 Jeanette B. Black
Jeanette B. Black Arthur R. Frampton
Arthur R. Frampton
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci. , 19 May 2023
Sec. Veterinary Infectious Diseases
Volume 10 - 2023 | https://doi.org/10.3389/fvets.2023.1165917
This article is part of the Research Topic Herpesviruses of Animals: Recent Advances and Updates View all 7 articles
Equine herpesvirus type 1 (EHV-1) is a highly transmissible pathogen that leads to a variety of clinical disease outcomes in infected horses. A major sequela that can occur after an EHV-1 infection is a neurological disease termed equine herpesvirus myeloencephalopathy (EHM). Clinical manifestations of EHM include fever, ataxia, incontinence, and partial to full paralysis, which may ultimately lead to the euthanization of the infected horse. To develop an effective treatment strategy for EHM, it is critical that the specific virus–host interactions that lead to EHM be investigated so that safe and effective therapeutic interventions can be developed and delivered. In this study, we examined the ability of four non-steroidal anti-inflammatory drugs (NSAIDs), a steroidal anti-inflammatory drug (dexamethasone), a Rho-kinase (ROCK) inhibitor, and a JAK/STAT inhibitor (AG490) to reduce EHV-1 virus yields and cell-to-cell spread. We show that the NSAID, flunixin meglumine (FM), and the JAK/STAT inhibitor, AG490, significantly reduced virus yields in endothelial and epithelial cell lines, and this inhibition was similar for two neurologic and two non-neurologic EHV-1 strains. In addition to reducing virus yields, AG490 and FM also significantly reduced the ability of EHV-1 to spread laterally from cell to cell.
Equine herpesvirus type 1 (EHV-1) is a ubiquitous virus that causes considerable morbidity and mortality in horses (1–3). Most horses are infected with EHV-1 in their first year of life and exhibit clinical signs that include respiratory distress, mucopurulent discharge, fever, and malaise. In addition to infection in the upper respiratory tract, EHV-1 can also cause abortigenic and/or neurologic disease. Disease outcomes or sequelae associated with EHV-1 can vary quite dramatically depending on various host and viral factors. One viral determinant that is positively correlated with a neurologic disease outcome is the presence of a point mutation within the viral DNA polymerase gene of the infecting strain (4–7). EHV-1 strains that contain a point mutation (A2254 > G2254) within the DNA polymerase (DNA Pol) gene have been classified as neurologic, while those that do not harbor this mutation are classified as non-neurologic. Data collected from various groups have shown that EHV-1 strains with this DNA Pol mutation are responsible for the majority of EHM cases (5, 8). EHV-1 strains that harbor this mutation replicate more efficiently in peripheral blood mononuclear cells (PBMCs) and establish a longer viremia, which may result in an increased ability of EHV-1 to reach and productively infect the vascular endothelium in the CNS (9–11).
The increased infection of the CNS endothelium leads to the induction and release of significant amounts of pro-inflammatory cytokines from endothelial cells which results in the recruitment of lymphocytes and natural killer (NK) cells to the infected site. This hyper-inflammation causes thrombosis, ischemia, and hypoxia in the CNS and damages neuronal cells (12–15). The increased incidence of EHM observed in horses infected with a neurologic strain led us to hypothesize that the neurologic strains would induce higher levels of damaging pro-inflammatory cytokines compared to the non-neurologic strains. However, in a previous study (16), when we examined the expression of 49 host immune genes in EHV-1 infected endothelial cells, we observed similar expression of these genes in cells infected with neurologic and non-neurologic strains. Therefore, these results indicate that the induction of pro-inflammatory cytokines is similar from one strain to the next and suggest that any EHV-1 strain, regardless of their designation as neurologic or non-neurologic, can cause inflammation that contributes to EHM when the virus can gain access to the CNS.
Due to the hyper-inflammation associated with EHV-1, various anti-inflammatory drugs have been used as part of a therapeutic regimen to treat EHV-1-infected horses (12). While these drugs were developed to target inflammation, studies have also shown that some anti-inflammatory drugs have the added benefit of blocking or reducing the replication and spread of multiple herpesviruses, such as pseudorabies virus (PRV) (17, 18), Marek's disease virus (MDV) (19), and cytomegalovirus (CMV) (20, 21). In addition to reducing replication and cell-to-cell spread, inhibition of the inflammatory mediator cyclooxygenase-2 (COX-2) also reduced the lytic reactivation of Epstein–Barr virus (EBV).
JAK/STAT inhibitors have also been shown to inhibit a variety of viruses. HIV replication (22), viral reservoir establishment (23), and encephalitis (24) have all been reduced by JAK/STAT inhibitors. In addition to HIV, Zika, and SARS-CoV-2, virus replication was also inhibited in the presence of JAK/STAT inhibitors (25, 26). Previous studies have also shown that the level of EHV-1 infection is negatively impacted by the host interferon response and that exogenous administration of recombinant equine interferon alpha reduced EHV-1 replication in primary equine respiratory epithelial cells (27). Together, these drug studies provide evidence that anti-inflammatory drugs, in addition to reducing inflammation, also have the added benefit of interfering with host cell factors that contribute to optimum virus infectivity, replication, and propagation. Based on data collected from these drug studies, we hypothesized that the administration of anti-inflammatory drugs and JAK/STAT inhibitors would reduce EHV-1 viral replication and cell-to-cell spread. To test this hypothesis, we used a cell culture model to investigate the ability of multiple compounds to reduce EHV-1 replication and cell-to-cell spread. We show that both the JAK/STAT pathway inhibitor, AG490, and the NSAID flunixin meglumine (FM) significantly inhibited virus replication and reduced cell-to-cell spread.
Rabbit kidney-13 (RK-13) cells were provided by Dennis O'Callaghan (Louisiana State University Health Sciences Center, Shreveport, Louisiana, USA), and equine cardiac artery endothelial (EE) cells were provided by Udeni Balasuriya (Louisiana State University, Baton Rouge, Lousiana, USA). All cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 2% of 10,000 U/ml penicillin and 10,000 μg/ml streptomycin in normal saline. Cells were cultured at 37°C in 5% CO2. EHV-1 strains T220, KyA, T953, and T967 were used in this study. T220 and KyA are non-neurologic, and T953 and T967 are neurologic strains based on the absence or presence, respectively, of the DNA polymerase mutation (A2254 > G2254). T220 and T967 were provided by Udeni Balasuriya. KyA was provided by Dennis O'Callaghan, and T953 (Ohio 2003) was provided by Gillian Perkins (Cornell University, Ithaca, New York, USA). Compounds tested include AG490 (JAK/STAT inhibitor), flunixin meglumine, ketoprofen, phenylbutazone, firocoxib (NSAIDs), and Y-27632 (Rho-kinase inhibitor). All compounds were purchased from Avantor (Avantor, Radnor, Pennsylvania, USA).
EE and RK-13 cells were seeded at 2.5 × 105 cells/well in a 24-well plate. At 24 h post-seeding, cells were treated with 100 μM each of AG490, flunixin meglumine, dexamethasone, ketoprofen, phenylbutazone, Y-27632, and firocoxib in DMEM containing 5% FBS. Control cells were treated with 1% DMSO. Cells were incubated for 24 h at 37°C and 5% CO2. Twenty-four hours after drug treatment, cells were infected with each EHV-1 strain at an MOI of 0.1 or 1 and incubated at 37°C and 5% CO2 for 24 h. Twenty-four hours post-infection, the supernatants were collected, and then 100 μl of trypsin was added to each well to detach any cells adhering to the well. The trypsin containing the cells was then added to the supernatants, and the samples were centrifuged at 13,000 RPM for 10 minutes. Supernatants were transferred to new 1.7-ml microcentrifuge tubes and stored on ice. The remaining cell pellets were re-suspended in 100 μl of their respective supernatant, and the samples were subjected to three freeze–thaw cycles between −80 and 37°C to release cell-associated virus. After three freeze–thaw cycles, the disrupted cell pellets were added back to their respective supernatants, and the samples were stored at −80°C.
Each sample was thawed in a 37°C water bath, and then 10-fold dilutions of each sample were made in DMEM. In total, 100 μl of each dilution was added to confluent monolayers of RK-13 cells in a 24-well plate, after which the titer plates were incubated at 37°C and 5% CO2. After a 1.5-h attachment period, cells were overlaid with 1 ml of semi-solid DMEM containing 1% methylcellulose and 10% FBS, and the cells were incubated for 72 h at 37°C and 5% CO2. After the 72 h incubation, the semi-solid medium was removed by aspiration, and the cells were stained with 1% crystal violet for 1 h. Crystal violet was rinsed off the cells, and the plates were inverted and allowed to dry overnight. The following day, plaques were counted, and the titers were determined for each sample and recorded as plaque-forming units per ml (pfu/ml). Three biological replicates were run for each condition. The mean titers of the three biological replicates of the drug-treated cells were compared to the non-drug-treated (1% DMSO) cells, and a Student's t-test was performed to determine whether the mean virus titers were significantly different between the two groups. p-values of ≤ 0.05 were considered statistically significant.
AG490 and FM were also added to RK-13 cells post-infection. RK-13 cells were seeded at 2.5 × 105 cells/well in a 24-well plate and then infected with each EHV-1 strain at an MOI of 0.1 for 24 h at 37°C and 5% CO2. Twenty-four hours post-infection, supernatants, and cell-associated viruses were collected and combined as described for the pre-drug treatment assays. Three biological replicates were run for each condition, and the mean titers of the three biological replicates of the drug-treated cells were compared to the non-drug-treated (1% DMSO) cells. A Student's t-test was performed to determine whether the mean virus titers were significantly different between the two groups. p-values of ≤ 0.05 were considered statistically significant.
RK-13 cells were seeded at 5 × 105 cells/well in 6-well plates which were then infected the next day with 100 pfu of T967, T220, T953, or KyA for 2 h. Following infection, media and virus were aspirated from each well, and a 1% methylcellulose solution containing 10% FBS was overlaid on the cell monolayers. Infected cells were incubated for 24 h at 37°C and 5% CO2, then each compound (100 μM) was mixed with the methylcellulose solution, added to the cells, and the cells were incubated for 24 h at 37°C and 5% CO2. After 24 h, another dose (100 μM) of the compound was added, and the cells were incubated for another 24 h at 37°C and 5% CO2. At 72 h post-infection, methylcellulose was aspirated from the wells, and the cells were fixed and stained with 1% crystal violet for 1 h and then rinsed with water. The wells were allowed to dry overnight, and the plaques were imaged using an Olympus Fluoview microscope. For each drug-treated group, a total of eight images (four quadrants per image) were processed using the Java-based image processing program, Image J (IJ), and ViralPlaque software. The size of the plaques in the drug-treated, infected cells was analyzed and compared with the size of the plaques that formed in the 1% DMSO control group. For each treatment, a total of 100 plaques were measured to determine the mean, standard deviation, and standard error.
Five anti-inflammatory drugs (flunixin meglumine, dexamethasone, firocoxib, ketoprofen, and phenylbutazone), a Rho-kinase (ROCK) inhibitor (Y-27632), and a JAK/STAT inhibitor (AG490) were investigated for their ability to reduce EHV-1 replication and cell-to-cell spread. These drugs were tested against four EHV-1 strains, on two cell lines (epithelial and endothelial), at two multiplicities of infection (MOIs). The EHV-1 strains consisted of two non-neurological strains, T220 and KyA, and two neurological strains, T953 (Ohio 2003) and T967. In the initial virus yield assay, equine endothelial (EE) and rabbit kidney (RK-13) epithelial cells were treated with each drug for 24 h and then infected with each EHV-1 strain at a multiplicity of infection (MOI) of 0.1 and 1 for 24 h; then, the total amount of virus produced (virus yield) was measured as described in the Materials and methods section. All experiments were run in triplicate (three biological runs), and the mean virus yields in each group were compared to the control cells that were only given the solvent DMSO and infected with EHV-1.
The measured virus yields are shown in Figures 1–4. Figure 1 shows the 24 h yields obtained from EE cells that were infected with an MOI of 0.1, and Figure 2 shows the 24 h yields obtained from EE cells infected at an MOI of 1. Figure 3 shows the 24 h yields obtained from RK-13 cells that were infected at an MOI of 0.1, and Figure 4 shows the 24 h yields obtained from RK-13 cells infected at an MOI of 1.
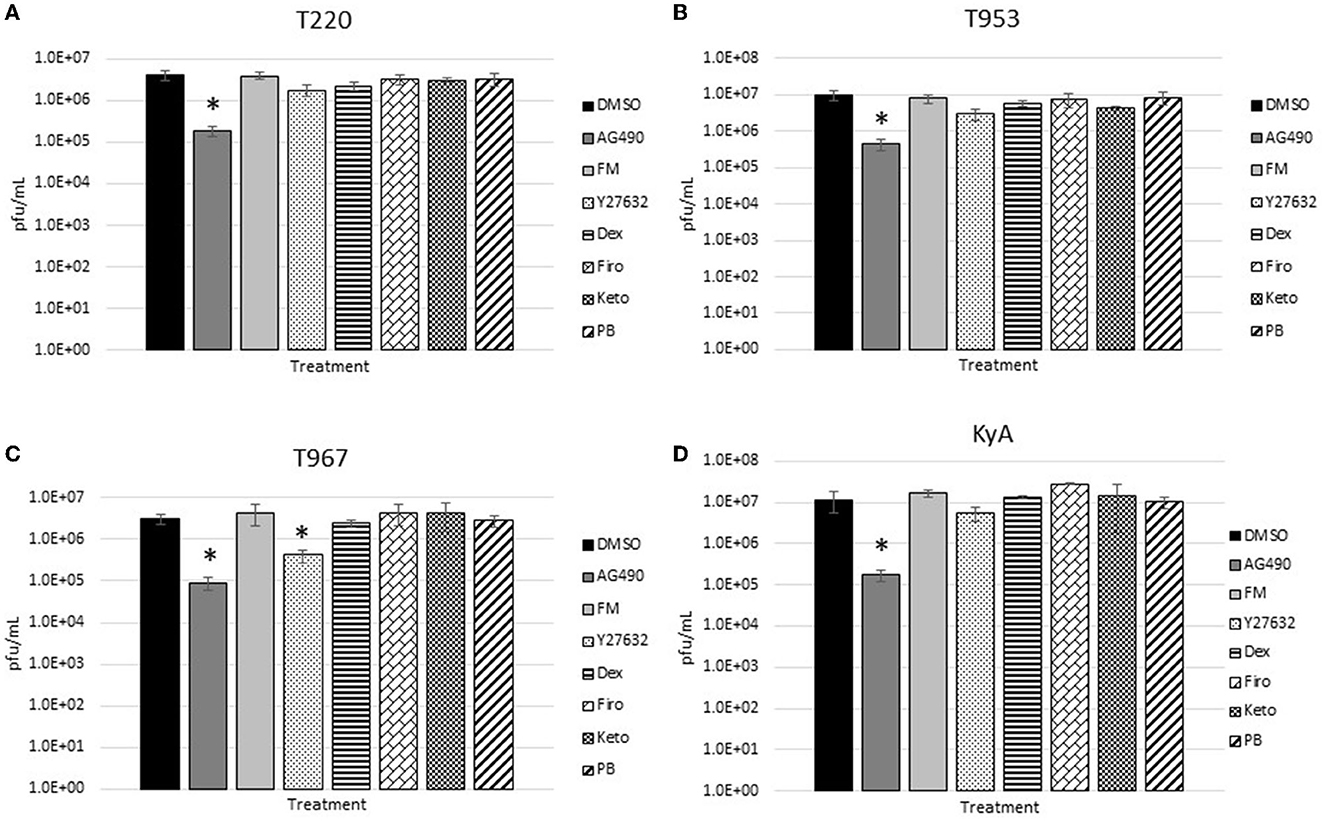
Figure 1. Equine endothelial (EE) cells (5 × 105 cells/well) were treated with drugs (100 μM) for 24 h and then infected with EHV-1 strains (A) T220, (B) T953, (C) T967, and (D) KyA at an MOI of 0.1 for 24 h in the presence of freshly added drug (100 μM). After 24 h from infection, the total virus was harvested and titered on rabbit kidney-13 (RK-13) cells. Three independent assays were run for each strain and drug, and the mean titers were calculated from the three biological assays. Virus yields in the drug-treated cells were compared to cells that were treated with the solvent, 1% DMSO, and infected with EHV-1. A Student's t-test was performed, and p-values of ≤0.05 were considered to be significant. *p-values ≤ 0.05.
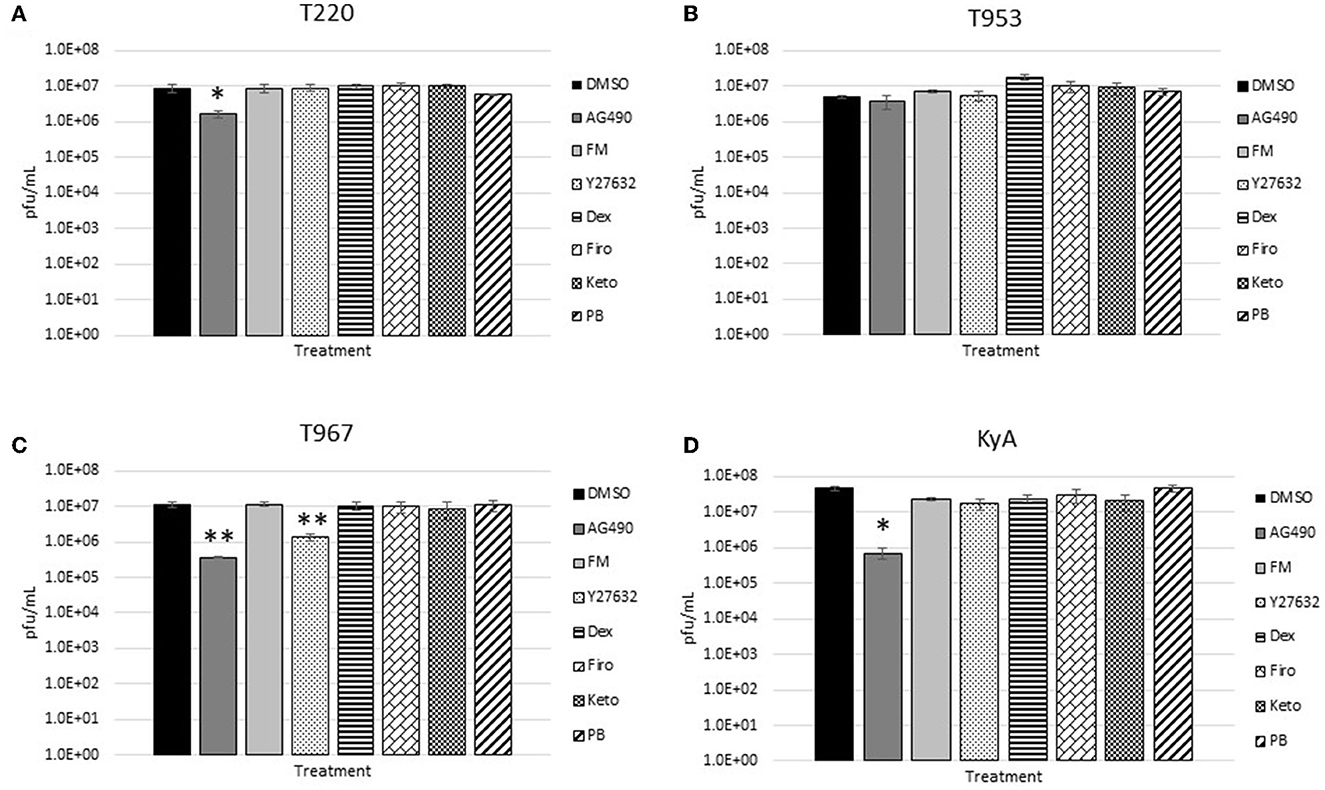
Figure 2. Equine endothelial (EE) cells (5 × 105 cells/well) were treated with drugs (100 μM) for 24 h and then infected with EHV-1 strains (A) T220, (B) T953, (C) T967, and (D) KyA at an MOI of 1 for 24 h in the presence of freshly added drug (100 μM). After 24 h from infection, the total virus was harvested and titered on rabbit kidney-13 (RK-13) cells. Three independent assays were run for each strain and drug, and the mean titers were calculated from the three biological assays. Virus yields in the drug-treated cells were compared to cells that were treated with the solvent, 1% DMSO, and infected with EHV-1. A Student's t-test was performed, and p-values of ≤0.05 were considered to be significant. *p-values ≤ 0.05 and **p-values ≤ 0.01.
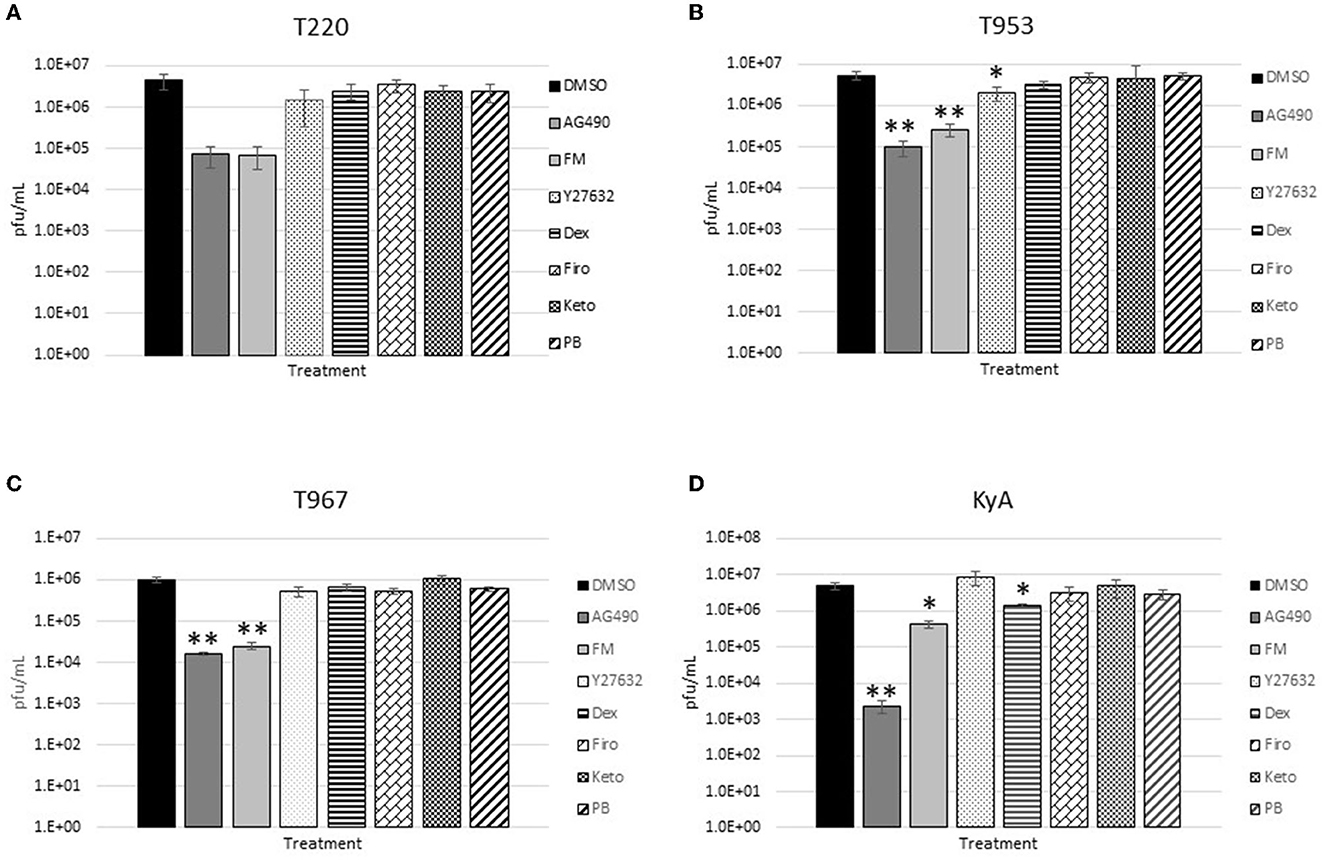
Figure 3. Equine endothelial (EE) cells (5 × 105 cells/well) were treated with drugs (100 μM) for 24 h and then infected with EHV-1 strains (A) T220, (B) T953, (C) T967, and (D) KyA at an MOI of 0.1 for 24 h in the presence of freshly added drug (100 μM). After 24 h from infection, the total virus was harvested and titered on rabbit kidney-13 (RK-13) cells. Three independent assays were run for each strain and drug, and the mean titers were calculated from the three biological assays. Virus yields in the drug-treated cells were compared to cells that were treated with the solvent, 1% DMSO, and infected with EHV-1. A Student's t-test was performed, and p-values of ≤0.05 were considered to be significant. *p-values ≤ 0.05 and **p-values ≤ 0.01.
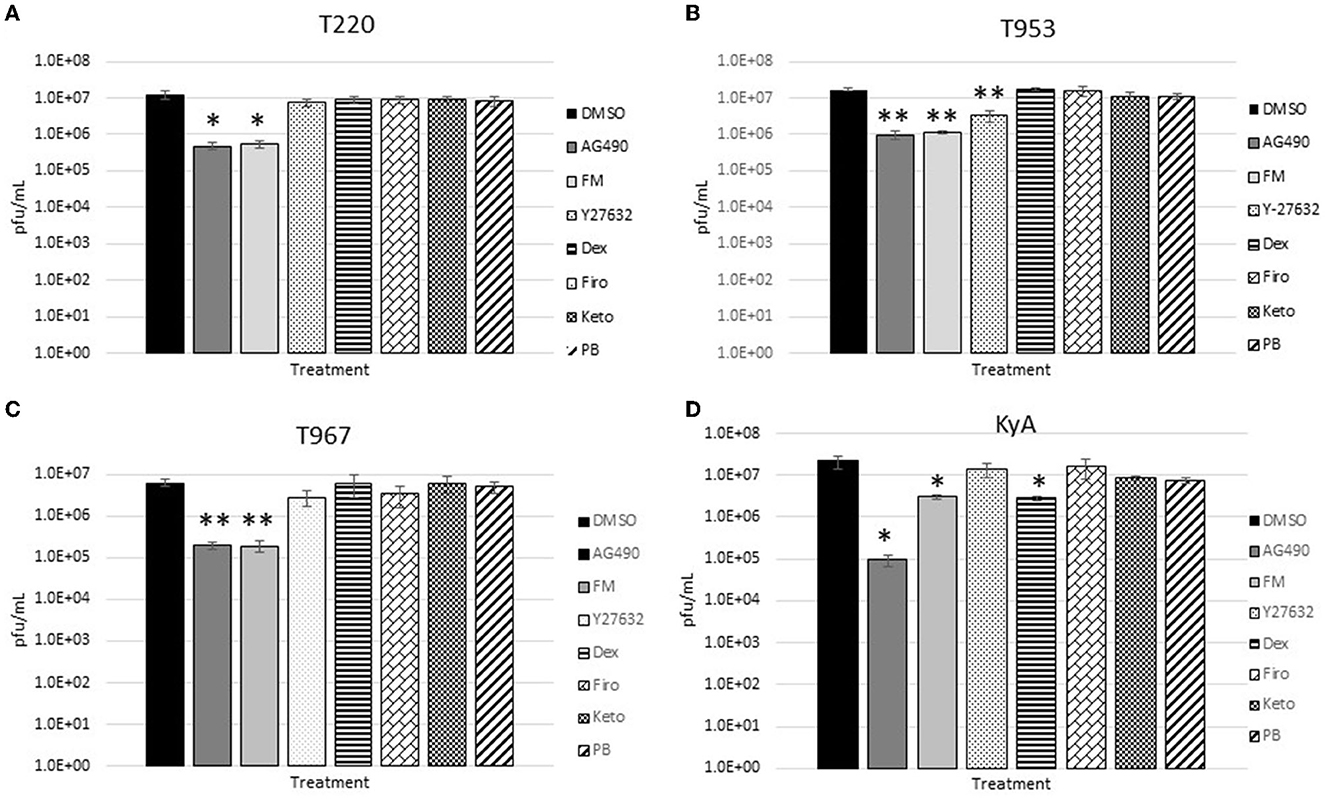
Figure 4. Rabbit kidney-13 (RK-13) epithelial cells (5 × 105 cells/well) were treated with drugs (100 μM) for 24 h and then infected with EHV-1 strains (A) T220, (B) T953, (C) T967, and (D) KyA at an MOI of 1 for 24 h in the presence of freshly added drug (100 μM). After 24 h from infection, the total virus was harvested and titered on rabbit kidney-13 (RK-13) cells. Three independent assays were run for each strain and drug, and the mean titers were calculated from the three biological assays. Virus yields in the drug-treated cells were compared to cells that were treated with the solvent, 1% DMSO, and infected with EHV-1. A Student's t-test was performed, and p-values of ≤0.05 were considered to be significant. *p-values ≤ 0.05 and **p-values ≤ 0.01.
Results obtained from the virus yield assays showed that the JAK/STAT inhibitor, AG490, was the only drug that demonstrated significant antiviral activity against all EHV-1 strains in equine endothelial (EE) cells. In EE cells infected at an MOI of 0.1 (Figure 1), AG490 reduced T220 virus yields by 23-fold, T953 virus yields by 22-fold, T967 virus yields by 35-fold, and KyA virus yields by 68-fold. At an MOI of 1 (Figure 2), AG490 reduced T220 yields by 5-fold, T967 yields by 31-fold, and KyA yields by 67-fold. Interestingly, we observed no difference in virus yields in AG490-treated EE cells infected with T953 at an MOI of 1. The Rho-kinase inhibitor, Y-27632, also exhibited significant antiviral activity against T967 at both MOIs.
In the RK-13 epithelial cell line, antiviral activity was also observed in cells treated with AG490. In these cells, the overall inhibitory activity was more robust compared to that observed in EE cells. In RK-13 cells infected at an MOI of 0.1 (Figure 3), AG490 reduced T953 virus yields by 53-fold, T967 by 61-fold, and KyA by 212-fold. A reduction was also observed in T220 virus yields at an MOI of 0.1, but the p-value was 0.08. In RK-13 cells infected at an MOI of 1 (Figure 4), AG490 reduced T220 virus yields by 25-fold, T953 by 18-fold, T967 by 31-fold, and KyA by 229-fold.
In addition to AG490, we also observed virus inhibition against all EHV-1 strains in RK-13 cells with the NSAID flunixin meglumine (FM). In RK-13 cells infected at an MOI of 0.1 (Figure 3), FM reduced T953 virus yields by 20-fold, T967 by 41-fold, and KyA by 12-fold. Similar to AG490, FM also reduced T220 virus yields at an MOI of 0.1, but the p-value was 0.08. In RK-13 cells infected at an MOI of 1 (Figure 4), FM-reduced T220 virus yields by 25-fold, T953 by 15-fold, T967 by 33-fold, and KyA by 7-fold. In addition to AG490 and FM, Y-27632 also significantly reduced T953 virus yields at an MOI of 0.1, and dexamethasone inhibited KyA virus yields at both MOIs.
Based on the ability of AG490 and FM to reduce virus yields in RK-13 epithelial cells when administered prior to infection, we investigated whether either of these compounds would be able to reduce EHV-1 yields when added post-infection. For these assays, RK-13 cells were infected with each EHV-1 strain at an MOI of 0.1 for 24 h; then, AG490 and FM were added to the cells for a further 24 h, after which the virus was harvested and titered to determine virus yields. As shown in Figure 5, post-infection administration of both drugs reduced virus yields in T220, T953, and T967-infected cells, while no inhibition was observed in KyA-infected cells. AG490 reduced T220 virus yields by 10-fold, T953 by 1.8-fold, and T967 by 4-fold. FM reduced T220 virus yields by 8-fold, T953 by 3-fold, and T967 by 5-fold.
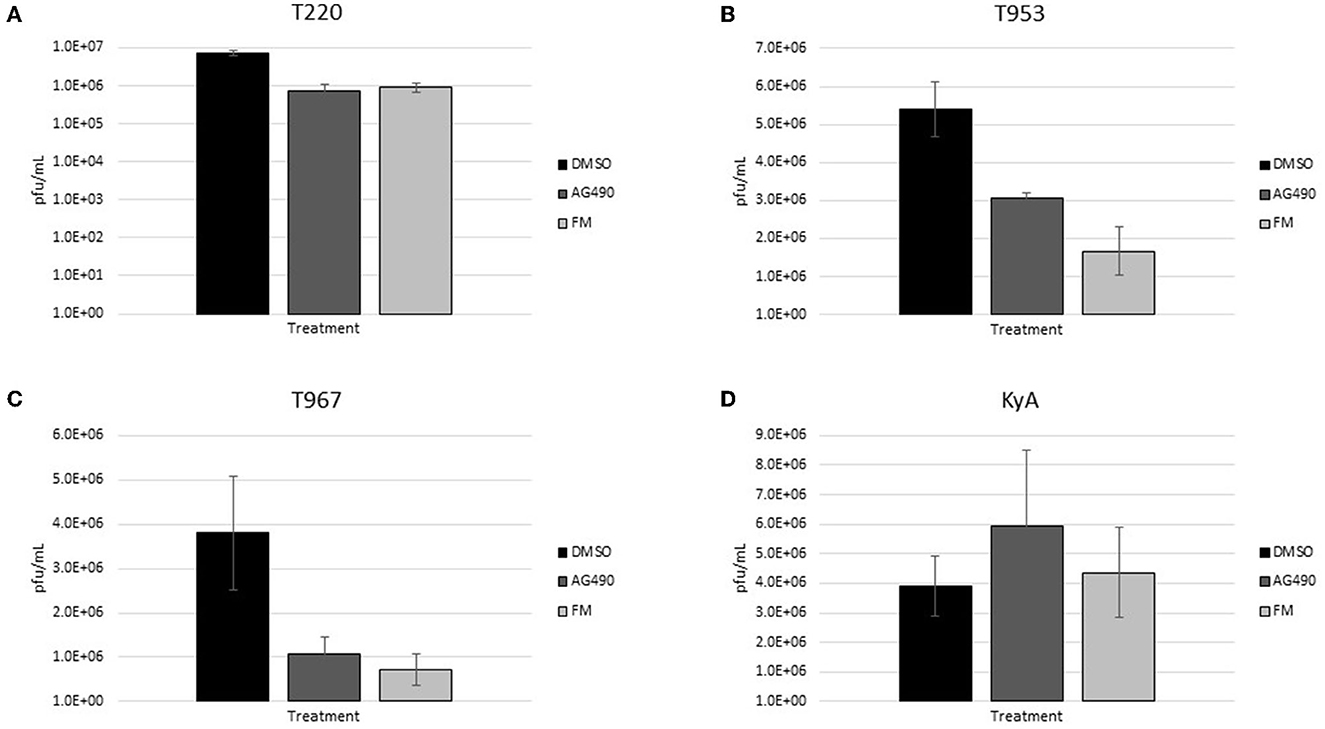
Figure 5. Rabbit kidney-13 (RK-13) epithelial cells (5 × 105 cells/well) were treated with drugs (100 μM) for 24 h and then infected with EHV-1 strains (A) T220, (B) T953, (C) T967, and (D) KyA at an MOI of 1 for 24 h in the presence of freshly added drug (100 μM). After 24 h from infection, the total virus was harvested and titered on rabbit kidney-13 (RK-13) cells. Three independent assays were run for each strain and drug, and the mean titers were calculated from the three biological assays. Virus yields in the drug-treated cells were compared to cells that were treated with the solvent, 1% DMSO, and infected with EHV-1. A Student's t-test was performed, and p-values of ≤0.05 were considered to be significant. *p-values ≤ 0.05 and **p-values ≤ 0.01.
In addition to evaluating drugs for their ability to reduce virus production, we also examined the ability of these drugs to reduce the amount of cell-to-cell spread by assessing the size of plaques that form in a cell monolayer (28, 29). Smaller plaques indicate that the virus is less able to spread from cell to cell. To test this hypothesis, we treated cells with the same panel of compounds used for the virus yield assays to test whether any of these compounds could effectively reduce cell-to-cell spread. As shown in Figure 6, AG490 significantly reduced the size of plaques formed by all four virus strains compared to the cells that were not treated with the drug (DMSO). We saw a similar but not as dramatic reduction in virus yields in response to the treatment of cells with flunixin meglumine after infection. A second NSAID, ketoprofen, significantly limited the cell-to-cell spread of one strain, T967. These data indicate that in addition to limiting virus replication and yield, FM, and AG490 also significantly limited the ability of each EHV-1 strain to spread from cell to cell, while the NSAID ketoprofen reduced the spread of strain T967.
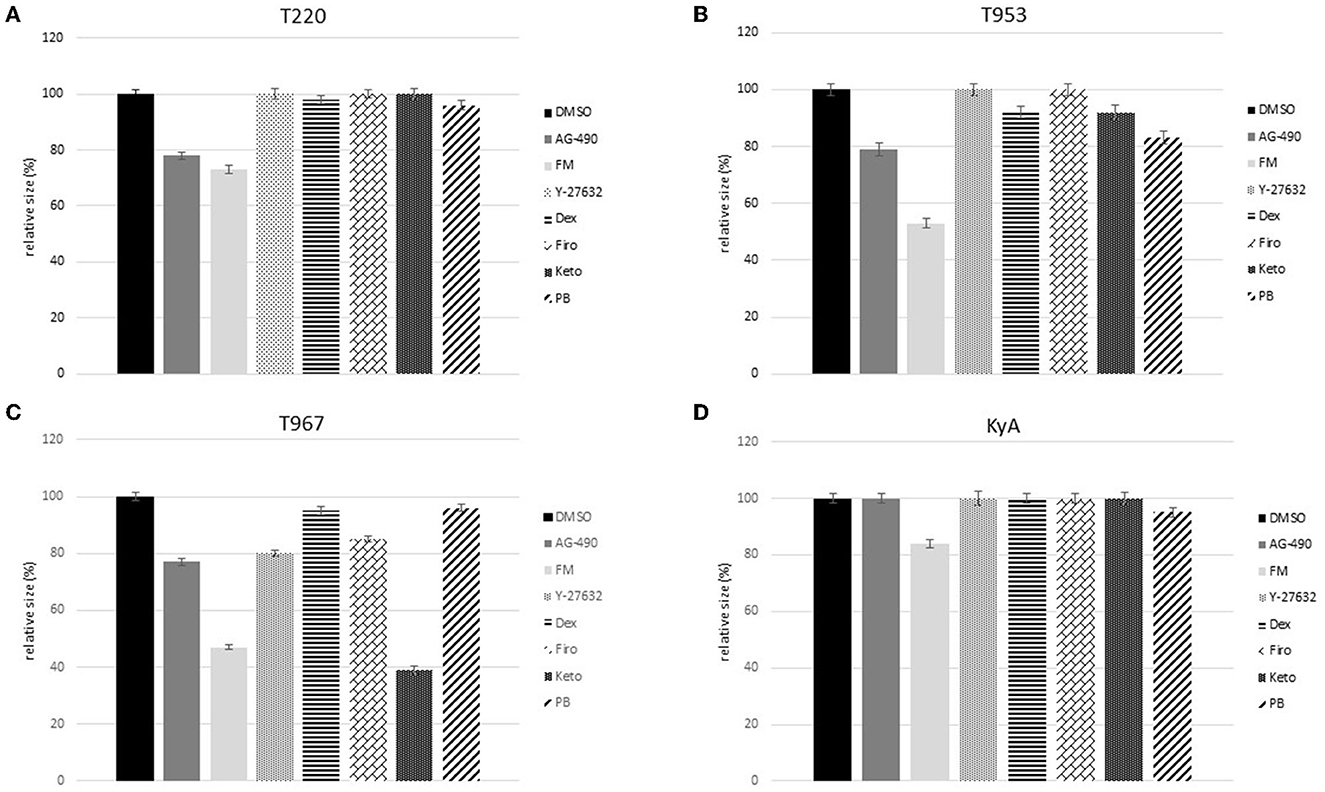
Figure 6. Rabbit kidney-13 (RK-13) epithelial cells (1 × 106 cells/well) were infected with 100 plaque-forming units (pfu) of (A) T220 (B) T953, (C) T967, and (D) KyA for 2 h. Following infection, media and virus were aspirated, and a 1% methylcellulose solution was overlaid on the cell monolayers. Infected cells were incubated for 24 h, then each drug (100 μM) was added, and the cells were incubated for a further 24 h, after which another dose (100 μM) of the drug was added, and the cells were incubated for an additional 24 h. At 72 h post-infection, the cells were fixed and stained with 1% crystal violet, and the size of the plaques in the drug-treated, infected cells was analyzed and compared to that of the plaques that formed in the control, 1% DMSO, treated, and infected groups. For each treatment, a total of 100 plaques were measured to determine the mean, standard deviation, and standard error.
Due to the hyper-inflammation observed in horses suffering from equine herpesvirus myeloencephalopathy (EHM), new treatment strategies that can be developed to reduce EHV-1 replication and subsequent inflammation are worth investigating. Multiple anti-inflammatory drugs have been shown to inhibit a wide array of viruses. Drugs that inhibit host cell mediators of inflammation have been reported to block or reduce the replication and/or spread of several herpesviruses, such as pseudorabies virus (PRV) (63, 64), Marek's disease virus (MDV) (65), and cytomegalovirus (66, 67). In addition to reducing replication and cell-to-cell spread, inhibition of the inflammatory mediator cyclooxygenase-2 (COX-2) also decreased the lytic reactivation of Epstein–Barr virus (EBV). JAK/STAT inhibitors have also been shown to exhibit antiviral activity against numerous viruses including HIV (22–24), Zika (25), and SARS-CoV-2 (26).
The ability of anti-inflammatory drugs to reduce inflammation, caused by a variety of stimuli in equines has been widely reported (30, 31). Non-steroidal anti-inflammatory drugs (NSAIDs), including ketoprofen, flunixin meglumine, firocoxib, and phenylbutazone, are commonly prescribed to treat inflammation in horses and have been shown to be very safe (12, 32). These drugs have been evaluated in numerous studies and have been shown to reduce the pathology observed in horses infected with other microbial pathogens including equine protozoal myeloencephalitis (EPM) (33). However, while NSAIDs have been used extensively in equines, they have not been rigorously evaluated for their ability to reduce virus replication and inflammation, which are positively correlated with EHM. Currently, there are very limited data on the drugs that have been administered to horses suffering from EHM. In one study, flunixin meglumine, firocoxib, and dexamethasone were shown to suppress the transfer of EHV-1 from infected peripheral blood monocytes (PBMC) to endothelial cells (34), but no data were collected on the ability of these drugs to reduce virus replication. Other studies have shown that the administration of drugs that enhance the host interferon response limits EHV-1 replication and spread (27, 35, 36). In addition, there have been no studies conducted to evaluate whether pre-exposure prophylactic treatment reduces EHV-1 infection or ameliorates EHV-1-induced pathology.
Equine herpesvirus myeloencephalopathy (EHM) is a disease primarily caused by severe inflammation within the CNS. Infection of endothelial cells as a result of cell-associated viremia results in the production of pro-inflammatory cytokines that leads to immune cell infiltration and localized thrombosis and hypoxia that damages adjacent neurons. The degree of CNS pathology determines the extent of EHM disease and ultimately whether a horse will recover or be euthanized due to paralysis (2, 24, 37, 38). Numerous studies have shown a positive correlation between the degree of viremia and negative disease outcomes following EHV-1 infection (10, 11). Therefore, any therapy that can effectively reduce the level of viremia should also decrease the risk of an EHV-1-infected horse developing EHM.
The data collected in this study show that specific compounds reduced the number of progeny virions produced in epithelial and endothelial cells. Data collected from the virus yield assays show that administration of both the JAK/STAT inhibitor, AG490, and the NSAID, flunixin meglumine (FM), significantly reduced the amount of virus produced in epithelial cells. In addition, AG490 reduced virus yields in both epithelial and endothelial cells while FM reduced virus production in epithelial cells but not in endothelial cells. The Rho-kinase inhibitor, Y-27632, which we have previously shown to inhibit viral movement to the nucleus (39, 40), also exhibited antiviral activity against T967 in endothelial cells and T953 in epithelial cells. The only other drug that reduced viral yields was dexamethasone, which had antiviral activity against KyA at both MOIs in epithelial cells.
To evaluate the ability of these drugs to reduce virus replication post-infection, FM and AG490 were administered 24 h after EHV-1 infection and viral yields were measured and compared to control-treated cells at 24 h post-drug administration. The results of these assays showed that both drugs exhibit antiviral activity even after the cells have been infected. Our data showing that these two drugs limit EHV-1 production both pre- and post-infection make them interesting candidates for further testing in horses, as they may prove to be an additional therapeutic option to consider during an EHV-1 outbreak. The ability of these two drugs, which have different mechanisms of action, to reduce the amount of virus produced in cells would be expected to result in a more positive outcome in an EHV-1-infected horse as the level of virus in the bloodstream (viremia), would be lowered, and previous studies have shown that the level of viremia is directly correlated with disease outcome (10, 11).
As with any model system, there are some limitations to the current study. First, the epithelial cells are a continuous rabbit kidney cell line. While these cells have been extensively used in EHV-1 research, data collected using these cells should be compared to data obtained from equine epithelial cells, ideally, equine cells isolated from the upper respiratory tract (URT). Mucosal explant/primary respiratory epithelial tissue culture systems have been developed (27, 41), and a similar study could be conducted using this system to see whether the reduction in EHV-1 replication and cell-to-cell spread with FM and AG490 is replicated in this system. Second, while a significant amount of data can be collected using continuous cell lines, an investigation of these drugs in the whole animal should be conducted. These studies could be performed either in a mouse model system like the one we previously developed (37) or ultimately in horses.
The results obtained in this study provide evidence that prophylactic and post-exposure administration of FM and AG490 may provide some protection against EHV-1 infection. In terms of practice, one could envision a scenario in which routine surveillance identifies a single or a few horses infected with EHV-1, which would trigger the prophylactic treatment of the remaining horses in the group. In addition, although we did not address this in our current study, other studies have shown that DNA chain-terminating drugs such as valacyclovir (38) and valganciclovir (42) are effective in reducing EHV-1 infection and viremia, and thus, it would be interesting to see if a synergistic reduction of EHV-1 would be observed in horses treated with a combination of a specific antiviral such as ganciclovir and FM or AG490.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
All authors contributed to developing the research questions, designing the experiments, conducted the research to collect the data, analyzed the data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding for this study was provided by the Grayson Jockey Club Research Foundation (GJCRF grant # 257).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1165917/full#supplementary-material
1. Allen GP, Bryans JT. Molecular epizootiology, pathogenesis, and prophylaxis of equine herpesvirus-1 infections. Prog Vet Microbiol Immunol. (1986) 2:78–144.
2. O'Callaghan DJ, Gentry GA, Randall CC. The equine herpesviruses. In:Roizman B, , editors. The Herpesviruses, Comprehensive Virology, Vol 2. New York, NY: Plenum Press (1983). p. 215–238
3. Ostlund EN. The equine herpesviruses. Vet Clin N Am Equine Pract. (1993) 9:283–94. doi: 10.1016/S0749-0739(17)30396-6
4. Goodman LB, Loregian A, Perkins GA, Nugent J, Buckles EL, Mercorelli B, et al. A point mutation in a herpesvirus polymerase determines neuropathogenicity. PLoS Pathog. (2007) 3:e160. doi: 10.1371/journal.ppat.0030160
5. Perkins GA, Goodman LB, Tsujimura K, Van de Walle GR, Kim SG, Dubovi EJ, et al. Investigation of the prevalence of neurologic equine herpes virus type 1 (EHV-1) in a 23-year retrospective analysis (1984-2007). Vet Microbiol. (2009) 139:375–8. doi: 10.1016/j.vetmic.2009.06.033
6. Nugent J, Birch-Machin I, Smith KC, Mumford JA, Swann Z, Newton JR, et al. Analysis of equid herpesvirus 1 strain variation reveals a point mutation of the DNA polymerase strongly associated with neuropathogenic versus nonneuropathogenic disease outbreaks. J Virol. (2006) 80:4047–60. doi: 10.1128/JVI.80.8.4047-4060.2006
7. Smith KL, Allen GP, Branscum AJ, Frank Cook R, Vickers ML, Timoney PJ, et al. The increased prevalence of neuropathogenic strains of EHV-1 in equine abortions. Vet Microbiol. (2010) 141:5–11. doi: 10.1016/j.vetmic.2009.07.030
8. Allen GP. Development of a real-time polymerase chain reaction assay for rapid diagnosis of neuropathogenic strains of equine herpesvirus-1. J Vet Diagn Invest. (2007) 19:69–72. doi: 10.1177/104063870701900110
9. Van de Walle GR, Goupil R, Wishon C, Damiani A, Perkins GA, Osterrieder N, et al. single-nucleotide polymorphism in a herpesvirus DNA polymerase is sufficient to cause lethal neurological disease. J Infect Dis. (2009) 200:20–5. doi: 10.1086/599316
10. Allen GP. Risk factors for development of neurologic disease after experimental exposure to equine herpesvirus-1 in horses. Am J Vet Res. (2008) 69:1595–600. doi: 10.2460/ajvr.69.12.1595
11. Allen GP, Breathnach CC. Quantification by real-time PCR of the magnitude and duration of leucocyte-associated viraemia in horses infected with neuropathogenic vs. non-neuropathogenic strains of EHV-1. Equine Vet J. (2006) 38:252–7. doi: 10.2746/042516406776866453
12. Pusterla N, Hussey GS. Equine herpesvirus 1 myeloencephalopathy. Vet Clin N Am Equine Pract. (2014) 30:489–506. doi: 10.1016/j.cveq.2014.08.006
13. Edington N, Bridges CG, Patel JR. Endothelial cell infection and thrombosis in paralysis caused by equid herpesvirus-1: equine stroke. Arch Virol. (1986) 90:111–24. doi: 10.1007/BF01314149
14. Wagner B, Wimer C, Freer H, Osterrieder N, Erb HN. Infection of peripheral blood mononuclear cells with neuropathogenic equine herpesvirus type-1 strain Ab4 reveals intact interferon-alpha induction and induces suppression of anti-inflammatory interleukin-10 responses in comparison to other viral strains. Vet Immunol Immunopathol. (2011) 143:116–24. doi: 10.1016/j.vetimm.2011.06.032
15. Kydd JH, Slater J, Osterrieder N, Lunn DP, Antczak DF, Azab W, et al. Third International Havemeyer Workshop on Equine Herpesvirus type 1. Equine Vet J. (2012) 44:513–7. doi: 10.1111/j.2042-3306.2012.00604.x
16. Johnstone S, Barsova J, Campos I, Frampton AR. Equine herpesvirus type 1 modulates inflammatory host immune response genes in equine endothelial cells. Vet Microbiol. (2016) 192:52–9. doi: 10.1016/j.vetmic.2016.06.012
17. Ray N, Bisher ME, Enquist LW. Cyclooxygenase-1 and−2 are required for production of infectious pseudorabies virus. J Virol. (2004) 78:12964–74. doi: 10.1128/JVI.78.23.12964-12974.2004
18. Zhao X, Tong W, Song X, Jia R, Li L, Zou Y, et al. Antiviral effect of resveratrol in piglets infected with virulent pseudorabies virus. Viruses. (2018) 10. doi: 10.3390/v10090457
19. Boodhoo N, Kamble N, Kaufer BB, Behboudi S. Replication of Marek's disease virus is dependent on synthesis of de novo fatty acid and prostaglandin E2. J Virol. (2019) 93. doi: 10.1128/JVI.00352-19
20. Schroer J, Shenk T. Inhibition of cyclooxygenase activity blocks cell-to-cell spread of human cytomegalovirus. Proc Natl Acad Sci USA. (2008) 105:19468–73. doi: 10.1073/pnas.0810740105
21. Zhu H, Cong JP Yu D, Bresnahan WA, Shenk TE. Inhibition of cyclooxygenase 2 blocks human cytomegalovirus replication. Proc Natl Acad Sci USA. (2002) 99:3932–7. doi: 10.1073/pnas.052713799
22. Haile WB, Gavegnano C, Tao S, Jiang Y, Schinazi RF, Tyor WR. The Janus kinase inhibitor ruxolitinib reduces HIV replication in human macrophages and ameliorates HIV encephalitis in a murine model. Neurobiol Dis. (2016) 92:137–43. doi: 10.1016/j.nbd.2016.02.007
23. Reece MD, Song C, Hancock SC, Pereira Ribeiro S, Kulpa DA, Gavegnano C. Repurposing BCL-2 and Jak 1/2 inhibitors: cure and treatment of HIV-1 and other viral infections. Front Immunol. (2022) 13:1033672. doi: 10.3389/fimmu.2022.1033672
24. Gavegnano C, Haile WB, Hurwitz S, Tao S, Jiang Y, Schinazi RF, et al. Baricitinib reverses HIV-associated neurocognitive disorders in a SCID mouse model and reservoir seeding in vitro. J Neuroinflammation. (2019) 16:182. doi: 10.1186/s12974-019-1565-6
25. Gavegnano C, Bassit LC, Cox BD, Hsiao HM, Johnson EL, Suthar M, et al. Jak inhibitors modulate production of replication-competent zika virus in human hofbauer, trophoblasts, and neuroblastoma cells. Pathog Immun. (2017) 2:199–218. doi: 10.20411/pai.v2i2.190
26. Stebbing J, Sanchez Nievas G, Falcone M, Youhanna S, Richardson P, et al. JAK inhibition reduces SARS-CoV-2 liver infectivity and modulates inflammatory responses to reduce morbidity and mortality. Sci Adv. (2021) 7. doi: 10.1126/sciadv.abe4724
27. Poelaert KCK, Van Cleemput J, Laval K, Favoreel HW, Soboll Hussey G, Maes RK, et al. Abortigenic but not neurotropic equine herpes virus 1 modulates the interferon antiviral defense. Front Cell Infect Microbiol. (2018) 8:312. doi: 10.3389/fcimb.2018.00312
28. Neubauer A, Osterrieder N. Equine herpesvirus type 1 (EHV-1) glycoprotein K is required for efficient cell-to-cell spread and virus egress. Virology. (2004) 329:18–32. doi: 10.1016/j.virol.2004.07.034
29. Frampton AR Jr, Smith PM, Zhang Y, Matsumura T, Osterrieder N, O'Callaghan DJ. Contribution of gene products encoded within the unique short segment of equine herpesvirus 1 to virulence in a murine model. Virus Res. (2002) 90:287–301. doi: 10.1016/S0168-1702(02)00245-9
30. Ziegler A, Fogle C, Blikslager A. Update on the use of cyclooxygenase-2-selective nonsteroidal anti-inflammatory drugs in horses. J Am Vet Med Assoc. (2017) 250:1271–4. doi: 10.2460/javma.250.11.1271
31. Knych HK. Nonsteroidal anti-inflammatory drug use in horses. Vet Clin N Am Equine Pract. (2017) 33:1–15. doi: 10.1016/j.cveq.2016.11.001
32. Castagnetti C, Mariella J. Anti-inflammatory drugs in equine neonatal medicine. Part II: corticosteroids. J Eq Vet Sci. (2015) 35:547–54. doi: 10.1016/j.jevs.2015.02.012
33. Reed SM, Furr M, Howe DK, Johnson AL, MacKay RJ, Morrow JK, et al. Equine protozoal myeloencephalitis: an updated consensus statement with a focus on parasite biology, diagnosis, treatment, and prevention. J Vet Int Med. (2016) 30:491–502. doi: 10.1111/jvim.13834
34. Goehring L, Brandes KM, Wittenburg L. Anti-inflammatory drugs decrease the rate of endothelial cell infection with EHV-1 in vitro. In: Proceedings of 2012 Veterinary Symposium. New Orleans, LA: American College of Veterinary Internal Medicine (2012).
35. Oladunni FS, Sarkar S, Reedy S, Balasuriya UBR, Horohov DW, Chambers TM. Equid herpesvirus 1 targets the sensitization and induction steps to inhibit the type I interferon response in equine endothelial cells. J Virol. (2019) 93. doi: 10.1128/JVI.01342-19
36. Sarkar S, Balasuriya UB, Horohov DW, Chambers TM. Equine herpesvirus-1 infection disrupts interferon regulatory factor-3 (IRF-3) signaling pathways in equine endothelial cells. Vet Immunol Immunopathol. (2016) 173:1–9. doi: 10.1016/j.vetimm.2016.03.009
37. Frampton AR Jr, Smith PM, Zhang Y, Grafton WD, Matsumura T, Osterrieder N, et al. Meningoencephalitis in mice infected with an equine herpesvirus 1 strain KyA recombinant expressing glycoprotein I and glycoprotein E. Virus Genes. (2004) 29:9–17. doi: 10.1023/B:VIRU.0000032785.19420.14
38. Maxwell LK, Bentz BG, Gilliam LL, Ritchey JW, Pusterla N, Eberle R, et al. Efficacy of the early administration of valacyclovir hydrochloride for the treatment of neuropathogenic equine herpesvirus type-1 infection in horses. Am J Vet Res. (2017) 78:1126–39. doi: 10.2460/ajvr.78.10.1126
39. Frampton AR Jr, Uchida H, von Einem J, Goins WF, Grandi P, Cohen JB, et al. Equine herpesvirus type 1 (EHV-1) utilizes microtubules, dynein, and ROCK1 to productively infect cells. Vet Microbiol. (2010) 141:12–21. doi: 10.1016/j.vetmic.2009.07.035
40. Frampton AR Jr, Stolz DB, Uchida H, Goins WF, Cohen JB, Glorioso JC. Equine herpesvirus 1 enters cells by two different pathways, and infection requires the activation of the cellular kinase ROCK1. J Virol. (2007) 81:10879–89. doi: 10.1128/JVI.00504-07
41. Poelaert KCK, Van Cleemput J, Laval K, Xie J, Favoreel HW, Nauwynck HJ. Equine herpesvirus 1 infection orchestrates the expression of chemokines in equine respiratory epithelial cells. J Gen Virol. (2019). doi: 10.1099/jgv.0.001317
Keywords: EHV-1, inflammation, anti-inflammatories, epithelial, endothelial
Citation: Black JB and Frampton AR (2023) Anti-inflammatory compounds reduce equine herpesvirus type 1 replication and cell-to-cell spread. Front. Vet. Sci. 10:1165917. doi: 10.3389/fvets.2023.1165917
Received: 14 February 2023; Accepted: 24 April 2023;
Published: 19 May 2023.
Edited by:
Pavulraj Selvaraj, Louisiana State University, United StatesReviewed by:
Harikrishnan Mohan, Louisiana State University, United StatesCopyright © 2023 Black and Frampton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arthur R. Frampton, ZnJhbXB0b25hQHVuY3cuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.