 Maria Butylina
Maria Butylina Ursula Föger-Samwald1
Ursula Föger-Samwald1 Peter Pietschmann
Peter Pietschmann Wolfgang Sipos
Wolfgang Sipos- 1Institute of Pathophysiology and Allergy Research, Center for Pathophysiology, Infectiology and Immunology, Medical University of Vienna, Vienna, Austria
- 2Clinical Department for Farm Animals, University of Veterinary Medicine Vienna, Vienna, Austria
Hypoparathyroidism is a relatively rare human and veterinary disease characterized by deficient or absent production of parathyroid hormone (PTH). PTH is known as a classical regulator of calcium and phosphorus homeostasis. Nevertheless, the hormone also appears to modulate immune functions. For example, increased CD4:CD8 T-cell ratios and elevated interleukin (IL)-6 and IL-17A levels were observed in patients with hyperparathyroidism, whereas gene expression of tumor necrosis factor-α (TNF-α) and granulocyte macrophage-colony stimulating factor (GM-CSF) was decreased in patients with chronic postsurgical hypoparathyroidism. Various immune cell populations are affected differently. So, there is a need for validated animal models for the further characterization of this disease for identifying targeted immune-modulatory therapies. In addition to genetically modified mouse models of hypoparathyroidism, there are surgical rodent models. Parathyroidectomy (PTX) can be well performed in rats—for pharmacological and associated osteoimmunological research and bone mechanical studies, a large animal model could be preferable, however. A major drawback for successfully performing total PTX in large animal species (pigs and sheep) is the presence of accessory glands, thus demanding to develop new approaches for real-time detection of all parathyroid tissues.
1. Parathyroid glands and parathyroid hormone—an introduction
The first description of the parathyroid gland is credited to Sir Richard Owen, who in 1862 published the findings of the autopsy of an Indian Rhinoceros (Rhinoceros unicornis) he dissected in the winter months of 1849/1850 (1). The term “parathyroid gland” was coined by Ivar Viktor Sandström, who in 1877 identified the gland in a dog and subsequently in cats, rabbits, horses, and humans (2). The pathologist Jakob Erdheim proved in a series of experiments with rats that total parathyroidectomy leads to tetany and for the first time related the parathyroid gland to calcium metabolism (3). The parathyroid glands are important organs, which are located in the neck posterior and inferior to the thyroid gland and emerge from the third and fourth pharyngeal pouches (Figures 1, 2).
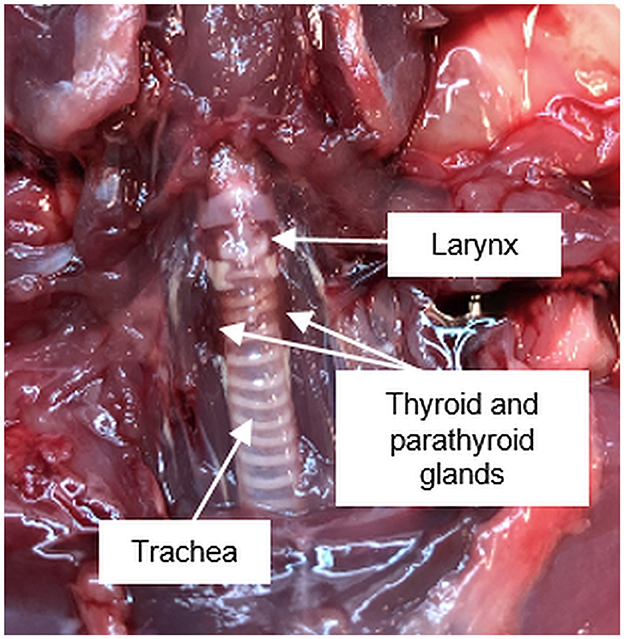
Figure 1. Anatomical position of the pharyngeal and ventral neck region of a Sprague Dawley rat.
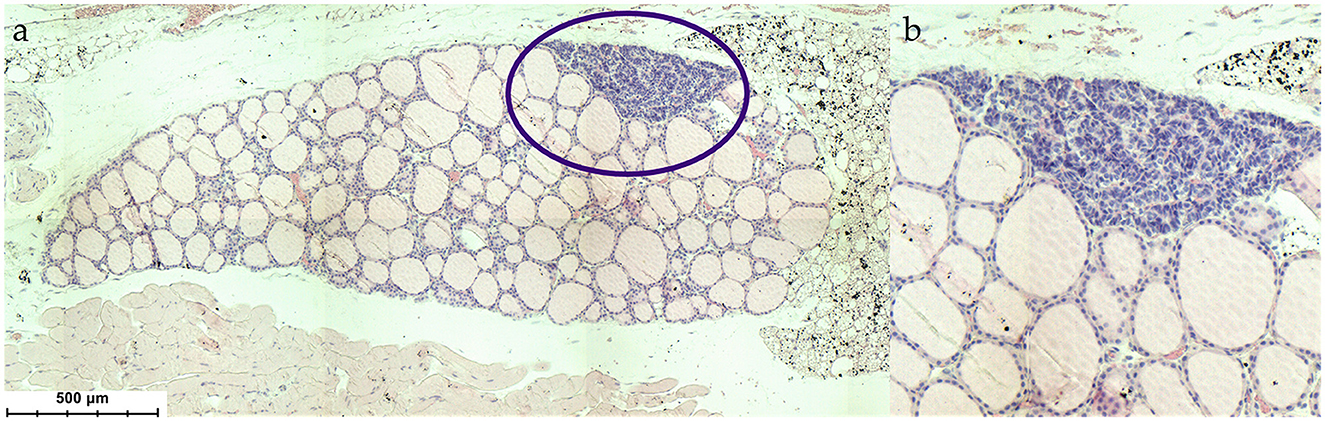
Figure 2. (a) Histological situs of the parathyroid gland within the thyroid (Mus musculus), (b) magnification of the parathyroid gland. HE stain.
In humans, there are usually four glands, which are crucial for the maintenance of blood calcium homeostasis, as parathyroid glands are responsible for parathyroid hormone (PTH) secretion. Furthermore, lower levels of PTH transcripts can also be determined in the thymus, pituitary, and hypothalamus (4). PTH is located in the secretory granules and is secreted from the chief cells in response to reduced circulating ionized calcium concentrations to maintain the normocalcemic state. Usually, a minimal proliferation of the parathyroid cells can be seen, but chronic hypocalcemia triggers an increase in size and number of the parathyroid cells (5). On the plasma membrane of the parathyroid cells, calcium-sensing receptors (CaSRs) are abundantly present, which are responsible for monitoring free calcium concentrations and binding Ca2+ (6–12). The CaSR belongs to the G-protein-coupled receptor superfamily, which has a calcium-binding element in the extracellular domain and signaling determinants in the cytoplasmic region (13). Outside the parathyroid gland, CaSRs play a crucial role in the kidneys in taking part in the regulation of urinary calcium excretion independently of PTH (14). Furthermore, CaSRs can be found in the intestine, vasculature, and lungs (13).
PTH is a peptide hormone consisting of 84 amino acids and is—together with vitamin D—essential for blood calcium homeostasis. It belongs, together with PTH-related peptide (PTHrP) and tuberoinfundibular peptide of 39 residues (TIP39), to the parathyroid hormone peptide subfamily (4). Additionally, a new member was discovered, PTH-like peptide (PTH-L), which is only existing in non-mammalian species like teleost fishes, chicken, or Xenopus (15). In mammals, PTH is first synthesized as a pre-pro-peptide consisting of 115 amino acids, but only the 84 amino acids full-length single-chain polypeptide is later secreted by the parathyroid glands (9). It is responsible for inducing the release of calcium and phosphate from the skeletal reservoir by bone resorption while simultaneously acting on the kidneys. High levels of extracellular calcium inhibit PTH secretion, while low serum calcium levels lead to an increase, which has an effect on the PTH receptor in the kidneys leading to higher resorption of tubular calcium and suppression of phosphate reabsorption (12, 13). Persistently decreased systemic calcium levels further lead to an upregulated PTH mRNA expression and an increased number of PTH-secreting parathyroid cells (16). As a consequence, there is a rise in the renal production of 1,25(OH)2VitD3, leading to enhanced intestinal calcium absorption, which is a negative regulator of PTH secretion (6, 12, 17). Decreased levels of VitD3 in turn lead to higher PTH production (18). Furthermore, PTH indirectly activates bone-resorbing cells, the osteoclasts, through the classical parathyroid hormone 1 receptor (PTH1R). This receptor is expressed mainly on osteoblasts and in the kidneys. Activated osteoblasts then activate osteoclasts via the RANKL (receptor activator of nuclear factor-κB ligand)–RANK axis, which results in increased net bone resorption (12, 19).
2. Pathophysiology and clinical aspects of hypoparathyroidism in humans
Hypoparathyroidism is a relatively rare disease characterized by deficient or absent production of PTH (with blood-serum PTH levels below the physiological reference range of 12–72 ng/L corresponding to 1.5–6.0 pmol/L), which leads to a disbalanced extracellular fluid calcium level. Low calcium levels can either have rapid onset or successively develop almost asymptomatically. On the contrary, serum phosphate levels typically are increased. In consequence, the calcium/phosphate ratio is decreased (but increased in primary hyperparathyroidism). The majority of human cases of hypoparathyroidism results from neck—in particular thyroid—surgery (20–22).
In addition to hypoparathyroidism resulting after neck surgery, non-surgical or genetic forms of hypoparathyroidism can be described. The most frequent genetic form is the DiGeorge syndrome, which affects approximately 60 % of children diagnosed with hypoparathyroidism (23). It emerges from a microdeletion in chromosome 22q11.2, leading to a lack of T box protein 1, which is crucial for the development of the thymus and parathyroid glands. Due to this deletion, cardiovascular malformations, thymus underdevelopment, and facial abnormalities arise (24, 25). Furthermore, affected children show symptoms like chronic infections, nasal regurgitation, hypocalcemia, feeding difficulties, and learning disabilities (26).
Another genetic cause for hypoparathyroidism is the autoimmune polyendocrine syndrome type 1 (APS-1), which is described as an autosomal recessive disorder and caused by a mutation in the autoimmune regulator gene AIRE on chromosome 21q22.3 (27, 28). This mutation gives rise to a lack of self-immunotolerance, leading to the destruction of the parathyroid, adrenal, and other endocrine glands. APS-1 usually develops during early childhood at an age of 2–5 years (29). For the diagnosis of autoimmune polyendocrine syndrome type 1, two of three major diseases must be present, including hypoparathyroidism, adrenal insufficiency, and mucocutaneous candidiasis (30). These diagnostic criteria are often met before an age of 20 years (31).
Typical symptoms occurring during hypoparathyroidism, often due to low calcium levels, are, tetanic spasms, which may be lethal (20). Regarding bone manifestations, affected persons show higher bone mineral densities than sex- and age-matched controls (32). Patients with hypoparathyroidism exhibit changes in bone metabolism, as low-normal values of bone turnover markers were detected in blood and urine (33). Consequently, hypoparathyroidism leads to a greater risk of developing fractures in the appendicular skeleton (34). Affected patients often show neuromotor manifestations, like, for example, that of parkinsonism, which partially improved after the treatment of hypocalcemia. Some patients also display increased anxiety, fatigue, difficulty to concentrate, and a decrease in memory (20). Furthermore, hypoparathyroidism is associated with heart failure with resistance to diuretics and other standard treatment options. Patients with acute hypocalcemia may also show hypotension, bradycardia, and arrhythmias. After the correction of hypocalcemia, patients with cardiac dysfunction showed improvement (35, 36). Regarding the gastrointestinal tract, patients often exhibit abdominal cramps and constipation (20). Moreover, hypoparathyroidism is often associated with intense photophobia, chronic conjunctivitis, and cataracts (37). Regarding cutaneous manifestations, patients suffer from dry skin, pustular psoriasis, or deformations of the nails (38). The most common dental manifestation of hypoparathyroidism is hypoplastic enamel followed by cemental hyperplasia (39).
To correct hypocalcemia, patients with hypoparathyroidism are treated with calcium supplements and vitamin D analogs (40). Nevertheless, despite (standard) treatment, the quality of life of many patients is impaired due to hypoparathyroidism-associated symptoms (41, 42). Recently, a novel treatment option with human recombinant PTH has become available (43–45) and has been associated with an improved quality of life (46). Probably due to the rarity of the disease, rigorous data on co-morbidities of hypoparathyroidism are relatively sparse. Patients with postsurgical hypoparathyroidism are at an increased risk of renal complications and hospitalization due to seizures (47). In a study based on the Danish National Patient Registry, Underbjerg and co-workers demonstrated that postsurgical hypoparathyroidism is associated with a significantly increased risk of hospitalization for infections (and depression/bipolar disorders) (48). This increased risk might be due to a compromised immune function (49). Interestingly, when patients with urinary tract infections (potentially resulting from urinary calcium deposition) were excluded from the analyses, the increased risk of hospitalization for infections persisted. In interpreting their results, the authors assumed that “PTH may impair the immunology response to infections” (47). In 2015, these authors extended their observation of an increased risk of hospitalization due to infections also to patients with non-surgical hypoparathyroidism (34). In a subsequent study, Underbjerg et al. investigated potential biochemical risk factors associated with infections. Persistent hyperphosphatemia, which develops due to a diminished renal excretion in hypoparathyroidism, was associated with increased mortality and risk of any infections (50).
3. Osteoimmunology of hypoparathyroidism
Osteoimmunology analyses the interplay between bone and immune cells. The main two types of bone cells are osteoblasts and osteoclasts. Osteoblasts are the so-called bone-forming cells, which are responsible for bone matrix formation and mineralization, whereas bone resorption is performed by osteoclasts. In addition to osteoblasts and osteoclasts, osteocytes and bone lining cells can be found in bones (Figure 3).
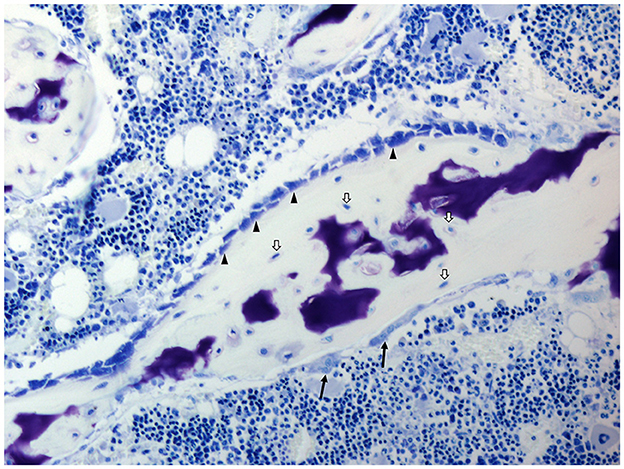
Figure 3. Histology of the bone of a domestic pig; toluidine blue stain. Osteoblasts (arrowheads), osteocytes (white arrows), and osteoclasts (black arrows). Cell types are identified due to their morphological characteristics and position within specific bone compartments. Porcine bone marrow-derived stromal cells as well as osteoblasts and osteoclasts exhibit similar in vitro growth characteristics and osteoimmunological properties as their rodent and human counterparts (51, 52).
Mature osteoblasts are the only cell type that is able to construct bones by secreting bone matrix proteins and guidance of mineralization. Bone-forming cells are cuboidal cells, which are present along the bone surface covering 4–6% of the total resident bone cells. Although mature osteoblasts are short-lived, a subset is differentiating into osteocytes that are encapsulated within the newly formed bone matrix. Osteocytes are characterized as the most abundant cell type (90%) present within the matrix or on bone surfaces, where they are responsible for supporting bone structure and metabolism. Furthermore, they are in charge of mechanosensation as they transduce stress signals from bending or stretching bone into biological activity. Those, which are not encapsulated, undergo apoptosis or become inactive flat-shaped bone lining cells, which cover the bone surface, where neither bone resorption nor formation occurs in humans. Bone lining cells prevent bone resorption by blocking the interaction between osteoclasts and the bone matrix, which should not be degraded. Another important function is the production of osteoprotegerin (OPG) and RANKL, which are crucial for osteoclast differentiation. Bone-resorbing osteoclasts are large multinucleated cells, which origin from the hematopoietic lineage. During resorption, osteoclasts secrete tartrate-resistant acid phosphatase (TRAP), cathepsin K, hydrogen ions, matrix metalloproteinase-9, and gelatinase, which are necessary for the digestion of the organic matrix. Dysregulations in the activity of osteoblasts can lead either to an increased or reduced bone mass (17, 53–56).
As mentioned before, PTH is a classical regulator of calcium and phosphorus homeostasis. Nevertheless, the hormone also appears to modulate immune functions. For instance, PTH receptors were found to be expressed by cells of the innate and acquired immune system [for review, see Geara et al. (57)]. Kotzmann et al. (58) described an increased CD4:CD8 T-cell ratio in patients with primary hyperparathyroidism, characterized by increased serum interleukin (IL)-6 and IL-17A levels (59, 60), and in mice, PTH augmented the production of tumor necrosis factor-α (TNF-α) by T cells. Moreover, in a recent study aiming at evaluating the immune function in patients with chronic postsurgical hypoparathyroidism, immune cell profiling revealed a decline in different immune cell populations including monocytes and regulatory, naïve, and total CD4+ lymphocytes. In addition, TNF-α and GM-CSF gene expression and circulating TNF-α levels were shown to be decreased in patients with chronic postsurgical hypoparathyroidism, whereas absolute numbers of total CD3−CD56+ natural killer cells were significantly increased (49). Collectively, these findings indicate that PTH induces proinflammatory cytokines and also nominates PTH as a regulator of the crosstalk between bone and the immune system, a field termed “osteoimmunology” as mentioned before (61, 62). Regarding this crosstalk, in addition to osteoclasts, also osteocytes are assumed to play an important role. In addition to their classical role as mechanosensors, osteocytes express several central regulators of bone and mineral metabolism and therefore can be regarded as endocrine cells (63). Osteocytes regulate bone formation by the expression of the Wnt antagonist sclerostin and dickkopf-1 (DKK-1), as well as osteopontin, a negative regulator of bone mineralization; fibroblast growth factor-23 (FGF-23) is an endocrine product of osteocytes that regulates phosphate homeostasis and 1,25(OH)2VitD3 synthesis (63–65). Osteocytes also express factors that determine osteoclast generation, namely RANKL, OPG, and proinflammatory cytokines such as IL-6, IL-17, and TNF-α (66, 67).
Responsiveness of osteocytes to PTH is well established in intermittent PTH administration causing net bone formation. This osteoanabolic effect is in part caused by decreasing sclerostin (68, 69). Given the well-established crosstalk between PTH and osteocytes on the one hand and the fact that osteocytes act as regulatory cells producing proinflammatory cytokines, among other substances, on the other hand, we expect that decreased PTH levels in hypoparathyroidism lead to decreased levels of proinflammatory cytokines produced by osteocytes, thereby contributing to bone effects seen in conjunction with hypoparathyroidism such as increased bone mineral density due to low bone turnover. Due to their inaccessible location, osteocytes are a challenging cell population to study; nevertheless, a number of osteocyte cell lines are available and have facilitated their investigation in vitro [for review, see Dallas et al. (70)]. As an alternative approach, for ex vivo studies, the advantage could be taken from the fact that osteocytes are by far the most abundant cell population in bone. In a recent publication, the protein expression of osteocytes in two different mouse strains was investigated immunohistochemically. It was evident that osteocytes express important proteins, such as sclerostin, DKK-1, or periostin, which are associated with bone formation (71).
In the genetic form of hypoparathyroidism (APS-1, see above), a mutation in the AIRE gene causes an impaired formation of the autoimmune regulator protein. The lack of this protein is associated with the decrease in autoantigen expression in the thymus and disruption of the negative selection of T-lymphocytes (28, 30). Many affected patients show autoantibodies against interferon (IFN)-α, IFN-ω, and IL-22 (72). As stated above, AIRE is known to play a crucial role in the induction of T-cell tolerance (73). 75 % of patients affected with DiGeorge syndrome, another form of genetic hypoparathyroidism, show immunodeficiencies including thymic hypoplasia and an impaired T-cell production, leading to a low T-cell count (26, 74).
4. Animal models of hypoparathyroidism
For the establishment of a valid biomedical model, it is of interest whether the targeted pathology occurs naturally in the respective species. Hypoparathyroidism, however, has only minor significance for veterinary species, and if yes, only pet animals are worth mentioning. There are descriptions of feline primary hypoparathyroidism (75), which resulted in a reversible myocardial failure due to excessive hypocalcemia in a patient (76) as well as primary hypocalcemia in dogs, which can be treated successfully with calcium and VitD3 supplementation (77). Whereas there exists a series of further case reports on canine and feline disease, there are only very few documentations on (primary) hypoparathyroidism in horses, which has a genetic background in this species (78), and a single report on a case of bovine disease (79). Remarkably, in pigs and sheep, there is no single report in the literature concerning hypoparathyroidism, which is in line with the findings of accessory parathyroid glands in these species. Interestingly, even in older pigs, such as minipigs, endocrinopathies (except for sexual ones) are nearly not encountered in the veterinary practice at all (80). No records on non-mammalian tetrapod hypoparathyroidism could be found.
Cats and dogs, although principally suited for parathyroidectomy (PTX) to induce hypoparathyroid conditions, have to be excluded in Austria due to legal regulations. Hence, large animal species worth considering as models are sheep and pigs. They are comparably easy to handle and inexpensive in housing. Moreover, they are frequently used in osteologic and osteoimmunological studies as they show anatomical and physiological similarities with humans in various organs including the bone compartment (81–83). Literature research and cadaveric feasibility studies regarding parathyroid gland anatomy and topography revealed that in sheep the superior parathyroid glands are easy to detect. However, the inferior parathyroid glands are deeply embedded in the thyroidal tissue and cannot be separated from the surroundings due to a missing encapsulation. Additionally, accessory parathyroid glands are disseminated over a large area of the ventral neck and total PTX is therefore impossible. This is underlined by reports in older literature of good tolerability of PTX in sheep, which points toward only a partial hypoparathyroid condition after surgery (84).
In swine, total PTX is difficult due to accessory glands, which can functionally replace primary parathyroid tissue. Moreover, the superior parathyroid glands are found in variable numbers and at variable sites. They are localized deeply in the massive cranio-ventral neck region in near vicinity to vulnerable structures and can easily be confused with other organs such as lymphatic tissue or thymus lobules. In recent literature, there is one report describing a new approach for PTX surgery in swine, however, without reporting on the effects of PTX (85). Another group described the effects of thyroparathyroidectomy (TPTX), but not isolated PTX, on bone development in unborn sheep (86). Taken together, both sheep and swine do not fulfill anatomical requirements for a suitable conventional surgical hypoparathyroidism model. Instead, these species ask for novel ways to visualize dispersed parathyroid tissues when choosing a surgical approach.
Existing genetically modified animal models of hypoparathyroidism include mice carrying parathyroid hormone (PTH)-null (87) or Glial Cells Missing Homolog 2 (GCM2)-null alleles (88). Whereas in the first model PTH levels are decreased by directly targeting the PTH gene, in the second model parathyroid gland development is impaired by targeting a transcription factor crucial for gland development. Unexpectedly, GCM2-deficient mice, despite their lack of parathyroid glands, displayed only a mildly abnormal bone phenotype with PTH levels that were identical to those in wild-type mice. Further studies revealed the thymus as an additional source of PTH compensating for impaired PTH output of the parathyroid gland (88). The use of these models for acquired hypoparathyroidism—the most common form of the disease (89)—is limited by an inherited chronic hypoparathyroid phenotype. Moreover, alterations in the development of organs affected by impairments of the hypothyroid gland (i.e., the skeleton) may occur. To overcome this limitation, Bi et al. established the PTHcre-iDTR mouse model, in which parathyroid cells selectively express the human diphtheria toxin receptors (DTR) (90). By systemic injection of diphtheria toxin, parathyroid cells can be ablated leading to low PTH levels and an acquired hypoparathyroid condition. There are also several genetic models mimicking related syndromes including impaired parathyroid gland development, such as the DiGeorge syndrome, the hypoparathyroidism–sensorineural deafness–renal dysplasia (HDR) syndrome, and the hypoparathyroidism–retardation–dysmorphism (HRD) syndrome [for review, see Garfield and Karaplis (91)]. Knockout mice are typically used as models, but there is also a zebra fish model mimicking the HRD syndrome (92). Given that these models failed to display conclusive symptoms of hypoparathyroidism (93–96), they are not suitable for use as models for that issue. Apart from this, in these syndromes, defects of the parathyroid gland are associated with other manifestations, making a comparison to hypoparathyroidism in humans difficult.
As an alternative to genetic models, surgical PTX is used to model acquired hypoparathyroidism. Mice, due to the small size of the parathyroid glands, are limited in their use for PTX. Hence, Bi et al. developed a mouse model in which green fluorescent protein (GFP) is selectively expressed in the parathyroid gland, thus facilitating a more precise PTX surgery (90). Another approach to overcome the limitation of small parathyroid glands in mice is the performance of TPTX (97). In this model, hormones of the thyroid gland (thyroid hormones and calcitonin) have to be supplemented, thereby reducing comparability to acquired hypoparathyroidism in humans. PTX was performed also in chickens and rabbits; however, most typically it is performed in rats (98–105). Comparable to mice, also in rats, fluorescence-based detection was used to enable precise excision of the parathyroid gland. To prevent animals from perishing, calcium must be supplemented. The ideal dietary calcium content in this model to mimic acquired hypoparathyroidism in humans was determined to be 0.5%. Serum calcium levels decreased, and phosphorus levels and bone volume increased (106, 107). Cadaveric studies performed by us revealed that rats are preferable to mice from a surgical perspective.
Thus, the seemingly most reliable model of hypoparathyroidism at the moment are Sprague Dawley rats being subject to PTX at the age of 8 weeks (106). As postsurgical hypoparathyroidism is more frequent in women than in men (54), female rats should be preferred. As an alternative for surgery, male Wistar rats treated with cinacalcet may be used for the development of a non-surgical rodent hypoparathyroid model. Cinacalcet suppresses the calcium levels, which was associated with a decline in PTH, and afterwards PTH 1-34 or a delayed-clearance PTH molecule (DC-PTH) were administered to reverse this effect (108). An advantage of this model is that there is no need to replace thyroid hormones (as in most surgical models). Nevertheless, it should be mentioned that long-term data are missing for this approach.
5. Future perspectives
Postsurgical hypoparathyroidism has been shown to be associated with a significantly increased risk of hospitalization due to infections. Deepening the knowledge on the osteoimmunological aspects of hypoparathyroidism will lead to a better understanding of the pathophysiological mechanisms behind this observation. In particular, the influence of decreased PTH levels on the ratio of pro- vs. anti-inflammatory cytokines and dynamics in responsible cell type populations should be assessed in vivo using rodent models as a first step. Moreover, by investigating the impact of PTH deficiency on the production of proinflammatory cytokines by osteocytes, a possible new role of osteocytes in linking PTH and hypoparathyroidism-related bone effects might be established. As mentioned before, lowered circulating TNF-α levels were observed in patients with chronic postsurgical hypoparathyroidism (49). We, therefore, expect not only a higher bone volume, as already described, but also a higher bone mineral content and increased cortical and trabecular thicknesses in PTX models and an altered cytokine profile of various immune cell populations and osteocytes. For future research, it would be an option to develop real-time detection methods for identifying accessory parathyroid tissue islets, thus being able to perform accurate total PTX also in large animal species, as these have advantages in further pharmacological studies.
6. Conclusion
Acquired hypoparathyroidism is the most common form of this rare endocrinopathy in humans with a reported range of prevalence of chronic hypoparathyroidism from 6.4 to 37/100,000 (109). Except for the PTHcre-iDTR mouse model (90), most genetic models available do not reflect acquired hypoparathyroidism satisfactorily. Moreover, concomitant alterations of immune functions that could interfere with the interpretation of results cannot be excluded, and PTHcre-iDTR mice are limited by high purchase costs. Hence, a surgical model of hypoparathyroidism is most suitable to meet clear criteria. Large animal models (sheep and pig)—although their immune system is also well characterized meanwhile (81, 110, 111), which is an argument in favor of their increased use in biomedical research in general, but also in osteological research in special (112)—do not fulfill the requirements of surgical feasibility and have thus to be excluded for such experiments till real-time detection methods for hypoparathyroid tissues exist. A model widely used for hypoparathyroidism and offering high availability of analytical reagents is PTX in mice and rats. Although the small size of mice limits the amount of tissue and biological fluids that can be harvested, PTX in rats seems to be the preferable option to date. One major limitation in all experimental models of hypoparathyroidism is the fact that over 80 % of respective human patients develop this condition as a complication of anterior neck surgery due to thyroid adenomas or related pathologies, meaning that data extrapolation of sole PTX (vs. TPTX) models needs to be done with caution.
Author contributions
PP and WS: conceptualization. MB, PP, and WS: writing—original draft preparation. MB, UF-S, KG, PP, and WS: writing—review and editing. PP: funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by an Investigator-Initiated Research grant from Takeda Austria GmbH (IIR-AUT-001855). The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kokenge F. Die Geschichte der Nebenschilddrüsen. Nuklearmediziner. (2019) 42:251–6. doi: 10.1055/a-0916-6441
2. Rowlands BC. Hyperparathyroidism: An early historical survey. Ann Royal Coll Surg Engl. (1972) 51:81.
3. Vermeulen AHM. The birth of endocrine pathology. Virchows Arch. (2010) 457:283–90. doi: 10.1007/s00428-010-0953-1
4. On On JSW, Chow BKC, Lee LTO. Evolution of parathyroid hormone receptor family and their ligands in vertebrate. Front Endocrinol. (2015) 6:149. doi: 10.3389/fendo.2015.00028
5. Cozzolino M, Brancaccio D, Gallieni M, Galassi A, Slatopolsky E, Dusso A. Pathogenesis of parathyroid hyperplasia in renal failure. J Nephrol. (2005) 18:5–8.
6. Potts JT. Parathyroid hormone: Past and present. J Endocrinol. (2005) 187:311–25. doi: 10.1677/joe.1.06057
7. Hannan FM, Babinsky VN, Thakker RV. Disorders of the calcium-sensing receptor and partner proteins: Insights into the molecular basis of calcium homeostasis. J Mol Endocrinol. (2016) 57:R127–42. doi: 10.1530/JME-16-0124
8. Brown SJ, Ruppe MD, Tabatabai LS. The parathyroid gland and heart disease. Methodist DeBakey Cardiovascular J. (2021) 13:49. doi: 10.14797/mdcj-13-2-49
9. Zajac JD, Danks JA. The development of the parathyroid gland: From fish to human. Curr Opin Nephrol Hypertens. (2008) 17:353–6. doi: 10.1097/MNH.0b013e328304651c
10. Åkerström G, Hellman PER, Hessman OLA, Segersten U, Westin G. Parathyroid glands in calcium regulation and human disease. Ann N Y Acad Sci. (2005) 1040:53–8. doi: 10.1196/annals.1327.005
11. Song L. Calcium and Bone Metabolism Indices. Adv Clin Chem. (2017) 82:1–46. doi: 10.1016/bs.acc.2017.06.005
12. Peacock M. Calcium metabolism in health and disease. Clin J Am Soc Nephrol. (2010) 5:S23–30. doi: 10.2215/CJN.05910809
13. Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. (2001) 81:239–97. doi: 10.1152/physrev.2001.81.1.239
14. Chang W, Tu C, Chen T-H, Bikle D, Shoback D. The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Sci Signal. (2008) 1:410. doi: 10.1126/scisignal.1159945
15. Guerreiro PM, Renfro JL, Power DM, Canario AVM. The parathyroid hormone family of peptides: Structure, tissue distribution, regulation, and potential functional roles in calcium and phosphate balance in fish. Am J Physiol Regul Integr Comp Physiol. (2007) 292:R679–96. doi: 10.1152/ajpregu.00480.2006
16. Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, et al. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature. (1993) 366:575–80. doi: 10.1038/366575a0
17. Grabowski P. Physiology of Bone. In:J. Allgrove and N. Shaw Calcium and bone disorders in children and adolescents 2nd, eds. Karger: Basel. (2015) 33–55. doi: 10.1159/000380991
18. Silver J, Russell J, Sherwood LM. Regulation by vitamin D metabolites of messenger ribonucleic acid for preproparathyroid hormone in isolated bovine parathyroid cells. Proc Natl Acad Sci USA. (1985) 82:4270–3. doi: 10.1073/pnas.82.12.4270
19. Datta NS, Abou-Samra AB. PTH and PTHrP signaling in osteoblasts. Cell Signal. (2009) 21:1245–54. doi: 10.1016/j.cellsig.2009.02.012
20. Giusti F, Brandi ML. Clinical Presentation of Hypoparathyroidism. In:M. L., Brandi, E., Ghigo, F., Guaraldi, , eds. Parathyroid Disorders: Focusing on Unmet Needs. Karger AG: Basel. (2018). p. 139–46. doi: 10.1159/000491044
21. Brandi ML, Bilezikian JP, Shoback D, Bouillon R, Clarke BL, Thakker RV, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metabol. (2016) 101:2273–83. doi: 10.1210/jc.2015-3907
22. Shoback DM, Bilezikian JP, Costa AG, Dempster D, Dralle H, Khan AA, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metabol. (2016) 101:2300–12. doi: 10.1210/jc.2015-3909
23. Kim JH, Shin Y-L, Yang S, Cheon CK, Cho JH, Lee BH, et al. Diverse genetic aetiologies and clinical outcomes of paediatric hypoparathyroidism. Clin Endocrinol. (2015) 83:790–6. doi: 10.1111/cen.12944
24. Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: The chromosome 22q112 deletion syndromes. Lancet. (2007) 370:1443–52. doi: 10.1016/S0140-6736(07)61601-8
25. Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q112 syndrome. Lancet. (2003) 362:1366–73. doi: 10.1016/S0140-6736(03)14632-6
26. McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JAS, et al. 22q112 deletion syndrome. Nat Rev Dis Primers. (2015) 1:907. doi: 10.1038/nrdp.2015.71
27. Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of aire control of T Cell tolerance. Immunity. (2005) 23:227–39. doi: 10.1016/j.immuni.2005.07.005
28. Ingelfinger JR, Husebye ES, Anderson MS, Kämpe O. Autoimmune polyendocrine syndromes. N Engl J Med. (2018) 378:1132–41. doi: 10.1056/NEJMra1713301
29. Weiler FG, Dias-da-Silva MR, Lazaretti-Castro M. Autoimmune polyendocrine syndrome type 1: Case report and review of literature. Arq Brasil Endocrinol Metabol. (2012) 56:54–66. doi: 10.1590/S0004-27302012000100009
30. Guo C-J, Leung PSC, Zhang W, Ma X, Gershwin ME. The immunobiology and clinical features of type 1 autoimmune polyglandular syndrome (APS-1). Autoimm Rev. (2018) 17:78–85. doi: 10.1016/j.autrev.2017.11.012
31. Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, et al. Diagnosis and treatment of primary adrenal insufficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metabol. (2016) 101:364–89. doi: 10.1210/jc.2015-1710
32. Sikjaer T, Rejnmark L, Rolighed L, Heickendorff L, Mosekilde L. The effect of adding PTH (1-84) to conventional treatment of hypoparathyroidism: A randomized, placebo-controlled study. J Bone Miner Res. (2011) 26:2358–70. doi: 10.1002/jbmr.470
33. Cusano NE, Nishiyama KK, Zhang C, Rubin MR, Boutroy S, McMahon DJ, et al. Noninvasive assessment of skeletal microstructure and estimated bone strength in hypoparathyroidism. J Bone Miner Res. (2016) 31:308–16. doi: 10.1002/jbmr.2609
34. Underbjerg L, Sikjaer T, Mosekilde L, Rejnmark L. The epidemiology of nonsurgical hypoparathyroidism in denmark: a nationwide case finding study. J Bone Miner Res. (2015) 30:1738–44. doi: 10.1002/jbmr.2501
35. Newman DB, Fidahussein SS, Kashiwagi DT, Kennel KA, Kashani KB, Wang Z, et al. Reversible cardiac dysfunction associated with hypocalcemia: A systematic review and meta-analysis of individual patient data. Heart Fail Rev. (2014) 19:199–205. doi: 10.1007/s10741-013-9371-1
36. Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metabol Clinics N A. (1993) 22:363–75. doi: 10.1016/S0889-8529(18)30171-3
37. Pohjola S. Ocular manifestations of idiopathic hypoparathyroidism. Acta Ophthalmol. (1962) 40:255–65. doi: 10.1111/j.1755-3768.1962.tb02365.x
38. Sarkar S, Mondal M, Das K, Shrimal A. Mucocutaneous manifestations of acquired hypoparathyroidism: An observational study. Indian J Endocr Metab. (2012) 16:819. doi: 10.4103/2230-8210.100637
39. Börglum Jensen S, Illum F, Dupont E. Nature and frequency of dental changes in idiopathic hypoparathyroidism and pseudohypoparathyroidism. Eur J Oral Sci. (1981) 89:26–37. doi: 10.1111/j.1600-0722.1981.tb01274.x
40. Bollerslev J, Rejnmark L, Marcocci C, Shoback DM, Sitges-Serra A, van Biesen W, et al. European Society of Endocrinology Clinical Guideline: Treatment of chronic hypoparathyroidism in adults. Eur J Endocrinol. (2015) 173:G1–G20. doi: 10.1530/EJE-15-0628
41. Astor MC, Løvås K, Debowska A, Eriksen EF, Evang JA, Fossum C, et al. Epidemiology and health-related quality of life in hypoparathyroidism in Norway. J Clin Endocrinol Metabol. (2016) 101:3045–53. doi: 10.1210/jc.2016-1477
42. Büttner M, Musholt TJ, Singer S. Quality of life in patients with hypoparathyroidism receiving standard treatment: A systematic review. Endocrine. (2017) 58:14–20. doi: 10.1007/s12020-017-1377-3
43. Mannstadt M, Clarke BL, Vokes T, Brandi ML, Ranganath L, Fraser WD, et al. Efficacy and safety of recombinant human parathyroid hormone (1-84) in hypoparathyroidism (REPLACE): A double-blind, placebo-controlled, randomised, phase 3 study. Lancet Diabetes Endocrinol. (2013) 1:275–83. doi: 10.1016/S2213-8587(13)70106-2
44. Mannstadt M, Bilezikian JP, Thakker RV, Hannan FM, Clarke BL, Rejnmark L, et al. Hypoparathyroidism. Nat Rev Dis Primers. (2017) 3:2317. doi: 10.1038/nrdp.2017.55
45. Bilezikian JP, Clarke BL, Mannstadt M, Rothman J, Vokes T, Lee H-M, et al. Safety and efficacy of recombinant human parathyroid hormone in adults with hypoparathyroidism randomly assigned to receive fixed 25-μg or 50-μg daily doses. Clin Therap. (2017) 39:2096–102. doi: 10.1016/j.clinthera.2017.08.011
46. Cusano NE, Rubin MR, McMahon DJ, Irani D, Anderson L, Levy E, et al. (1–84) is Associated with improved quality of life in hypoparathyroidism through 5 years of therapy. J Clin Endocrinol Metabol. (2014) 99:3694–9. doi: 10.1210/jc.2014-2267
47. Underbjerg L, Sikjaer T, Mosekilde L, Rejnmark L. Cardiovascular and renal complications to postsurgical hypoparathyroidism: A Danish nationwide controlled historic follow-up study. J Bone Miner Res. (2013) 28:2277–85. doi: 10.1002/jbmr.1979
48. Underbjerg L, Sikjaer T, Mosekilde L, Rejnmark L. Postsurgical hypoparathyroidism-risk of fractures, psychiatric diseases, cancer, cataract, and infections. J Bone Miner Res. (2014) 29:2504–10. doi: 10.1002/jbmr.2273
49. Puliani G, Hasenmajer V, Sciarra F, Barbagallo F, Sbardella E, Pofi R, et al. Impaired immune function in patients with chronic postsurgical hypoparathyroidism: results of the EMPATHY study. J Clin Endocrinol Metabol. (2021) 106:e2215–27. doi: 10.1210/clinem/dgab038
50. Underbjerg L, Sikjaer T, Rejnmark L. Long-term complications in patients with hypoparathyroidism evaluated by biochemical findings: a case-control study. J Bone Miner Res. (2018) 33:822–31. doi: 10.1002/jbmr.3368
51. Sipos W, Duvigneau JC, Schmoll F, Exel B, Hofbauer G, Baravalle G, et al. Characterization of the cytokine pattern of porcine bone marrow-derived cells treated with 1α,25(OH)2D3. J Vet Med A Physiol Pathol Clin Med. (2005) 52:382–7. doi: 10.1111/j.1439-0442.2005.00755.x
52. Sipos W, Zysset P, Kostenuik P, Mayrhofer E, Bogdan C, Rauner M, et al. OPG-Fc treatment in growing pigs leads to rapid reductions in bone resorption markers, serum calcium, and bone formation markers. Horm Metab Res. (2011) 43:944–9. doi: 10.1055/s-0031-1295463
53. Clarke B. Normal bone anatomy and physiology. Clin J Am Soc Nephrol. (2008) 3:S131–S139. doi: 10.2215/CJN.04151206
54. Le B, Nurcombe V, Cool S, van Blitterswijk C, de Boer J, LaPointe V. The components of bone and what they can teach us about regeneration. Materials. (2018) 11:14. doi: 10.3390/ma11010014
55. Bellido T, Plotkin LI, Bruzzaniti A. Bone cells. In:D.B. Burr and M. R.Allen, eds. Basic and Applied Bone Biology. Academic Press, Elsevier (2019). p. 37–55. doi: 10.1016/B978-0-12-813259-3.00003-8
56. Florencio-Silva R, Sasso GRdS, Sasso-Cerri E, Simões MJ, Cerri PS. Biology of bone tissue: structure, function, and factors that influence bone cells. BioMed Res Inter. (2015) 2015:421746. doi: 10.1155/2015/421746
57. Geara AS, Castellanos MR, Bassil C, Schuller-Levis G, Park E, Smith M, et al. Effects of parathyroid hormone on immune function. Clin Dev Immunol. (2010) 2010:418695. doi: 10.1155/2010/418695
58. Kotzmann H, Köller M, Abela C, Clodi M, Riedl M, Graninger W, et al. Effects of parathyroid hormone and serum calcium on the phenotype and function of mononuclear cells in patients with primary hyperparathyroidism. Eur J Clin Invest. (1998) 28:353–8. doi: 10.1046/j.1365-2362.1998.00283.x
59. Guo CY, Holland PA, Jackson BF, Hannon RA, Rogers A, Harrison BJ, et al. Immediate changes in biochemical markers of bone turnover and circulating interleukin-6 after parathyroidectomy for primary hyperparathyroidism. Eur J Endocrinol. (2000) 142:451–9. doi: 10.1530/eje.0.1420451
60. Li J-Y, D'Amelio P, Robinson J, Walker LD, Vaccaro C, Luo T, et al. IL-17A is increased in humans with primary hyperparathyroidism and mediates PTH-Induced bone loss in mice. Cell Metabol. (2015) 22:799–810. doi: 10.1016/j.cmet.2015.09.012
61. Rauner M, Sipos W, Thiele S, Pietschmann P. Advances in osteoimmunology: pathophysiologic concepts and treatment opportunities. Int Arch Allergy Immunol. (2013) 160:114–25. doi: 10.1159/000342426
62. Pietschmann P, Föger-Samwald U, Butylina M, Sipos W. Evolution and history of osteoimmunology. Osteologie. (2021) 30:286–91. doi: 10.1055/a-1561-3426
63. Pietschmann P, Skalicky M, Kneissel M, Rauner M, Hofbauer G, Stupphann D, et al. Bone structure and metabolism in a rodent model of male senile osteoporosis. Exp Gerontol. (2007) 42:1099–108. doi: 10.1016/j.exger.2007.08.008
64. Sipos W, Rauner M, Skalicky M, Viidik A, Hofbauer G, Schett G, et al. Running has a negative effect on bone metabolism and proinflammatory status in male aged rats. Exp Gerontol. (2008) 43:578–83. doi: 10.1016/j.exger.2008.03.008
65. Jaschke N, Sipos W, Hofbauer LC, Rachner TD, Rauner M. Skeletal endocrinology: Where evolutionary advantage meets disease. Bone Res. (2021) 9:1–10. doi: 10.1038/s41413-021-00149-x
66. Bonucci E, Ballanti P. Osteoporosis—Bone Remodeling and Animal Models. Toxicol Pathol. (2014) 42:957–69. doi: 10.1177/0192623313512428
67. Oheim R, Schinke T, Amling M, Pogoda P. Can we induce osteoporosis in animals comparable to the human situation? Injury. (2016) 47:S3–9. doi: 10.1016/S0020-1383(16)30002-X
68. Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. (2005) 37:148–58. doi: 10.1016/j.bone.2005.03.018
69. Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O'Brien CA, et al. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. (2005) 146:4577–83. doi: 10.1210/en.2005-0239
70. Dallas SL, Prideaux M, Bonewald LF. The osteocyte: an endocrine cell … and more. Endocr Rev. (2013) 34:658–90. doi: 10.1210/er.2012-1026
71. Kerschan-Schindl K, Schramek V, Butylina M, Föger-Samwald U, Pietschmann P. Differential expression of dickkopf 1 and periostin in mouse strains with high and low bone mass. Biology. (2022) 11:1840. doi: 10.3390/biology11121840
72. Savvateeva EN, Yukina MY, Nuralieva NF, Filippova MA, Gryadunov DA, Troshina EA. Multiplex autoantibody detection in patients with autoimmune polyglandular syndromes. Int J Mol Sci. (2021) 22:5502. doi: 10.3390/ijms22115502
73. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self-shadow within the thymus by the aire protein. Science. (2002) 298:1395–401. doi: 10.1126/science.1075958
74. Lavi RF, Kamchaisatian W, Sleasman JW, Martin DP, Haraguchi S, Day NK, et al. Thymic output markers indicate immune dysfunction in DiGeorge syndrome. J Allergy Clin Immunol. (2006) 118:1184–6. doi: 10.1016/j.jaci.2006.07.052
75. Gunn-Moore D. Feline endocrinopathies. Vet Clin North Am Small Anim Pract. (2005) 35:171–210. doi: 10.1016/j.cvsm.2004.09.002
76. Lie AR, MacDonald KA. Reversible myocardial failure in a cat with primary hypoparathyroidism. J Feline Med Surg. (2013) 15:932–40. doi: 10.1177/1098612X13480982
77. Russell NJ, Bond KA, Robertson ID, Parry BW, Irwin PJ. Primary hypoparathyroidism in dogs: A retrospective study of 17 cases. Australian Vet J. (2006) 84:285–90. doi: 10.1111/j.1751-0813.2006.00015.x
78. Rivas VN, Magdesian KG, Fagan S, Slovis NM, Luethy D, Javsicas LH, et al. Nonsense variant in Rap Guanine Nucleotide Exchange Factor 5 (RAPGEF5) is associated with equine familial isolated hypoparathyroidism in Thoroughbred foals. PLoS Genet. (2020) 16:e1009028. doi: 10.1371/journal.pgen.1009028
79. Villarroel A, Hustace JL, Piripi SA. Progressive alopecia associated with hypoparathyroidism in a Jersey heifer. Vet Dermatol. (2013) 24:470–e112. doi: 10.1111/vde.12036
80. Sipos W, Schmoll F, Stumpf I. Minipigs and Potbellied Pigs as Pets in the Veterinary Practice – A Retrospective Study. J Vet Med A. (2007) 54:504–11. doi: 10.1111/j.1439-0442.2007.00968.x
81. Sipos W. RANKL inhibition: preclinical data. In:Pietschmann P, (, ed.) Principles of Osteoimmunology: Molecular Mechanisms and Clinical Applications. Springer-Verlag Wien (2012). p. 197–215. doi: 10.1007/978-3-7091-0520-7_9
82. Sipos W, Föger-Samwald U, Pietschmann P. Supporting apparatus of vertebrates: skeleton and bones. In:Jensen-Jarolim E, (, ed.) Comparative Medicine. Springer-Verlag Wien (2013). p. 35–44. doi: 10.1007/978-3-7091-1559-6_3
83. Sipos W, Föger-Samwald U, Pietschmann P. Comparing two major bone pathologies in humans and companion animals. In:Jensen-Jarolim E, (, ed.) Comparative Medicine. Springer-Verlag Wien (2017). p. 87–96. doi: 10.1007/978-3-319-47007-8_6
84. Joest E. Handbuch der speziellen pathologischen Anatomie der Haustiere. Paul Parey (1967). p. 104–105.
85. Karakas E, Steinfeldt T, Gockel A, Westermann R, Kiefer A, Bartsch DK. Transoral thyroid and parathyroid surgery. Surg Endosc. (2010) 24:1261–7. doi: 10.1007/s00464-009-0757-z
86. Aaron JE, Abbas SK, Colwell A, Eastell R, Oakley BA, Russell RGG, et al. Parathyroid gland hormones in the skeletal development of the ovine foetus: The effect of parathyroidectomy with calcium and phosphate infusion. Bone Miner. (1992) 16:121–9. doi: 10.1016/0169-6009(92)90882-E
87. Miao D, He B, Lanske B, Bai X-Y, Tong X-K, Hendy GN, et al. Skeletal abnormalities in Pth-null mice are influenced by dietary calcium. Endocrinology. (2004) 145:2046–53. doi: 10.1210/en.2003-1097
88. Günther T, Chen Z-F, Kim J, Priemel M, Rueger JM, Amling M, et al. Genetic ablation of parathyroid glands reveals another source of parathyroid hormone. Nature. (2000) 406:199–203. doi: 10.1038/35018111
89. Clarke BL, Brown EM, Collins MT, Jüppner H, Lakatos P, Levine MA, et al. Epidemiology and diagnosis of hypoparathyroidism. J Clin Endocrinol Metabol. (2016) 101:2284–99. doi: 10.1210/jc.2015-3908
90. Bi R, Fan Y, Lauter K, Hu J, Watanabe T, Cradock J, et al. Diphtheria Toxin- and GFP-based mouse models of acquired hypoparathyroidism and treatment with a long-acting parathyroid hormone analog. J Bone Miner Res. (2016) 31:975–84. doi: 10.1002/jbmr.2769
91. Garfield N, Karaplis AC. Genetics and animal models of hypoparathyroidism. Trends Endocrinol Metabol. (2001) 12:288–94. doi: 10.1016/S1043-2760(01)00435-0
92. Sheehan-Rooney K, Swartz ME, Zhao F, Liu D, Eberhart JK. Ahsa1 and Hsp90 activity confers more severe craniofacial phenotypes in a zebrafish model of hypoparathyroidism, sensorineural deafness and renal dysplasia (HDR). Dis Model Mech. (2013) 19:87. doi: 10.1242/dmm.011965
93. Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah Y-C, Rosenblatt HM, et al. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. (1999) 401:379–83. doi: 10.1038/43900
94. Schinke M, Izumo S. Deconstructing DiGeorge syndrome. Nat Genet. (2001) 27:238–40. doi: 10.1038/85784
95. Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. (2001) 27:286–91. doi: 10.1038/85845
96. Pandolfi PP, Roth ME, Karis A, Leonard MW, Dzierzak E, Grosveld FG, et al. Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nat Genet. (1995) 11:40–4. doi: 10.1038/ng0995-40
97. Sakai A, Mori T, Sakuma-Zenke M, Takeda T, Nakai K, Katae Y, et al. Osteoclast development in immobilized bone is suppressed by parathyroidectomy in mice. J Bone Miner Metab. (2005) 23:8–14. doi: 10.1007/s00774-004-0534-y
98. Cole JA, Forte LR, Thorne PK, Poelling RE, Krause WJ. Autotransplantation of avian parathyroid glands: An animal model for studying parathyroid function. Gen Comp Endocrinol. (1989) 76:451–60. doi: 10.1016/0016-6480(89)90142-1
99. Gates FL, Grant JHB. Experimental observations on irradiated, normal, and partially parathyroidectomized rabbits. J Exp Med. (1927) 45:139–50. doi: 10.1084/jem.45.1.139
100. Sebastian EM, Suva LJ, Friedman PA. Differential effects of intermittent PTH (1–34) and PTH (7–34) on bone microarchitecture and aortic calcification in experimental renal failure. Bone. (2008) 43:1022–30. doi: 10.1016/j.bone.2008.07.250
101. Berdud I, Martin-Malo A, Almaden Y, Aljama P, Rodriguez M, Felsenfeld AJ. The PTH-calcium relationship during a range of infused PTH doses in the parathyroidectomized rat. Calc Tiss Int. (1998) 62:457–61. doi: 10.1007/s002239900460
102. Katsumata S-I, Masuyama R, Koshihara M, Matsuzaki H, Uehara M, Suzuki K. Effect of high phosphorus diet on phosphorus metabolism in parathyroidectomized rats. BioFactors. (2004) 22:33–7. doi: 10.1002/biof.5520220106
103. Chou F-F, Huang S-C, Chang S-F, Liaw J, Hung P-H. Oral gene therapy for hypoparathyroidism: a rat model. Hum Gene Ther. (2009) 20:1344–50. doi: 10.1089/hum.2009.015
104. Ferreira JC, Ferrari GO, Neves KR, Cavallari RT, Dominguez WV, dos Reis LM, et al. Effects of dietary phosphate on adynamic bone disease in rats with chronic kidney disease – role of sclerostin? PLoS One. (2013) 8:e79721. doi: 10.1371/journal.pone.0079721
105. Liao H-W, Hung P-H, Hsiao C-Y, Liou H-H, Lin H-S, Huang T-H, et al. Relationship between fibroblast growth factor 23 and biochemical and bone histomorphometric alterations in a chronic kidney disease rat model undergoing parathyroidectomy. PLoS ONE. (2015) 10:e0133278. doi: 10.1371/journal.pone.0133278
106. Jung SY, Kim HY, Park HS, Yin XY, Chung SM, Kim HS, et al. Standardization of a physiologic hypoparathyroidism animal model. PLoS ONE. (2016) 11:e0163911. doi: 10.1371/journal.pone.0163911
107. Park HS, Jung SY, Kim HY, Kim DY, Kim MS, Chung SM, et al. Development of hypoparathyroidism animal model and the feasibility of small intestinal submucosa application on the parathyroid autotransplantation. Eur Arch Otorhinolaryngol. (2015) 272:2969–77. doi: 10.1007/s00405-014-3262-5
108. Ramezanipour N, Esfahani SHZ, Eastell R, Newell-Price J, Trevitt G, Ross RJ, et al. Development of a hypoparathyroid male rodent model for testing delayed-clearance PTH molecules. Endocrinology. (2022) 163:1–9. doi: 10.1210/endocr/bqab239
109. Bjornsdottir S, Ing S, Mitchell DM, Sikjaer T, Underbjerg L, Hassan-Smith Z, et al. Epidemiology and financial burden of adult chronic hypoparathyroidism. J Bone Miner Res. (2022) 37:2602–14. doi: 10.1002/jbmr.4675
110. Sipos W, Duvigneau CJ, Hartl RT, Schwendenwein I. Exploratory reference intervals on hematology and cellular immune system of multiparous Large White sows. Vet Immunol Immunopathol. (2011) 141:307–11. doi: 10.1016/j.vetimm.2011.03.007
111. Sipos W. Shifts in porcine PBMC populations from adolescence to adulthood. Vet Immunol Immunopathol. (2019) 211:35–7. doi: 10.1016/j.vetimm.2019.04.002
Keywords: animal models, bone, PTH, T cells, interleukins, TNF-α
Citation: Butylina M, Föger-Samwald U, Gelles K, Pietschmann P and Sipos W (2023) Challenges in establishing animal models for studying osteoimmunology of hypoparathyroidism. Front. Vet. Sci. 10:1163903. doi: 10.3389/fvets.2023.1163903
Received: 11 February 2023; Accepted: 30 March 2023;
Published: 26 April 2023.
Edited by:
Ping Yang, Nanjing Agricultural University, ChinaReviewed by:
Hong Dong, The Ohio State University, United StatesWaseem Ali Vistro, Sindh Agriculture University, Pakistan
Copyright © 2023 Butylina, Föger-Samwald, Gelles, Pietschmann and Sipos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wolfgang Sipos, d29sZmdhbmcuc2lwb3NAdmV0bWVkdW5pLmFjLmF0