 Jayedul Hassan1*
Jayedul Hassan1* Md. Abdus Sattar Bag1
Md. Abdus Sattar Bag1 Md. Wohab Ali1
Md. Wohab Ali1 Ajran Kabir1
Ajran Kabir1 M. Nazmul Hoque2
M. Nazmul Hoque2 Muhammad Maqsud Hossain3,4
Muhammad Maqsud Hossain3,4 Md. Tanvir Rahman1
Md. Tanvir Rahman1 Md. Shafiqul Islam1
Md. Shafiqul Islam1 Md. Shahidur Rahman Khan1
Md. Shahidur Rahman Khan1- 1Department of Microbiology and Hygiene, Faculty of Veterinary Science, Bangladesh Agricultural University, Mymensingh, Bangladesh
- 2Department of Gynecology, Obstetrics and Reproductive Health, Faculty of Veterinary Medicine and Animal Science, Bangabandhu Sheikh Mujibur Rahman Agricultural University, Gazipur, Bangladesh
- 3Department of Biochemistry and Microbiology, North South University, Dhaka, Bangladesh
- 4NSU Genome Research Institute (NGRI), North South University, Dhaka, Bangladesh
Introduction: Streptococci are the major etiology in mastitis in dairy cattle, a cause of huge economic losses in the dairy industries. This study was aimed to determine the diversity of Streptococcus spp. isolated from clinical mastitis of cattle reared in Bangladesh.
Methods: A total of 843 lactating cattle reared in four prominent dairy farms and one dairy community were purposively included in this study where 80 cattle were positive to clinical mastitis (CM) based on gross changes in the udder (redness, swelling, and sensitive udder) and/or milk (flakes and/or clots). Milk samples were collected from all the eighty cattle with clinical mastitis (CCM) and twenty five apparently healthy cattle (AHC). Samples were enriched in Luria Bertani broth (LB) and one hundred microliter of the enrichment culture was spread onto selective media for the isolation of Staphylococcus spp., Streptococcus spp., Enterococcus spp., Escherichia coli and Corynebacterium spp., the major pathogen associated with mastitis. Isolates recovered from culture were further confirmed by species specific PCR.
Results and Discussion: Out of 105 samples examined 56.2% (59/105), 17.14% (18/105), 9.52% (10/105) and 22.9% (24/105) samples were positive for Staphylococcus, Streptococcus, Enterococcus faecalis and E. coli, respectively. This study was then directed to the determination of diversity of Streptococcus spp. through the sequencing of 16S rRNA. A total of eighteen of the samples from CCM (22.5%) but none from the AHC were positive for Streptococcus spp. by cultural and molecular examination. Sequencing and phylogenetic analysis of 16S rRNA identified 55.6, 33.3, 5.6 and 5.6% of the Streptococcus isolates as Streptococcus uberis, Streptococcus agalactiae, Streptococcus hyovaginalis and Streptococcus urinalis, respectively. Considering the high prevalence and worldwide increasing trend of S. uberis in mastitis, in-depth molecular characterization of S. uberis was performed through whole genome sequencing. Five of the S. uberis strain isolated in this study were subjected to WGS and on analysis two novel ST types of S. uberis were identified, indicating the presence of at least two different genotypes of S. uberis in the study areas. On virulence profiling, all the isolates harbored at least 35 virulence and putative virulence genes probably associated with intramammary infection (IMI) indicating all the S. uberis isolated in this study are potential mastitis pathogen. Overall findings suggest that Streptococcus encountered in bovine mastitis is diverse and S. uberis might be predominantly associated with CM in the study areas. The S. uberis genome carries an array of putative virulence factors that need to be investigated genotypically and phenotypically to identify a specific trait governing the virulence and fitness of this bacterium. Moreover, the genomic information could be used for the development of new genomic tools for virulence gene profiling of S. uberis.
Introduction
Bovine mastitis is the major hindering factor in the dairy industry responsible for huge economic losses worldwide. Bovine mastitis was estimated to cost €16–26 billion per annum worldwide (1) and contributed to the reduction of milk yield and quality, culling cows and veterinary care (2). Bacteria are the main cause of this disease among 130 well-known pathogens of bovine mastitis (3). Genus Streptococcus is major among the bacterial pathogens and account for 23%–50% of the total mastitis cases worldwide (4). Mastitis is classified into three distinct forms based on the degree of inflammation, sub-clinical, clinical and chronic (5), and the occurrence of Streptococcus have been reported in all three forms of this disease (6–8).
Streptococcus agalactiae, Streptococcus dysagalactiae, and Streptococcus uberis are most commonly responsible for bovine mastitis with varying global frequencies among Streptococcus spp. (4, 9). S. agalactiae was found in 90% of mastitis cases in the past; however, due to their contagious nature, the incidence of S. agalactiae was drastically reduced by implementing a five-point hygiene plan in the 1960s (3, 10, 11). In contrast, the majority of the causes of mastitis cases shifted towards environmental mastitis (3, 12). As a result, the prevalence of environmental mastitis pathogens such as S. uberis has increased remarkably (4, 13). However, the prevalence of the major streptococci was variable based on geographical locations (4). In Bangladesh, information on Streptococcus and their antibiotic susceptibility in mastitis are minimal (14–16), and to the best of our knowledge, no research has been documented on the occurrence, diversity and molecular characteristics of Streptococcus in clinical mastitis of cattle in Bangladesh. In Bangladesh, dairying is a growing industry and mostly dominated by small holding farms producing 70-80% of the country’s total milk (17). In recent years, dairy farming is going intensification and new large scales farm units (farms with ⁓200 dairy cows) are emerging; however, most of the large dairy farms belonged to the government and private sectors.1 Although mastitis is frequently reported from different corner of the country, reaching them and collecting information on therapeutic interventions are quite difficult and lacks authentications. Thus, the present study aimed to determine the diversity of Streptococcus spp. in clinical mastitis of cattle in the major dairy production units (farms/community) in Bangladesh and their antibiotic susceptibility pattern. In addition, the research was further extended to the genomic characterization and virulence profiling of the S. uberis isolated in this study through whole genome sequencing and subsequent bioinformatic analysis considering the increased prevalence of this pathogen in bovine mastitis.
Materials and methods
Ethical statement
This study was approved by the Animal Welfare and Experimentation Ethics Committee (AWEEC), Bangladesh Agricultural University, Mymensingh-2,202, Bangladesh [Approval No. AWEEC/BAU/2020(45)]. In addition, verbal as well as written consent of the farm owner or respective authority was taken before collecting samples.
Sample collection and enrichment
Milk samples were collected from cattle with clinical mastitis (CCM) from four major dairy farms and a community (consisting several small holding dairy farms) at different locations in Bangladesh during the period from January 2019 to June 2021. Dairy farms and the community was purposively selected as they were well managed and major dairy units located in distinct geographical locations including Dhaka (23.8105° N, 90.3372° E), Mymensingh (24.7539° N, 90.4073° E) and Sirajganj (24.3141° N, 89.5700° E) districts of Bangladesh. A total of 2,731 dairy cattle comprising 843 lactating cows were included in this study (Supplementary Table S1) and clinical mastitis was diagnosed by the field or residential veterinarian through the observation of gross changes in the udder (redness, swelling, and sensitive udder) and/or milk (flakes and/or clots). A total of 105 milk samples comprising 80 from the cattle with clinical mastitis (CCM) and 25 from apparently healthy cattle (AHC) were collected at the study period. Strict hygienic conditions including sterile hand gloves, collection tubes [50 mL Falcon tubes (SPL Life Sciences, Gyeonggi-do, Korea)], were used during sample collection. In addition, the udder was cleaned with potable water, wiped with paper towel until the fecal materials completely removed and care was taken to avoid contact of the collection tube with the skin and/or teat of the mammary gland during sample collection. Furthermore, first few squirts of milk from the udder were discarded before collecting the original sample. A 10 mL composite milk sample was collected directly from the udder of a cow and carried to the laboratory in ice box for microbiological analysis. In the laboratory, 0.5 mL of the milk was enriched in 4.5 mL LB broth overnight at 37°C following the procedure described earlier by different authors (18–20).
Isolation and identification of bacteria
One hundred microlitres of enriched culture was spread onto different selective media such as Mannitol salt agar (Himedia, Thane, Maharashtra, India) for Staphylococcus spp., modified Edwards medium (MEM) (Himedia) for Streptococcus and Enterococcus spp., MacConkey agar (Liofilchem, Roseto d. Abruzzi, Italy) for Escherichia coli, and brain heart infusion agar (Himedia) supplemented with 0.25 g/L potassium tellurite (Himedia) and 1.0% Tween 80 (Smart Lab, Tangerang, Indonesia) for Corynebacterium spp. After an overnight incubation at 37°C at least 10 colonies characteristic of the respective bacterium were screened by Gram’s staining followed by purification to single isolated colonies with uniform morphologies by repetitive culture onto selective media and Gram’s staining. Genomic DNA was extracted from the isolated colonies using the boiling method (21) and confirmed by species specific PCR for Staphylococcus spp., Streptococcus spp. (S. agalactiae, S. dysagalactiae and S. uberis), Enterococcus spp. (E. faecalis, E. faecium), E. coli and Corynebacterium spp. (22–28). Primers and PCR conditions followed for the identification of bacterial species are enlisted in the Supplementary Table S2. Streptococcus isolates were subjected to amplification and sequencing of 16S rRNA using 8F (5′-AGAGTTTGATCMTGGC-3′) and 1492R (5′-TACCTTG TTACGACTT-3′) primers to further confirmation and discrimination (29). Briefly, amplification of 16S rRNA was performed in 50 μL volume with 25 μL of 2X Go Taq® G2 Green Mater Mix (Promega, WI, United States), 2.5 μL of each primer (10 pmol/μL) and 3 μL of DNA template. The PCR product was sent for sequencing commercially (Macrogen, Seoul, Korea). Alignment and phylogenetic analysis of the 16S rRNA sequences were performed on CLC genomic workbench 22 (Qiagen, Germany).
Antibiotic susceptibility testing of Streptococcus spp.
Susceptibility of Streptococcus spp. against antibiotics commonly used in mastitis treatment was performed by the disc diffusion method (30). A total of 14 antimicrobials of different classes were purchased and stored according to the manufacturer’s instructions before use (Oxoid, United Kingdom). The antimicrobials includes β-lactams [amoxicillin 10 μg (AMX), ampicillin 10 μg (AMP), ceftazidime 30 μg (CAZ), ceftriaxone 30 μg (CTR), cephalexin 30 μg (CN), and penicillin G 10 μg (P)], aminoglycosides [kanamycin 30 μg (K), gentamicin 30 μg (GEN), and streptomycin 10 μg (S), glycopeptides (vancomycin 30 μg (VA)], macrolides [azithromycin 15 μg (AZM)], phenicols [chloramphenicol 30 μg (C)], polypeptides [bacitracin 10 units (B)], and tetracyclines [tetracycline 30 μg (TE)] was used in this study. E. coli strain ATCC25922 was used as the control strain and the results were interpreted according to the Clinical and Laboratory Standards Institute (CLSI) guidelines (31). Isolates exhibiting resistance to three or more antibiotic classes were considered as multidrug-resistant (MDR) (32).
Whole genome sequencing and annotation
Five of ten S. uberis isolated in this study were subjected to whole genome sequencing (WGS). For isolate selection no definite criteria were followed; however, from each S. uberis positive farm at least one isolate was selected for WGS (Supplementary Table S3). WGS was performed on an Illumina Nextseq 550 platform (Illumina, San Diego, CA, United States) in the commercial facility of Invent Technologies Ltd., Dhaka, Bangladesh. Briefly, pure isolated colony from LB agar plate was inoculated in LB broth and incubated overnight at 37°C. Genomic DNA was extracted from the overnight culture using QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany) and the quantity was measured with a NanoDrop 2000 UV–Vis Spectrophotometer (Thermo Fisher, Waltham, MA, United States). Libraries were prepared from 1 ng of DNA using Nextera™ DNA Flex Library Prep Kit (Illumina, San Diego, United States) for short-read sequencing. Libraries were cleaned (AMPure XP beads; Beckman Coulter) and sequencing was performed on an Illumina Nextseq 550 platform (Illumina, CA, United States) using a 150 bp pairedend sequencing protocol. Quality of the FASTQ reads and trimming of low-quality residues was performed on Trimmomatic (Galaxy Version 0.38). Trimmed reads were assembled on a hybrid assembler Unicycler (Galaxy Version 0.4.8.0). Assembled contigs were annotated using Prokka (Galaxy Version 1.14.6), RAST (Rapid Annotation using Subsystem Technology),2 and NCBI prokaryotic genome annotation pipeline (PGAP) to identify the functional features. The annotated data were used for antibiotic resistance gene profiling, plasmid identification, sequence typing and virulence genes profiling.
Antibiotic resistance genes were identified using the Comprehensive Antibiotic Resistance Database (CARD),3 and for virulence gene detection, the S. uberis Putative Virulence Database suPVDB (33) and virulence factor database (VFDB)4 was explored. Further, the known virulence related genes (34, 35); were identified through search on NCBI blastn [Nucleotide BLAST: Search nucleotide databases using a nucleotide query (nih.gov)] and blastx [blastx: search protein databases using a translated nucleotide query (nih.gov)] platforms.
Average nucleotide identity (ANI) of the S. uberis sequences along with genome assemblies downloaded from the NCBI [Streptococcus uberis—Assembly—NCBI (nih.gov)] was calculated using a perl program GET_HOMOLOGUES (36). The genome sequences were further analyzed using the Roary pan genome pipeline (37) to identify orthologous genes in S. uberis and construction of a tree by clustering the core and accessory genes in a matrix. In addition, core genome based whole genome phylogeny and tree visualization were performed on CLC Genomic workbench (Qiagen) and iTOL v6 [iTOL: Interactive Tree Of Life (embl.de)], respectively. The comparative genomic feature of the study strains against the S. uberis strain 0140J (GenBank accession no.: AM946015.1) reference genome was visualized using BLAST Ring Image Generator (BRIG) version 0.95 (38). Pathways and subsystems for the strains under study were obtained from the Kyoto Encyclopedia of Genes and Genomes orthology (KEGG) (39), and the SEED databases (40), respectively. CRISPRCasFinder 4.2.20 software at Proksee tools;5 was used for rapid identification and classification of clustered regularly interspaced short palindromic Repeats (CRISPRs) and the PHASTER web server6 was used to identify a prophage sequence. The S. uberis genome sequences were examined for their sequence type (ST) through PubMLST website.7 Concatenated ST sequences were downloaded from PubMLST8 and the minimum spanning tree was constructed using the goeBurst algorithm of Phyloviz9 to identify the closest neighbors.
Results
Occurrence and diversity of Streptococcus spp.
Cultural and PCR based identification revealed 56.2% (59/105), 17.14% (18/105), 9.52% (10/105) and 22.9% (24/105) of the samples (n = 105) were positive for Staphylococcus spp., Streptococcus spp., E. faecalis and E. coli, respectively. Streptococcus spp. was detected in 18 of 80 (22.5%) milk samples examined from CCM (Table 1). However, none of the samples from apparently healthy cattle carried Streptococcus spp. Out of 18 Streptococcus positive cattle 5 (27.8%) carried E. coli, 6 (33.3%) carried Staphylococcus, 1 (5.6%) carried both E. coli and Staphylococcus (5.6%), indicated that 66.67% Streptococcus positive cattle carried mixed infection. Streptococcus spp. were sequenced targeting 16S rRNA and identified as S. agalactiae (33.3%), S. uberis (55.6%), S. hyovaginalis (5.6%), and S. urinalis (5.6%) (Figure 1) through homology search, and phylogenetic analysis (Accession no. OL581668 to OL581684, and ON935775). S. uberis was most prevalent followed by S. agalactiae (Table 1). None of the samples carried S. dysagalactiae.
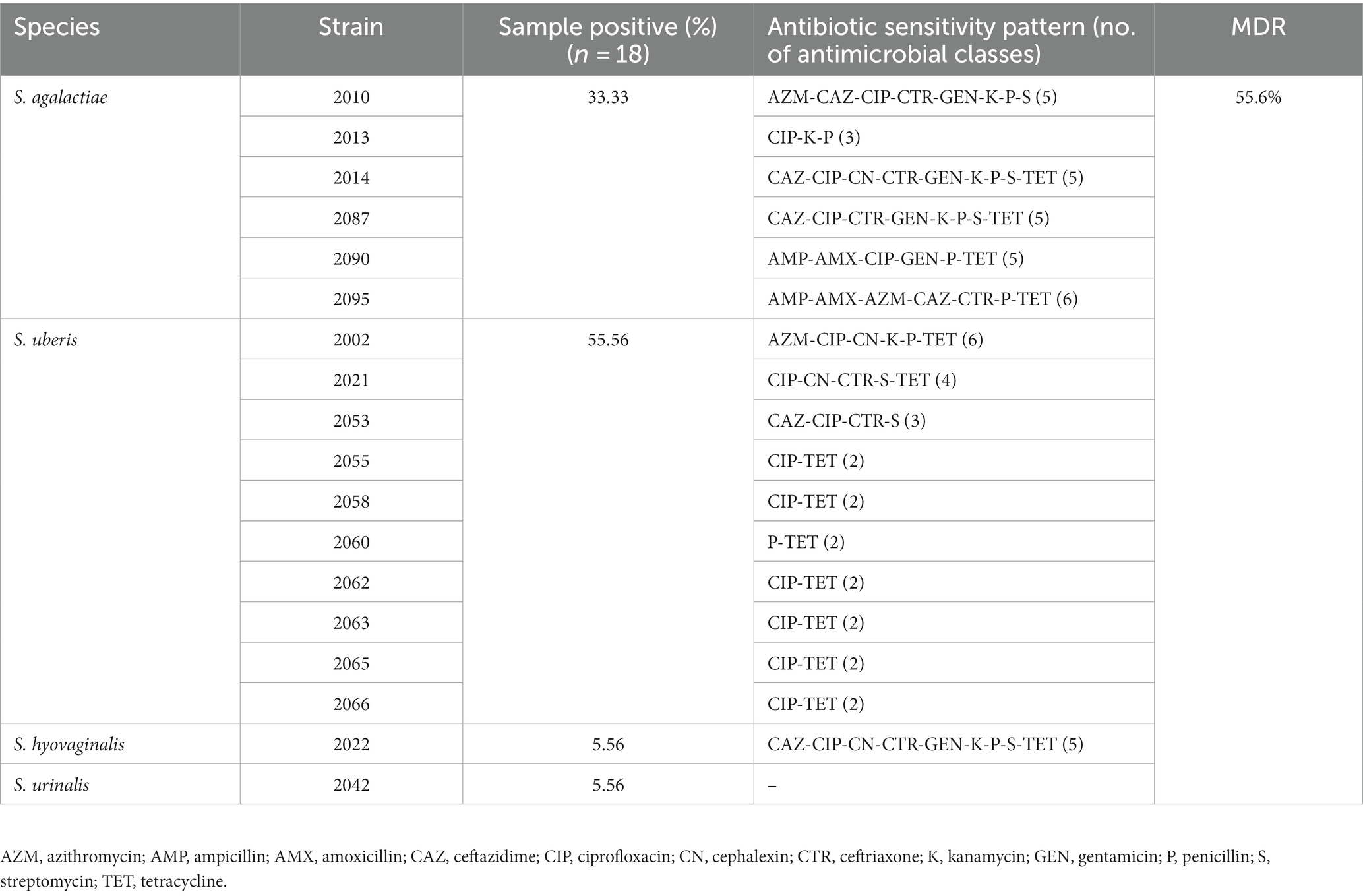
Table 1. Frequency of isolation and antibiotic resistance pattern of the Streptococcus spp. isolated in this study.
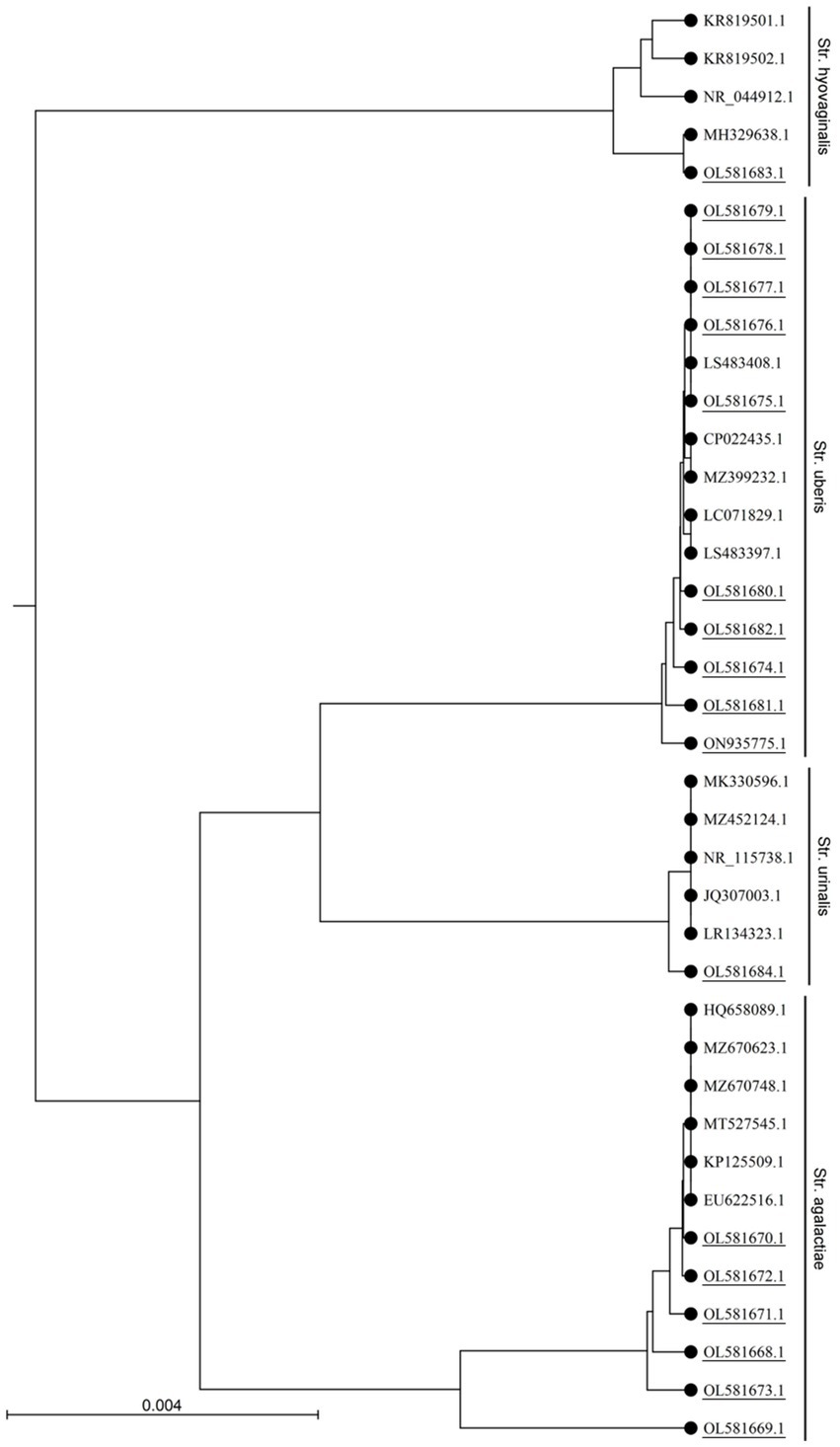
Figure 1. Phylogenetic analysis of the 16S rRNA gene sequences of Streptococcus sp. isolated in this study. The UPGMA tree was prepared using CLC sequence viewer 8.0 and the nucleotide distances were calculated using Kimura 80 model. Accession numbers with underlines denotes sequences obtained in this study.
Antibiotic susceptibility of Streptococcus isolates
Fifty-five percent (55.6%, 10/18) of the Streptococcus isolated in this study showed multidrug resistance (MDR), with 100% of the S. agalactiae and S. hyovaginalis. Whereas, S. urinalis was susceptible to all the antimicrobials evaluated in this study (Table 1). On the other hand, only 30% of the S. uberis showed MDR, and they were susceptible to most of the antibiotics used in this study (Table 1). Among the antibiotics tested, the highest resistance was encountered against ciprofloxacin (83.3%) followed by tetracycline (77.8%). All (100%) of the Streptococcus isolates were sensitive to chloramphenicol and vancomycin (Table 1).
General features of the Streptococcus uberis genomes
Five S. uberis isolated in this study were sequenced and assembled to elucidate their genomic features. The assembled genome length ranged from 18,86,469 to 20,33,655 bp with GC contents of 36.4% to 36.5% (Table 2). The raw sequences and draft genomes were submitted to NCBI-bioproject (Accession numbers: PRJNA856652, PRJNA856654, PRJNA856664, PRJNA856665, PRJNA856669), GenBank (Accession numbers: JANLBK000000000–JANLBO000000000) and sequence read archive (SRA) (SRR21812789, SRR21813362, SRR21813574, SRR21813756, SRR21814019). The draft genome of strain 2053 was the largest and strain 2055 being the smallest among the genomes examined in this study. Strains 2002, 2021, 2055 and 2062 carried 100% identical nucleotide sequences, but 2053 shared 99.04% sequence identity with them (Figure 2). The isolates carried 1632 core genes and 626 shell genes (Figure 3A). Gene clustering based on the presence and absence of genes in S. uberis revealed identical patterns of the genome 2002, 2021, 2055 and 2062 on a pangenome matrix (Figure 3B) and clustered close to the pathogenic strain 0140J, whereas the strain 2053 clustered close to the non-pathogenic strain EF20 described from the UK (34) (Figure 3B). Similar pattern of clustering was evident on a phylogenetic tree constructed with core genome sequences of S. uberis genomes described throughout the world (Figure 3C).
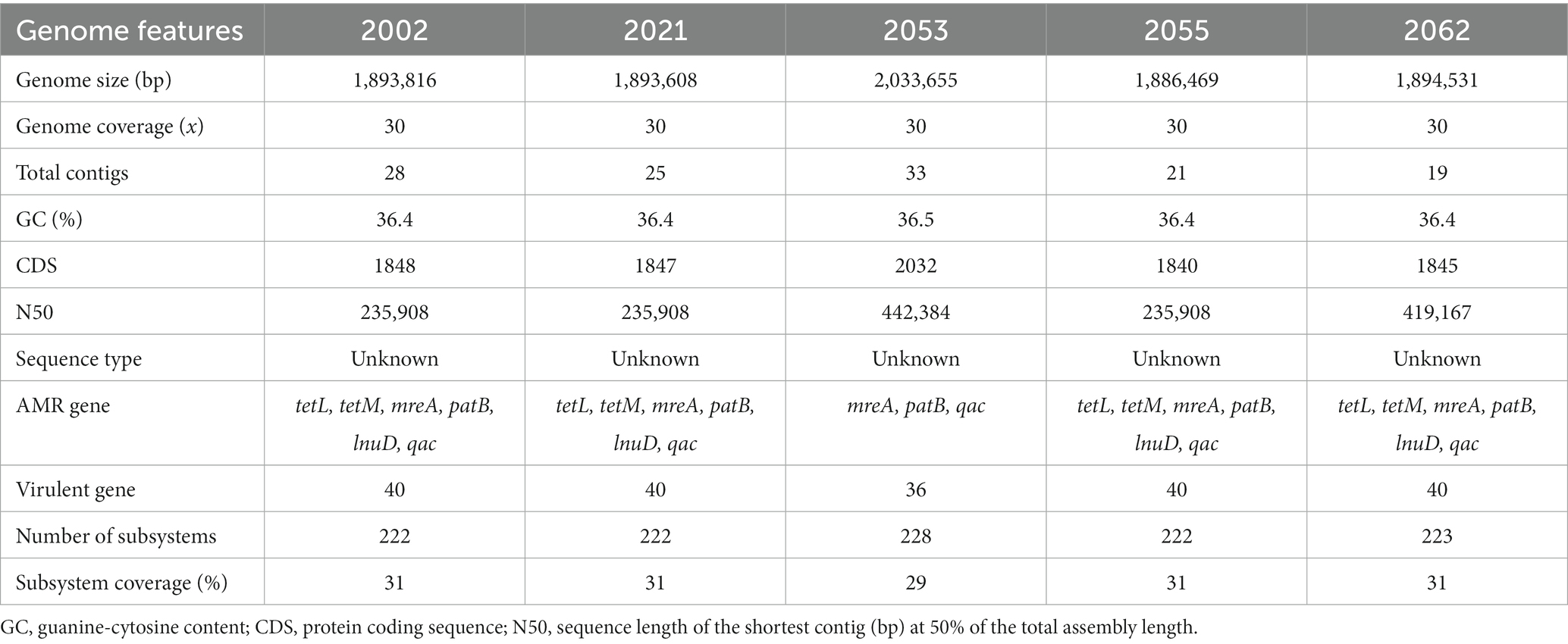
Table 2. The genomic features of five Streptococcus uberis strains isolated from clinical mastitis milk of dairy cows.
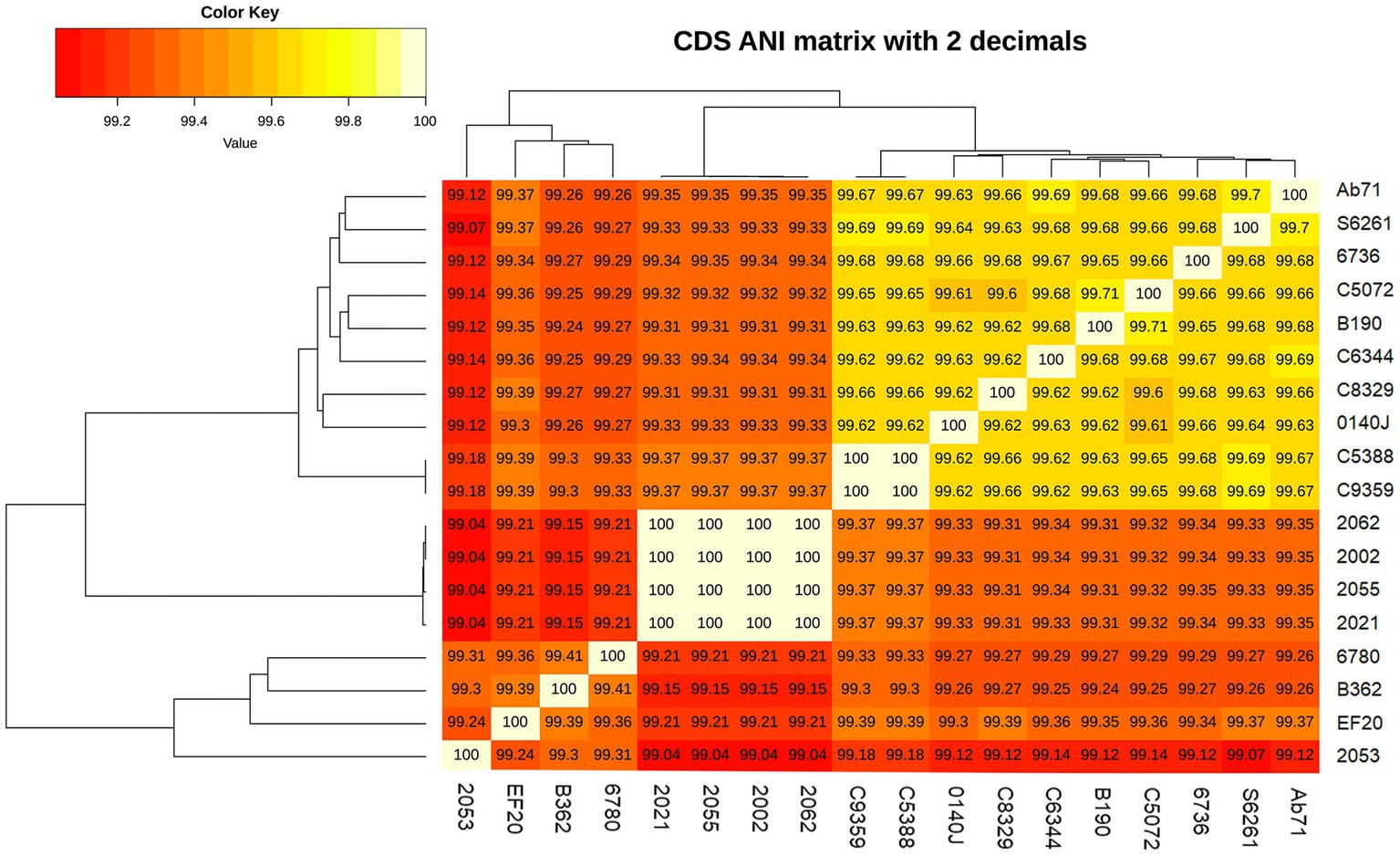
Figure 2. Average nucleotide identity (ANI) of Streptococcus uberis genomes described in this study and related strains from UK (34). The nucleotide identity was calculated with get_homologue.pl.
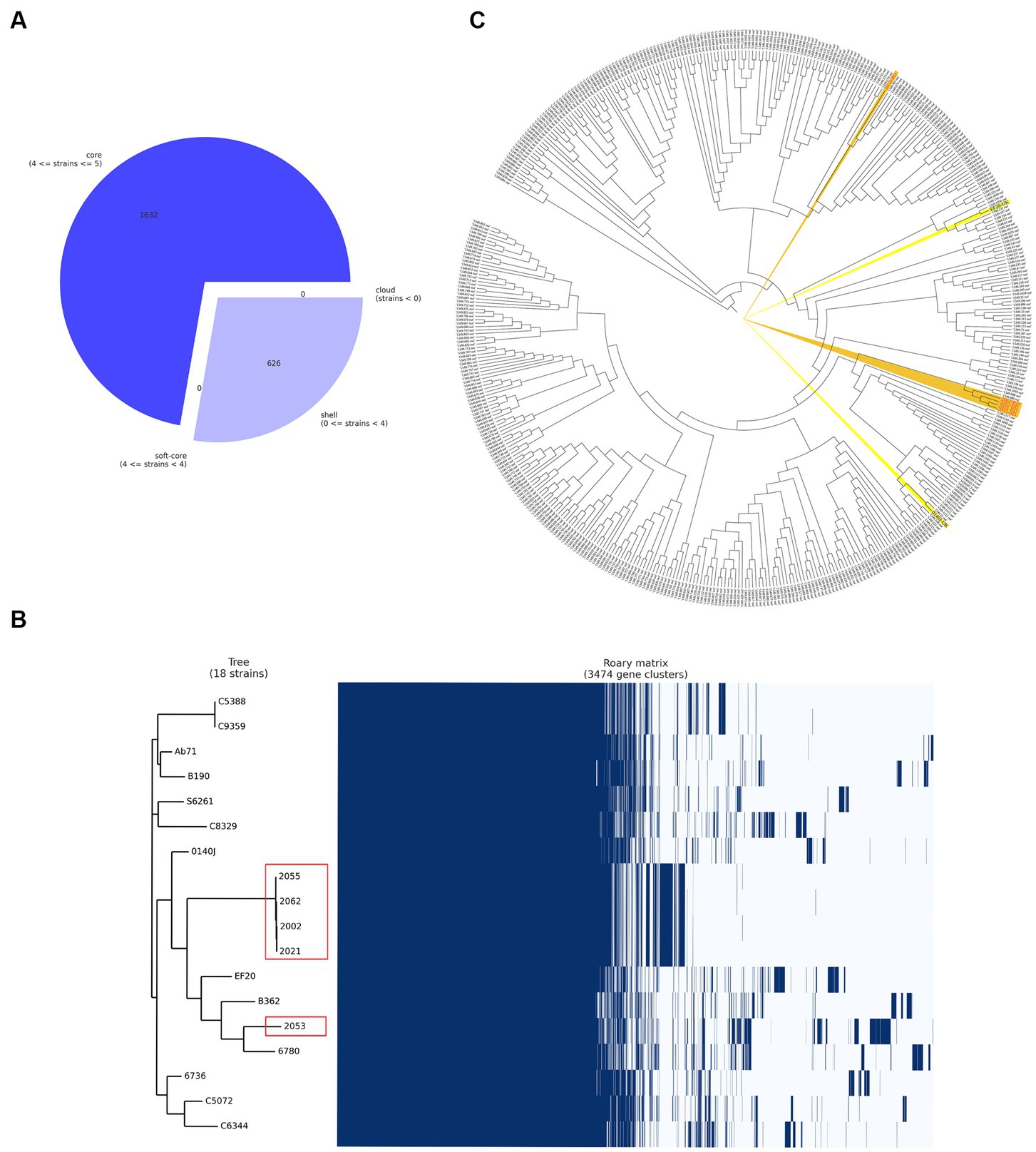
Figure 3. Pangenome analysis of the S. uberis isolated from Bangladesh (BD). (A) Breakdown of genes in S. uberis isolates from BD, (B) pangenome based (gene presence and absence) gene clustering matrix of BD (enclosed in red boxes) and UK isolates and (C) core genome-based phylogeny with S. uberis genomes described throughout the world. The figures were prepared based on the data acquired from Roary pangenome analysis using roary_plots.py script.
A comparative genomic analysis was performed with the study strains and the pathogenic close relative S. uberis strain 0140J genome (GenBank accession no.: AM946015.1). The circular genome map generated by the BRIG shows that five S. uberis strains share high similarities among themselves. In addition, the genomic map obtained from the BRIG comparison revealed that multiple regions were absent in the study genomes in comparison to reference genome and vice versa (Figure 4), suggesting the possible acquisition and/or loss of several additional genes and recombination which may have evolutionarily favored their competitiveness in mastitis pathogenesis. Moreover, some of the predicted pathogenicity and resistance islands overlap with part of those regions (Figure 4).
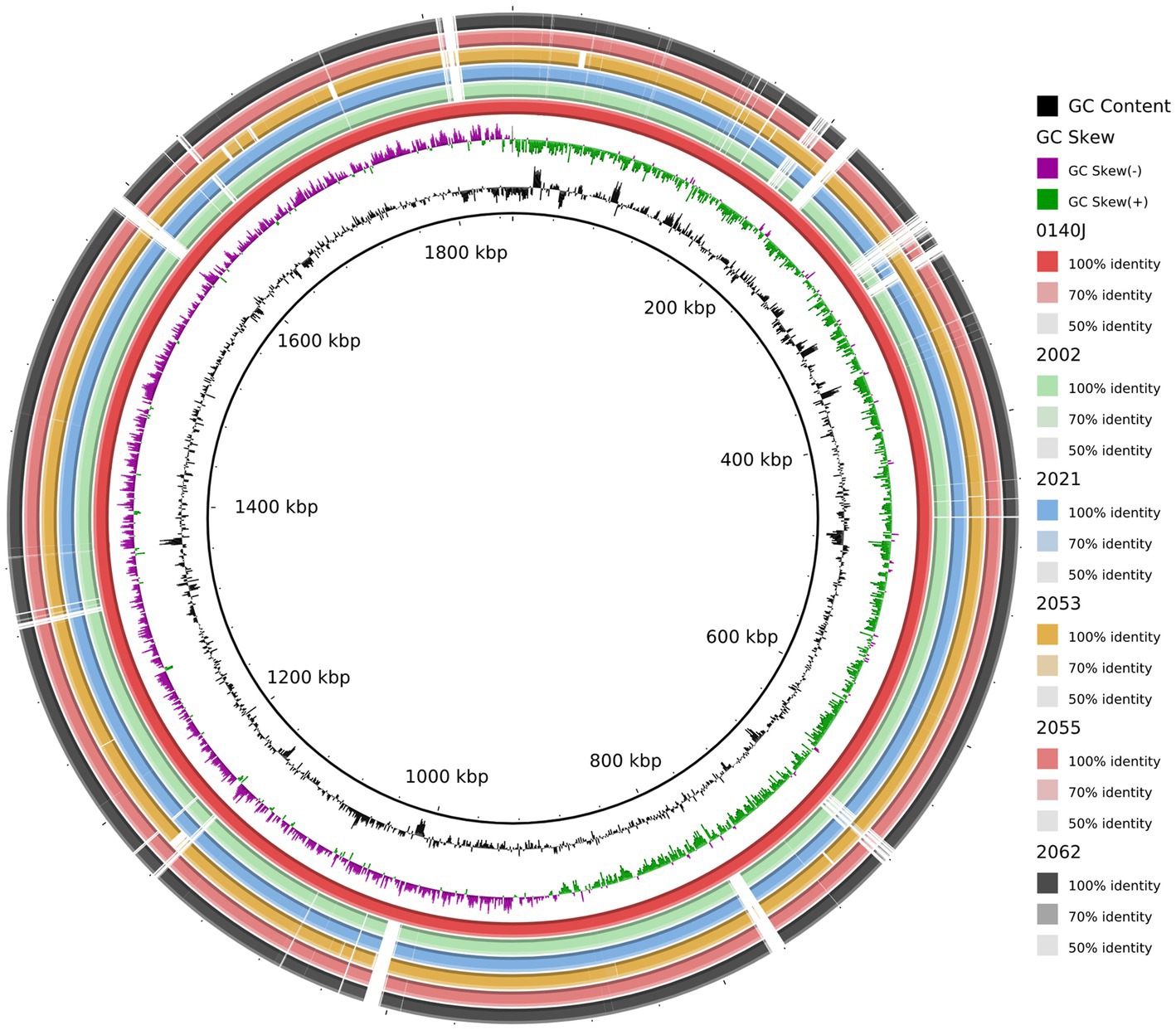
Figure 4. Circular representation of the S. uberis complete genomes. Circles (from inside to outside) 1 and 2 (GC content; black line and GC skew; magenta and green lines), circle 3 (S. uberis strain 0140J; maroon circle); circle 4 (mapped S. uberis strain 2002 genome with S. uberis strain 0140J genome; light green circle); circle 5 (mapped S. uberis strain 2021 genome with S. uberis strain 0140J genome; deep sky circle); circle 6 (mapped S. uberis strain 2053 genome with S. uberis strain 0140J genome; orange circle); circle 7 (mapped S. uberis strain 2055 genome with S. uberis strain 0140J genome; brown circle), and circle 8 (mapped S. uberis strain 2062 genome with S. uberis strain 0140J genome; black circle). BRIG 0.95 was used to build the circular representation. Mapping studies were done using BLASTn with an e-value cut-off 1e-5.
The coding sequences (CDSs) of the five S. uberis strains were annotated using the KEGG pathways and SEED subsystems which classified CDSs into six subcategories including metabolism, cofactors, vitamins and pigments, genetic information processing, cellular processing, stress response and virulence and defenses (Figure 5). The strains 2002, 2021, 2053, 2055 and 2062 were found to conserve 1848, 1847, 2032, 1840 and 1845 CDSs, respectively, that were further divided into 27 functional KEGG subcategories and subsystems according to the aforementioned six categories (Figure 5; Supplementary Table S4). Notably, all of the strains contained a significantly higher number of genes related to genetic information processing (1,149 CDSs), followed by metabolism (883 CDSs), virulence and defenses (531 CDSs), cellular processing (346 CDSs), stress response (243 CDSs) and cofactors, vitamins and pigments (100 CDSs) metabolism (Figure 5; Supplementary Table S4). On an average, S. uberis strain 2062 harbors higher number of CDSs (n = 767) for KEGG subcategories and SEED subsystems followed by S. uberis strain 2053 (n = 733), S. uberis strain 2001 (n = 682), S. uberis strain 2055 (n = 663), and S. uberis strain 2021 (n = 510) (Supplementary Table S4).
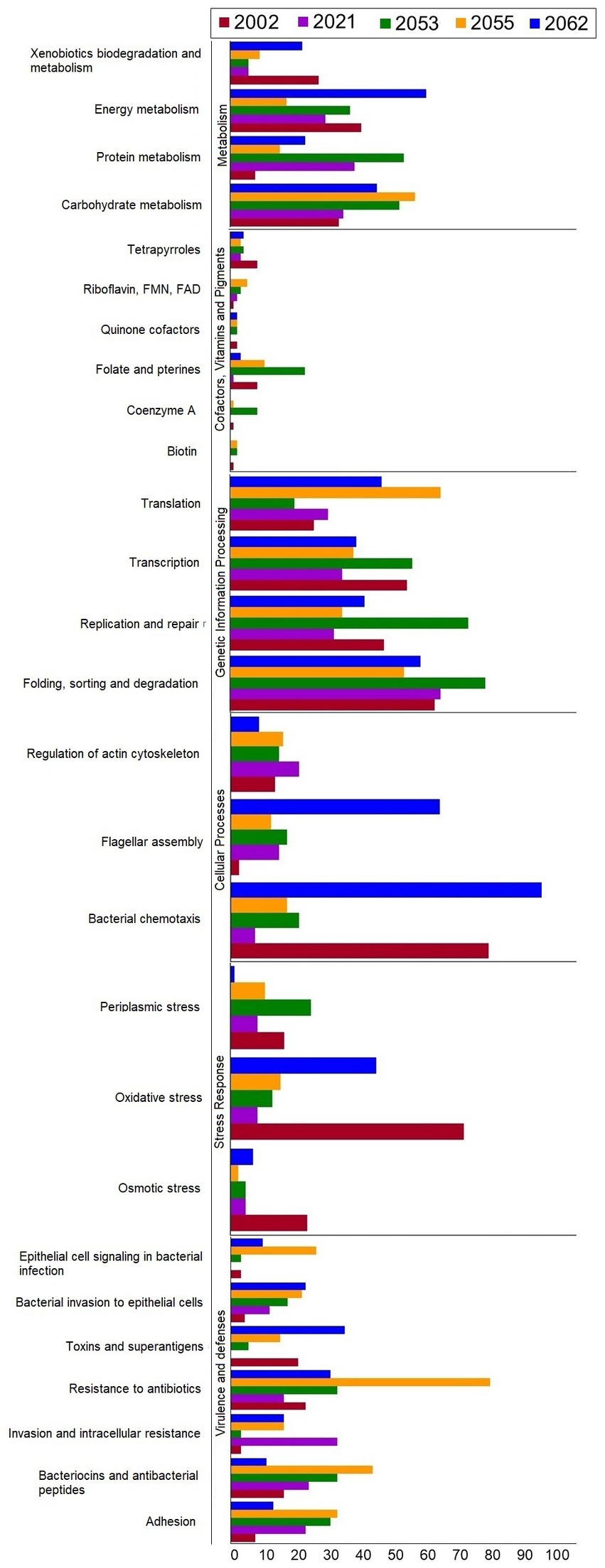
Figure 5. The number of genes categorized by KEGG functional pathways and SEED subsystems annotation of S. uberis strains 2002, 2021, 2053, 2055 and 2062.
Sequence type of the Streptococcus uberis isolates
Whole genome-based sequence typing through the PubMLST website revealed the existence of two novel ST (unknown according to PubMLST) sequences in the S. uberis isolated from Bangladesh (BD). One ST is close to ST474 (isolates 2002, 2021, 2055, and 2063), and the other (2053) was close to several ST sequences of ST152, ST133, ST82 and ST968. Phylogeny of the concatenated ST sequences clustered the former group with sequences described from India, Thailand and China. On the other hand, ST sequences of 2053 closely clustered with isolates from Ireland, UK and Australia (Figure 6).
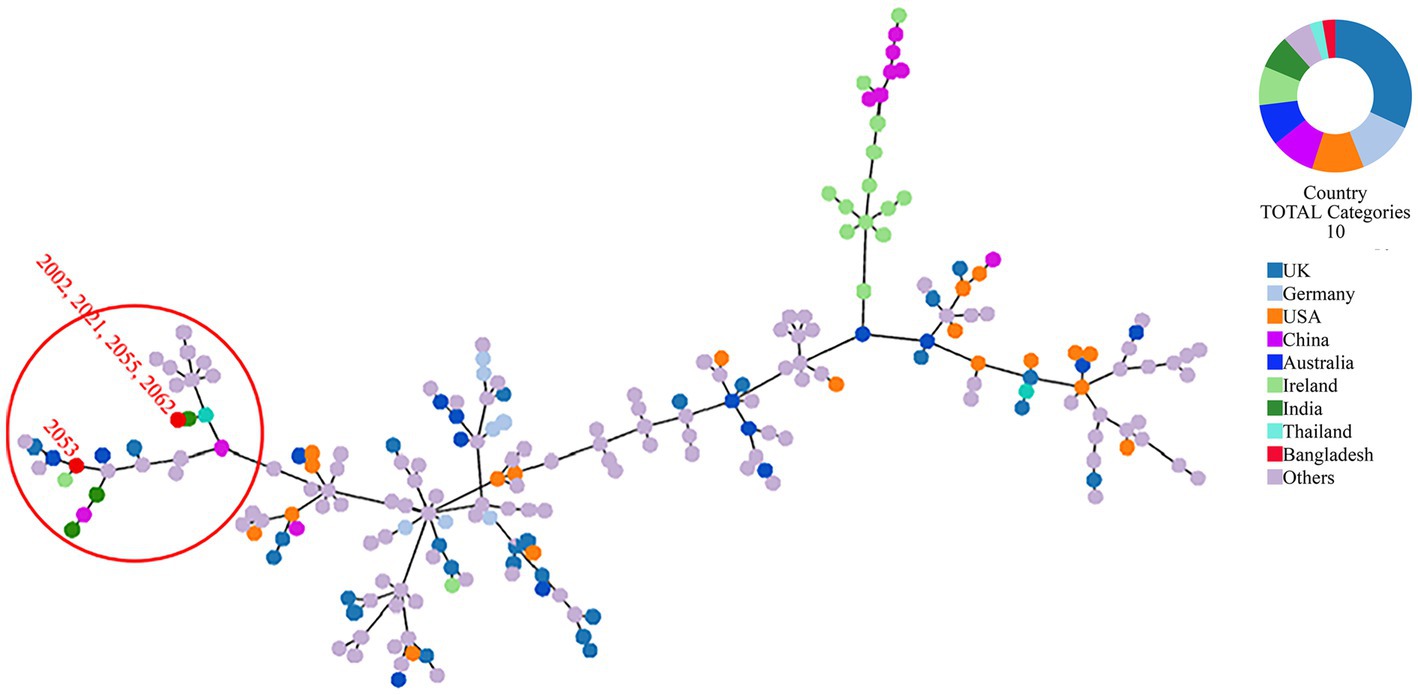
Figure 6. MLST phylogeny of S. uberis sequences obtained in this study. Concatenated ST sequences of S. uberis were downloaded from PubMLST (https://pubmlst.org/organisms/streptococcus-uberis) and the minimum spanning tree was constructed using goeBurst algorithm of Phyloviz (PHYLOViZ Online). Red dot inside the circle denotes isolates described in this study from Bangladesh.
Virulence factor and antibiotic resistance in the Streptococcus uberis isolates
The S. uberis genomes were examined for the presence of sixty-one (61) putative virulence genes described so far. The virulence factors and corresponding orthologs identified in the S. uberis genomes are provided in Figure 7 and Supplementary Table S5. All the isolates recovered in this study carried thirty-five (35) of the virulence genes examined. In addition, strains 2002, 2021, 2055 and 2062 carried five virulence genes such as cps4E/pglC, cpsM, hasA, hasB, and uberolysin encoding genes which were absent in strain 2053. On the other hand, 2053 carried one putative virulence factor, “collagen-like surface-anchored protein,” which was absent in the 4 other isolates. Analysis of the has operon revealed that all four positive isolates carried hasA and hasB gene in chronicles, but the hasC gene was located 3.9 kb downstream of the hasAB and in the opposite direction. In addition, upstream of the VMHJH_06810, VMHJH1_07130, VMHJH2_06655, BAUJH3_05795, BAUJH4_06250 locus of the sequences described in this study, which were homologous to vru (SUB0144) of strain 0140J was analyzed for a four bp deletion region as described earlier (34). Unlikely 0140J, a five bp (TATAA) deletion was observed in the position-75 to-79 in all the S. uberis isolates described in this study (Figure 8).
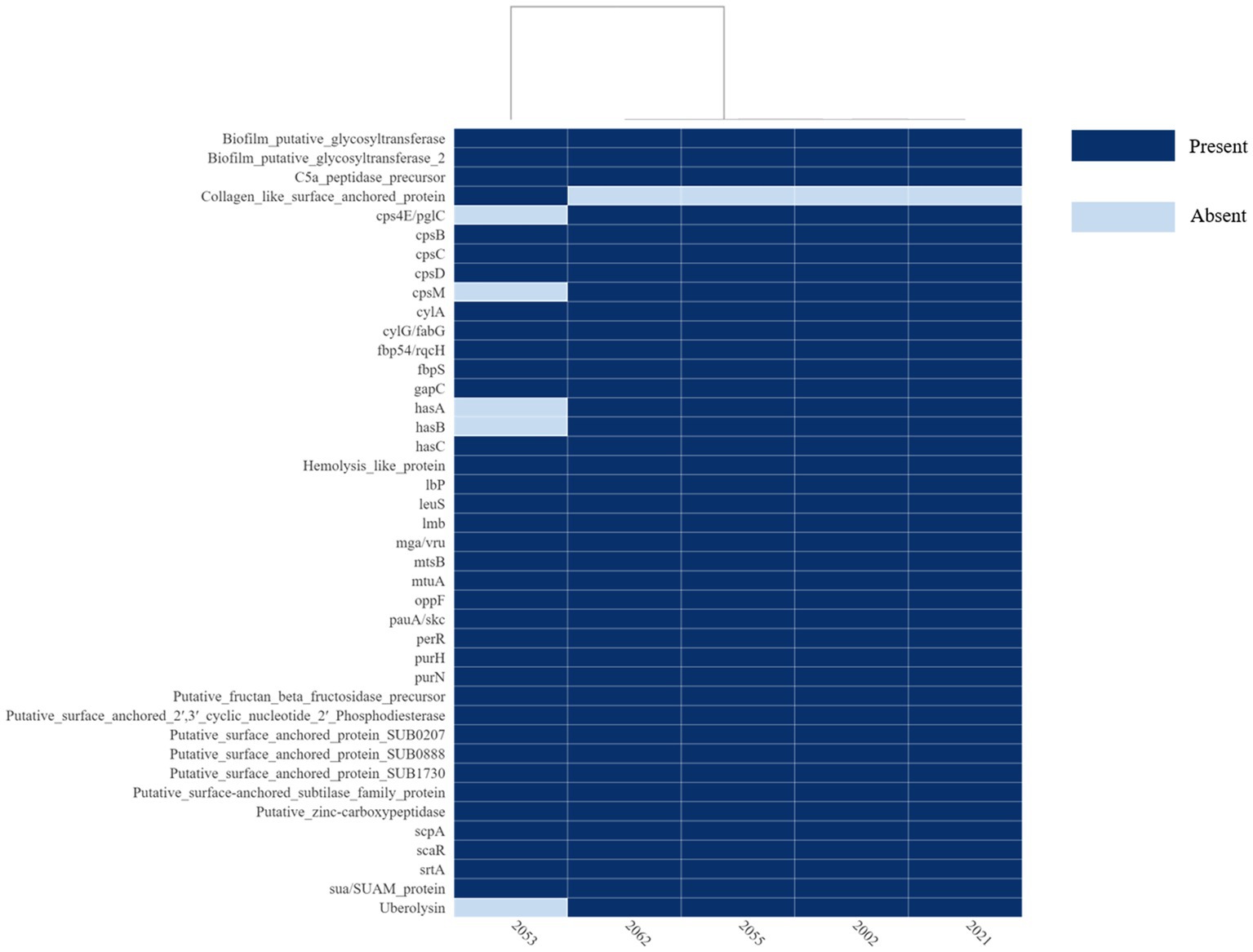
Figure 7. Heatmap of the virulence or putative virulence genes/factors present or absent in the S. uberis genomes from Bangladesh. The isolate ID is in the x-axis and the virulence genes/factors are in the y-axis. The figure was prepared on DISPLAYR (https://southeastasia.displayr.com/) using default parameters and dendrogram appearance.

Figure 8. Analysis of the upstream of vru gene homologues in the S. uberis isolated in this study. Upstream of the vru was identified according to the criteria described (34) and the alignment was performed on CLC Genomic Workbench 22.0. The initiation of the vru homologue is indicated with arrow. Matching residues were replaced with dots and the deleted residues are marked with dashes. Deletion of 5 pb TATAA was observed in all the strain (2002, 2021, 2053, 2055, 2062) described in this study.
Analysis of the S. uberis genome through CARD revealed the presence of tetL, tetM, patA, patB and lnuD genes in all but in strain 2053 only the patA and patB genes were evident. The genes tetL and tetM confer resistance to tetracyclines, patA and patB to fluoroquinolones and lnuD to lincosamide antibiotics. The antibiotic resistance genotype correlates with the phenotypic ciprofloxacin and tetracycline resistance pattern of the isolates described in this study (Table 1).
Analysis of the CRISPR-Cas regions
Streptococcus uberis isolated in this study were examined for CRISPR-Cas regions where the region was found deleted in all the strains except 2053 when compared to that with reference strain 0140J. In strain 2053 a Type II CRISPR/Cas system was identified comprising cas9_cas1_cas2_csn2-st genetic arrangement (Figure 9).
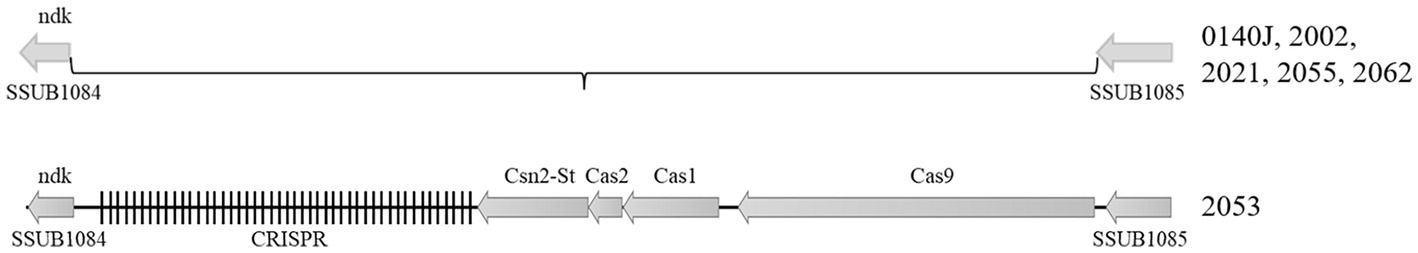
Figure 9. Type-II CRISPR region in the S. uberis isolates. Figure showing the relative position of the CRISPR-Cas genes in the S. uberis genome. S. uberis strain 0140J was used as the reference genome. The CRISPR-Cas system was detected only in strain 2053 among the five S. uberis isolates sequenced in this study.
Prophage and other mobile genetic elements
Prophages are significant in the evolution of bacteria and their virulence. The S. uberis genomes were examined for the presence of prophages through the PHASTER website. All the isolates carried two to four prophages; however, intact prophages were detected only in strain 2053 (Table 3). The strain 2053 carried two intact prophages of 44.8 kb (locus_tag: VMHJH2_06750–07050) and 33.7 kb (locus_tag: VMHJH2_09380–09605) sizes with 39.17% and 37.43% GC contents, respectively. Prophages of 8.6 to 11Kb sizes were present in the other 4 isolates, but they were incomplete (Table 3). In further analysis, the intact phages contained fifty-seven and forty-four protein sequences, respectively, where most of the proteins were phage-related (forty-seven and thirty-five sequences, respectively). No known virulence related genes were found in the prophage sequences. The S. uberis strains did not carry any plasmids but carried a number of insertion sequences (Table 4). None of the insertion sequences was also associated with virulence determinants.

Table 3. Distribution of prophages in the S. uberis isolated in this study.
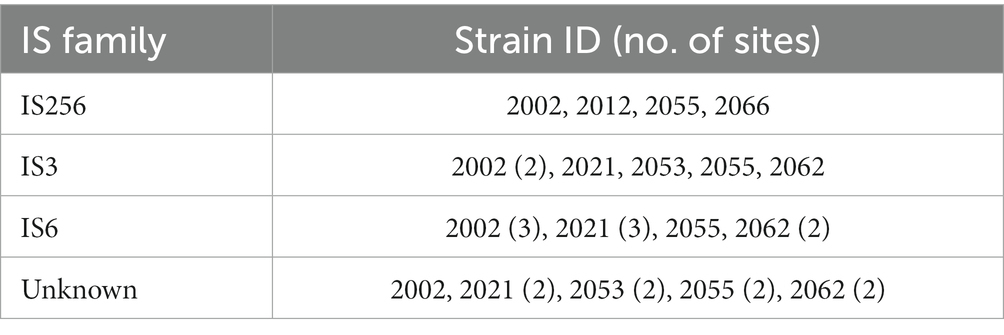
Table 4. Transposon and insertion sequences in the S. uberis isolated in this study.
Discussion
The present study was aimed to the determination of occurrence of Streptococcus spp. and get genetic insight into the S. uberis genome isolated from clinical mastitis of cattle in Bangladesh. Milk samples from clinical mastitis (CM) were collected from major dairy farms and a dairy community in Dhaka, Mymensingh and Sirajganj district of Bangladesh. On cultural examination and sequencing of 16S rRNA gene, milk from 22.5% of the CCM was found positive for Streptococcus spp. The frequency of isolation of Streptococcus recorded in this study is in consent with the worldwide prevalence of Streptococcus spp. in mastitis (4). In this study, four different species of Streptococcus, including S. agalactiae, S. uberis, S. hyovaginalis and S. urinalis with the highest frequency of S. uberis (55.56%) followed by S. agalactiae (33.33%) was identified. The increased occurrence of S. uberis is in consent with current research throughout the world but differs with that reported from Asia where S. agalactiae was predominantly associated with mastitis (4). The difference in the occurrence of S. uberis in mastitis is not impossible; however, to reveal the actual scenario and make a generalized statement, country-wide study with more samples is necessary. Besides, we have encountered two new species S. hyovaginalis and S. urinalis from clinical mastitis for the first time in the world. Although the pathogenicity or association of these two species with mastitis was not studied, they were the sole Streptococcus spp. isolated from the respective milk samples, as well as none of the other samples from CCM or apparently healthy cattle carried these two Streptococcus spp. indicating their possible association with mastitis in the respective cows; however, as we have used composite milk sample their non-pathogenic presence in the unaffected quarter also cannot be excluded. Moreover, aseptic sample collection from mastitis is pretty critical and enrichment of sample might give advantages for contaminants to be selected if the media does not allow growth of the bacteria that cause clinical mastitis. Thus, further studies on their virulence and possible association with mastitis are under investigation in our laboratory.
Antimicrobial resistance (AMR) is a critical problem making mastitis control challenging through systemic or intramammary therapies. Moreover, extensive use of antimicrobials in mastitis control resulted in antimicrobial residue in milk and entering the human body through the food chain (41). In this study, we found that 100% of the S. agalactiae and S. hyovaginalis were MDR, whereas antibiotic resistance was observed in 30% of the S. uberis and none of the S. urinalis. The highest resistance was observed against ciprofloxacin and tetracycline (83.3%), which correlates with the antibiotic uses in the study farms described in our previous study (18) as well as follows the global trends of antibiotic resistance in Streptococcus spp. (42). Interestingly, S. uberis was sensitive to most of the antibiotics tested in this study with 100% sensitivity to ampicillin, amoxicillin, bacitracin, chloramphenicol, gentamicin, vancomycin; 90% to ceftazidime and 80% to penicillin. On the other hand, S. agalactiae showed resistance to penicillin (100%), ceftazidime and gentamicin (66.7%), ampicillin and amoxicillin (33.3%). Increased resistance in S. agalactiae is not unusual, and their increased resistance in comparison to S. uberis and S. dysagalactiae has been reported earlier (4). The overall findings represents a variable resistance pattern of Streptococcus spp., warning for an antibiotic susceptibility testing before prescribing any antibiotic to treat streptococcal mastitis to ensure prudent use and prevent subsequent threat of transferring antibiotic residue to human through milk.
In this study, the prevalence of S. uberis was documented highest (55.6%). Considering the worldwide increasing trend of S. uberis in bovine mastitis (4, 13), this study was further extended to characterize the S. uberis isolates through WGS. Sequencing of the five representative isolates followed by assembly revealed the genome sizes ranging from 1,886,469 bp (strain 2055) to 2,033,655 bp (strain 2053) with 36.4 to 36.5% GC contents, respectively. The genome sizes correspond to the S. uberis genomes reported earlier (33, 34). The variation in genome was investigated and annotation revealed the presence of phages and insertion sequence of variable lengths. The genome of strain 2053 carried two complete phages and a total of around 140 kb phage-related sequences that were absent in the other four genomes, which might have greatly contributed to its increased genome size. Pan-genome analysis, whole genome phylogeny and MLST revealed the presence of two unknown sequence types (STs) of S. uberis in the study populations. Similarly, novel ST types have been reported earlier from different countries (4, 33, 34, 43), which might be related to the limited databases of S. uberis genomes. The clustering of four out of five isolates with India, Thailand and China indicated the circulation of similar STs in this subcontinent. Moreover, the S. uberis strains isolated and sequenced in this study was found to sharing more common regions of genetic variation with the reference genomes 0140J at different sites on the genome. The great genomic resemblance between the study strains and the S. uberis strain 0140J, another mastitis clone from the European continent indicates the possible common ancestry between these lineages, despite the wide geographical distance where the strains were originated. However, with the few isolates analyzed in this study an overall diversity of Streptococcus could not be completely described; thus, further country wide studies with more samples and isolates are recommended to elucidate the genomic diversity of S. uberis circulating in Bangladesh as well as predominance of certain STs in this sub-continent.
Streptococcus uberis is known to establish a successful intramammary infection through the exploitation of different virulence factors associated with invasion of mammary epithelium, evasion of host immunity, binding antimicrobial compounds, biofilm formation through the acquisition of amino acids by breaking milk protein casein (4). Although the virulence determinant in S. uberis is not completely understood; several putative virulence determinants have been described previously (4, 33, 34, 43). Considering the previous reports and S. uberis putative virulence database (suPVDB), a total of sixty-one (61) putative virulence factors were investigated, where forty-one (41) virulence factors were identified in the studied genomes. Among the virulence factors thirty-five virulence factors were common to all, including genes encoding for major virulence factors relating to the adhesion to epithelial cells (srtA, fbp54/rqcH and fbps), colonization (lmb), invasion/penetration and dissemination (pauA/skc), immuno invasion through the synthesis of hyalurinic acid capsule (hasC) and delayed accumulation of lymphocytes (scpA and C5a peptidase), biofilm formation (cpsBCD), hemolytic activity (cylAG and hemolysis like protein) and virulence regulator (mga/vru) which is known to regulate a number of phenotypes such as growth, biofilm formation and resistance to phagocytosis (33, 44, 45).
In addition, five (cps4E/pglC, cpsM, hasA, hasB and uberolysin encoding gene) were native to the strains 2002, 20,021, 2055 and 2066. HasABC is associated with the formation of the hyaluronic acid capsule, which was required for the prevention of opsonization and phagocytosis as well as escaping neutrophil traps (46); however, some authors claimed they are non-essential or play a minor role in the induction of mastitis by S. uberis (33, 47, 48). Similarly, bacteriocin provides competitive benefits to the bacterium for surviving in a complex environment by inhibiting the growth of other bacteria. The genome 2053 carried five different genes associated with bacteriocin production, similar to the other genome in this study except for uberolysin. The absence of uberolysin might not render strain 2053 avirulent but may give additional benefits or fitness to the other isolates carrying the gene; however, further investigations are necessary to establish the associations.
The vru gene was reported to regulate a number of virulence genes, and inactivation of this gene was responsible for reduced mammary gland colonization and clinical signs (49). A four bp deletion at the upstream (−76 to −79) was speculated to play an important role in the regulatory function of vru gene and subsequently influence host-pathogen interactions (34). In this study, all five genomes had a five bp deletion (TATAA) at −75 to −79 positions similar to that reported in EF20, a non-virulent S. uberis strain reported from the UK (34). Considering the putative virulence gene makeup, although strain 2053 closely clustered with EF20, they are genetically different, and the deletion at upstream of the vru gene might not be critical to the virulence of the isolates. However, as we did not perform phenotypic studies, making any concrete statement from our findings might be misleading. Thus, further phenotypic experiments with isolates having different variations in the vru upstream would provide comprehensive information on its association with the regulation of vru gene and virulence of S. uberis in bovine mastitis.
We further sought to determine potential metabolic functions of the five S. uberis strains described in this study. The KEGG pathways and SEED subsystems analysis uncovered significant differences in metabolic functions among the study strains. All KEGG and SEED subsystem characterized proteins in five strains were assigned into six different categories representing 27 subcategories. Of the identified subcategories, genes associated with the carbohydrate, protein and energy metabolism, translation, transcription, bacterial chemotaxis, flagellar assembly and oxidative stress remained highest in the study genomes, which might be associated with host-pathogen interactions during the progression of mastitis. These findings corroborated with many of the earlier studies on bovine mastitis pathogens (50–52). Further work is needed to verify these findings and metabolic gene reservoirs and the mechanism of transfer across different strains and/or with their hosts.
Conclusion
Genomic information of S. uberis generated in this study revealed two novel STs and possible virulence makeup that will significantly contribute to the development of new tools for virulence profiling and ST typing of S. uberis. The sequence data generated in this study could also be used for the exploration of the genomic background behind the evolution of this pathogen and the story behind its enhanced environmental fitness over time.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The animal study was reviewed and approved by Animal Welfare and Experimentation Ethics Committee (AWEEC), Bangladesh Agricultural University, Mymensingh-2,202, Bangladesh Approval No. AWEEC/BAU/2020(45). Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
JH and MSK received the project funds, planned research and manage laboratory activities. Lab work was conducted by JH, MAB, and MWA. Analysis of genomic data and curation was performed by JH, MMH, MNH, and AK. The manuscript was drafted and revised by JH, AK, MTR, MSI, and MNH. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by Bangladesh Agricultural University Research System (BAURES) (Project no. 2021/21/BAU).
Acknowledgments
The authors extend their thanks to bioRxiv for publishing preprint of this manuscript. The preprint is available at https://www.biorxiv.org/content/10.1101/2022.12.19.521080v1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1198393/full#supplementary-material
Footnotes
1. ^https://en.banglapedia.org/index.php/Dairy_Farm
2. ^https://rast.nmpdr.org/rast.cgi
References
1. Phys.Org. (2022). Potential biomarkers of mastitis in dairy cattle milk identified. (Accessed June 7, 2023).
2. Kebede, N, and Tilahun, A. Review on dairy cow mastitis and its economic impact. Int J Adv Res Biol Sci. (2023) 10:109–25. doi: 10.22192/ijarbs
3. Bradley, AJ. Bovine mastitis: an evolving disease. Vet J. (2002) 164:116–28. doi: 10.1053/TVJL.2002.0724
4. Kabelitz, T, Aubry, E, van Vorst, K, Amon, T, and Fulde, M. The role of Streptococcus spp. in bovine mastitis. Microorganisms. (2021) 9:1497. doi: 10.3390/MICROORGANISMS9071497
5. Cheng, WN, and Han, SG. Bovine mastitis: risk factors, therapeutic strategies, and alternative treatments—a review. Asian-Australas J Anim Sci. (2020) 33:1699–713. doi: 10.5713/AJAS.20.0156
6. Cojkic, A, Cobanovic, N, Suvajdzic, B, Savic, M, Petrujkic, B, and Dimitrijevic, V. Chronic mastitis in cows caused by Streptococcus dysgalactiae: case report. Vet Glas. (2015) 69:303–14. doi: 10.2298/VETGL1504303C
7. Jain, B, Tewari, A, Bhandari, BB, and Jhala, MK. Antibiotic resistance and virulence genes in Streptococcus agalactiae isolated from cases of bovine subclinical mastitis. Vet Arh. (2012) 82:423–32.
8. Nikolova, M, Urumova, V, and Liuzkanov, M. Streptococcus spp. as etiological agent of subclinical and clinical mastitis of dairy cows in Republic of Bulgaria. Trakia J Sci. (2022) 20:113–8. doi: 10.15547/TJS.2022.02.005
9. Alnakip, MEA, Rhouma, NR, Abd-Elfatah, EN, Quintela-Baluja, M, Böhme, K, Fernández-No, I, et al. Discrimination of major and minor streptococci incriminated in bovine mastitis by MALDI-TOF MS fingerprinting and 16S rRNA gene sequencing. Res Vet Sci. (2020) 132:426–38. doi: 10.1016/J.RVSC.2020.07.027
10. Benić, M, Maćešić, N, Cvetnić, L, Habrun, B, Cvetnić, Ž, Turk, R, et al. Bovine mastitis: a persistent and evolving problem requiring novel approaches for its control-a review. Vet Arh. (2018) 88:535–57. doi: 10.24099/vet.arhiv.0116
11. Hillerton, JE, and Berry, EA. The management and treatment of environmental streptococcal mastitis. Vet Clin North Am Food Anim Pract. (2003) 19:157–69. doi: 10.1016/S0749-0720(02)00069-5
12. Kalińska, A, Gołębiewski, M, and Wójcik, A. Mastitis pathogens in dairy cattle—a review. World Sci News. (2017) 89:22–31.
13. Cvetnic, L, Samardžija, M, Habrun, B, Kompes, G, and Benić, M. Microbiological monitoring of mastitis pathogens in the control of udder health in dairy cows. Slov Vet Res. (2016) 53:131–40.
14. Hasan, MT, Islam, MR, Runa, NS, Hasan, MN, Uddin, AHMM, and Singh, SK. Study on bovine sub-clinical mastitis on farm condition with special emphasis on antibiogram of the causative bacteria. Bangladesh J Vet Med. (2017) 14:161–6. doi: 10.3329/BJVM.V14I2.31386
15. Rahman, MM, Islam, M, Uddin, M, and Aktaruzzaman, M. Prevalence of subclinical mastitis in dairy cows reared in Sylhet district of Bangladesh. Int J Biol Res. (2010) 1:23–8.
16. Rahman, MT, Islam, MS, and Hasan, M. Isolation and identification of bacterial agents causing clinical mastitis in cattle in Mymensingh and their antibiogram profile. Microbes Heal. (2013) 2:19–21. doi: 10.3329/mh.v2i1.17258
17. Uddin, MM, Sultana, MN, Bruermer, B, and Peters, KJ. Assessing the impact of dairy policies on farm-level profits in dairy farms in Bangladesh: benchmarking for rural livelihoods improvement policy. J Rev Glob Econ. (2012) 1:124–38. doi: 10.6000/1929-7092.2012.01.11
18. Bag, MAS, Khan, MSR, Sami, MDH, Begum, F, Islam, MS, Rahman, MM, et al. Virulence determinants and antimicrobial resistance of E. coli isolated from bovine clinical mastitis in some selected dairy farms of Bangladesh. Saudi J Biol Sci. (2021) 28:6317–23. doi: 10.1016/J.SJBS.2021.06.099
19. Huma, ZI, Sharma, N, Kour, S, and Lee, SJ. Phenotypic and molecular characterization of bovine mastitis milk origin bacteria and linkage of intramammary infection with milk quality. Front Vet Sci. (2022) 9:565. doi: 10.3389/FVETS.2022.885134/XML/NLM
20. Saed, HAEMR, and Ibrahim, HMM. Antimicrobial profile of multidrug-resistant Streptococcus spp. isolated from dairy cows with clinical mastitis. J Adv Vet Anim Res. (2020) 7:186–97. doi: 10.5455/JAVAR.2020.G409
21. Bag, MAS, Arif, M, Riaz, S, Khan, MSR, Shafiqul, IM, Punom, SA, et al. Antimicrobial resistance, virulence profiles, and public health significance of Enterococcus faecalis isolated from clinical mastitis of cattle in Bangladesh. Biomed Res Int. (2022) 2022:1–8. doi: 10.1155/2022/8101866
22. Alves-Barroco, C, Roma-Rodrigues, C, Raposo, LR, Brás, C, Diniz, M, Caço, J, et al. Streptococcus dysagalactiae subsp. dysagalactiae isolated from milk of the bovine udder as emerging pathogens: in vitro and in vivo infection of human cells and zebrafish as biological models. Microbiology. (2019) 8:e00623. doi: 10.1002/mbo3.623
23. Baron, F, Cochet, MF, Pellerin, JL, Ben Zakour, N, Lebon, A, Navarro, A, et al. Development of a PCR test to differentiate between Staphylococcus aureus and Staphylococcus intermedius. J Food Prot. (2004) 67:2302–5. doi: 10.4315/0362-028x-67.10.2302
24. Dutka-Malen, S, Evers, S, and Courvalin, P. Detection of glycopeptide resistance genotypes and identification to the species level of clinically relevant enterococci by PCR. J Clin Microbiol. (1995) 33:24–7. doi: 10.1128/jcm.33.1.24-27.1995
25. Hassan, AA, Khan, UI, Abdulmawjood, A, and Lammler, C. Evaluation of PCR methods for rapid identification and differentiation of Streptococcus uberis and Streptococcus parauberis. J Clin Microbiol. (2001) 39:1618–21. doi: 10.1128/JCM.39.4.1618-1621.2001
26. Khamis, A, Raoult, D, and La Scola, B. RpoB gene sequencing for identification of Corynebacterium species. J Clin Microbiol. (2004) 42:3925–31. doi: 10.1128/JCM.42.9.3925-3931.2004
27. Martinez, G, Harel, J, and Gottschalk, M. Specific detection by PCR of Streptococcus agalactiae in milk. Can J Vet Res. (2001) 65:68–72.
28. Wang, RF, Cao, WW, and Cerniglia, CE. PCR detection and quantitation of predominant anaerobic bacteria in human and animal fecal samples. Appl Environ Microbiol. (1996) 62:1242–7. https://doi:10.1128/aem.62.4.1242-1247.1996. doi: 10.1128/aem.62.4.1242–1247.1996
29. Hahne, J, Kloster, T, Rathmann, S, Weber, M, and Lipski, A. Isolation and characterization of Corynebacterium spp. from bulk tank raw cow’s milk of different dairy farms in Germany. PLoS One. (2018) 13:e0194365. doi: 10.1371/JOURNAL.PONE.0194365
30. Bauer, A, Kirby, W, Sherris, J, and Turch, M. Antibiotic susceptibility testing by a standardized single disk method. Am J Clin Pathol. (1966) 45:493–6. doi: 10.1093/ajcp/45.4_ts.493
31. CLSI. Performance standards for antimi-crobial susceptibility testing, vol. 40. 30th ed CLSI Supplement M100 Clinical and Laboratory Standards Institute (2020). 2020 p. Available at: https://webstore.ansi.org/Standards/CLSI/CLSIM100Ed30
32. Magiorakos, AP, Srinivasan, A, Carey, RB, Carmeli, Y, Falagas, ME, Giske, CG, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. (2012) 18:268–81. doi: 10.1111/j.1469-0691.2011.03570.x
33. Vezina, B, Al-harbi, H, Ramay, HR, Soust, M, Moore, RJ, Olchowy, TWJ, et al. Sequence characterisation and novel insights into bovine mastitis-associated Streptococcus uberis in dairy herds. Sci Rep. (2021) 11:3046. doi: 10.1038/S41598-021-82357-3
34. Hossain, M, Egan, SA, Coffey, T, Ward, PN, Wilson, R, Leigh, JA, et al. Virulence related sequences; insights provided by comparative genomics of Streptococcus uberis of differing virulence. BMC Genomics. (2015) 16:334. doi: 10.1186/S12864-015-1512-6
35. Kaczorek, E, Małaczewska, J, Wójcik, R, and Siwicki, AK. Biofilm production and other virulence factors in Streptococcus spp. isolated from clinical cases of bovine mastitis in Poland. BMC Vet Res. (2017) 13:398. doi: 10.1186/S12917-017-1322-Y
36. Contreras-Moreira, B, and Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol. (2013) 79:7696–701. doi: 10.1128/AEM.02411-13
37. Page, AJ, Cummins, CA, Hunt, M, Wong, VK, Reuter, S, Holden, MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. (2015) 31:3691–3. doi: 10.1093/BIOINFORMATICS/BTV421
38. Alikhan, NF, Petty, NK, Ben Zakour, NL, and Beatson, SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. (2011) 12:402. doi: 10.1186/1471-2164-12-402
39. Kanehisa, M, and Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. (2000) 28:27–30. doi: 10.1093/nar/28.1.27
40. Overbeek, R, Olson, R, Pusch, GD, Olsen, GJ, Davis, JJ, Disz, T, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. (2014) 42:D206–14. doi: 10.1093/nar/gkt1226
41. Gomes, F, and Henriques, M. Control of bovine mastitis: old and recent therapeutic approaches. Curr Microbiol. (2016) 72:377–82. doi: 10.1007/S00284-015-0958-8
42. Haenni, M, Lupo, A, and Madec, JY. Antimicrobial resistance in Streptococcus spp. Microbiol Spectr. (2018) 6. doi: 10.1128/MICROBIOLSPEC.ARBA-0008-2017
43. Silva, NCC, Yang, Y, Rodrigues, MX, Tomazi, T, and Bicalho, RC. Whole-genome sequencing reveals high genetic diversity of Streptococcus uberis isolated from cows with mastitis. BMC Vet Res. (2021) 17:321. doi: 10.1186/S12917-021-03031-4
44. Cho, KH, and Caparon, MG. Patterns of virulence gene expression differ between biofilm and tissue communities of Streptococcus pyogenes. Mol Microbiol. (2005) 57:1545–56. doi: 10.1111/J.1365-2958.2005.04786.X
45. Le Breton, Y, Belew, AT, Freiberg, JA, Sundar, GS, Islam, E, Lieberman, J, et al. Genome-wide discovery of novel M1T1 group a streptococcal determinants important for fitness and virulence during soft-tissue infection. PLoS Pathog. (2017) 13:e1006584. doi: 10.1371/JOURNAL.PPAT.1006584
46. Cole, JN, Pence, MA, von Köckritz-Blickwede, M, Hollands, A, Gallo, RL, Walker, MJ, et al. M protein and hyaluronic acid capsule are essential for in vivo selection of cov RS mutations characteristic of invasive serotype M1T1 group a Streptococcus. mBio. (2010) 1:e00191. doi: 10.1128/MBIO.00191-10
47. Field, TR, Ward, PN, Pedersen, LH, and Leigh, JA. The hyaluronic acid capsule of Streptococcus uberis is not required for the development of infection and clinical mastitis. Infect Immun. (2003) 71:132–9. doi: 10.1128/IAI.71.1.132-139.2003
48. Ward, PN, Holden, MTG, Leigh, JA, Lennard, N, Bignell, A, Barron, A, et al. Evidence for niche adaptation in the genome of the bovine pathogen Streptococcus uberis. BMC Genomics. (2009) 10:54. doi: 10.1186/1471-2164-10-54
49. Flores, AR, Olsen, RJ, Wunsche, A, Kumaraswami, M, Shelburne, SA, Carroll, RK, et al. Natural variation in the promoter of the gene encoding the Mga regulator alters host-pathogen interactions in group a Streptococcus carrier strains. Infect Immun. (2013) 81:4128–38. doi: 10.1128/IAI.00405-13
50. Hoque, MN, Istiaq, A, Clement, RA, Sultana, M, Crandall, KA, Siddiki, AZ, et al. Metagenomic deep sequencing reveals association of microbiome signature with functional biases in bovine mastitis. Sci Rep. (2019) 9:13536. doi: 10.1038/s41598-019-49468-4
51. Lee, Y, Nguyen, TL, Kim, A, Kim, N, Roh, HJ, Han, HJ, et al. Complete genome sequence of multiple-antibiotic-resistant Streptococcus parauberis strain SPOF3K, isolated from diseased olive flounder (Paralichthys olivaceus). Genome Announc. (2018) 6:e00248–18. doi: 10.1128/genomeA.00248-18
Keywords: Streptococcus uberis, diversity, clinical mastitis, cattle, WGS
Citation: Hassan J, Bag MAS, Ali MW, Kabir A, Hoque MN, Hossain MM, Rahman MT, Islam MS and Khan MSR (2023) Diversity of Streptococcus spp. and genomic characteristics of Streptococcus uberis isolated from clinical mastitis of cattle in Bangladesh. Front. Vet. Sci. 10:1198393. doi: 10.3389/fvets.2023.1198393
Edited by:
Getahun E. Agga, Agricultural Research Service (USDA), United StatesReviewed by:
Yasmine Hasanine Tartor, Zagazig University, EgyptOudessa Kerro Dego, The University of Tennessee, United States
Copyright © 2023 Hassan, Bag, Ali, Kabir, Hoque, Hossain, Rahman, Islam and Khan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jayedul Hassan, ZHJfamFoaWRAYmF1LmVkdS5iZA==