 Yangkai Liu
Yangkai Liu Yanan Mu3
Yanan Mu3 Wenxiang Wang
Wenxiang Wang Zulfiqar Ahmed
Zulfiqar Ahmed Chuzhao Lei
Chuzhao Lei- 1Plateau Livestock Genetic Resources Protection and Innovative Utilization Key Laboratory of Qinghai Province, Academy of Animal Science and Veterinary Medicine, Qinghai University, Xining, China
- 2Key Laboratory of Animal Genetics and Breeding on Tibet Plateau, Ministry of Agriculture and Rural Affairs, Xining, China
- 3Key Laboratory of Animal Genetics, Breeding and Reproduction of Shaanxi Province, College of Animal Science and Technology, Northwest A&F University, Xianyang, China
- 4Faculty of Veterinary and Animal Sciences, University of Poonch Rawalakot, Rawalakot, Pakistan
Qaidam cattle (CDM) are indigenous breed inhabiting Northwest China. In the present study, we newly sequenced 20 Qaidam cattle to investigate the copy number variants (CNVs) based on the ARS-UMD1.2 reference genome. We generated the CNV region (CNVR) datasets to explore the genomic CNV diversity and population stratification. The other four cattle breeds (Xizang cattle, XZ; Kazakh cattle, HSK; Mongolian cattle, MG; and Yanbian cattle, YB) from the regions of North China embracing 43 genomic sequences were collected and are distinguished from each of the other diverse populations by deletions and duplications. We also observed that the number of duplications was significantly more than deletions in the genome, which may be less harmful to gene formation and function. At the same time, only 1.15% of CNVRs overlapped with the exon region. Population differential CNVRs and functional annotations between the Qaidam cattle population and other cattle breeds revealed the functional genes related to immunity (MUC6), growth (ADAMTSL3), and adaptability (EBF2). Our analysis has provided numerous genomic characteristics of some Chinese cattle breeds, which are valuable as customized biological molecular markers in cattle breeding and production.
1. Introduction
Domestic cattle are one of the important animals that have been used as a source of materials for production and development by human civilization. Approximately 850,000 years ago, domestic cattle diverged into two groups, namely, humpless taurine (Bos Taurus) and humped indicine (Bos Indicus) (1, 2). Moreover, environmental factors, geographical isolation, and human activities also contributed to the development of present-day cattle. Through a long period of domestication, megabases (Mb) of DNA gradually enriched the genomic diversity among cattle breeds (3).
As of 2021 (4), there are already 55 Chinese indigenous breeds. The Qaidam cattle (CDM) is one of the 55 breeds reared in Northwest China (36°21'−39°23' N, 90°30'−99°30' E, Qinghai Province, China), where the drought (annual precipitation <200 mm) and high altitude (2,600–3,000 m) environment is predominant, and these conditions made the Qaidam cattle breed have more stress resistance, rough feeding tolerance, and environmental adaptability. During the Yuan dynasty (AD 1271–1638) period, the Mongolian army introduced the Mongolian cattle (Bostaurus) into the present-day Qinghai and Gansu Provinces of China during a southward invasion, which might have influenced the breeding herds of the present Qaidam cattle. Paternal and maternal diversity studies indicated that Qaidam cattle included two lineages (1, 5). The autosomal genetic evidence suggests that the Qaidam cattle was closer to Mongolian cattle, which is a hybrid of Bos Taurus × Bos Indicus (1). The purebred Qaidam cattle have not been effectively protected for their low economic returns. There was a 47.80% decrease in the Qaidam cattle population by 2006 compared to the 1981 Qaidam cattle population (6).
The copy number variations (CNVs) are defined as the deletion or duplication of a genome copy number, ranging from 50 bp to several Mb in length (7). As compared to SNP mutations, the CNV fragments are large in length and cover a wider range of genomes that have broader prospects in studying animal genetics and breeding application. Recently, next-generation genome sequencing technologies have been continuously used to detect the genome-wide CNVs of livestock (8, 9). However, numerous genomic studies exploring CNVs in commercial cattle breeds have underestimated the role of native breeds in the adaptation process (10, 11).
In the present study, we performed a genome-wide CNV analysis using genomic resequencing data in six Chinese cattle breeds. The purpose was to generate a comprehensive CNV landscape in Qaidam cattle to investigate and compare the diversity and population–genetic properties of the CNV regions (CNVRs) among them and to explore the diverse selection patterns involved with the CNV genes for local adaptation in Chinese native cattle.
2. Materials and methods
2.1. Genome resequencing and samples collection
Qaidam Basin is the highest basin in China with an altitude of 2,600–3,000 m and is located in the northwest region of the Qinghai Province and the northeast region of the Qinghai–Tibet Plateau. The climate of the basin is characterized as extremely dry and cold, with an annual average precipitation of <200 mm and an annual average temperature of ~3.0–6.5°C. To reflect the sample representativeness of the Qaidam cattle, 20 samples were collected from five different counties/cities (Dulan, Golmud, Mangya, Wulan, and Dachaidan) in the Qaidam Basin (Supplementary Table 1, Figure 2A).
The ear tissues of selected samples were used for DNA extraction by the standard phenol–chloroform protocol. Genomic DNA was constructed into 350-bp libraries and sequenced using Illumina NovaSeq at Novogene Bioinformatics Institute (Beijing, China). Moreover, 42 publicly available data of four Chinese cattle breeds were downloaded in this study (10 Mongolian cattle, MG; 9 Xizang cattle, XZ; 15 Yanbian cattle, YB; and 8 Kazakh cattle, HSK) (Supplementary Table 2). It is worth noticing that the resequencing data of one Xizang cattle (Sample ID: Xizang9) was offered by the Key Laboratory of Animal Genetics, Breeding and Reproduction of Northwest A&F University (Supplementary Table 2).
2.2. Genome data generation and CNV calling
Read pairs were aligned to the B. taurus reference assembly (ARS-UCD1.2) using the Burrows–Wheeler Aligner (BWA) program with default parameters (12). Then, CNVcaller (13) was applied to call the CNV in each individual. First, to create a B. taurus reference database, the ARS-UCD1.2 was split and the overlapping windows were recommended as 800 bp (13). Second, the reads number in each window was calculated, and high similarity (≧97%) reads were merged into segments of the autosomes. Third, the GC bias was used to standardize the copy number in each window, and it was used to classify the different genotypes of each sample. Finally, various steps of CNVcaller filtering parameters were carried out: -f 0.1 -h 3 -r 0.1; a Silhouette score of > 0.6; the length of CNVR of ≤ 50 kb (deletion and both), with the length of CNVR of <500 kb (duplication) (15).
2.3. Breed/population differentiation
Principal component analysis (PCA) was used to stratify and cluster the close breeds/populations, which plays a positive role in understanding the genetic differences among cattle subpopulations. According to the smartPCA module of EIGENSOFT (Program 2006), the PCA calculation was performed based on the four different CNVR datasets.
2.4. Differential CNVR identification
We calculated VST (15) between Qaidam cattle and the other four cattle breeds (XZ, YB, HSK, and MG) to identify the differential CNVRs. The VST is a method to calculate selection between populations similar to the FST method. The formula is VST = (VT – VS)/VT, where VT represents the variance among all the unrelated individuals and VS is the average variance within each population, weighted for population size (16). Finally, the top 1% gene cluster of the VST method was kept out by the outlier method.
The ANNOVAR was applied to annotate the CNVRs in our results (14). Further, the Kyoto Encyclopedia of Genes and Genomes (KEGG) and gene ontology (GO) analysis were performed on the candidate CNV genes by KOBAS 3.0. Since the enriched terms were retained with a p-value of <0.05, we preferred showing some of the top pathways; for more information, please see Supplementary Tables.
3. Results
3.1. CNV discovery and CNVR set statistics
We collected 63 Chinese cattle whole genomes representing five breeds from the north and northwest regions of China, including 20 Qaidam cattle, 10 Mongolian cattle, 15 Yanbian cattle, nine Kazakh cattle, and nine Xizang cattle (Figure 2A, Supplementary Table 2). The mean sequencing depth was performed to 12-fold coverage of the Bos taurus genome (Supplementary Table 2). Among the 63 genomes, we newly sequenced 20 Qaidam samples and one Xizang sample at ~9-fold coverage each (Supplementary Table 2), and the other 42 genomic sequences were available online.
We applied a read–depth-based bio-software (CNVcaller) to discover autosomal CNVs among individuals relative to the ARS-UCD1.2 reference genome. We generated the CNVR datasets from each cattle breed. The CNVR set contained 10,178 CNVRs, which were detected from 63 cattle genome datasets. There were 5,743 duplication CNVRs; 4,187 deletion CNVRs; and 248 both duplication and deletion CNVRs (Supplementary Table 3). Here, 10,178 CNVRs (duplication, deletion, and both duplication and deletion) were divided into different length groups (Figure 1A). The CNVRs annotation showed that the number of CNVRs was 5,398 (53.04%), which were detected in 2–5 kb size. It was observed that 55.92% CNVRs were located in the intergenic region followed by the intron region (35.77%). However, only 1.16 % CNVRs were detected in the coding exonic region (Figure 1B). And the CNVRs distribute randomly in the chromosome both in number and length (Figure 1C).
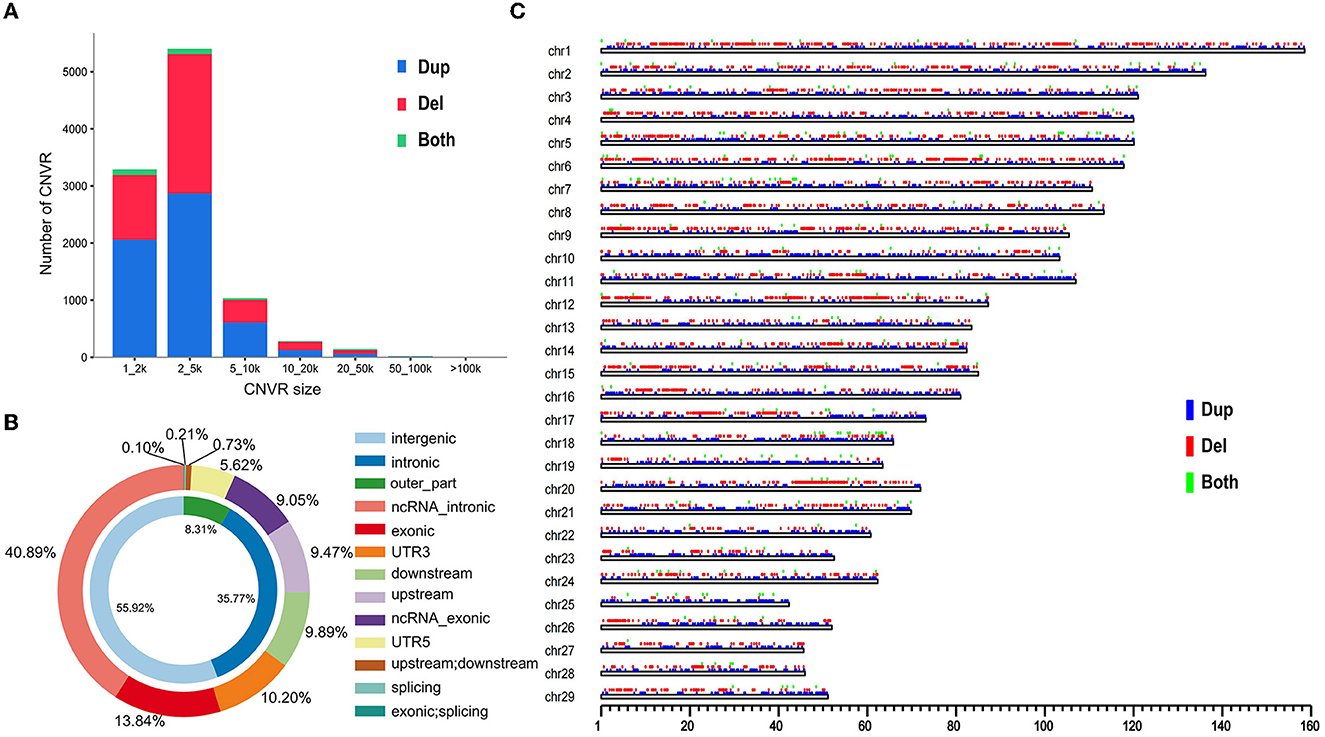
Figure 1. Genomic diversity and distribution of CNVRs. (A) The number of the detected CNVR. (B) Annotation of CNVRs with various genomic features. The inner circle indicates the intronic region, intergenic region, and the remaining set of functional regions (outer_part). The outer circle includes ncRNA intronic; exonic; UTR3; downstream; upstream; ncRNA exonic; UTR6; splicing; upstream and downstream; exonic and splicing. (C) The autosomal distribution of CNVRs. The location with different colors represent duplication (blue), deletion (red), and both duplication and deletion (green).
3.2. Population structure
With the effect of balancing selection, abundant polymorphisms of the genomic copy number variation are found in Chinese Bos taurus. A principal component analysis (PCA) was carried out with an obvious distinction from deletions (Figure 2B), duplications (Figure 2C), and total CNVRs datasets (Figure 2D). Qaidam cattle population is broadly distinguished from Mongolian, Kazakh, and Xizang breeds and closely clustered with the Yanbian breed. The PC1 explained ~30.13–50.86% of the genetic variation. For deletions, PC1 (30.13% of the variance) could separate Qaidam and Yanbian breeds from the other breeds, and PC2 (3.52% of the variance) could distinguish Xizang cattle (Figure 2B) from the other breeds (Kazakh and Mongolian cattle). Compared to deletions, duplications separated Qaidam cattle from all other breeds, in general, as shown in the PCA, but its clustering had less accuracy (Figure 2C). The effect of the PCA using both CNVR types was not optimistic in the clustering populations (Supplementary Figure 1). Interestingly, Kazakh and Mongolian cattle populations showed greater separation within these breeds by duplication. Unlike Qaidam, Xizang, and Yanbian cattle, Kazakh and Mongolian cattle may have less pressure of selection, which caused numerous meaningless duplications. These data suggest that artifical selection has shaped the CNVR diversity of each cattle breed during animal domestication.
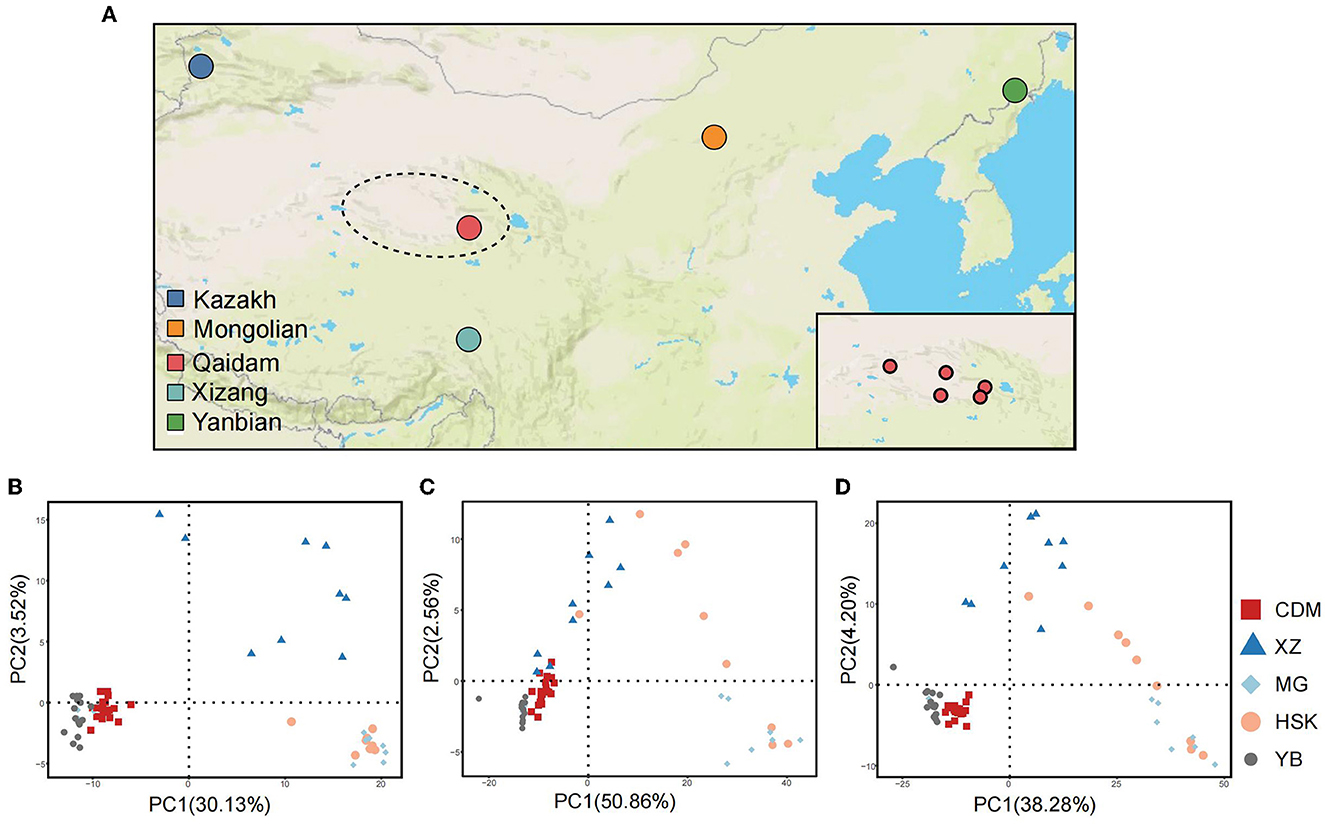
Figure 2. Geographic distribution and population stratification in five Chinese cattle. (A) The major geographical locations of the five Chinese cattle breeds. The dotted circle represents the Qaidam Basin, which is the main geographical distribution area of Qaidam cattle. In this study, Qaidam cattle were selected from five geographical locations along the Qaidam Basin, as shown in the lower right corner, which are Dulan, Golmud, Mangya, Wulan, and Dachaidan. (B) The principal component analysis (PCA) was derived from CNVRs (deletions). (C) PCA derived from CNVRs (duplications). (D) PCA derived from all CNVRs in five population genomes.
3.3. Differentiated CNVRs between Qaidam cattle and other cattle breeds
We calculated the VST between Qaidam cattle (CDM) and other cattle breeds from the regions of North China (XZ, HSK, YB, and MG) (Supplementary Figure 2, Supplementary Tables 4–7). First, the top 1% signal value regions were kept out; then, it was annotated by the cattle reference genome (ARS-UCD1.2).
The selection signals between CDM and YB were enriched to “MAPK signaling pathway” (p = 4.49 × 10−7), “Pi3k-akt signaling pathway” (p = 1.82 × 10−8), “mTOR signaling pathway” (p = 3.22 × 10−6), “HIF-1 signaling pathway” (p = 4.95 × 10−5), and “aldosterone-regulated sodium reabsorption” (p = 5.91 × 10−4) (Figure 3A, Supplementary Table 8). There were five candidate genes (MUC6, WDR25, CNNM4, MGAM, and GFRA2) in the study (Figure 4A, Supplementary Table 4). The selection signals between CDM and MG were enriched to “ErbB signaling pathway” (p =2.73 × 10−4), “calcium signaling pathway” (p =2.69 × 10−3), “GnRH signaling pathway” (p = 0.01122), and “insulin signaling pathway” (p = 0.04588) (Figure 3B, Supplementary Table 9). Among these annotated genes, four genes (PTPRT, BOLL, PLIN4, and ADGRL3) deserved more attention in copy number between CDM and MG (Figure 4B, Supplementary Table 5). The selection signals between CDM and XZ were enriched to “axon guidance” (p = 5.48 × 10−6) and “ErbB signaling pathway” (p = 7.49 × 10−5) (Figure 3C, Supplementary Table 10). Among these annotated genes, five genes (PLIN4, CDH13, SYCP1, PTPRC, and ADAMTSL3) were noteworthy in copy number between CDM and XZ (Figure 4C, Supplementary Table 6). There was a difference in copy numbers between CDM and HSK. The selection signal enrichment pathways between CDM and HSK include “bacterial invasion of epithelial cells” (p = 1.10 × 10−3), “Salmonella infection” (p = 0.02195), and “human immunodeficiency virus 1 infection” (p = 0.02446) (Figure 3D, Supplementary Table 11). Among these annotated genes, three genes (KHDRBS2, THRDE, and EBF2) were notable in copy number between CDM and HSK (Figure 4D, Supplementary Table 7).
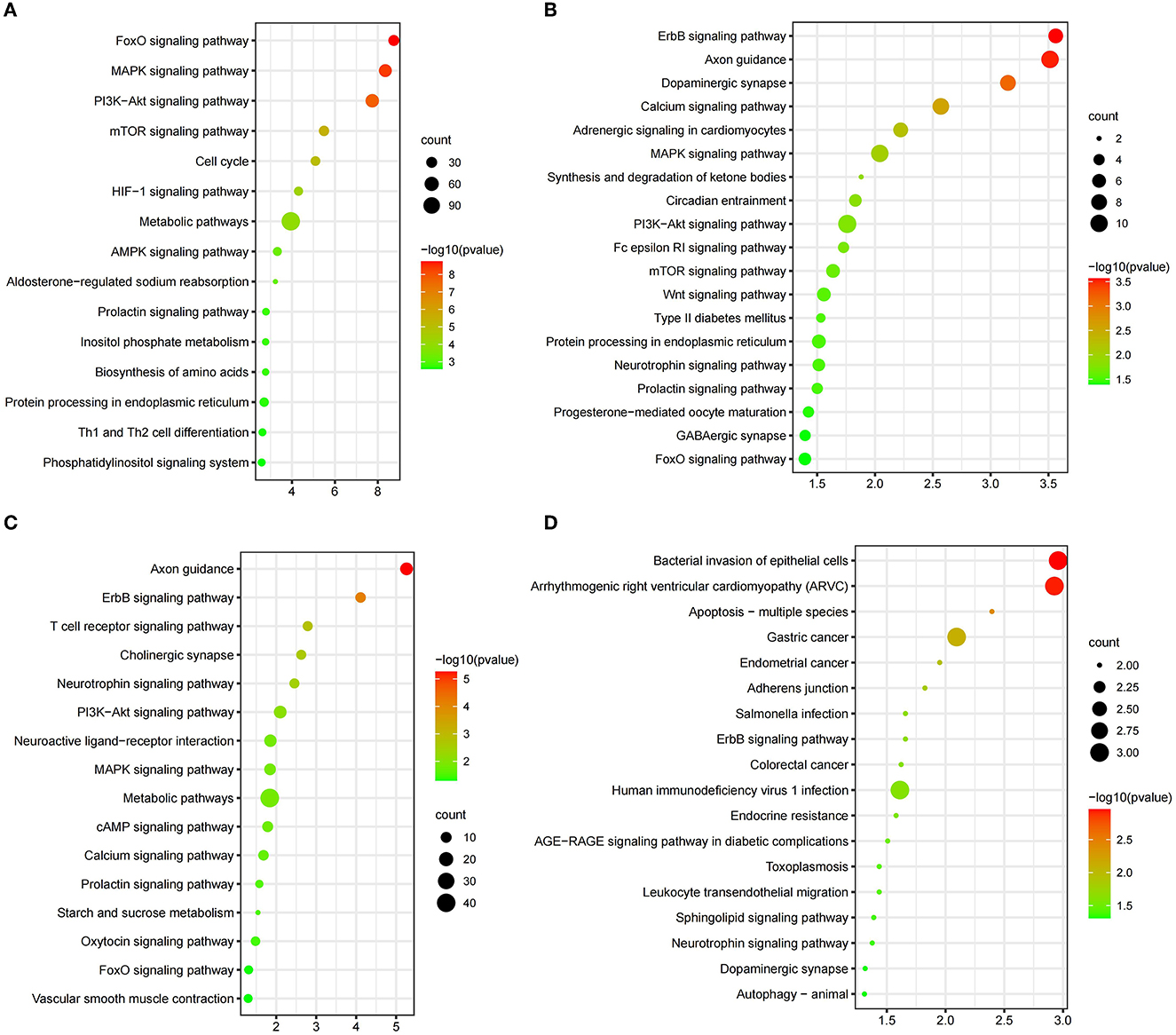
Figure 3. KEGG pathways from the enrichment analysis. (A) CDM vs. YB; (B) CDM vs. MG; (C) CDM vs. XZ; (D) CDM vs. HSK) (p < 0.05). We preferred showing part of the pathways; for more information, please see Supplementary Tables 8–11.
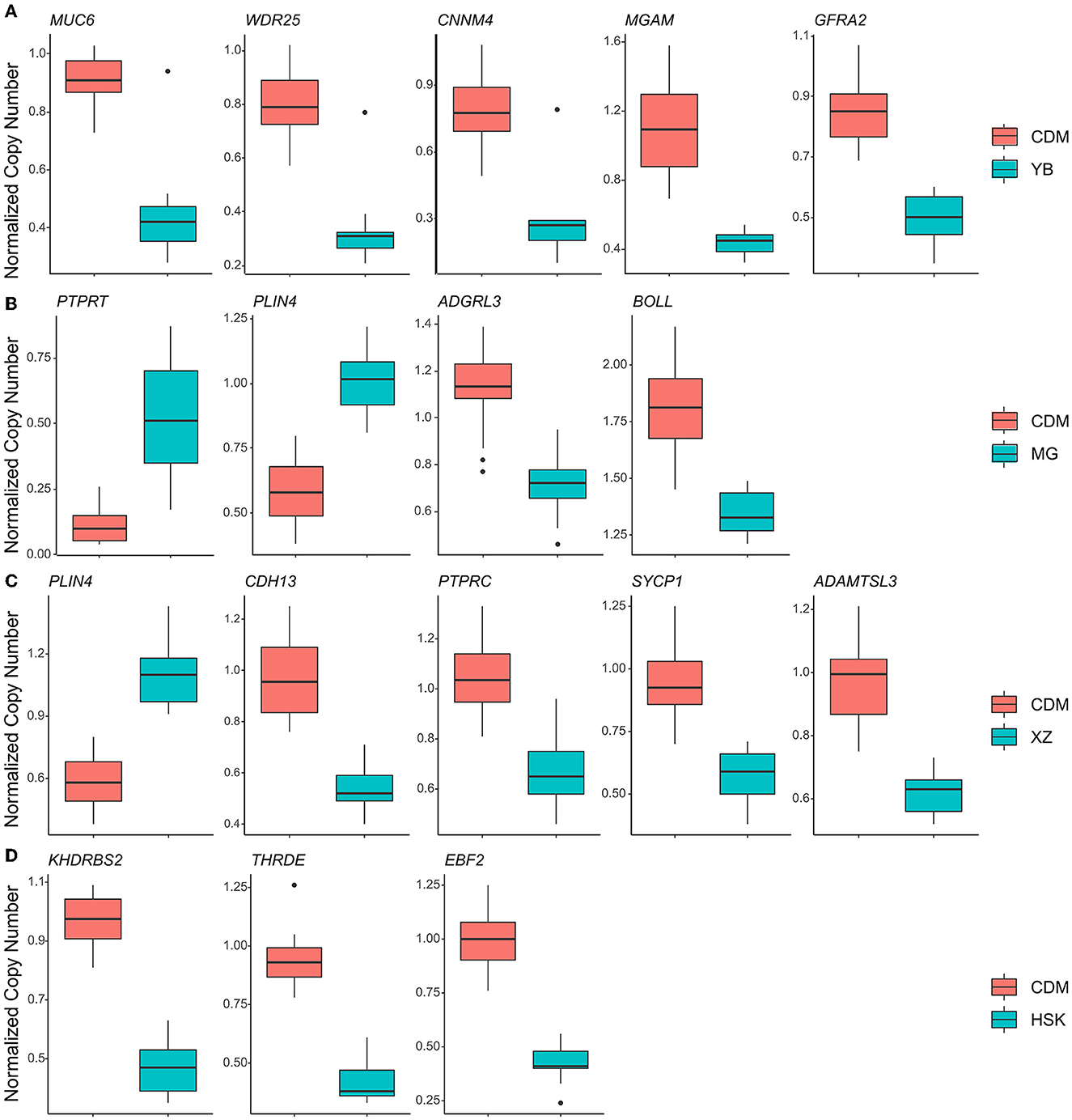
Figure 4. Normalized copy number comparison of the top 1% VST genes between CDM and other populations. (A) CDM compared with the YB population. (B) CDM compared with the MG population. (C) CDM compared with the XZ population. (D) CDM compared with the HSK population.
4. Discussion
During domestication and diversification, the frequency of copy number variation in the species' genome responds to selective pressure. Considerable effort has been applied to identify the causal mutations and genes. However, screening the selected genomic copy number genetic markers is complex. Over the past decades, high-throughput sequencing techniques and bioinformatics tools have been increasingly used to construct genome-wide CNV maps (1, 15, 17, 18). The diversity of CNVs has been extensively explored in Bos Taurus, Bos Indicus, and their crossing populations.
In our study, we investigated the CNV of 20 newly resequenced Qaidam cattle genomes based on the ARS_UCD 1.2 cattle reference genome. It improved the reliability of screening CNVs more than through UMD 3.1 assembly (19, 20). A total of 10,178 CNVRs were detected in five Chinese indigenous cattle breeds, and more than 99.9% CNVRs in length ranged from 1 to 100 kb. It was suggested that CNVs were widespread in Chinese cattle and may have been caused by the rapid adaptation during population expansion. For better statistics, variants were divided into three categories: duplication, deletion, and both duplication and deletion. The duplication was higher than deletions in number (Supplementary Table 3). And most of the CNVRs ranging from 2 to 5 kb in length (Figure 1A). In addition, the location of CNVRs is not uniformly distributed in the cattle genome (Figure 1C), and they are also not randomly distributed on chromosomes. The annotation uncovered that CNVRs are mostly annotated in the intergenic or intronic regions in the cattle genome. A previous study has also supported that many CNVRs are located on highly variable genes (15).
Compared to the analysis of the genome CNV in Qaidam cattle (18) for the first time, the role that CNVs have in the evolution of Qaidam cattle is becoming clear through our present study. The Qaidam cattle have strong adaptability to the arid environment, exhibiting dry, hypoxia, low air pressure, and large diurnal temperature difference (relative humidity 29–42%, precipitation 140–210.4 mm). Interestingly, YB cattle have almost opposite living conditions (relative humidity 68.6%; precipitation 500–700 mm) than Qaidam cattle. By consulting scientific articles, we found that EBF1 and ZNF521 related to fat development (21, 22) and VEGFA, EGLN2, and ENO3 were associated with high altitude hypoxic adaptation (23–25). In the enrichment analysis, the MGAM gene was significantly enriched in the “metabolic pathways (bta01100, P-value = 0.000113)” (Figure 3A), and was also clustered in the “carbohydrate metabolic process (GO:0005975, P-value = 0.014482)” (Supplementary Table 8). A previous study reported on the CNVR overlapping with the MAGM gene, and that it was related to starch digestion (26). Specifically, we found that MUC6 in Qaidam cattle was a normal-type CNVR, but it is a deletion CNVR in the YB cattle genome (Figure 4A). A previous study found CNV polymorphism in the MUC6 gene of domestic sheep, and this CNVR presents normal or duplication under arid environments, and deletion in warm and humid environments (27). Structurally, large numbers of tandem repeats rich in Pro, Thr, and Ser residues in MUC6 can affect the covalent attachment of O-glycans (28). In ruminants, such as sheep and cattle, the MUC6 gene has been associated with gastrointestinal parasite resistance (29, 30). Therefore, we hypothesized that the copy number difference of the MUC6 gene may influence the ability of antiparasitic immunity in Qaidam cattle and YB cattle.
High-quality beef is the breeding target of Qaidam cattle. In the comparison between Qaidam cattle and MG cattle (Supplementary Figure 2B), we observed that the PRKCA, CAMK2D, PHKB, and GRID2 genes (Supplementary Table 5) (VST value > 0.43) were related to muscle growth and development by searching previous research studies (31–34). Moreover, we identified PTPRT, BOLL, PLIN4, and ADGRL3 gene regions in the CNVRs of the top VST values which have obvious copy number differences between Qaidam cattle and MG cattle (Figure 4B). The ADGRL3 gene is associated with the nervous system of the Fuzhong buffalo (34). In addition, PTPRT (Chr13: s17021.1) was associated with body weight for pre-weaning growth in Esme sheep (35). In addition, the functional enrichment analysis of candidate genes with top 1% signal VST values revealed that the “GnRH signaling pathway” and “calcium signaling pathway” were significantly overrepresented. These results imply that the selected genes might contribute to the characteristics of growth rate and meat quality in Qaidam cattle.
Body size is one of the important traits in the evaluation of beef selection. In this study, we identified ADAMTSL3, PLIN4, CDH13, SYCP1, and PTPRC genes of the top 1% signal regions between Qaidam and XZ cattle (Supplementary Table 6). According to previous research, ADAMTSL3 plays an important role in chondrogenesis, morphogenesis, and skeletal growth in humans (36). A previous study reported that the bovine ADAMTSL3 gene has specific polymorphisms in individuals and the SNPs (T1532C and C1899T) were significantly associated with body size traits (37). Our results further suggested that copy number in the ADAMTSL3 gene may be one of the reasons for the difference in body size between XZ cattle and Qaidam cattle.
By comparing the copy number differences between the HSK and Qaidam breeds on the genome (Supplementary Figure 2C), we identified CNVRs with significant differences including KHDRBS2, THRDE, and EBF2 (Figure 4D, Supplementary Table 7). One of the eye-catching genes was EBF2, which has copy number polymorphism and showed a normal type in Qaidam cattle but a deletion type in HSK cattle (Figure 4D). Previous studies showed that EBF2 promotes brown adipocyte differentiation (38) and that its loss in mouse adipocytes abrogates brown adipose tissue (BAT) characteristics and function, leading to cold intolerance (39, 40). The cold tolerance of Qaidam cattle is an essential characteristic and it was speculated to be related to the copy number variation of EBF2.
5. Conclusion
Based on the high-quality Bos taurus reference genome, we constructed a CNV map of Northern Chinese Qaidam cattle using whole-genome resequencing data. Moreover, there are many copy number differences between Qaidam cattle and other cattle breeds from the regions of North China. It may play a crucial role in understanding the Qaidam cattle's adaptability, growth, and developmental characteristics In conclusion, these results provide a wealth of CNVR information to explore the valuable molecular markers in the Qaidam cattle genome.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Qinghai Academy of Animal Science and Veterinary Medicine, Qinghai University. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
YL drafted the manuscript and took part in the analysis of genome data. ZM, CL, and XW contributed to the sample collection. WW and YM performed the primary analysis of genome data. ZA and ZM revised the writing. ZM and CL designed the experiment and provided the funding for this research. All authors read and approved the final manuscript.
Funding
This work was supported by the Natural Science Foundation of Qinghai Province of China (2021-ZJ-914), Kunlun Talent. High-end Innovation and Entrepreneurship Talents Program of Qinghai Province and the earmarked fund for China Agriculture Research System of MOF and MARA (CARS-37).
Acknowledgments
The authors would like to thank the High-Performance Computing (HPC) of Northwest A&F University (NWAFU), China for providing computing resources.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1148070/full#supplementary-material
References
1. Chen N, Cai Y, Chen Q, Li R, Wang K, Huang Y, et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat Commun. (2018) 9:2337. doi: 10.1038/s41467-018-04737-0
2. Loftus RT, MacHugh DE, Bradley DG, Sharp PM, Cunningham P. Evidence for two independent domestications of cattle. Proc Nat Acad Sci. (1994) 91:2757–61. doi: 10.1073/pnas.91.7.2757
3. Lye ZN, Purugganan MD. Copy number variation in domestication. Trends Plant Sci. (2019) 24:352–65. doi: 10.1016/j.tplants.2019.01.003
4. National Committee of Animal Genetic Resources (2021). National List of Livestock and Poultry Genetic Resources in China. Beijing, China. Available online at: http://www.moa.gov.cn/govpublic/nybzzj1/202101/t20210114_6359937.htm.
5. Ma Z, Li R, Xia X, Luo J, Xie Y, Sun Y, et al. Y-SNPs genetic diversity, population genetic structure and paternal origin of Qaidamcattle. Genomics Appl Biol. (2018) 37:1920–5. doi: 10.13417/j.gab.037.001920
6. Zhao XZ, Zhao LX. Conservation status and development strategy of livestock and poultry genetic material in Qinghai Province. Qinghai J Animal Husbandry Vet Med. (2022) 52:65–8.
7. Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. (2011) 470:59–65. doi: 10.1038/nature09708
8. He Y, Hong Q, Zhou D, Wang S, Yang B, Yuan Y, et al. Genome-wide selective detection of Mile red-bone goat using next-generation sequencing technology. Ecol Evol. (2021) 11:14805–12. doi: 10.1002/ece3.8165
9. Yuan X, Li J, Bai J, Xi J. A local outlier factor-based detection of copy number variations from NGS data. IEEE/ACM Trans Comput Biol Bioinform. (2021) 18:1811–20. doi: 10.1109/TCBB.2019.2961886
10. Zhang Y, Hu Y, Wang X, Jiang Q, Zhao H, Wang J, et al. Population structure, and selection signatures underlying high-altitude adaptation inferred from genome-wide copy number variations in Chinese indigenous cattle. Front Genet. (2019) 10:1404. doi: 10.3389/fgene.2019.01404
11. Yang L, Niu Q, Zhang T, Zhao G, Zhu B, Chen Y, et al. Genomic sequencing analysis reveals copy number variations and their associations with economically important traits in beef cattle. Genomics. (2021) 113: 812–20. doi: 10.1016/j.ygeno.2020.10.012
12. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
13. Wang X, Zheng Z, Cai Y, Chen T, Li C, Fu W, et al. CNVcaller: highly efficient and widely applicable software for detecting copy number variations in large populations. Gigascience. (2017) 6:1–12. doi: 10.1093/gigascience/gix115
14. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
15. Huang Y, Li Y, Wang X, Yu J, Cai Y, Zheng Z, et al. An atlas of CNV maps in cattle, goat and sheep. Sci China Life Sci. (2021) 64:1747–64. doi: 10.1007/s11427-020-1850-x
16. Yang L, Xu L, Zhu B, Niu H, Zhang W, Miao J, et al. Genome-wide analysis reveals differential selection involved with copy number variation in diverse Chinese Cattle. Sci Rep. (2017) 7:14299. doi: 10.1038/s41598-017-14768-0
17. Zhou B, Ho SS, Zhang X, Pattni R, Haraksingh RR, Urban AE, et al. Whole-genome sequencing analysis of CNV using low-coverage and paired-end strategies is efficient and outperforms array-based CNV analysis. J Med Genet. (2018) 55:735. doi: 10.1136/jmedgenet-2018-105272
18. Guo S, Wu X, Pei J, Wang X, Bao P, Xiong L, et al. Genome-wide CNV analysis reveals variants associated with high-altitude adaptation and meat traits in Qaidam cattle. Electron J Biotechnol. (2021) 54:8–16. doi: 10.1016/j.ejbt.2021.07.006
19. Zhou J, Liu L, Reynolds E, Huang X, Garrick D, Shi Y, et al. Discovering copy number variation in dual-purpose Xinjiang brown cattle. Front Genet. (2022) 12:747431. doi: 10.3389/fgene.2021.747431
20. Lloret-Villas A, Bhati M, Kadri NK, Fries R, Pausch H. Investigating the impact of reference assembly choice on genomic analyses in a cattle breed. BMC Genom. (2021) 22:363. doi: 10.1186/s12864-021-07554-w
21. Chiarella E, Aloisio A, Codispoti B, Nappo G, Scicchitano S, Lucchino V, et al. ZNF521 Has an inhibitory effect on the adipogenic differentiation of human adipose-derived mesenchymal stem cells. Stem Cell Rev Rep. (2018) 14:901–14. doi: 10.1007/s12015-018-9830-0
22. Dang TN, Taylor JL, Kilroy G, Yu Y, Burk DH, Floyd ZE, et al. SIAH2 is Expressed in adipocyte precursor cells and interacts with EBF1 and ZFP521 to promote adipogenesis. Obesity. (2021) 29:98–107. doi: 10.1002/oby.23013
23. Wu DD, Ding XD, Wang S, Wójcik JM, Zhang Y, Tokarska M, et al. Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat Ecol Evol. (2018) 2:1139–45. doi: 10.1038/s41559-018-0562-y
24. Droma Y, Hanaoka M, Kinjo T, Kobayashi N, Yasuo M, Kitaguchi Y, et al. The blunted vascular endothelial growth factor-A (VEGF-A) response to high-altitude hypoxia and genetic variants in the promoter region of the VEGFA gene in Sherpa highlanders. Peer J. (2022) 10:e13893. doi: 10.7717/peerj.13893
25. Zhang B, Chamba Y, Shang P, Wang Z, Ma J, Wang L, et al. Comparative transcriptomic and proteomic analyses provide insights into the key genes involved in high-altitude adaptation in the Tibetan pig. Sci Rep. (2017) 7:3654. doi: 10.1038/s41598-017-03976-3
26. Lee Y, Bosse M, Mullaart E, Groenen MAM, Veerkamp RF, Bouwman AC, et al. Functional and population genetic features of copy number variations in two dairy cattle populations. BMC Genom. (2020) 21:89. doi: 10.1186/s12864-020-6496-1
27. Zheng Z, Wang X, Li M, Li Y, Yang Z, Wang X, et al. The origin of domestication genes in goats. Sci Adv. (2020) 6:z5216. doi: 10.1126/sciadv.aaz5216
28. Moniaux N, Escande F, Porchet N, Aubert JP, Batra SK. Structural organization and classification of the human mucin genes. Front Biosci. (2001) 6:D1192–206. doi: 10.2741/A579
29. Simpson HV, Umair S, Hoang VC, Savoian MS. Histochemical study of the effects on abomasal mucins of Haemonchus contortus or Teladorsagia circumcincta infection in lambs. Vet Parasitol. (2016) 226:210–21. doi: 10.1016/j.vetpar.2016.06.026
30. Rinaldi M, Dreesen L, Hoorens PR, Li RW, Claerebout E, Goddeeris B, et al. Infection with the gastrointestinal nematode Ostertagia ostertagi in cattle affects mucus biosynthesis in the abomasum. Vet Res. (2011) 42:61. doi: 10.1186/1297-9716-42-61
31. Luo, R., Dai, X., Zhang, L., Li, G., and Zheng, Z. (2022). Genome-wide DNA methylation patterns of muscle and tail-fat in dairymeade sheep and mongolian sheep. Animals. 12:1399. doi: 10.3390/ani12111399
32. Dou D, Shen L, Zhou J, Cao Z, Luan P, Li Y, et al. Genome-wide association studies for growth traits in broilers. BMC Genom Data. (2022) 23:1. doi: 10.1186/s12863-021-01017-7
33. Smith JL, Wilson ML, Nilson SM, Rowan TN, Schnabel RD, Decker JE, et al. Genome-wide association and genotype by environment interactions for growth traits in U.S. Red Angus cattle. BMC Genom. (2022) 23:517. doi: 10.1186/s12864-022-08667-6
34. Sun T, Huang GY, Wang ZH, Teng SH, Cao YH, Sun JL, et al. Selection signatures of Fuzhong Buffalo based on whole-genome sequences. BMC Genom. (2020) 21:674. doi: 10.1186/s12864-020-07095-8
35. Yilmaz O, Kizilaslan M, Arzik Y, Behrem S, Ata N, Karaca O, et al. Genome-wide association studies of preweaning growth and in vivo carcass composition traits in Esme sheep. J Animal Breed Genet. (2022) 139:26–39. doi: 10.1111/jbg.12640
36. Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet. (2008) 40:575–83. doi: 10.1038/ng.121
37. Liu Y, Zan L, Zhao S, Xin Y, Jiao Y, Li K, et al. Molecular characterization, expression pattern, polymorphism and association analysis of bovine ADAMTSL3 gene. Mol Biol Rep. (2012) 39:1551–60. doi: 10.1007/s11033-011-0894-z
38. Rajakumari S, Wu J, Ishibashi J, Lim HW, Giang AH, Won KJ, et al. EBF2 determines and maintains brown adipocyte identity. Cell Metab. (2013) 17:562–74. doi: 10.1016/j.cmet.2013.01.015
39. Angueira AR, Shapira SN, Ishibashi J, Sampat S, Sostre-Colón J, Emmett MJ, et al. Early B cell factor activity controls developmental and adaptive thermogenic gene programming in adipocytes. Cell Rep. (2020) 30:2869–78. doi: 10.1016/j.celrep.2020.02.023
Keywords: Qaidam cattle, whole genome resequencing, copy number variation (CNV), genome selection, population structure
Citation: Liu Y, Mu Y, Wang W, Ahmed Z, Wei X, Lei C and Ma Z (2023) Analysis of genomic copy number variations through whole-genome scan in Chinese Qaidam cattle. Front. Vet. Sci. 10:1148070. doi: 10.3389/fvets.2023.1148070
Received: 19 January 2023; Accepted: 01 March 2023;
Published: 31 March 2023.
Edited by:
Zexi Cai, Aarhus University, DenmarkReviewed by:
Zhixin Chai, Southwest Minzu University, ChinaWenlin Bai, Shenyang Agricultural University, China
Copyright © 2023 Liu, Mu, Wang, Ahmed, Wei, Lei and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhijie Ma, emhpamllbWFAMTI2LmNvbQ==; Chuzhao Lei, bGVpY2h1emhhbzExMThAMTI2LmNvbQ==