. Markby GR, Macrae VE, Corcoran BM, Summers KM. Comparative transcriptomic profiling of myxomatous mitral valve disease in the cavalier King Charles spaniel. BMC Vet Res. (2020) 16:350. doi: 10.1186/s12917-020-02542-w
14. Markby GR, Macrae VE, Summers KM, Corcoran BM. Disease severity-associated gene expression in canine myxomatous mitral valve disease is dominated by TGFbeta signaling. Front Genet. (2020) 11:372. doi: 10.3389/fgene.2020.00372
15. Oyama MA, Chittur SV. Genomic expression patterns of mitral valve tissues from dogs with degenerative mitral valve disease. Am J Vet Res. (2006) 67:1307–18. doi: 10.2460/ajvr.67.8.1307
16. Tan K, Markby G, Muirhead R, Blake R, Bergeron L, Fici G, et al. Evaluation of canine 2D cell cultures as models of myxomatous mitral valve degeneration. PLoS ONE. (2019) 14:e0221126. doi: 10.1371/journal.pone.0221126
17. Connolly JM, Bakay MA, Fulmer JT, Gorman RC, Gorman JH III, Oyama MA, et al. Fenfluramine disrupts the mitral valve interstitial cell response to serotonin. Am J Pathol. (2009) 175:988–97. doi: 10.2353/ajpath.2009.081101
18. Lacerda CMR, Disatian S, Orton EC. Differential protein expression between normal, early-stage, adn late-stage myxomatous mitral valves from dogs. Proteomics Clin Appl. (2009) 3:1422–9. doi: 10.1002/prca.200900066
19. Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. (2004) 114:1586–92. doi: 10.1172/JCI200422715
20. Geirsson A, Singh M, Ali R, Abbas H, Li W, Sanchez JA, et al. Modulation of transforming growth factor-beta signaling and extracellular matrix production in myxomatous mitral valves by angiotensin II receptor blockers. Circulation. (2012) 126(11 Suppl. 1):S189–97. doi: 10.1161/CIRCULATIONAHA.111.082610
21. Hagler MA, Hadley TM, Zhang H, Mehra K, Roos CM, Schaff HV, et al. TGF-beta signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves. Cardiovasc Res. (2013) 99:175–84. doi: 10.1093/cvr/cvt083
22. Hulin A, Deroanne C, Lambert C, Defraigne JO, Nusgens B, Radermecker M, et al. Emerging pathogenic mechanisms in human myxomatous mitral valve: lessons from past and novel data. Cardiovasc Pathol. (2013) 22:245–50. doi: 10.1016/j.carpath.2012.11.001
23. Hulin A, Moore V, James JM, Yutzey KE. Loss of Axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease. Cardiovasc Res. (2017) 113:40–51. doi: 10.1093/cvr/cvw229
24. Toomer SK, Fulmer D, Guo L, Moore K, Stairley R, Sauls K, et al. Filamin-A as a balance between Erk/Smad activities during cardiac valve development. Anat Rec. (2019) 302:117–24. doi: 10.1002/ar.23911
25. Prakash S, Borreguero LJJ, Sylva M, Flores Ruiz L, Rezai F, Gunst QD, et al. Deletion of Fstl1 (follistatin-like 1) from the endocardial/endothelial lineage causes mitral valve disease. Arterioscler Thromb Vasc Biol. (2017) 37:e116–30. doi: 10.1161/ATVBAHA.117.309089
26. Thalji NM, Hagler MA, Zhang H, Casaclang-Verzosa G, Nair AA, Suri RM, et al. Nonbiased molecular screening identifies novel molecular regulators of fibrogenic and proliferative signaling in myxomatous mitral valve disease. Circ Cardiovasc Genet. (2015) 8:516–28. doi: 10.1161/CIRCGENETICS.114.000921
27. Zhang L, Zhou F, ten Dijke P. Signaling interplay between transforming growth factor-beta receptor and PI3K/AKT pathways in cancer. Trends Biochem Sci. (2013) 38:612–20. doi: 10.1016/j.tibs.2013.10.001
28. Tang B, Zhu B, Liang Y, Bi L, Hu Z, Chen B, et al. Asiaticoside suppresses collagen expression and TGF-β/Smad signaling through inducing Smad7 and inhibiting TGF-βRI and TGF-βRII in keloid fibroblasts. Arch Dermatol Res. (2011) 303:563–72. doi: 10.1007/s00403-010-1114-8
29. McNair AJ, Wilson KS, Martin PE, Welsh DJ, Dempsie Y. Connexin 43 plays a role in proliferation and migration of pulmonary arterial fibroblasts in response to hypoxia. Pulm Circ. (2020) 10:2045894020937134. doi: 10.1177/2045894020937134
30. Das F, Ghosh-Choudhury N, Lee DY, Gorin Y, Kasinath BS, Choudhury GG. Akt2 causes TGFbeta-induced deptor downregulation facilitating mTOR to drive podocyte hypertrophy and matrix protein expression. PLoS ONE. (2018) 13:e0207285. doi: 10.1371/journal.pone.0207285
31. Lan A, Qi Y, Du J. Akt2 mediates TGF-beta1-induced epithelial to mesenchymal transition by deactivating GSK3beta/snail signaling pathway in renal tubular epithelial cells. Cell Physiol Biochem. (2014) 34:368–82. doi: 10.1159/000363006
32. Valderrama-Carvajal H, Cocolakis E, Lacerte A, Lee EH, Krystal G, Ali S, et al. Activin/TGF-beta induce apoptosis through Smad-dependent expression of the lipid phosphatase SHIP. Nat Cell Biol. (2002) 4:963–9. doi: 10.1038/ncb885
33. Shin I, Bakin AV, Rodeck U, Brunet A, Arteaga CL. Transforming growth factor beta enhances epithelial cell survival via Akt-dependent regulation of FKHRL1. Mol Biol Cell. (2001) 12:3328–39. doi: 10.1091/mbc.12.11.3328
34. Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. (2007) 25:903–15. doi: 10.1016/j.molcel.2007.03.003
35. Hers I, Tavare JM. Mechanism of feedback regulation of insulin receptor substrate-1 phosphorylation in primary adipocytes. Biochem J. (2005) 388 (Pt 2):713–20. doi: 10.1042/BJ20041531
36. Carneiro BA, Kaplan JB, Altman JK, Giles FJ, Platanias LC. Targeting mTOR signaling pathways and related negative feedback loops for the treatment of acute myeloid leukemia. Cancer Biol Ther. (2015) 16:648–56. doi: 10.1080/15384047.2015.1026510
37. Thedieck K, Polak P, Kim ML, Molle KD, Cohen A, Jeno P, et al. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS ONE. (2007) 2:e1217. doi: 10.1371/journal.pone.0001217
38. Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. (2010) 120:2805–16. doi: 10.1172/JCI43008
39. Blake RR, Markby GR, Culshaw GJ, Martinez-Pereira Y, Lu CC, Corcoran BM. Survival of activated myofibroblasts in canine myxomatous mitral valve disease and the role of apoptosis. Res Vet Sci. (2020) 128:99–106. doi: 10.1016/j.rvsc.2019.11.004
40. Surachetpong S, Jiranantasak T, Rungsipipat A, Orton EC. Apoptosis and abundance of Bcl-2 family and transforming growth factor beta1 signaling proteins in canine myxomatous mitral valves. J Vet Cardiol. (2013) 15:171–80. doi: 10.1016/j.jvc.2013.02.005
41. Tang Q, Markby GR, MacNair AJ, Tang K, Tkacz M, Parys M, et al. TGF-beta-induced PI3K/AKT/mTOR pathway controls myofibroblast differentiation and secretory phenotype of valvular interstitial cells through the modulation of cellular senescence in a naturally occurring in vitro canine model of myxomatous mitral valve disease. Cell Prolif . (2023) 56:e13435. doi: 10.1111/cpr.13435
42. Disatian S, Orton EC. Autocrine serotonin and transforming growth factor beta 1 signaling mediates spontaneous myxomatous mitral valve disease. J Heart Valve Dis. (2009) 18:44–51. doi: 10.1096/fasebj.23.1_supplement.928.3
43. Driesbaugh KH, Branchetti E, Grau JB, Keeney SJ, Glass K, Oyama MA, et al. Serotonin receptor 2B signaling with interstitial cell activation and leaflet remodeling in degenerative mitral regurgitation. J Mol Cell Cardiol. (2018) 115:94–103. doi: 10.1016/j.yjmcc.2017.12.014
44. Lee CM, Han JI, Kang MH, Kim SG, Park HM. Polymorphism in the serotonin transporter protein gene in Maltese dogs with degenerative mitral valve disease. J Vet Sci. (2018) 19:129–35. doi: 10.4142/jvs.2018.19.1.129
45. Cremer SE, Moesgaard SG, Rasmussen CE, Zois NE, Falk T, Reimann MJ, et al. Alpha-smooth muscle actin and serotonin receptors 2A and 2B in dogs with myxomatous mitral valve disease. Res Vet Sci. (2015) 100:197–206. doi: 10.1016/j.rvsc.2015.03.020
46. Hutcheson JD, Setola V, Roth BL, Merryman WD. Serotonin receptors and heart valve disease–it was meant 2B. Pharmacol Ther. (2011) 132:146–57. doi: 10.1016/j.pharmthera.2011.03.008
47. Oyama MA, Levy RJ. Insights into serotonin signaling mechanisms associated with canine degenerative mitral valve disease. J Vet Intern Med. (2010) 24:27–36. doi: 10.1111/j.1939-1676.2009.0411.x
48. Babu GJ, Lalli MJ, Sussman MA, Sadoshima J, Periasamy M. Phosphorylation of elk-1 by MEK/ERK pathway is necessary for c-fos gene activation during cardiac myocyte hypertrophy. J Mol Cell Cardiol. (2000) 32:1447–57. doi: 10.1006/jmcc.2000.1185
49. Koga Y, Tsurumaki H, Aoki-Saito H, Sato M, Yatomi M, Takehara K, et al. Roles of cyclic AMP response element binding activation in the ERK1/2 and p38 MAPK signaling pathway in central nervous system, cardiovascular system, osteoclast differentiation and mucin and cytokine production. Int J Mol Sci. (2019) 20:1346. doi: 10.3390/ijms20061346
50. Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle. (2009) 8:1168–75. doi: 10.4161/cc.8.8.8147
51. Munjal C, Jegga AG, Opoka AM, Stoilov I, Norris RA, Thomas CJ, et al. Inhibition of MAPK-Erk pathway in vivo attenuates aortic valve disease processes in Emilin1-deficient mouse model. Physiol Rep. (2017) 5:e13152. doi: 10.14814/phy2.13152
52. Melzer C, Hass R. von der Ohe J, Lehnert H, Ungefroren H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun Signal. (2017) 15:19. doi: 10.1186/s12964-017-0175-0
53. Lu CC, Liu MM, Clinton M, Culshaw G, Argyle DJ, Corcoran BM. Developmental pathways and endothelial to mesenchymal transition in canine myxomatous mitral valve disease. Vet J. (2015) 206:377–84. doi: 10.1016/j.tvjl.2015.08.011
54. Markby GR, Summers KM, MacRae VE, Corcoran BM. Comparative transcriptomic profiling and gene expression for myxomatous mitral valve disease in the dog and human. Vet Sci. (2017) 4:34. doi: 10.3390/vetsci4030034
55. Parichatikanond W, Luangmonkong T, Mangmool S, Kurose H. Therapeutic targets for the treatment of cardiac fibrosis and cancer: focusing on TGF-beta signaling. Front Cardiovasc Med. (2020) 7:34. doi: 10.3389/fcvm.2020.00034
56. Bjornstad JL, Skrbic B, Marstein HS, Hasic A, Sjaastad I, Louch WE, et al. Inhibition of SMAD2 phosphorylation preserves cardiac function during pressure overload. Cardiovasc Res. (2012) 93:100–10. doi: 10.1093/cvr/cvr294
Keywords: myxomatous mitral valve disease, transforming growth factor-beta, canine, SMAD2, PI3K/AKT/mTOR
Citation: McNair AJ, Markby GR, Tang Q, MacRae VE and Corcoran BM (2023) TGF-β phospho antibody array identifies altered SMAD2, PI3K/AKT/SMAD, and RAC signaling contribute to the pathogenesis of myxomatous mitral valve disease. Front. Vet. Sci. 10:1202001. doi: 10.3389/fvets.2023.1202001
Received: 07 April 2023; Accepted: 28 August 2023;
Published: 16 October 2023.
Edited by:
Sidharth Mehan, Indo-Soviet Friendship College of Pharmacy, IndiaReviewed by:
Frank Van Steenbeek, Utrecht University, NetherlandsCopyright © 2023 McNair, Markby, Tang, MacRae and Corcoran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brendan M. Corcoran, YnJlbmRhbi5jb3Jjb3JhbkBlZC5hYy51aw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.