 Hu Xu1†
Hu Xu1† Lirun Xiang2†
Lirun Xiang2† Yan-Dong Tang2†
Yan-Dong Tang2† Chao Li2Jing Zhao2Bangjun Gong2Qi Sun2Chaoliang Leng3Jinmei Peng2
Chao Li2Jing Zhao2Bangjun Gong2Qi Sun2Chaoliang Leng3Jinmei Peng2 Qian Wang2Guohui Zhou2
Qian Wang2Guohui Zhou2 Tongqing An2Xuehui Cai2Zhi-Jun Tian2
Tongqing An2Xuehui Cai2Zhi-Jun Tian2 Hongliang Zhang2*Mingxin Song1*
Hongliang Zhang2*Mingxin Song1*- 1College of Veterinary Medicine, Northeast Agricultural University, Harbin, China
- 2State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Harbin, China
- 3Henan Key Laboratory of Insect Biology in Funiu Mountain, Henan Provincial Engineering Laboratory of Insect Bioreactors, China-UK-NYNU-RRes Joint Laboratory of Insect Biology, Nanyang Normal University, Nanyang, China
In the last decade, the emergence of QYYZ-like porcine reproductive and respiratory syndrome virus (PRRSV) has attracted increasing attention due to the high incidence of PRRSV mutation and recombination. However, the endemic status and genomic characteristics of the QYYZ-like strains are unclear. From 2018 to October 2021, 24 QYYZ-like PRRSV isolates were obtained from 787 PRRSV-positive clinical samples. Only one QYYZ-like positive sample was from a northern province, and the rest were from central and southern provinces. We selected 9 samples for whole-genome sequencing, revealing genome lengths of 15,008–15,316 nt. We retrieved all the available whole-genome sequences of QYYZ-like PRRSVs isolated in China from 2010 to 2021 (n = 28) from GenBank and analyzed them together with the new whole-genome sequences (n = 9). Phylogenetic tree analysis based on the ORF5 gene showed that all QYYZ-like PRRSV strains belonged to sublineage 3.5 but were clustered into three lineages (sublineage 1.8, sublineage 3.5, and sublineage 8.7) based on whole-genome sequences. Genomic sequence alignment showed that QYYZ-like strains, have characteristic amino acids insertions or deletions in the Nsp2 region (same as NADC30, JXA1 and QYYZ) and that thirteen strains also had additional amino acid deletions, mostly between 468 and 518 aa. Moreover, QYYZ-like strains (sublineage 3.5) have seven identical characteristic amino acid mutations in ORF5. Recombination analysis revealed that almost all QYYZ-like complete genome sequences (36/37) were products of recombination and mainly provided structural protein fragments (GP2-N) for the recombinant strains. Overall, QYYZ-like strains were mainly prevalent in central and southern China from 2018 to 2021, and these strains provided recombinant fragments in the PRRSV epidemic in China.
Introduction
Porcine respiratory and reproductive syndrome (PRRS) is a major disease in the pig industry that causes huge economic losses to the swine industry worldwide (1). The causative agent, porcine reproductive and respiratory syndrome virus (PRRSV), is an enveloped, positive-sense, single-stranded RNA virus belonging to the genus Betaarterivirus and family Arteriviridae of order Nidovirales (2). Due to its high degree of genetic diversity, PRRSV has been further divided into two species, PRRSV-1 (formerly known as European genotype 1) and PRRSV-2 (formerly known as North American genotype 2) (3). Since PRRSV was first discovered in China in 1996, the PRRSV-2 strain has been the main circulating strain in China (4).
Phylogenetic analyses of ORF5 of PRRSV-2 strains showed that PRRSV-2 could be divided into nine distinct lineages, with each lineage containing several sublineages (5, 6). Four different PRRSV-2 lineages have become widespread in China, including lineage 8 (JXA1-like/CH-1a-like), lineage 5 (VR-2332-like), lineage 1 (NADC30-like/NADC34-like), and lineage 3 (QYYZ-like) (7). The first PRRSV-2 strain CH-1a of China was isolated in 1996 in lineage 8 (8). HP-PRRSV (JXA1-like) was recognized in 2006 and originated from CH-1a-like strains (9). In 2013, some NADC30-like strains were isolated in China, and they have gradually become the dominant strains in recent years (10). NADC34-like strains were first reported in China in 2017 and became one of the major epidemic strains in 2020 (7, 11). Lineage 3 strains were initially reported in Taiwan and have emerged in Hong Kong (12). The FJ-1 strain was the first lineage 3 PRRSV detected in mainland China in 2005. The representative isolate of lineage 3 was QYYZ, which was identified in 2010 in mainland China and gradually became prevalent in southern China (12, 13). More importantly, lineage 3 viruses with greater virulence have been reported in southern China, and these viruses, recombining with lineage 1 and 8 PRRSV, pose a great threat to the Chinese pig industry (14–17).
Several studies have summarized the origin, classification, epidemic history and population dynamics of QYYZ-like strains based on the ORF5 gene (18, 19). However, the prevalence and genomic characteristics of these strains in recent years remain unclear. In this study, we carried out molecular epidemiological investigations for QYYZ-like PRRSV surveillance from 2018 to 2021. Meanwhile, the genome-wide characteristics of QYYZ-like strains and the role of these strains in the PRRSV epidemic were explored by comparing the latest strains and all reported genome-wide sequences.
Materials and Methods
Sample Collection and Genome Sequencing
From 2018 to 2021, we collected 1,803 clinical samples (including lung, lymph node and serum samples) of suspected PRRSV infection from different pig farms in 16 provinces in China (Heilongjiang, Jilin, Liaoning, Shandong, Henan, Guangdong, Guangxi, Zhejiang, Hebei, Hubei, Xinjiang, Inner Mongolia, Tianjin, Sichuan, Jiangxi and Jiangsu). Tissue sample processing, RNA extraction, cDNA preparation, RT–PCR and genome sequencing were performed as described in previous reports (8, 11). The primers used to detect PRRSV and amplify entire gene sequences were reported previously (20).
Sequence Analysis and Phylogenetic Analysis
Sequence analysis was performed with DNASTAR (version 7.1) software. Phylogenetic trees and molecular evolutionary analyses were conducted by MEGA 7 software using the neighbor-joining method with 1,000 bootstrap replications (21). The generated phylogenetic tree was annotated using the online software ITOL (https://itol.embl.de/) (22).
Recombination Analysis
To determine whether recombination screening occurred in the generation of QYYZ-like PRRSV strains, recombination events were considered only when supported by at least four of seven recombination detection algorithms (RDP, GENECONV, BootScan, MaxChi, Chimera, SiScan and 3Seq) in the Recombination Detection Program version 4.8 (RDP v.4.8). Finally, the pictures of recombination events were drawn by SimPlot v3.5.1 within a 500-bp window sliding along the genome alignment (20-bp step size).
Results and Discussion
From 2018 to 2021, 1,803 clinical samples were collected from 16 provinces of China; 787 (43.64%) tested positive for PRRSV according to RT–PCR. Of the 787 positive samples, 191 were from central or southern provinces (Henan, Guangdong, Guangxi, Zhejiang, Hubei, Sichuan, Jiangsu, and Jiangxi), and the remaining 596 were from northern provinces (Heilongjiang, Jilin, Liaoning, Shandong, Hebei, Xinjiang, Inner Mongolia, and Tianjin) (Figure 1). Through ORF5 phylogenetic analysis, 24 samples were confirmed to have QYYZ-like PRRSV. The results showed that QYYZ-like PRRSV did not cause a pandemic in China but persisted during 2018–2021. Interestingly, almost all samples with QYYZ-like PRRSV were from central and southern provinces, including Henan (4), Guangdong (9), Zhejiang (1) and Guangxi (9), and only one QYYZ-like strain came from Heilongjiang Province (Table 1). The QYYZ-like strains accounted for approximately 12% (23/191) of the cases in the central and southern provinces (Figure 1). Therefore, QYYZ-like PRRSV was mainly prevalent in central and southern China. To further study the complete genome characteristics of QYYZ-like PRRSV in China, we selected 9 strains (HNLCL15-1903, GXXNF10-1803, GXXNF53-1805, GDXNF60-1805, GXXNF74-1806, GXXNF78-1806, GDXNF229-1811, HNTZJ1714-2011, GXTZJ2325-2112) from 24 newly identified QYYZ-like isolates based on large homology differences and different branches of an ORF5 phylogenetic tree for whole-genome sequencing. The genomes of these isolates were 15,008–15,316 nt in length, excluding 3′ poly (A) tails. The significant difference in gene length between the newly QYYZ-like PRRSV strains may be due to the different deletion or insertion patterns in the Nsp2 region.
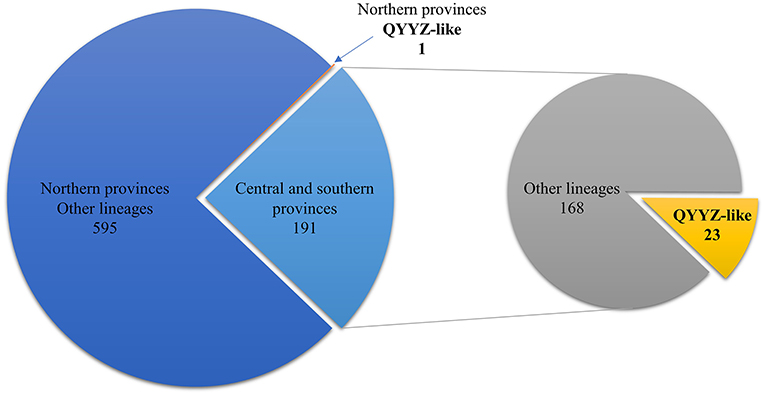
Figure 1. Number of QYYZ-like strains detected in different regions from 2018 to 2021. Among the 596 PRRSV-positive samples from northern provinces, 1 was a QYYZ-like strain. The remaining 191 PRRSV-positive samples were from central and southern provinces, of which 23 were QYYZ-like strains.
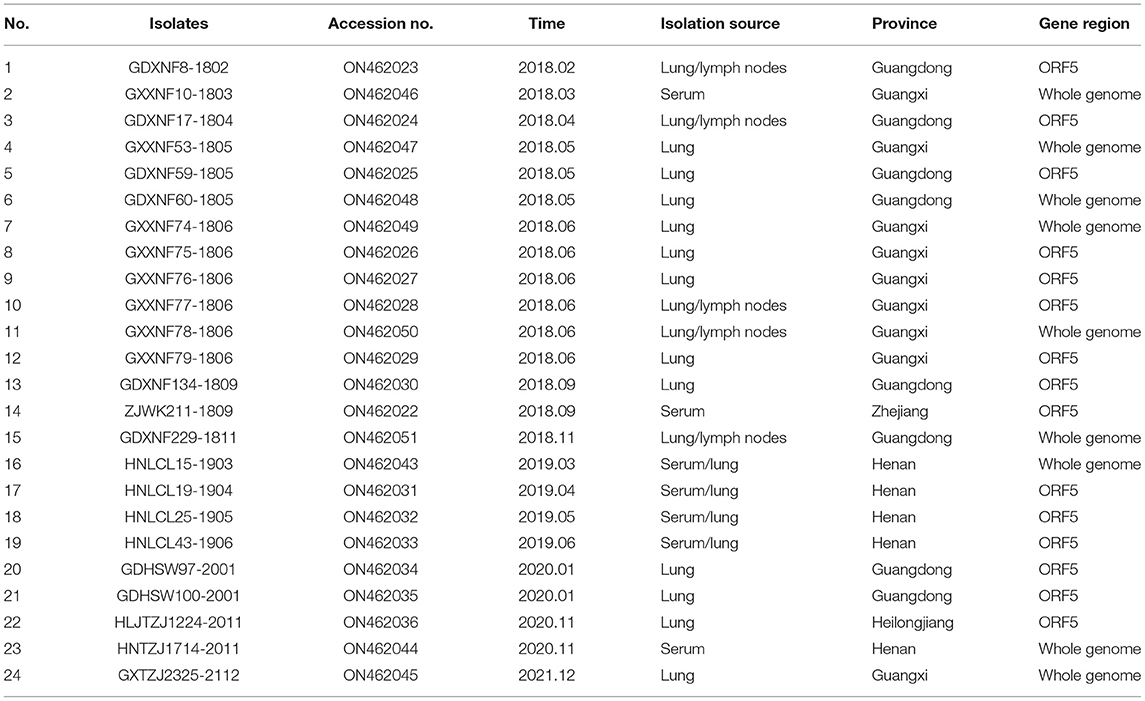
Table 1. Information on 24 strains of the newly identified QYYZ-like PRRSV.
To evaluate the genomic characteristics of the newly identified QYYZ-like PRRSV strains, the genomes of the novel PRRSV isolates were compared with those of different lineage viruses, including JXA1 (lineage 8), NADC30 (lineage 1) and QYYZ (lineage 3) (Table 2). Genome alignment revealed that the ORF5 gene of 24 QYYZ-like PRRSV strains shared 91.2–96.0% nucleotide homology with that of QYYZ, which was higher than the homology shared with that of JXA1 (81.9–85.7%) and NADC30 (82.2–85.2%). The homology among the 24 QYYZ-like strains was 86.4–100% (Table 2), and most of the QYYZ-like PRRSV ORF5 genes had low homology. The complete genome results also showed that the 9 newly identified QYYZ-like PRRSV strains shared 84.6–96.1% identity with JXA1, 82.3–90.8% identity with NADC30, and 83.6–93.8% identity with QYYZ (Table 2). Among them, the isolates GXXNF53-1805, GDXNF60-1805, GXXNF78-1806 and HNTZJ1714-2011 showed the highest identity (93.2, 95.4, 96.1, and 89.0%) with JXA1; the isolates GXXNF10-1803, GXXNF74-1806, GDXNF229-1811 and GXTZJ2325-2112 displayed the highest identity (87.8, 93.8, 91.0, and 86.3%) with QYYZ; and HNLCL15-1903 shared the highest identity with NADC30, but the homology was only 90.8%, suggesting that the newly identified QYYZ-like PRRSVs may have undergone large variation or recombination.
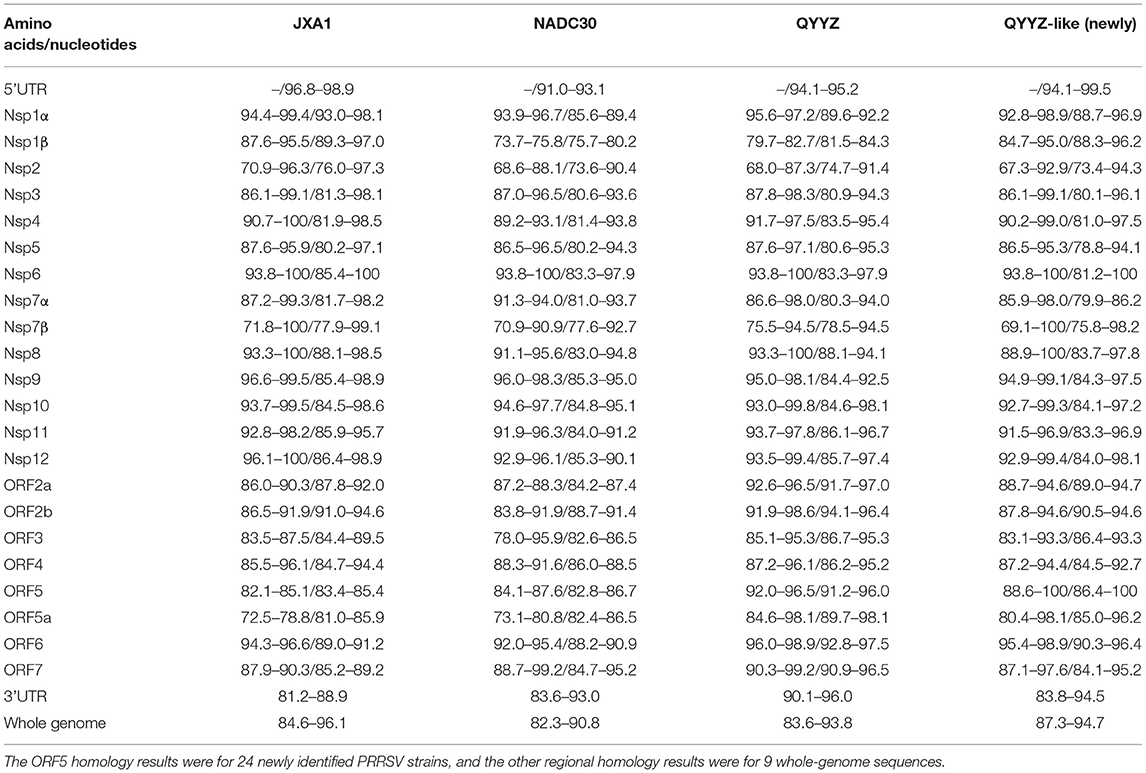
Table 2. Nucleotide and amino acid sequence similarity between the 24 new QYYZ-like PRRSVs and the reference strain.
To establish genetic relationships between Chinese QYYZ-like PRRSV strains and other PRRSV isolates, we constructed phylogenetic trees based on both the ORF5 gene and complete genomic sequence. Phylogenetic analysis based on ORF5 gene sequences demonstrated that all 24 new QYYZ-like PRRSV strains could be classified into sublineage 3.5 (Figure 2A). To analyze the genetic diversity of the novel QYYZ-like PRRSV in as much detail as possible, we expanded a data set (n = 470) including all ORF5 sequences of lineage 3 collected from GenBank and submitted before December 2021. As shown in Figure 2B, all 24 newly identified QYYZ-like strains in China (deep red labeled) clustered deeply within sublineage 3.5 from mainland China and did not form a separate clade. They are distantly related to sublineages 3.1–3.3 circulating in Taiwan and sublineage 3.4 circulating in Hong Kong (Figure 2B). To understand the genome-wide characteristics of QYYZ-like strains in China, we collected the whole-genome sequences of all QYYZ-like strains (ORF5 classified into sublineage 3.5) from GenBank (n = 28) (Table 3) and submitted before December 2021 and constructed phylogenetic trees together with the new strains (n = 9). Since the homology of QY2010 (Accession: JQ743666.1) and QYYZ (Accession: JQ308798.1) was up to 100% and the collection time of the two strains was very close, we regarded them as one strain and expressed them as QYYZ (Table 3). As shown in Figure 2C, 37 isolates were clustered into several branches with viruses belonging to different lineages. A total of 21 strains (GXXNF53-1805, GDXNF60-1805, GXXNF78-1806, HNTZJ1714-2011, etc.) were closely related to JXA1 and Hun4 (HP-PRRSV sublineage 8.7), while ten strains (GXXNF10-1803, GXXNF74-1806, GDXNF229-1811, etc.) were clustered into a separate branch close to QYYZ (sublineage 3.5). Additionally, HNLCL15-1903, GXTZJ2325-2112, SCya18 and FJLIUY-2017 were closer to NADC30 (sublineage 1.8). There was a large difference between the ORF5 gene-based and whole genome-based phylogenetic analyses. The epidemiology of PRRSV has been investigated largely by sequencing the ORF5 gene and classifying virus lineages based on ORF5 phylogenetic analysis (5, 6, 33). With the increasing number of recombinant strains, it is necessary to sequence the whole genome of key strains in order to classify them.
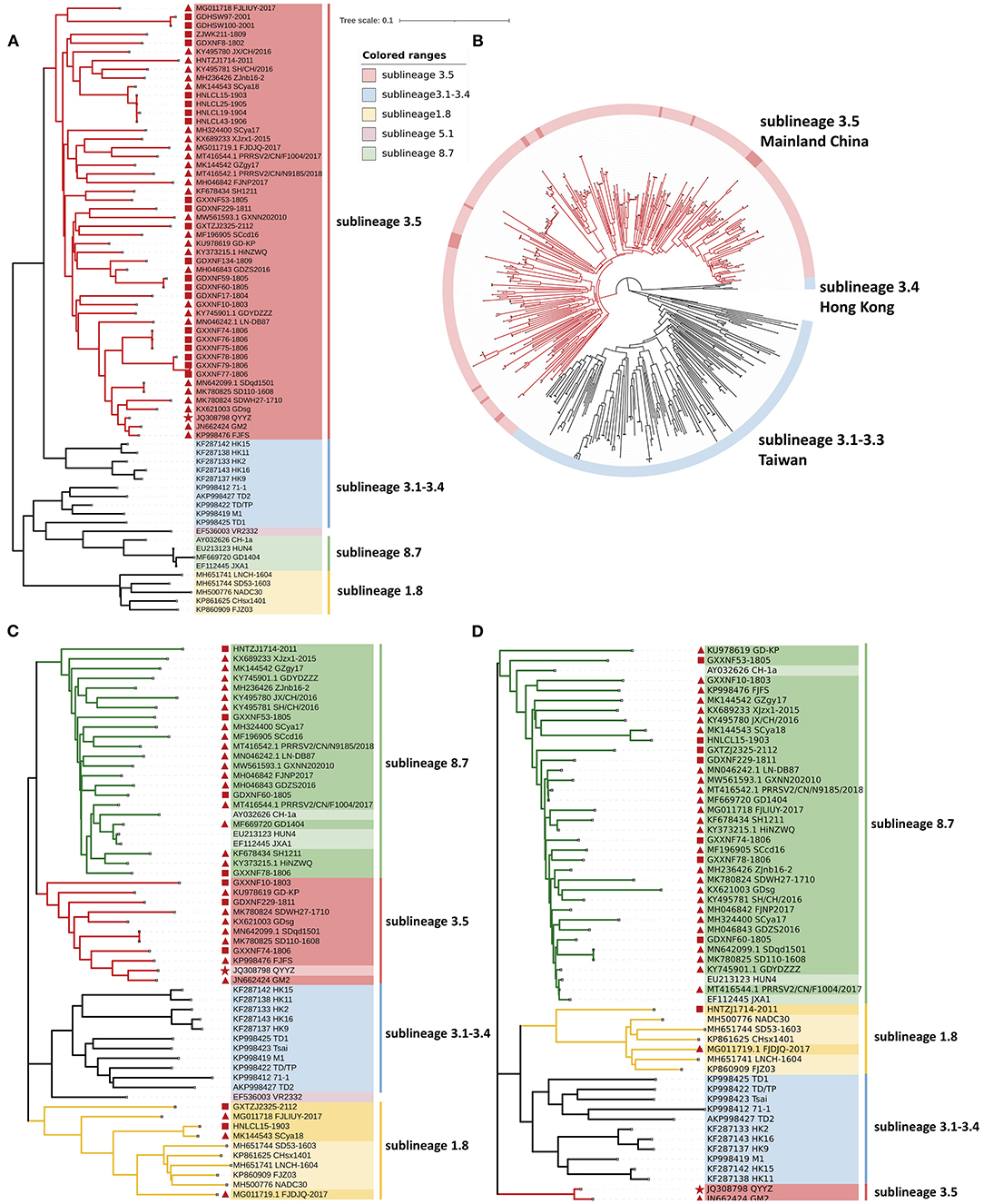
Figure 2. Phylogenetic analysis of QYYZ-like PRRSVs. (A) Phylogenetic tree constructed based on the ORF5 gene of novel QYYZ-like PRRSV isolates and reference PRRSV strains from each lineage. (B) Phylogenetic tree reconstructed based on the global data set (n = 470) of the lineage 3 PRRSV ORF5 genome. (C) Phylogenetic tree constructed based on full-length genomes of 37 QYYZ-like PRRSV isolates and reference PRRSV strains from each lineage. (D) Phylogenetic tree constructed based on the Nsp1 gene of 37 QYYZ-like PRRSV isolates and reference PRRSV strains from each lineage. The QYYZ-like prototype strain QYYZ is labeled with a red star ( ). Newly obtained sequences in this study are labeled with red squares (
). Newly obtained sequences in this study are labeled with red squares ( ). The QYYZ-like strains with complete genome sequences obtained from GenBank are labeled with red triangles (
). The QYYZ-like strains with complete genome sequences obtained from GenBank are labeled with red triangles ( ).
).
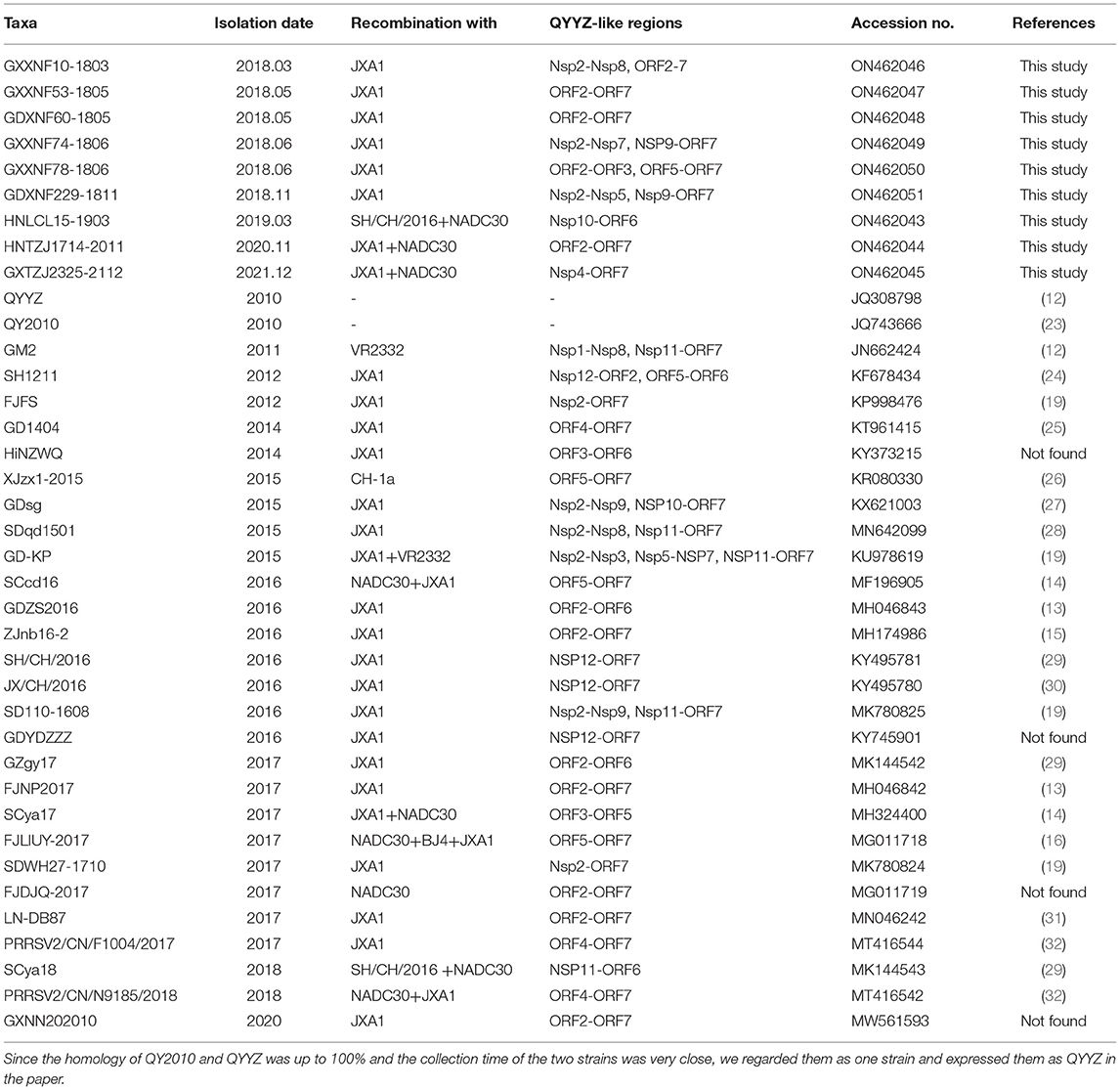
Table 3. Information on the whole genomes of all reported QYYZ-like PRRSV strains in China.
Nsp2 is the most significant variable region, with remarkable mutations, insertions and deletions, and is recognized as an important target gene for analyzing the genetic variation and molecular epidemiology of PRRSV (34). Partial Nsp2 sequence alignment showed that 37 Nsp2 sequences (from complete genome sequences of sublineage 3.5 PRRSVs) were divided into 5 large patterns (Figure 3A). Compared to Nsp2 of VR2332, the Nsp2 proteins of pattern A strains had a deletion pattern that was identical to that of JXA1 (1 aa +29 aa). Pattern B strains not only have the same 30-amino acid (aa) deletion in Nsp2 as JXA1 (1 aa +29 aa) but also have a 36-aa insertion at position 817-852, which was the same as QYYZ (Figure 3A). Pattern C strains also have the same 36-aa insertion in Nsp2 as QYYZ (Figure 3A). Additionally, Pattern D strains contained the discontinuous 131-aa deletion in Nsp2 identical to that in NADC30 (111 + 1 + 19 aa) (Figure 3A). In addition, 13 of the 37 Chinese lineage 3 PRRSV strains (XJzx12015, HiNZWQ, GXXNF78-1806, SH/CH/2016, GZgy17, JX/CH/2016, GD-KP, FJFS, GXXNF10-1803, GXXNF74-1806, GDXNF229-1811, GDsg, and SDWH27-1710) also have special amino acid deletions in the Nsp2 region, and they all have different deletion patterns (Figure 3A). Interestingly, 12 out of 13 strains with special amino acid deletions showed an amino acid deletion at positions 468-518 (Figure 3A). QYYZ-like PRRSV strains appeared to have more amino acid insertions or deletions in the Nsp2 region than other PRRSV strains. They seem to be more prone to amino acid deficiencies at position 468-518 of Nsp2. The Nsp2 hypervariable region (323-521) not only plays an important regulatory role in maintaining the balance of different viral mRNA species but also regulates PRRSV tropism to primary porcine alveolar macrophages (PAMs) (35). Therefore, these amino acid deletions in the Nsp2 region may alter the cellular tropism of the strains.
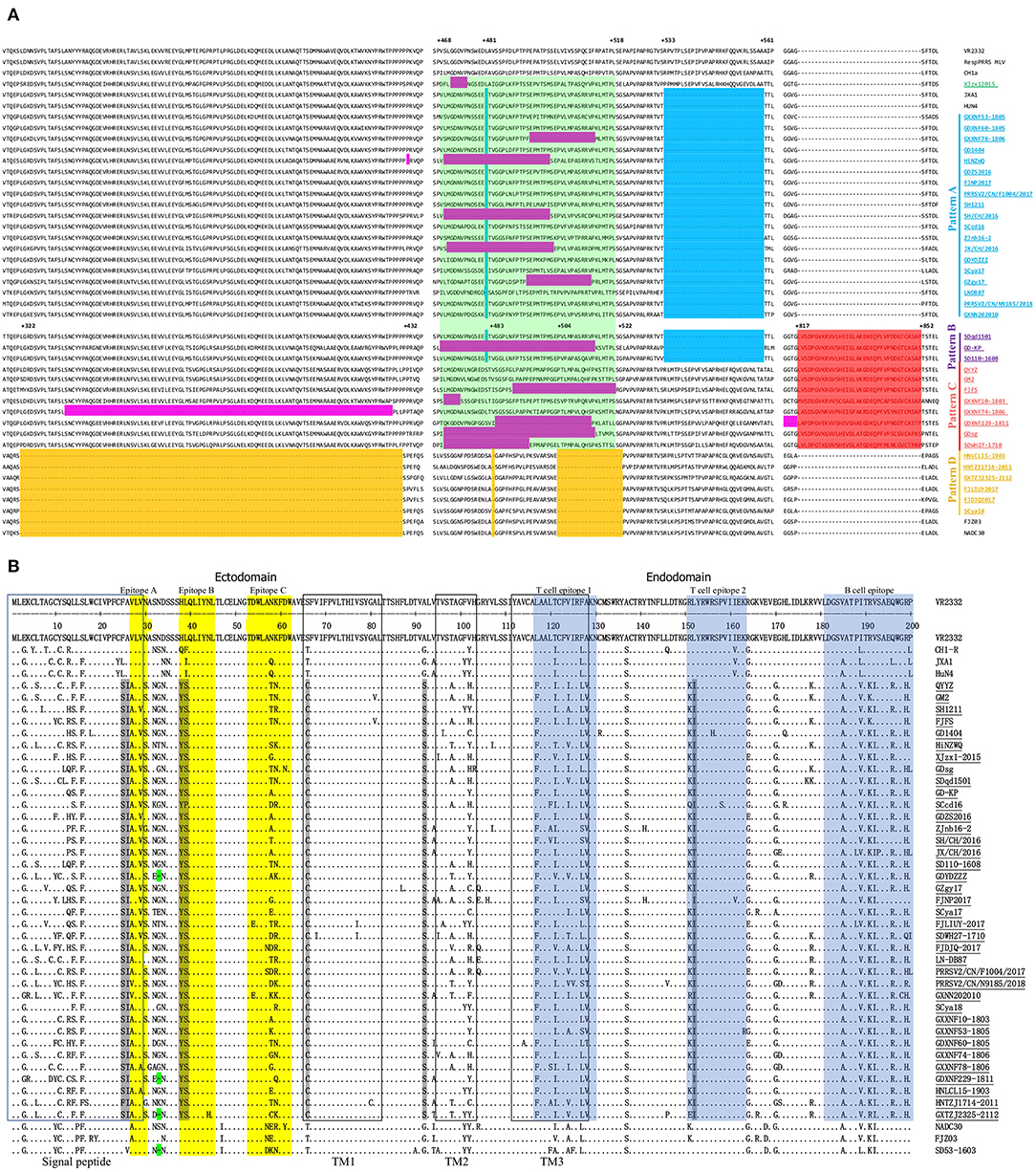
Figure 3. Alignment of the deduced amino acid sequences among QYYZ-like PRRSVs. (A) The positions marked in the figure represent positions of the Nsp2 amino acid sequence and refer to the position of VR-2332. Red indicates the QYYZ strain 36-aa characteristic continuous insertion; yellow indicates the NADC30-like PRRSV 131-aa characteristic discontinuous deletion; blue indicates the HP-PRRSV 30-aa characteristic discontinuous deletion; and purple indicates some additional a deletions in QYYZ-like PRRSVs. (B) Alignment of the deduced amino acid sequence based on the ORF5 gene. The signal peptide and transmembrane (TM) regions are shown in blue and black boxes, respectively. The linear antigenic epitopes and cellular epitopes are indicated in yellow and blue, respectively. The seven amino acids characteristic of QYYZ-like strains are shown in gray.
GP5 is the major envelope protein of PRRSV and is responsible for the lack of immunological cross-protection among different PRRSV strains due to its hypervariability (36–38). Comparison analyses of the amino acid sequence of QYYZ-like GP5 with those of the other representative strains showed that most strains encode 201 aa, but GDXNF229-1811, GDYDZZZ, and GXTZJ2325-2112 have a 1-aa deletion at residue 33 in VR2332, which is identical to the mutation in SD53-1603 (Figure 3B). Seven unique amino acid substitutions, namely, F25 → S25, A26 → I26, H38→Y38, L39 → S39, T66 → C66, A92 → S92, and L152 → I152, compared with the VR2332 strain. These amino acid mutations were identified only in all QYYZ-like strains but no other representative PRRSV strains (Figure 3B). Interestingly, although the QYYZ-like strains have low homology among them, they still showed consistent molecular features, which may be used as molecular markers to distinguish QYYZ-like strains from other type 2 PRRSV strains in China. GP5 is an envelope protein essential for viral infection, and at least three B-cell and two T-cell epitopes were identified for GP5 (39, 40). Two characteristic mutations of QYYZ-like strains, at positions 38(Y38) and 39(S39), are in epitope B (37-45 aa), which is a highly conserved peptide sequence that presumably functions as a major target for broadly neutralizing antibodies (37, 41). At the same time, there was one amino acid substitution in T-cell epitope 2 (I152), compared to PRRSV strains in other lineages (42). We then analyzed the functional domains in GP5 of QYYZ-like strains, including the signal peptide and transmembrane (TM) domain (43). Two residues (S25 and I26) resided within the signal peptide (aa 1-31). Moreover, there was a unique amino acid mutation at position 66 in the TM region of the QYYZ-like strains, substituting T66 to C66. Briefly, we found 7 unique amino acid mutations in the GP5 protein, and some mutation sites were located in cell epitopes, the signal peptide and the TM region. These amino acid substitutions might lead to the failure of receptor recognition and thus result in the failure of vaccines.
Recombination is a pervasive phenomenon among PRRSV isolates, and there are an increasing number of reports about the recombination of QYYZ-like PRRSVs (14, 16, 44). In recent years, many recombinant QYYZ-like PRRSVs have reportedly reemerged with increased pathogenicity (12, 18, 34, 45, 46). To identify possible recombination events of the new QYYZ-like PRRSVs in China, RDP4 and SimPlot software were used to assess possible recombinant events (Table 4 and Supplementary Figure 1). This analysis demonstrated that all nine new QYYZ-like isolates were recombinant viruses. Six recombinant isolates (GXXNF10-1803, GXXNF53-1805, GDXNF60-1805, GXXNF74-1806, GXXNF78-1806, and GDXNF229-1811) emerged from recombination events between HP-PRRSV isolates and QYYZ-like virus; three recombinant isolates (HNLCL15-1903, HNTZJ1714-2011, and GXTZJ2325-2112) were also derived from NADC30-like PRRSV, HP-PRRSV isolates, and QYYZ-like PRRSV (Table 4 and Supplementary Figure 1). These putative recombination events were further supported by statistically incongruent phylogenetic trees (Supplementary Figure 1). According to a similarity plot, these nine strains have an extremely complex recombination pattern (Supplementary Figure 1). Interestingly, the HNLCL15-1903 strain has almost the same recombination pattern as the previous strain SCya18 (MK144543.1) and high homology (BLAST analysis: 97.99%). SCya18 was isolated in Sichuan Province in 2018 (29), while HNLCL15-1903 was isolated in Henan Province in 2019. The two provinces are not adjacent, and under the background of low homology and complex recombination of QYYZ-like strains, the emergence of two strains with high similarity is noteworthy.
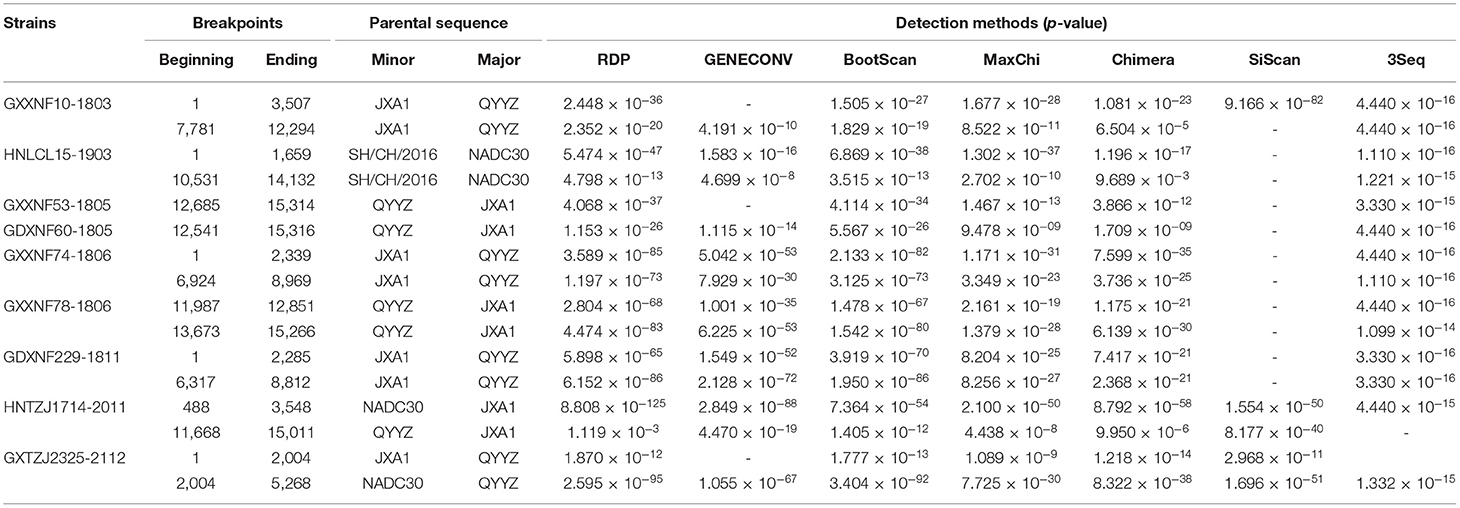
Table 4. Information on recombination events of QYYZ-like PRRSV isolates detected by RDP4 software.
To better explore the recombination characteristics of QYYZ-like strains in China (Table 3), we summarized the reported genome-wide recombination of all QYYZ-like strains (ORF5 classified into sublineage 3.5). Interestingly, all 37 QYYZ-like PRRSV strains except for QYYZ (the QYYZ-like original strain) have undergone recombination (Table 3 and Supplementary Figure 2B). The distribution of recombination breakpoints is dispersed and complex, there is no obvious recombination hotspot, and there are relatively many recombination breakpoints located in Nsp12(7), Nsp2(13) and GP2(13) (Supplementary Figure 2B). In a previous study in China, it was found that between 2014 and 2018, the high-frequency interlineage (mainly lineage 1 and lineage 8) recombination regions were located in Nsp9 and GP2 to GP3 (20, 31). Obviously, the recombination hot spots of QYYZ-like strains (sublineage 3.5) are not exactly the same as those of lineage 1 and lineage 8 strains. At the same time, it can be clearly seen that QYYZ-like strains mainly provide fragments of structural protein regions (GP2-N) for recombinant strains (Supplementary Figure 2A), while in the Nsp1 region of the sequence, only GM2 is provided by the QYYZ strain (Table 3). We used the Nsp1 region of QYYZ-like strains to construct a phylogenetic tree for verification (Figure 2D). The majority (33/37) of the QYYZ-like strains were grouped into sublineage 8.7 (JXA1-like), and FJLIUY-2017 and HNTZJ1714-2011 were grouped into sublineage 1.8 (NADC30-like). Only GM2 and QYYZ were classified as sublineage 3.5 (QYYZ-like). To obtain better information about the QYYZ-like strains, we suggest that researchers add a pair of primers to sequence the Nsp1 region of the virus when it is identified as a QYYZ-like strain by ORF5 sequencing.
Now, an overwhelming majority of the PRRSV-2 strains in China can be classified into JXA1-like/CH-1a-like (sublineage 8.7), VR2332-like (sublineage 5.1), NADC30-like/NADC34-like (lineage 1), and QYYZ-like (sublineage 3.5). The CH-1a-like strains first appeared in China and then gradually evolved into JXA1-like (HP-PRRSV) strains (45). The JXA1-like strains are also widespread in Cambodia, Thailand, Vietnam and many other Asian countries (6, 47–49). VR2332-like strains containing the Ingelvac PRRS MLV vaccine sequence are the most widespread, with viruses introduced to more than 10 countries (5). Lineage 1 (NADC30-like and NADC34-like) strains are also globally epidemic and have spread to South America, North America and Asia (7, 33, 50–52). The lineage 3 strains originated in Taiwan. Interestingly, after 20 years of transmission and evolution, these strains are prevalent only in greater China (mainland China, Taiwan, and Hong Kong) (18). Moreover, the QYYZ-like strains (sublineage 3.5) were prevalent only in mainland China. According to the results of this study and previous studies, QYYZ-like strains have been occasionally reported in northern China (19, 26, 28, 53) but are mainly prevalent in southern and central China (12–16, 23–25, 27, 29, 30, 32). Although recombination of PRRSV is very common, many non-recombination strains of JXA1-like (54), VR2332-like (28) and NADC30-like/NADC34-like PRRSV (7, 55) have been reported after long-term evolution in China. All QYYZ-like strains were recombined with other lineages of PRRSV except the prototype strain QYYZ. Thus, we speculated that the non-recombination QYYZ-like strains are no longer circulating and that they played a role as a provider of recombination fragments in the PRRSV epidemic in China.
Conclusion
In summary, QYYZ-like PRRSV strains did not have a large-scale epidemic status but persisted in central and southern China during 2018–2021. QYYZ-like strains have low homology and extremely complex amino acid insertion and deletion patterns in the Nsp2 region. However, they have seven identical amino acid mutations in the GP5 protein. These strains all underwent complex recombination except the prototype strain QYYZ and mainly provided structural protein fragments (GP2-N) for the recombinant strains. These results will help us to understand the overall genomic characteristics of QYYZ-like PRRSV, which is useful for the prevention and control of this virus.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by Sampling procedures were performed in accordance with the guidelines of the Animal Ethics Committee of the School of Harbin Veterinary Research Institute of the Chinese Academy of Agricultural Sciences. The Animal Ethics Committee Approval Number was SYXK(Hei) 2011022.
Author Contributions
Conceived and designed the experiments: MS, Y-DT, HZ, and Z-JT. Performed the experiments: HX and LX. Contributed reagents or materials and assisted in some experiments: CLi, JZ, BG, QS, JP, QW, GZ, TA, and XC. Analyzed the data: LX, CLe, and Z-JT. Contributed to the writing of the manuscript: HX, LX, and HZ. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Parasitic Resources Center (NPRC-2019-194-30), the National Natural Science Foundation of China (Grant no. 32172890 and 32002315), and the China Postdoctoral Fund (Grant no. 2020M680788).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.945381/full#supplementary-material
Supplementary Figure 1. Recombination analysis of GXXNF10-1803, GXXNF53-1805, GDXNF60-1805, GXXNF74-1806, GXXNF78-1806, GDXNF229-1811, HNLCL15-1903, HNTZJ1714-2011, and GXTZJ2325-2112. Phylogenic trees were constructed based on different parent regions. Blue represents HP-PRRSV, yellow represents NADC30-like PRRSV, and red represents QYYZ-like PRRSV.
Supplementary Figure 2. Summary of recombination breakpoints of all reported QYYZ strains in China. (A) The number of recombinant fragments provided by QYYZ in each region of 37 recombinant strains. The ORF5 of all 37 recombinant strains was provided by the QYYZ strain, while the Nsp1 region was provided by the QYYZ strain in only two strains. (B) Recombination breakpoints identified across the full-length genome. The putative recombinants are listed in the key, with the red triangles matching each corresponding breakpoint. Most breakpoints were localized in Nsp2 and GP2, and the backgrounds of the two regions are highlighted in green.
References
1. Nathues H, Alarcon P, Rushton J, Jolie R, Fiebig K, Jimenez M, et al. Cost of porcine reproductive and respiratory syndrome virus at individual farm level - An economic disease model. Prev Vet Med. (2017) 142:16–29. doi: 10.1016/j.prevetmed.2017.04.006
2. Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, et al. Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses. Arch Virol. (2017) 162:2505–38. doi: 10.1007/s00705-017-3358-5
3. Kuhn JH, Lauck M, Bailey AL, Shchetinin AM, Vishnevskaya TV, Bao Y, et al. Reorganization and expansion of the nidoviral family Arteriviridae. Arch Virol. (2016) 161:755–68. doi: 10.1007/s00705-015-2672-z
4. An TQ, Zhou YJ, Liu GQ, Tian ZJ, Li J, Qiu HJ, et al. Genetic diversity and phylogenetic analysis of glycoprotein 5 of PRRSV isolates in mainland China from 1996 to 2006: coexistence of two NA-subgenotypes with great diversity. Vet Microbiol. (2007) 123:43–52. doi: 10.1016/j.vetmic.2007.02.025
5. Shi M, Lam TT, Hon CC, Hui RK, Faaberg KS, Wennblom T, et al. Molecular epidemiology of PRRSV: a phylogenetic perspective. Virus Res. (2010) 154:7–17. doi: 10.1016/j.virusres.2010.08.014
6. Shi M, Lam TT, Hon CC, Murtaugh MP, Davies PR, Hui RK, et al. Phylogeny-based evolutionary, demographical, and geographical dissection of North American type 2 porcine reproductive and respiratory syndrome viruses. J Virol. (2010) 84:8700–11. doi: 10.1128/JVI.02551-09
7. Xu H, Li C, Li W, Zhao J, Gong B, Sun Q, et al. Novel characteristics of Chinese NADC34-like PRRSV during 2020-2021. Transbound Emerg Dis. (2022). doi: 10.1111/tbed.14485. [Epub ahead of print].
8. Leng CL, Tian ZJ, Zhang WC, Zhang HL, Zhai HY, An TQ, et al. Characterization of two newly emerged isolates of porcine reproductive and respiratory syndrome virus from Northeast China in 2013. Vet Microbiol. (2014) 171:41–52. doi: 10.1016/j.vetmic.2014.03.005
9. An TQ, Tian ZJ, Xiao Y, Li R, Peng JM, Wei TC, et al. Origin of highly pathogenic porcine reproductive and respiratory syndrome virus, China. Emerg Infect Dis. (2010) 16:365–7. doi: 10.3201/eid1602.090005
10. Guo Z, Chen XX, Li X, Qiao S, Deng R, Zhang G. Prevalence and genetic characteristics of porcine reproductive and respiratory syndrome virus in central China during 2016-2017: NADC30-like PRRSVs are predominant. Microb Pathog. (2019) 135:103657. doi: 10.1016/j.micpath.2019.103657
11. Zhang HL, Zhang WL, Xiang LR, Leng CL, Tian ZJ, Tang YD, et al. Emergence of novel porcine reproductive and respiratory syndrome viruses (ORF5 RFLP 1-7-4 viruses) in China. Vet Microbiol. (2018) 222:105–8. doi: 10.1016/j.vetmic.2018.06.017
12. Lu WH, Tun HM, Sun BL, Mo J, Zhou QF, Deng YX, et al. Re-emerging of porcine respiratory and reproductive syndrome virus (lineage 3) and increased pathogenicity after genomic recombination with vaccine variant. Vet Microbiol. (2015) 175:332–40. doi: 10.1016/j.vetmic.2014.11.016
13. Sun YK, Li Q, Yu ZQ, Han XL, Wei YF, Ji CH, et al. Emergence of novel recombination lineage 3 of porcine reproductive and respiratory syndrome viruses in Southern China. Transbound Emerg Dis. (2019) 66:578–87. doi: 10.1111/tbed.13067
14. Zhou L, Kang R, Zhang Y, Ding M, Xie B, Tian Y, et al. Whole genome analysis of two novel Type 2 porcine reproductive and respiratory syndrome viruses with complex genome recombination between lineage 8, 3, and 1 strains identified in Southwestern China. Viruses. (2018) 10:60328. doi: 10.3390/v10060328
15. Han G, Xu H, Wang K, He F. Emergence of Two different recombinant PRRSV strains with low neutralizing antibody susceptibility in China. Sci Rep. (2019) 9:2490. doi: 10.1038/s41598-019-39059-8
16. Liu J, Wei C, Lin Z, Fan J, Xia W, Dai A, et al. Recombination in lineage 1, 3, 5 and 8 of porcine reproductive and respiratory syndrome viruses in China. Infect Genet Evol. (2019) 68:119–26. doi: 10.1016/j.meegid.2018.12.006
17. Zhao J, Xu L, Xu Z, Deng H, Li F, Sun X, et al. Emergence and spread of NADC34-like PRRSV in Southwest China. Transbound Emerg Dis. (2022) 69:254–67. doi: 10.1111/tbed.14463
18. Sun YK, Han XL, Wei YF, Yu ZQ, Ji CH, Li Q, et al. Phylogeography, phylodynamics and the recent outbreak of lineage 3 porcine reproductive and respiratory syndrome viruses in China. Transbound Emerg Dis. (2019) 66:2152–62. doi: 10.1111/tbed.13269
19. Zhang WL, Zhang HL, Xu H, Tang YD, Leng CL, Peng JM, et al. Two novel recombinant porcine reproductive and respiratory syndrome viruses belong to sublineage 3.5 originating from sublineage 3.2. Transbound Emerg Dis. (2019) 66:2592–600. doi: 10.1111/tbed.13320
20. Zhang H, Leng C, Ding Y, Zhai H, Li Z, Xiang L, et al. Characterization of newly emerged NADC30-like strains of porcine reproductive and respiratory syndrome virus in China. Arch Virol. (2019) 164:401–11. doi: 10.1007/s00705-018-4080-7
21. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. (2016) 33:1870–4. doi: 10.1093/molbev/msw054
22. Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. (2021) 49:W293–6. doi: 10.1093/nar/gkab301
23. Deng Y, Pan Y, Wang D, Zhou Q, Bi Y, Chen F, et al. Complete genome sequence of porcine reproductive and respiratory syndrome virus strain QY2010 reveals a novel subgroup emerging in China. J Virol. (2012) 86:7719–20. doi: 10.1128/JVI.00977-12
24. Fan B, Wang H, Bai J, Zhang L, Jiang P. A novel isolate with deletion in GP3 gene of porcine reproductive and respiratory syndrome virus from mid-eastern China. Biomed Res Int. (2014) 2014:306130. doi: 10.1155/2014/306130
25. Zhang Q, Bai J, Hou H, Song Z, Zhao Y, Jiang P. A novel recombinant porcine reproductive and respiratory syndrome virus with significant variation in cell adaption and pathogenicity. Vet Microbiol. (2017) 208:150–8. doi: 10.1016/j.vetmic.2017.07.028
26. Zhang X, Li Y, Xiao S, Yang X, Chen X, Wu P, et al. High-frequency mutation and recombination are responsible for the emergence of novel porcine reproductive and respiratory syndrome virus in northwest China. Arch Virol. (2019) 164:2725–33. doi: 10.1007/s00705-019-04373-z
27. Dong JG, Yu LY, Wang PP, Zhang LY, Liu YL, Liang PS, et al. A new recombined porcine reproductive and respiratory syndrome virus virulent strain in China. J Vet Sci. (2018) 19:89–98. doi: 10.4142/jvs.2018.19.1.89
28. Zhang Z, Qu X, Zhang H, Tang X, Bian T, Sun Y, et al. Evolutionary and recombination analysis of porcine reproductive and respiratory syndrome isolates in China. Virus Genes. (2020) 56:354–60. doi: 10.1007/s11262-020-01751-7
29. Zhou L, Kang R, Zhang Y, Yu J, Xie B, Chen C, et al. Emergence of two novel recombinant porcine reproductive and respiratory syndrome viruses 2 (lineage 3) in Southwestern China. Vet Microbiol. (2019) 232:30–41. doi: 10.1016/j.vetmic.2019.01.026
30. Zhang F, Song D, Ye Y, Guo N, Zhang M, Li H, et al. Complete genome sequence of a novel subgenotype of porcine reproductive and respiratory syndrome virus strain JX/CH/2016, isolated in Jiangxi, China. Genome Announc. (2017) 5:e00980-17. doi: 10.1128/genomeA.00980-17
31. Yu F, Yan Y, Shi M, Liu HZ, Zhang HL, Yang YB, et al. Phylogenetics, genomic recombination, and NSP2 Polymorphic patterns of porcine reproductive and respiratory syndrome virus in China and the United States in 2014-2018. J Virol. (2020) 94:19. doi: 10.1128/JVI.01813-19
32. Liu J, Xu Y, Lin Z, Fan J, Dai A, Deng X, et al. Epidemiology investigation of PRRSV discharged by faecal and genetic variation of ORF5. Transbound Emerg Dis. (2021) 68:2334–44. doi: 10.1111/tbed.13894
33. Kim SC, Moon SH, Jeong CG, Park GS, Park JY, Jeoung HY, et al. Whole-genome sequencing and genetic characteristics of representative porcine reproductive and respiratory syndrome virus (PRRSV) isolates in Korea. Virol J. (2022) 19:66. doi: 10.1186/s12985-022-01790-6
34. Han G, Lei K, Xu H, He F. Genetic characterization of a novel recombinant PRRSV2 from lineage 8, 1 and 3 in China with significant variation in replication efficiency and cytopathic effects. Transbound Emerg Dis. (2020) 67:1574–84. doi: 10.1111/tbed.13491
35. Song J, Gao P, Kong C, Zhou L, Ge X, Guo X, et al. The nsp2 hypervariable region of porcine reproductive and respiratory syndrome virus strain JXwn06 is associated with viral cellular tropism to primary porcine alveolar macrophages. J Virol. (2019) 93:e01436-19. doi: 10.1128/JVI.01436-19
36. Veit M, Matczuk AK, Sinhadri BC, Krause E, Thaa B. Membrane proteins of arterivirus particles: structure, topology, processing and function. Virus Res. (2014) 194:16–36. doi: 10.1016/j.virusres.2014.09.010
37. Popescu LN, Trible BR, Chen N, Rowland RRR. GP5 of porcine reproductive and respiratory syndrome virus (PRRSV) as a target for homologous and broadly neutralizing antibodies. Vet Microbiol. (2017) 209:90–96. doi: 10.1016/j.vetmic.2017.04.016
38. Sui X, Guo X, Jia H, Wang X, Lin W, Li M, et al. Genomic sequence and virulence of a novel NADC30-like porcine reproductive and respiratory syndrome virus isolate from the Hebei province of China. Microb Pathog. (2018) 125:349–60. doi: 10.1016/j.micpath.2018.08.048
39. de Lima M, Pattnaik AK, Flores EF, Osorio FA. Serologic marker candidates identified among B-cell linear epitopes of Nsp2 and structural proteins of a North American strain of porcine reproductive and respiratory syndrome virus. Virology. (2006) 353:410–21. doi: 10.1016/j.virol.2006.05.036
40. Vashisht K, Goldberg TL, Husmann RJ, Schnitzlein W, Zuckermann FA. Identification of immunodominant T-cell epitopes present in glycoprotein 5 of the North American genotype of porcine reproductive and respiratory syndrome virus. Vaccine. (2008) 26:4747–53. doi: 10.1016/j.vaccine.2008.06.047
41. Ostrowski M, Galeota JA, Jar AM, Platt KB, Osorio FA, Lopez OJ. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. J Virol. (2002) 76:4241–50. doi: 10.1128/jvi.76.9.4241-4250.2002
42. Zhao K, Ye C, Chang XB, Jiang CG, Wang SJ, Cai XH, et al. Importation and recombination are responsible for the latest emergence of highly pathogenic porcine reproductive and respiratory syndrome virus in China. J Virol. (2015) 89:10712–6. doi: 10.1128/JVI.01446-15
43. Zhu Z, Yuan L, Hu D, Lian Z, Yao X, Liu P, et al. Isolation and genomic characterization of a Chinese NADC34-like PRRSV isolated from Jiangsu province. Transbound Emerg Dis. (2021) 69:254–267. doi: 10.1111/tbed.14392
44. Kappes MA, Faaberg KS. PRRSV structure, replication and recombination: Origin of phenotype and genotype diversity. Virology. (2015) 479–480:475–86. doi: 10.1016/j.virol.2015.02.012
45. Guo Z, Chen XX, Li R, Qiao S, Zhang G. The prevalent status and genetic diversity of porcine reproductive and respiratory syndrome virus in China: a molecular epidemiological perspective. Virol J. (2018) 15:2. doi: 10.1186/s12985-017-0910-6
46. Hou FH, Lee WC, Liao JW, Chien MS, Kuo CJ, Chung HP, et al. Evaluation of a type 2 modified live porcine reproductive and respiratory syndrome vaccine against heterologous challenge of a lineage 3 highly virulent isolate in pigs. PeerJ. (2020) 8:e8840. doi: 10.7717/peerj.8840
47. Lager KM, Schlink SN, Brockmeier SL, Miller LC, Henningson JN, Kappes MA, et al. Efficacy of Type 2 PRRSV vaccine against Chinese and Vietnamese HP-PRRSV challenge in pigs. Vaccine. (2014) 32:6457–62. doi: 10.1016/j.vaccine.2014.09.046
48. Jantafong T, Sangtong P, Saenglub W, Mungkundar C, Romlamduan N, Lekchareonsuk C, et al. Genetic diversity of porcine reproductive and respiratory syndrome virus in Thailand and Southeast Asia from 2008 to 2013. Vet Microbiol. (2015) 176:229–38. doi: 10.1016/j.vetmic.2015.01.017
49. Fukunaga W, Hayakawa-Sugaya Y, Koike F, Van Diep N, Kojima I, Yoshida Y, et al. Newly-designed primer pairs for the detection of type 2 porcine reproductive and respiratory syndrome virus genes. J Virol Methods. (2021) 291:114071. doi: 10.1016/j.jviromet.2021.114071
50. Ramirez M, Bauermann FV, Navarro D, Rojas M, Manchego A, Nelson EA, et al. Detection of porcine reproductive and respiratory syndrome virus (PRRSV) 1-7-4-type strains in Peru. Transbound Emerg Dis. (2019) 66:1107–13. doi: 10.1111/tbed.13134
51. Kim SC, Jeong CG, Park GS, Park JY, Jeoung HY, Shin GE, et al. Temporal lineage dynamics of the ORF5 gene of porcine reproductive and respiratory syndrome virus in Korea in 2014-2019. Arch Virol. (2021) 166:2803–15. doi: 10.1007/s00705-021-05169-w
52. Paploski IAD, Pamornchainavakul N, Makau DN, Rovira A, Corzo CA, Schroeder DC, et al. Phylogenetic structure and sequential dominance of sub-lineages of PRRSV type-2 lineage 1 in the United States. Vaccines. (2021) 9:9060608. doi: 10.3390/vaccines9060608
53. Sun YF, Liu Y, Yang J, Li WZ, Yu XX, Wang SY, et al. Recombination between NADC34-like and QYYZ-like strain of porcine reproductive and respiratory syndrome virus with high pathogenicity for piglets in China. Transbound Emerg Dis. (2022). doi: 10.1111/tbed.14471. [Epub ahead of print].
54. Ding Y, Wubshet AK, Ding X, Zhang Z, Li Q, Dai J, et al. Evaluation of four commercial vaccines for the protection of piglets against the highly pathogenic porcine reproductive and respiratory syndrome virus (hp-PRRSV) QH-08 strain. Vaccines. (2021) 9:1020. doi: 10.3390/vaccines9091020
Keywords: QYYZ-like PRRSV, whole-genome analysis, complex patterns of recombination, recombination hotspots, epidemiological characteristics
Citation: Xu H, Xiang L, Tang Y-D, Li C, Zhao J, Gong B, Sun Q, Leng C, Peng J, Wang Q, Zhou G, An T, Cai X, Tian Z-J, Zhang H and Song M (2022) Genome-Wide Characterization of QYYZ-Like PRRSV During 2018–2021. Front. Vet. Sci. 9:945381. doi: 10.3389/fvets.2022.945381
Received: 17 May 2022; Accepted: 06 June 2022;
Published: 30 June 2022.
Edited by:
Zhou Mo, Heilongjiang University, ChinaReviewed by:
Jianguo Dong, Xinyang Agriculture and Forestry University, ChinaYifeng Jiang, Shanghai Veterinary Research Institute (CAAS), China
Copyright © 2022 Xu, Xiang, Tang, Li, Zhao, Gong, Sun, Leng, Peng, Wang, Zhou, An, Cai, Tian, Zhang and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingxin Song, c29uZ214QG5lYXUuZWR1LmNu; Hongliang Zhang, emhhbmdob25nbGlhbmcwMUBjYWFzLmNu
†These authors have contributed equally to this work and share first authorship