 Kaichuang Shi
Kaichuang Shi Huixin Liu2†
Huixin Liu2† Hongbin Si
Hongbin Si- 1Guangxi Center for Animal Disease Control and Prevention, Nanning, China
- 2College of Animal Science and Technology, Guangxi University, Nanning, China
African swine fever virus (ASFV) causes contagious hemorrhagic disease of pigs with high morbidity and mortality. To identify the molecular characteristics of ASFV strains circulating in Guangxi province, southern China, a total of 336 tissue samples collected from 336 domestic pigs that died as a result of severe hemorrhagic disease during 2019–2020 were tested for ASFV. Furthermore, 66 ASFV strains were genetically characterized by sequence analysis of the C-terminal region of B646L (p72) gene, the complete E183L (p54) gene, the variable region of EP402R (CD2v) gene, the central variable region (CVR) of B602L gene, the full MGF505-2R gene, and the tandem repeat sequence (TRS) within intergenic region (IGR) between the I73R and I329L (I73R/I329L) genes. Phylogenetic analysis revealed that the ASFV strains from Guangxi province belonged to genotypes I and II based on the B646L (p72) and E183L (p54) genes, and there were eight different tetrameric TRS variants based on the CVR of B602L gene. Phylogenetic analysis of the EP402R (CD2v) gene revealed that these ASFV strains belonged to serogroups 4 and 8. Eight of the 66 strains belonged to genotype I and serogroup 4, and showed deletion of whole MGF505-2R gene. The sequence analysis of the IGR between the I73R/I329L genes showed that IGR II and III variants were co-circulating in Guangxi province. The results indicated that ASFV strains circulating in Guangxi province during 2019–2020 outbreaks showed high genetic diversity, of which genotypes I and II, as well as serogroups 4 and 8, were simultaneously circulating in Guangxi province, and there existed wild-type and naturally gene-deleted strains in the field. This is the first detailed report on the molecular characterization of the ASFV strains circulating in southern China, and serogroup 4 in China.
Introduction
African swine fever virus (ASFV) is an enveloped double-stranded DNA virus of the genus Asfivirus in the family Asfarviridae (1). ASFV causes African swine fever (ASF), a devastating hemorrhagic disease infecting domestic pigs and wild boars, with high mortality rate up to 100% (2). ASF was first identified in Kenya in the 1920s, Europe in 1957, the Caucasus region and southern Russia in 2007 (3, 4), China in August 2018 (5), and other Asian countries, such as Mongolia, Korea, Vietnam, Laos, Cambodia, the Philippines, and Indonesia, since the end of 2018 (6, 7). Currently, the outbreaks of ASF are still ongoing in Africa, the trans-Caucasus region, eastern Europe, Russian Federation, Asia, and Latin America, which pose a huge challenge to the swine industry in these regions (8, 9). ASF has caused huge economic losses to the swine industry worldwide since the 1920s, and is a notifiable disease to the World Organization for Animal Health (OIE).
The ASFV genome varies by ~170–193 kb and encodes over 168 kinds of protein (10). Based on the partial B646L (p72) gene sequences, ASFV strains from different countries are currently classified into 24 genotypes (11, 12). Of all the 24 genotypes of ASFV found in Africa, only genotypes I and II have outbroken outside Africa until now (13, 14). Genotype I first occurred in Portugal in 1957, and then in other European countries, southern America, and Caribbean island countries (15). Genotype II entered Georgia from southern Africa in 2007 (16), later spread to eastern Europe and westward into western Europe arriving in Belgium in September 2018 (17), and introduced into Asia arriving in China in August 2018 (18). Since then, genotype II strains have spiraled out of control, spreading to multiple Chinese provinces and reaching other Asian countries, such as Vietnam, Cambodia, Laos, and Korea (8, 9).
Since China raises more than 50% of the world's pigs every year, it is very important to master the molecular characterization of ASFV strains circulating in China. ASF was first identified in China in August 2018 (5), where it rapidly spread to most provinces in China within a short time (19). To date, genotypes I and II ASFV strains have been reported in China (20, 21), and the wild-type and gene-deleted ASFV strains have been identified in several provinces in China (22, 23). However, the molecular characterization of ASFV strains circulating in China has been limited, thus the ASFV genotypes mapping in the country is incomplete until now. In this study, the tissue samples, which were collected in the field during the ASF outbreaks from 2019 to 2020 in Guangxi province, southern China, were tested for ASFV by real-time quantitative polymerase chain reaction (qPCR). The positive samples were randomly selected to amplify and sequence the C-terminal region of B646L gene encoding protein 72 (p72), the complete E183L gene encoding protein 54 (p54), the central variable region (CVR) of B602L gene, the full MGF505-2R gene, and the tandem repeat sequence (TRS) of intergenic region (IGR) between the I73R and I329L genes (I73R/I329L genes), and analyze the genetic characteristics and variations of ASFV strains circulating in Guangxi province. Furthermore, the serogroups of ASFV strains were determined by sequence analysis of the variable region of EP402R gene encoding protein CD2v (pCD2v). This is the first detailed report on the molecular characterization of the ASFV strains circulating in southern China.
Materials and Methods
Sample Collection and Detection of ASFV Genome
From January 2019 to December 2020, a total of 336 tissue samples from 336 domestic pigs were collected from 86 different pig farms in 14 regions of Guangxi province, southern China in this study. The pig farms with high fever, hemorrhagic pig herds suspected of ASF were selected, and three to five dead pigs form each farm were selected randomly to collect clinical samples. The tissue samples, including lung, liver, spleen and lymph nodes from each dead pig, were collected, transported immediately at ≤4°C to the laboratory, and stored at −70°C until used. The tissue samples were homogenized (10%, W/V) in phosphate-buffered saline (PBS, pH7.2) using a blender (Tianlong, Xi'an, China), followed by shaking with small glass beads for 5 min, and then freeze-thawed three times, centrifuged at 12, 000 × g at 4°C for 5 min. Total DNA was extracted from the supernatants using MiniBEST RNA/DNA Extraction Kit (TaKaRa, Beijing, China) and confirmed the presence of ASFV by a real-time qPCR targeting the B646L (p72) gene by the following specific primers and probe: ASFV-U: GGCGTATAAAAAGTCCAGGAAATTC, ASFV-D: TTCGGCGAGCGCTTTATC, ASFV-P: Texas Red-TCACCAAATCCTTTTGCGATGCAAGCT-BHQ2. The parameters of real-time qPCR were as follows: 10 μl of Premix Ex Taq (TaKaRa, Beijing, China), 0.4 μl of each of ASFV primers (20 pmol/μl), 0.5 μl of ASFV probe (20 pmol/μl), 2 μl of total DNA as templates and distilled water to a total volume of 20 μl. The amplification parameters were as follows: 95°C for 1 min; and then 40 cycles of 95°C for 5 s, 59°C for 34 s. The fluorescent signals were determined at the end of each cycle. The ASFV positive samples were randomly selected for genetic analysis.
Amplification and Sequencing of the Targeted Genes
The positive samples were randomly selected for amplifying the C-terminal region of the B646L (p72) gene, the E183L (p54) gene, the CVR of the B602L gene, the EP402R (CD2v) gene, the full MGF505-2R gene, and the TRS within IGR between the I73R/I329L genes, using six different pairs of primers (Table 1) according to the previous reports (24–29) with some modifications. The targeted genes were amplified by PCR using the Tks Gflex™ DNA Polymerase kit (TaKaRa, Beijing, China), and the products were electrophoresed in a 1.2% agarose gel and visualized using an imaging system (UVITEC, France). The amplified fragments were purified and cloned into a pMD18-T vector (TaKaRa, Beijing, China), transferred into E. coli DH5α competent cells (TaKaRa, Beijing, China) and sequenced with an ABI 3730XL sequencer (ABI, Los Angeles, CA, United States).
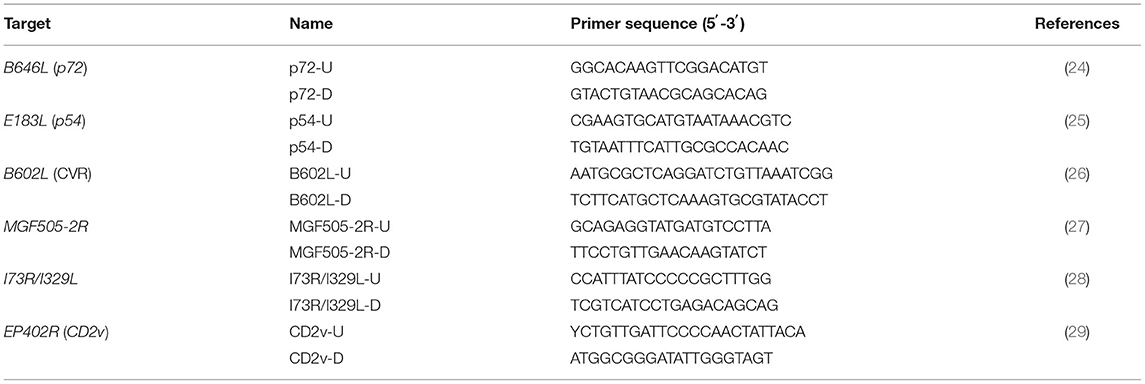
Table 1. Primers used for evaluation and amplification of ASFV in this study.
Genomic Analysis
Nucleotide and amino acid alignments were carried out using the EditSeq and Megalign program in the DNAstar package (DNAstar, Madison, WI, United States). All the reference sequences of ASFV were downloaded from the GenBank in the National Center for Biotechnology Information (NCBI) (Supplementary Tables S1–S3). Phylogenetic analysis based on the B646L (p72) gene and the E183L (p54) gene nucleotide sequences, respectively, were conducted using MEGA X, and the genotypes of ASFV strains were determined as previously described (30–32). The serogroups of the obtained ASFV strains were achieved by phylogenetic analysis of the EP402R (CD2v) gene sequences using MEGA X as previously described (33, 34). The maximum likelihood (ML) and the best-fit model methods were used to compare the differences between the two typing methods and analyze gene sequences. Phylogeny was inferred following 1,000 bootstrap replications.
The nucleotides of the CVR of the B602L gene and the TRS within the IGR between I73R/I329L genes were first translated into amino acids and searched by similarity analysis according to the ASFV amino acid sequences using BLASTp (v2.2.2.29). The CVR amino acid tetramers were matched with those previously reported codes (31, 34, 35).
Results
Detection of ASFV Genome in Clinical Samples
Of all 336 clinical samples evaluated by real-time qPCR using primers ASFV-U/D and probe ASFV-P, 192 samples were positive for ASFV, with a positive rate of 57.14% (192/336). Sixty-six samples were randomly selected from these 192 positive samples and further confirmed by real-time qPCR using a commercial ASFV qPCR detection kit (Lijian, China) which targeted the B646L (p72), EP402R (CD2v) and MGF505-2R genes, before they were used for gene amplification, sequencing and analysis. The results showed that all the 66 samples were positive for ASFV based on the B646L (p72) and EP402R (CD2v) genes, of which eight samples lacked MGF505-2R gene. Then, these samples were further amplified and sequenced to verify the deletion of MGF505-2R gene.
Amplification of the Targeted Genes of ASFV
A total of 66 positive samples were randomly selected for amplifying six targeted genes, including B646L (p72), E183L (p54), EP402R (CD2v), B602L (CVR), MGF505-2R and IGR between I73R/I329L genes. The results showed that the B646L (p72), E183L (p54), EP402R (CD2v), B602L (CVR), and IGR between I73R/I329L genes were amplified from all the 66 positive samples. However, the MGF505-2R gene fragments could be amplified from 58 of the 66 samples. The targeted fragments were purified, cloned and sequenced. The generated sequences of the 66 strains from Guangxi province have been deposited in GenBank under accession numbers OM986132-OM986197 for the B646L (p72) gene, OM986396-OM986461 for the E183L (p54) gene, OM986264-OM986329 for the EP402R (CD2v) gene, OM986198-OM986263 for the B602L (CVR) gene, OM986330-OM986395 for the IGR between I73R/I329L genes, and OM986074-OM986131 for the MGF505-2R gene (Table 2).
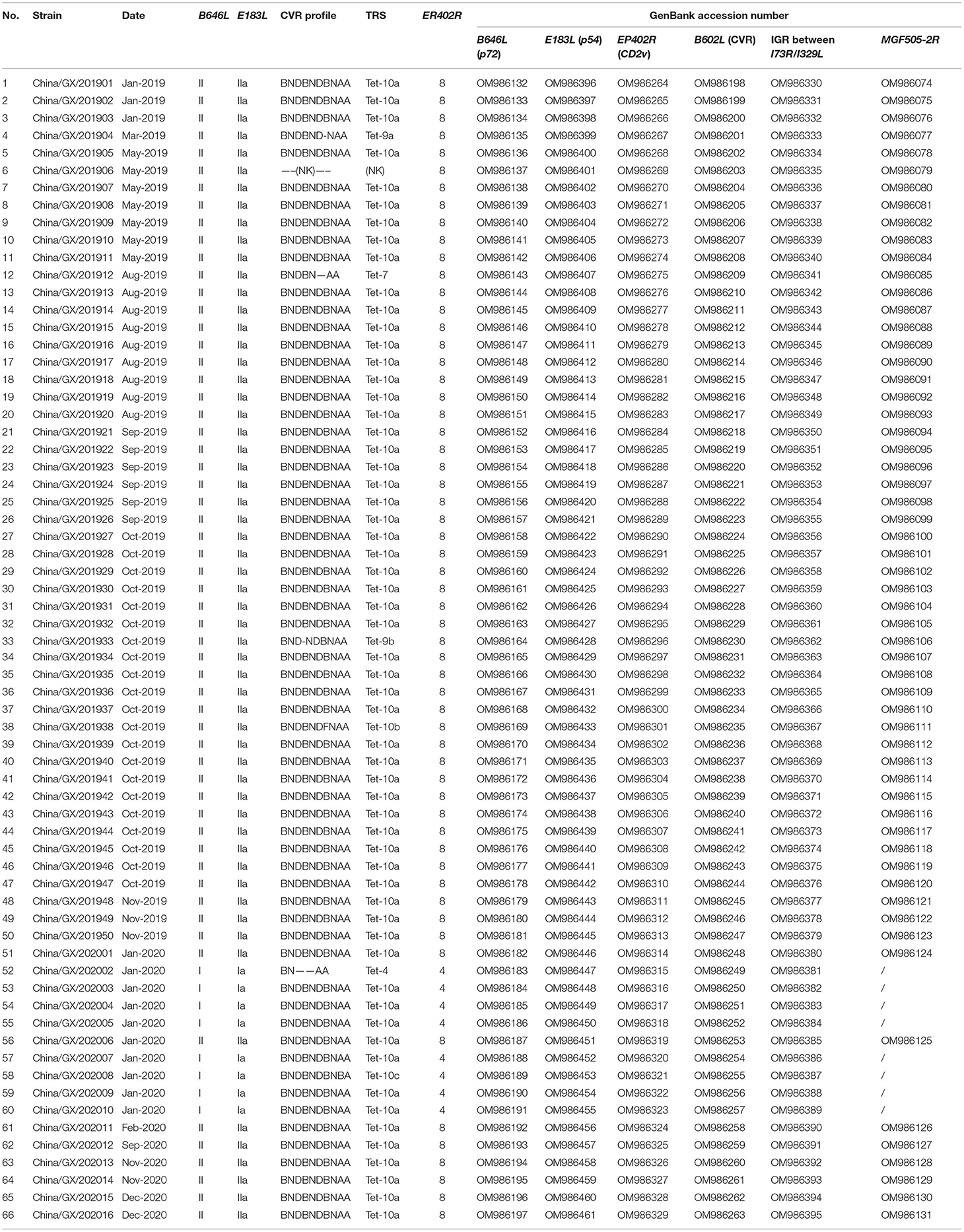
Table 2. Summary of the 66 ASFV strains from domestic pigs in Guangxi province in this study.
Sequence Alignment of the MGF505-2R Gene
The evaluation of ASFV positive samples using a commercial qPCR detection kit (Lijian, China) suggested that eight samples lacked MGF505-2R gene. To verify this result, the MGF505-2R genes of all the 66 samples were amplified, sequenced and aligned. The results confirmed the deletion of the MGF505-2R gene in eight samples (Figure 1). The further analysis showed that these 8 strains belonged to genotype I based on the phylogenetic analysis of the B646L (p72) gene (Figure 2A) and serogroup 4 based on the phylogenetic analysis of the EP402R (CD2v) gene (Figure 2B).

Figure 1. Nucleotide sequence alignment of the strains lacking the MGF505-2R gene. Eight clinical samples lacking the MGF505-2R gene by the commercial qPCR detection kit (Lijian, China) were further verified by sequence alignment, and confirmed to be lacking the partial MGF360-9R, whole MGF360-10R~MGF360-14R, MGF505-1R and MGF505-2R, and partial MGF505-3R genes with a total deletion of 10,134 bp.
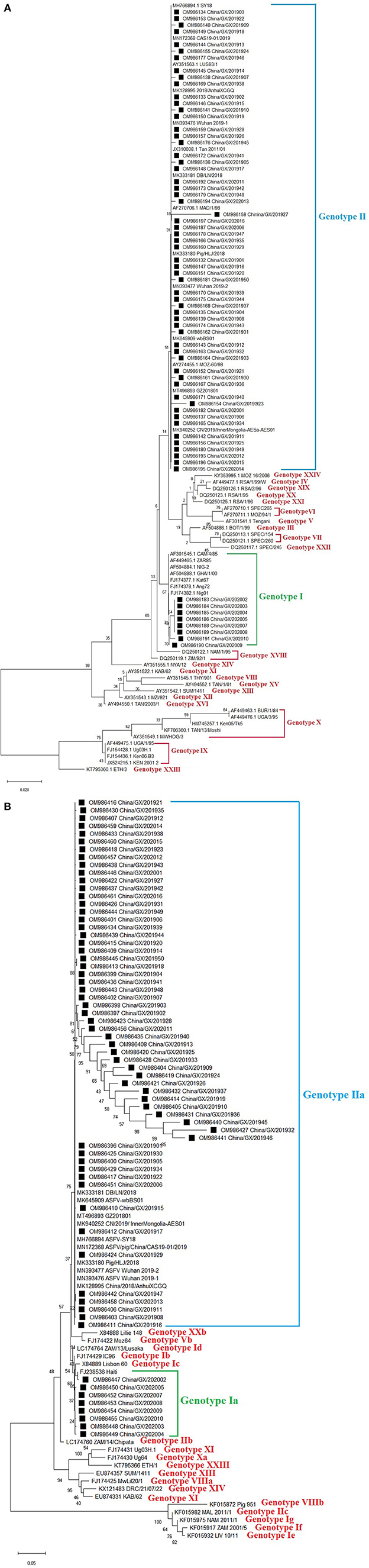
Figure 2. Phylogenetic trees based on nucleotide sequences of the B646L (p72) (A) and E183L (p54) (B) genes. The evolutionary history was inferred by the maximum likelihood method based on the Kimura 2-parameter model [B646L (p72) gene] and the Kimura 2-parameter + G model [E183L (p54) gene]. The phylogeny was inferred following 1,000 bootstrap replications, and node values showed percentage bootstrap support. (A) Phylogenetic analysis of the B646L (p72) gene. The ASFV strains from Guangxi province were clustered into genotypes I and II. (B) Phylogenetic analysis of the E183L (p54) gene. The ASFV strains from Guangxi province were clustered into genotypes Ia and IIa. The strains from this study were highlighted with black squares (■) before the names.
Phylogenetic Analysis of the B646L (p72) Gene
The B646L (p72) gene was amplified, sequenced and analyzed. Phylogenetic analysis of B646L (p72) nucleotide sequences revealed that all ASFV strains, including the 66 strains obtained in this study (Table 2) and the reference strains downloaded from GenBank (Supplementary Table 1), could be clustered into 24 genotypes. The 66 strains from this study distributed in genotypes I and II, of which 8 strains belonged to genotype I and 58 strains belonged to genotype II (Figure 2A).
Phylogenetic Analysis of the E183L (p54) Gene
The E183L (p54) gene was amplified, sequenced and analyzed. Since all the 66 strains from Guangxi province belonged to genotypes I and II based on phylogenetic analysis of the B646L (p72) gene, we wanted to know whether these strains also belonged to genotypes I and II based on phylogenetic analysis of the E183L (p54) gene. The result revealed that all the 66 strains from this study (Table 2) and the reference strains downloaded from GenBank (Supplementary Table 2) could be clustered into different genotypes, and further into different sub-genotypes. Of all the 66 strains from this study, 8 strains belonged to sub-genotype Ia, and the other 58 strains belonged to sub-genotype IIa which could be further divided into different branches (Figure 2B).
Phylogenetic Analysis of the EP402R (CD2v) Gene
The EP402R (CD2v) gene was amplified, sequenced and analyzed. Phylogenetic analysis of EP402R (CD2v) nucleotide sequences revealed that all ASFV strains, including the 66 strains from this study (Table 2) and the reference strains downloaded from GenBank (Supplementary Table 3), could be clustered into eight serogroups. Of the 66 strains from this study, eight strains of genotype I belonged to serogroup four, and the other 58 strains of genotype II belonged to serogroup eight (Figure 3).
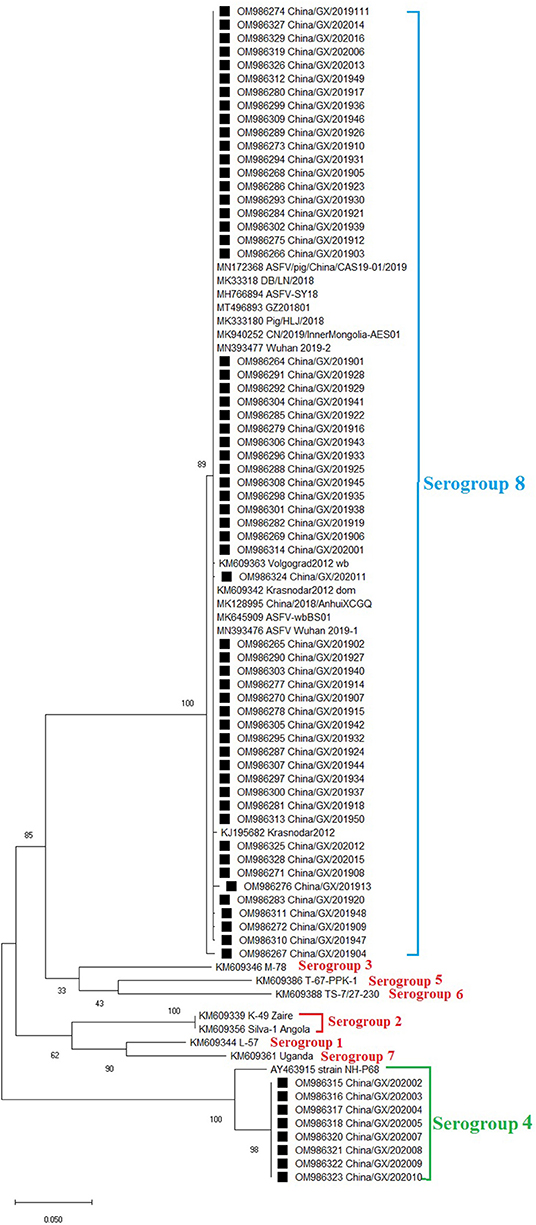
Figure 3. Phylogenetic tree based on nucleotide sequences of the ER402R (CD2v) gene. The evolutionary history was inferred by the maximum likelihood method based on the Tamura 3-parameter + G model. The phylogeny was inferred following 1,000 bootstrap replications, and node values showed percentage bootstrap support. The ASFV strains from Guangxi province were clustered into serogroups 4 and 8. The strains from this study were highlighted with black squares (■) before the names.
Analysis of the CVR of the B602L Gene
The CVR of the B602L gene is a highly variable region and is often used for intragenotypic resolution of viruses belonging to the same B646L (p72) and E183L (p54) genotypes. Therefore, the CVR of the B602L gene of Guangxi ASFV strains was amplified, sequenced and analyzed. The results showed that the 66 strains from this study had different nucleotide lengths from 68 to 178 bp. The nucleotides were transformed into amino acids and analyzed. The sequencing analysis based on amino acids obtained eight different TRS variants from the circulating strains in Guangxi province (Table 2). Of the eight TRS variants, three variants (Tet-4, Tet-10a, and Tet-10c) belonged to genotype I, and six variants (NK, Tet-7, Tet-9a, Tet-9b, Tet-10a, and Tet-10b) belonged to genotype II, of which Tet-10a was found in both genotypes I and II. The tetrameric TRS of BNDBNDBNAA (Tet-10a) was the most dominant variant in this study. Sequence alignment revealed that four new tetrameric TRSs were found due to the mutation of a single amino acid (Figure 4), indicating the presence of single nucleotide polymorphism (SNP) in CVR variants. The similarity analysis of the tetrameric TRSs on NCBI database showed that, except for the sequence of BNDBNDBNAA (Tet-10a), the other seven TRS variants did not find 100% identical sequence. Furthermore, it was found that the CVR of B602L gene of the 66 strains was not affected by the region of source, the time of collection and the genotype of the strains.

Figure 4. Amino acid sequence alignment of the tetrameric tandem repeat sequences (TRSs) of the central variable region (CVR) of the B602L gene. The ASFV strains from Guangxi province generated 8 different TRS variants. Only one representative strain of each variant was presented in this figure. Codes were as follows and as previously described: (CAST, CVST, CTST, CASI = A) (CADT, CADI, CTDT, CAGT, CVDT = B) (GAST, GANT = C) (CASM = D) (CANT, CAAT = F) (CTNT = G) (RAST = H) (GTDT = J) (CTSP = K) (YTNT = L) (NEDT = M) (NVDT, NVGT, NVDI = N) (NANI, NADI, NASI = O) (SAST = S) (NVNT = T) (NIDT, NTDT = U) (NAGT, NAST, NAVT, NADT, NANT = V) (SADT, SVDT = W) (NTDI = X).
Analysis of the IGR Between the I73R/I329L Genes
The IGR between the I73R/I329L genes was amplified, sequenced and characterized by the presence of TRSs. The results showed that all the 66 strains from Guangxi province in this study belonged to IGR II variant with an TRS insertion of the sequence GGAATATATA compared to the Georgia 2007/1 (GenBank accession no. FR682468.1), while one previously reported strain (China/Guangxi/2019/domestic pig, GenBank accession no. MK670729) from Guangxi province (36) belonged to IGR III variant with two TRS insertions, indicating that two types of variants (IGR II and IGR III variants) were co-circulating in Guangxi province during 2019–2020 outbreaks (Figure 5).
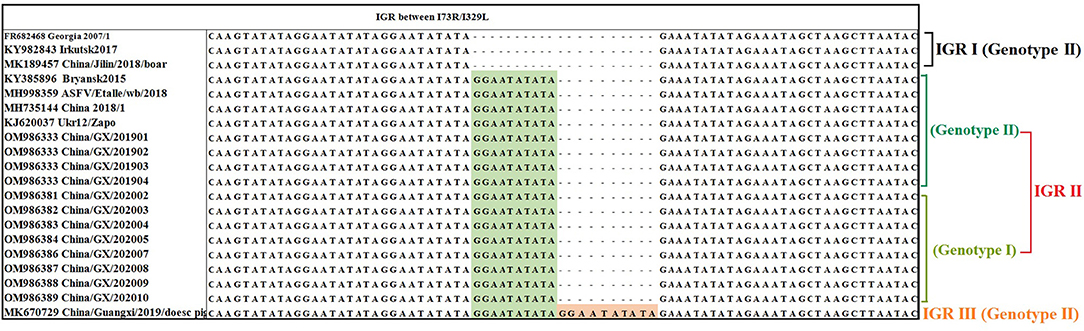
Figure 5. Nucleotide sequence alignment of the intergenic region (IGR) between I73R/I329L genes. The ASFV strains from Guangxi province belonged to IGR II with one insertion of 10 nucleotides (GGAATATATA) and IGR III with two insertions of 10 nucleotides (GGAATATATA). All the strains from this study belonged to IGR II and only partial strains were presented in this figure.
China/GX/202010 strain (GenBank accession no. OM986389) of genotype I and China/GX/201904 strain (GenBank accession no. OM986333) of genotype II from Guangxi province were selected as the representative strains for sequence similarity retrieval by BLAST software in the NCBI database (Table 3). The genotype II strain had 100% identity of the IGR between I73R/I329L genes with some strains from Eurasian countries, including Belarus, Ukraine, Russia, Belgium and Poland in Europe, and Mongolia, Vietnam, Indonesia and China in Asia. In addition, the IGR between I73R/I329L genes of the genotype I strain could not found a strain with 100% similarity in NCBI database, and the highest similar strains were the Portuguese strain OURT88/3 and the Spanish strain NHV with a similarity of 99.74%.
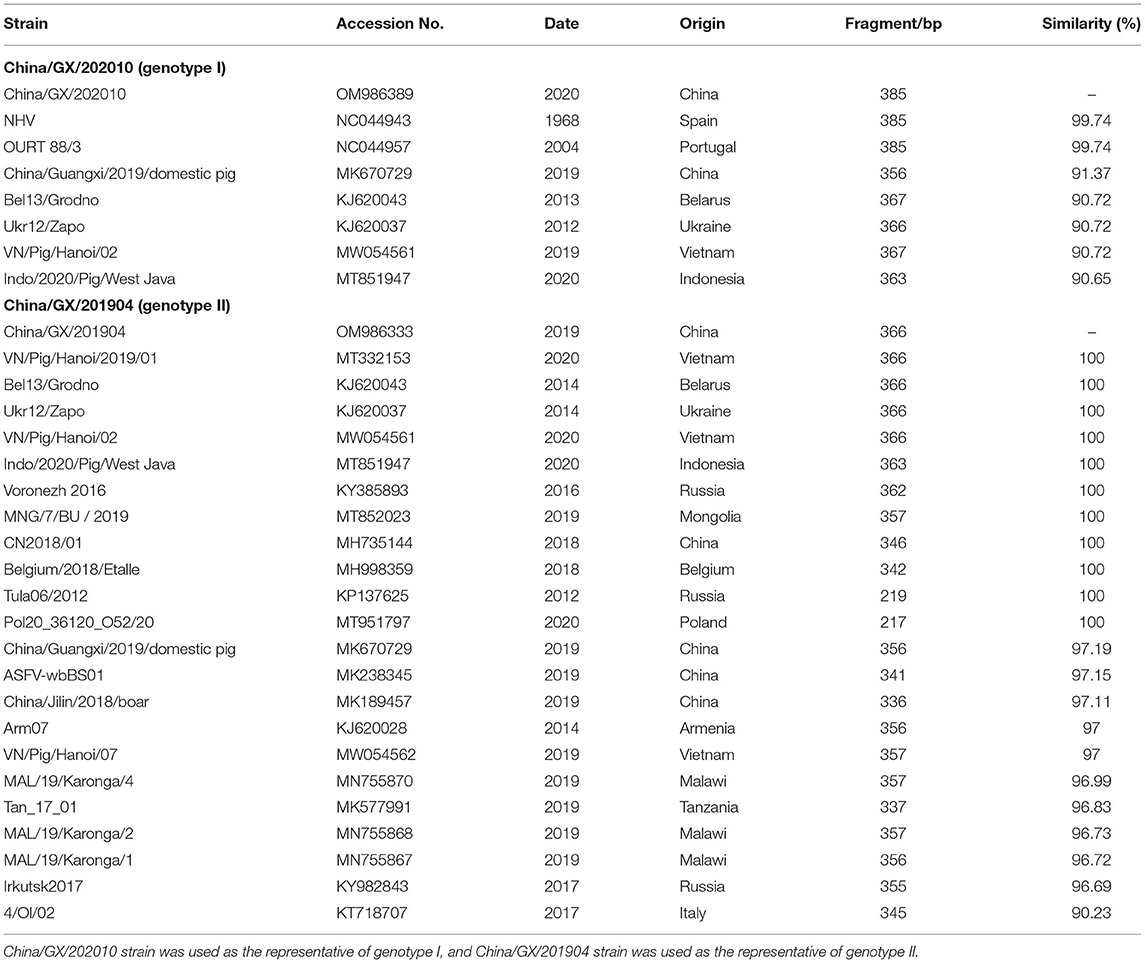
Table 3. Similarity analysis of the tandem repeat sequence (TRS) between I73R/I329L genes.
Discussion
ASF was first reported in China on August 3, 2018 (5), and then rapidly spread to most provinces in China within a short time (19). According to a report from the Ministry of Agriculture and Rural Affairs of China, by August 2019, ASF had caused a loss of 190.9 million pigs, including 29.4 million sows with a 40.5% decrease in China (37), showing that ASF had caused huge losses to the pig industry within a very short time. It has been reported that genotypes I and II of ASFV were found to be simultaneously epidemic in the pig herds in China (20, 21), and the wild-type ASFV strains with different virulence and the naturally gene-deleted ASFV strains with decreased virulence were identified in several provinces in China (22, 23), indicating that many different ASFV strains with high genetic diversity were circulating in China. Therefore, the molecular characterization of ASFV strains circulating in China, which has not been reported in detail until now, is necessary to further study.
The tissue samples from dead pigs collected from 86 pig farms during 2019–2020 in Guangxi province, southern China, were tested for ASFV, and a high positive rate of 57.14% (192/336) was found, indicating that ASF occurred seriously in these pig farms. To further analyze the molecular characterization of the circulating ASFVs, 66 positive samples were randomly selected for gene sequencing and analysis. These samples were also tested by a commercial ASFV qPCR detection kit (Lijian, China), and 8 ASFV strains lacked MGF505-2R gene. These situations were further confirmed by the sequence alignment. Recently, there were reports on ASFV strains with different levels of virulence causing a broad range of clinical symptoms in susceptible animals. Per-acute and acute ASFV infections might lead to mortality rate up to 100% in naïve domestic pig populations (5, 38, 39), while pigs infected with low virulence strains could develop resistance to ASFV leading to an increased number of chronic or subclinical infections (20, 22, 40, 41). Some researchers have reported that the gene-deleted ASFV strains showed decreased virulence and low pathogenicity, such as the non-pathogenic strain OURT88/3 in Portugal lacking the MGF505-2R gene (42), and the attenuated strain BA71 in Spain losing its EP402R (CD2v) gene (43). The naturally gene-deleted ASFV strains with decreased virulence has been reported in several provinces in eastern China (22, 23), and the man-made gene-deleted strains with decreased virulence have also been reported in China (44). Of the 66 strains in this study, eight strains lacked MGF505-2R gene. Therefore, the wild-type and the gene-deleted ASFV strains were simultaneously circulating during 2019–2020 in Guangxi province, southern China, and their harm to the pig industry needs to be further evaluated.
ASF was first reported in Kenya in the 1920s, introduced into Georgia in 2007 and has subsequently spread to Russia, eastern Europe, Asia and Latin America with devastating socioeconomic consequences (3, 8, 9, 45, 46). To date, two of the 24 currently described ASFV genotypes defined by the B646L (p72) gene, namely genotypes I and II, have been reported outside Africa, with genotype II being responsible for the ongoing ASF pandemic (13, 14). In this study, the positive clinical samples, collected during 2019–2020 outbreaks from Guangxi province, were randomly selected for gene sequence analysis. The B646L (p72) and E183L (p54) gene sequences were used to determine the genotype of the viruses by phylogenetic analysis, while the EP402R (CD2v) gene sequence was analyzed to determine the serogroup. Phylogenetic analysis of the B646L (p72) gene revealed that all 66 strains from Guangxi province belonged to genotypes I (8/66) and II (58/66), and phylogenetic analysis of the E183L (p54) gene revealed that all 66 strains from Guangxi province belonged to genotypes Ia (8/66) and IIa (58/66), indicating that the genotyping by the B646L (p72) and E183L (p54) nucleotide sequences generated similar genotypes for the circulating ASFV strains in Guangxi province. Phylogenetic analysis of the EP402R (CD2v) gene revealed that the strains from Guangxi province could be clustered into two serogroups, serogroups 4 (8/66) and 8 (58/66). Traditionally, genotyping of ASFV strains depended on the phylogenetic analysis of the B646L (p72) and E183L (p54) gene sequences, and the analysis of the E183L (p54) gene sequences could improve the identification of each genotype (30, 32, 33, 47, 48). However, some scholars proposed that the B646L (p72) gene could not accurately define the serogroup of ASFV, distinguish viruses with different virulence, or predict the effectiveness of specific ASFV vaccines, so they advocated to determine the serogroup of ASFVs based on the phylogenetic analysis of the EP402R (CD2v) gene (34, 49, 50). Therefore, it is recommended that genotyping should be combined with serotyping for epidemiological investigation of ASFV.
Many reports have used the genetic characteristics of the CVR in the B602L gene for genotyping and subgrouping closely related ASFV strains (25, 30–35, 48–52). In this study, the ASFV strains circulating in Guangxi province from 2019 to 2020 presented a multi-complex profile with a high degree of heterogeneity that could be clustered into 8 different TRS variants, and three variants belonged to genotype I and six variants belonged to genotype II (Table 2). To better track the source of these strains, the similarity analysis between these 8 TRS variants and the strains from GenBank database showed that only the TRS variant BNDBNDBNAA had 100% similar sequences, and the other seven variants could not find a completely consistent sequence. In addition, the situation that the TRSs within CVR of some genotype I strains was identical to those of the genotype II strains was also reported previously (50). The results demonstrated that the CVR in the B602L gene is a hypervariable genetic marker for high-resolution discrimination of viruses that are identical based on their B646L (p72) and E183L (p54) genotypes.
The CVR of the B602L gene has been widely used to distinguish closely related ASFVs, but the relatively low CVR genetic variability necessitates further research on alternative and more informative gene regions, so the sequence analysis of the IGR between I73R/I329L genes is used to determine the relationship within the relevant genotypes and origin of circulating ASFV strains (28, 29, 32, 34, 36, 49, 52–55). According to the results in this study, the IGR II and IGR III variants were simultaneously circulating in Guangxi province (Figure 5). Furthermore, the circulating genotype II strains in Guangxi province had 100% identity with some strains from Eurasian countries (Table 3). In addition, the IGR between I73R/I329L genes of the circulating genotype I strains in Guangxi province did not find a 100% similar sequence on NCBI database, and the most similar strain was the Portuguese OURT88/3 strain and Spanish NHV strain with 99.74% similarity. Therefore, Chinese ASFV strains, including the strains form Guangxi province, might be derived from Europe, and more genome sequences of ASFV strains circulating in pig herds will help precisely trace the origin of ASFV strains in China.
In conclusion, this study first reported that ASFV strains of two genotypes (genotypes I and II based on the B646L (p72) and E183L (p54) genes) and two serogroups (serogroups 4 and 8 based on the EP402R (CD2v) gene) were simultaneously circulating during 2019–2020 outbreaks in Guangxi province, southern China. There existed wild-type and naturally gene-deleted ASFV strains in the field. These strains generated eight different CVR variants within the B602L gene and belonged to IGR II and III variants between I73R/I329L genes. To our knowledge, this is the first detailed report on the molecular characterization of ASFV strains circulating in southern China, and serogroup 4 ASFV strains in China.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
KS contributed to study design, laboratory supervision, and manuscript writing and editing. HL contributed to experiments, data analysis, and manuscript drafting. YY, FL, and SF contributed to sample collection and data analysis. HS contributed to study design and manuscript editing. All authors have read and approved the submitted manuscript.
Funding
This study was supported by the Key Research and Development Program (AB21238003) and the Science and Technology Major Project (AA17204057) of Guangxi Science and Technology Bureau, China, and the Agricultural Science and Technology Program (Z201954, Z202031) of Guangxi Agricultural and Rural Bureau, China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank to Guangxi Center for Animal Disease Control and Prevention (CADC) for providing all the clinical samples used in this study. Guangxi CADC was approved by the Ministry of Agriculture and Rural Affairs, China for collection and detection of ASFV in clinical samples (Approval Number: 2018-154-25).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.912224/full#supplementary-material
References
1. Alonso C, Borca M, Dixon L, Revilla Y, Rodriguez F, Escribano JM, et al. ICTV virus taxonomy profile: Asfarviridae. J Gen Virol. (2018) 99:613–14. doi: 10.1099/jgv.0.001049
2. Dixon LK, Sun H, Roberts H. African swine fever. Antiviral Res. (2019) 165:34–41. doi: 10.1016/j.antiviral.2019.02.018
3. Cwynar P, Stojkov J, Wlazlak K. African swine fever status in Europe. Viruses. (2019) 11:310. doi: 10.3390/v11040310
4. Sánchez-Vizcaíno JM, Mur L, Martínez-López B. African swine fever: an epidemiological update. Transbound Emerg Dis. (2012) 59:27–35. doi: 10.1111/j.1865-1682.2011.01293.x
5. Zhou X, Li N, Luo Y, Liu Y, Miao F, Chen T, et al. Emergence of African swine fever in China, 2018. Transbound Emerg Dis. (2018) 65:1482–84. doi: 10.1111/tbed.12989
6. Mighell E, Ward MP. African swine fever spread across Asia, 2018–2019. Transbound Emerg Dis. (2021) 68:2722–32. doi: 10.1111/tbed.14039
7. Sánchez-Cordón PJ, Nunez A, Neimanis A, Wikström-Lassa E, Montoya M, Crooke H, et al. African swine fever: disease dynamics in wild boar experimentally infected with ASFV strains belonging to genotype I and II. Viruses. (2019) 11:852. doi: 10.3390/v11090852
8. Danzetta ML, Marenzoni ML, Iannetti S, Tizzani P, Calistri P, Feliziani F. African swine fever: lessons to learn from past eradication experiences: a systematic review. Front Vet Sci. (2020) 7:296. doi: 10.3389/fvets.2020.00296
9. Gaudreault NN, Madden DW, Wilson WC, Trujillo JD, Richt JA. African swine fever virus: an emerging DNA arbovirus. Front Vet Sci. (2020) 7:215. doi: 10.3389/fvets.2020.00215
10. Wang G, Xie M, Wu W, Chen Z. Structures and functional diversities of ASFV proteins. Viruses. (2021) 13:2124. doi: 10.3390/v13112124
11. Achenbach JE, Gallardo C, Nieto-Pelegrín E, Rivera-Arroyo B, Degefa-Negi T, Arias M, et al. Identification of a new genotype of African swine fever virus in domestic pigs from Ethiopia. Transbound Emerg Dis. (2017) 64:1393–404. doi: 10.1111/tbed.12511
12. Quembo CJ, Jori F, Vosloo W, Heath L. Genetic characterization of African swine fever virus strains from soft ticks at the wildlife/domestic interface in Mozambique and identification of a novel genotype. Transbound Emerg Dis. (2018) 65:420–31. doi: 10.1111/tbed.12700
13. Njau EP, Domelevo Entfellner JB, Machuka EM, Bochere EN, Cleaveland S, Shirima GM, et al. The first genotype II African swine fever virus isolated in Africa provides insight into the current Eurasian pandemic. Sci Rep. (2021) 11:13081. doi: 10.1038/s41598-021-92593-2
14. Simulundu E, Lubaba CH, van Heerden J, Kajihara M, Mataa L, Chambaro HM, et al. The epidemiology of African swine fever in “non-endemic” regions of Zambia (1989–2015): implications for disease prevention and control. Viruses. (2017) 9:236. doi: 10.3390/v9090236
15. Costard S, Wieland B, de Glanville W, Jori F, Rowlands R, Vosloo W, et al. African swine fever: how can global spread be prevented? Philos Trans R Soc Lond B Biol Sci. (2009) 364:2683–96. doi: 10.1098/rstb.2009.0098
16. Sánchez-Cordón PJ, Montoya M, Reis AL, Dixon LK. African swine fever: a re-emerging viral disease threatening the global pig industry. Vet J. (2018) 233:41–8. doi: 10.1016/j.tvjl.2017.12.025
17. Linden A, Licoppe A, Volpe R, Paternostre J, Lesenfants C, Cassart D, et al. Summer 2018: African swine fever virus hits north-western Europe. Transbound Emerg Dis. (2019) 66:54–5. doi: 10.1111/tbed.13047
18. Ge S, Li J, Fan X, Liu F, Li L, Wang Q, et al. Molecular characterization of African swine fever virus, China, 2018. Emerg Infect Dis. (2018) 24:2131–33. doi: 10.3201/eid2411.181274
19. Tao D, Sun D, Liu Y, Wei S, Yang Z, An T, et al. One year of African swine fever outbreak in China. Acta Trop. (2020) 211:105602. doi: 10.1016/j.actatropica.2020.105602
20. Sun E, Huang L, Zhang X, Zhang J, Shen D, Zhang Z, et al. Genotype I African swine fever viruses emerged in domestic pigs in China and caused chronic infection. Emerg Microbes Infect. (2021) 10:2183–93. doi: 10.1080/22221751.2021.1999779
21. Liu H, Shi K, Zhao J, Yin Y, Chen Y, Si H, et al. Development of a one-step multiplex qRT-PCR assay for the detection of African swine fever virus, classical swine fever virus and atypical porcine pestivirus. BMC Vet Res. (2022) 18:43. doi: 10.1186/s12917-022-03144-4
22. Sun E, Zhang Z, Wang Z, He X, Zhang X, Wang L, et al. Emergence and prevalence of naturally occurring lower virulent African swine fever viruses in domestic pigs in China in 2020. Sci China Life Sci. (2021) 64:752–65. doi: 10.1007/s11427-021-1904-4
23. Wang X, Wang X, Zhang X, He S, Chen Y, Liu X, et al. Genetic characterization and variation of African swine fever virus China/GD/2019 strain in domestic pigs. Pathogens. (2022) 11:97. doi: 10.3390/pathogens11010097
24. Bastos AD, Penrith ML, Cruciere C, Edrich JL, Hutchings G, Roger F, et al. Genotyping field strains of African swine fever virus by partial p72 gene characterization. Arch Virol. (2003) 148:693–706. doi: 10.1007/s00705-002-0946-8
25. Gallardo C, Mwaengo DM, Macharia JM, Arias M, Taracha EA, Soler A, et al. Enhanced discrimination of African swine fever virus strains through nucleotide sequencing of the p54, p72, and pB602L (CVR) genes. Virus Genes. (2009) 38:85–95. doi: 10.1007/s11262-008-0293-2
26. Nix RJ, Gallardo C, Hutchings G, Blanco E, Dixon LK. Molecular epidemiology of African swine fever virus studied by analysis of four variable genome regions. Arch Virol. (2006) 151:2475–94. doi: 10.1007/s00705-006-0794-z
27. Fraczyk M, Wozniakowski G, Kowalczyk A, Bocian Ł, Kozak E, Niemczuk K, et al. Evolution of African swine fever virus genes related to evasion of host immune response. Vet Microbiol. (2016) 193:133–44. doi: 10.1016/j.vetmic.2016.08.018
28. Gallardo C, Fernández-Pinero J, Pelayo V, Gazaev I, Markowska-Daniel I, Pridotkas G, et al. Genetic variation among African swine fever genotype II viruses, eastern and central Europe. Emerg Infect Dis. (2014) 20:1544–47. doi: 10.3201/eid2009.140554
29. Sanna G, Dei Giudici S, Bacciu D, Angioi PP, Giammarioli M, De Mia GM, et al. Improved strategy for molecular characterization of African swine fever viruses from Sardinia, based on analysis of p30, CD2V and I73R/I329L variable regions. Transbound Emerg Dis. (2017) 64:1280–86. doi: 10.1111/tbed.12504
30. Wade A, Achenbach JE, Gallardo C, Settypalli TBK, Souley A, Djonwe G, et al. Genetic characterization of African swine fever virus in Cameroon, 2010–2018. J Microbiol. (2019) 57:316–24. doi: 10.1007/s12275-019-8457-4
31. Luka PD, Achenbach JE, Mwiine FN, Lamien CE, Shamaki D, Unger H, et al. Genetic characterization of circulating African swine fever viruses in Nigeria (2007–2015). Transbound Emerg Dis. (2017) 64:1598–609. doi: 10.1111/tbed.12553
32. Chang'a JS, Mayenga C, Settypalli TBK, Achenbach JE, Mwanandota JJ, Magidanga B, et al. Symptomatic and asymptomatic cases of African swine fever in Tanzania. Transbound Emerg Dis. (2019) 66:2402–10. doi: 10.1111/tbed.13298
33. Molini U, Mushonga B, Settypalli TBK, Dundon WG, Khaiseb S, Jago M, et al. Molecular characterization of African swine fever virus from outbreaks in Namibia in 2018. Transbound Emerg Dis. (2020) 67:1008–14. doi: 10.1111/tbed.13399
34. Mai NTA, Vu XD, Nguyen TTH, Nguyen VT, Trinh TBN, Kim YJ, et al. Molecular profile of African swine fever virus (ASFV) circulating in Vietnam during 2019–2020 outbreaks. Arch Virol. (2021) 166:885–90. doi: 10.1007/s00705-020-04936-5
35. Simulundu E, Chambaro HM, Sinkala Y, Kajihara M, Ogawa H, Mori A, et al. Co-circulation of multiple genotypes of African swine fever viruses among domestic pigs in Zambia (2013–2015). Transbound Emerg Dis. (2018) 65:114–22. doi: 10.1111/tbed.12635
36. Ge S, Liu Y, Li L, Wang Q, Li J, Ren W, et al. An extra insertion of tandem repeat sequence in African swine fever virus, China, 2019. Virus Genes. (2019) 55:843–47. doi: 10.1007/s11262-019-01704-9
37. Ma M, Wang HH, Hua Y, Qin F, Yang J. African swine fever in China: impacts, responses, and policy implications. Food Policy. (2021) 192:102065. doi: 10.1016/j.foodpol.2021.102065
38. Ebwanga EJ, Ghogomu SM, Paeshuyse J. African swine fever in Cameroon: a review. Pathogens. (2021) 10:421. doi: 10.3390/pathogens10040421
39. Misinzo G, Magambo J, Masambu J, Yongolo MG, Van Doorsselaere J, Nauwynck HJ. Genetic characterization of African swine fever viruses from a 2008 outbreak in Tanzania. Transbound Emerg Dis. (2011) 58:86–92. doi: 10.1111/j.1865-1682.2010.01177.x
40. Gallardo C. Sole, A, Nurmoja I, Cano-Gómez C, Cvetkova S, Frant M, et al. Dynamics of African swine fever virus (ASFV) infection in domestic pigs infected with virulent, moderate virulent and attenuated genotype II ASFV European strains. Transbound Emerg Dis. (2021) 68:2826–41. doi: 10.1111/tbed.14222
41. Atuhaire DK, Afayoa M, Ochwo S, Mwesigwa S, Mwiine FN, Okuni JB, et al. Prevalence of African swine fever virus in apparently healthy domestic pigs in Uganda. BMC Vet Res. (2013) 9:263. doi: 10.1186/1746-6148-9-263
42. Chapman DAG, Tcherepanov V, Upton C, Dixon LK. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus strains. J Gen Virol. (2008) 89(Pt 2):397–408. doi: 10.1099/vir.0.83343-0
43. Monteagudo PL, Lacasta A, López E, Bosch L, Collado J, Pina-Pedrero S, et al. BA71ΔCD2: a new recombinant live attenuated African swine fever virus with cross-protective capabilities. J Virol. (2017) 91:e01058–17. doi: 10.1128/JVI.01058-17
44. Chen W, Zhao D, He X, Liu R, Wang Z, Zhang X, et al. A seven-gene-deleted African swine fever virus is safe and effective as a live attenuated vaccine in pigs. Sci China Life Sci. (2020) 63:623–34. doi: 10.1007/s11427-020-1657-9
45. Gogin A, Gerasimov V, Malogolovkin A, Kolbasov D. African swine fever in the North Caucasus region and the Russian Federation in years 2007–2012. Virus Res. (2013) 173:198–203. doi: 10.1016/j.virusres.2012.12.007
46. Gonzales W, Moreno C, Duran U, Henao N, Bencosme M, Lora P, et al. African swine fever in the Dominican Republic. Transbound Emerg Dis. (2021) 68:3018–19. doi: 10.1111/tbed.14341
47. Sidi M, Zerbo HL, Ouoba BL, Settypalli TBK, Bazimo G, Ouandaogo HS, et al. Molecular characterization of African swine fever viruses from Burkina Faso, 2018. BMC Vet Res. (2022) 18:69. doi: 10.1186/s12917-022-03166-y
48. Rajukumar K, Senthilkumar D, Venkatesh G, Singh F, Patil VP, Kombiah S, et al. Genetic characterization of African swine fever virus from domestic pigs in India. Transbound Emerg Dis. (2021) 68:2687–92. doi: 10.1111/tbed.13986
49. Qu H, Ge S, Zhang Y, Wu X, Wang Z. A systematic review of genotypes and serogroups of African swine fever virus. Virus Genes. (2022) 58:77–87. doi: 10.1007/s11262-021-01879-0
50. Mulumba-Mfumu LK, Achenbach JE, Mauldin MR, Dixon LK, Tshilenge CG, Thiry E, et al. Genetic assessment of African swine fever strains involved in outbreaks in the Democratic Republic of Congo between 2005 and 2012 reveals co-circulation of p72 genotypes I, IX and XIV, including 19 variants. Viruses. (2017) 9:31. doi: 10.3390/v9020031
51. Bisimwa PN, Ishara LK, Wasso DS, Bantuzeko F, Tonui R, Bwihangane AB. Detection and genetic characterization of African swine fever virus (ASFV) in clinically infected pigs in two districts in South Kivu province, Democratic Republic Congo. Heliyon. (2021) 7:e06419. doi: 10.1016/j.heliyon.2021.e06419
52. Hakizimana JN, Kamwendo G, Chulu JLC, Kamana O, Nauwynck HJ, Misinzo G. Genetic profile of African swine fever virus responsible for the 2019 outbreak in northern Malawi. BMC Vet Res. (2020) 16:316. doi: 10.1186/s12917-020-02536-8
53. Li L, Ren Z, Wang Q, Ge S, Liu Y, Liu C, et al. Infection of African swine fever in wild boar, China, 2018. Transbound Emerg Dis. (2019) 66:1395–98. doi: 10.1111/tbed.13114
54. Kim SH, Lee SI, Jeong HG, Yoo J, Jeong H, Choi Y, et al. Rapid emergence of African swine fever virus variants with different numbers of a tandem repeat sequence in South Korea. Transbound Emerg Dis. (2021) 68:1726–30. doi: 10.1111/tbed.13867
Keywords: African swine fever virus, B646L (p72) gene, E183L (p54) gene, EP402R (CD2v) gene, genotype, serogroup, phylogenetic analysis
Citation: Shi K, Liu H, Yin Y, Si H, Long F and Feng S (2022) Molecular Characterization of African Swine Fever Virus From 2019-2020 Outbreaks in Guangxi Province, Southern China. Front. Vet. Sci. 9:912224. doi: 10.3389/fvets.2022.912224
Received: 04 April 2022; Accepted: 23 May 2022;
Published: 15 June 2022.
Edited by:
Carlos Juan Perfumo, National University of La Plata, ArgentinaReviewed by:
Jean Nepomuscene Hakizimana, Sokoine University of Agriculture, TanzaniaJuan Manuel Sanhueza, Catholic University of Temuco, Chile
Copyright © 2022 Shi, Liu, Yin, Si, Long and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaichuang Shi, c2hpa2FpY2h1YW5nQDEyNi5jb20=; Hongbin Si, c2hiMjAwOUBneHUuZWR1LmNu
†These authors have contributed equally to this work